

Abstract
Background
Pyroptosis and oxidative stress play pivotal roles in cardiomyocyte loss after myocardial infarction. NF-κB is associated with oxidative stress and gasdermin D (GSDMD), the effector molecule of pyroptosis. However, the exact relationship between oxidative stress and cardiomyocyte pyroptosis remains unknown.
Material/Methods
We measured inflammasome-mediated cardiomyocyte pyroptosis in vivo via membrane pore formation, lactate dehydrogenase (LDH) release, and expression of caspase-1, cleaved caspase-1, NACHT, LRR and PYD domains-containing protein 3 (NLRP3), and apoptosis-associated speck-like protein containing a CARD (ASC). Furthermore, we induced pyroptosis in vitro by oxygen-glucose deprivation (OGD) in H9C2 cells. NLRP3 inflammasome-mediated pyroptosis was confirmed by LDH assay kit and Western blot. Oxidative stress was evaluated by reactive oxygen species (ROS) and superoxide dismutase (SOD) activity. We suppressed oxidative stress with N-acetyl-cysteine (NAC) and measured subsequent changes to the NF-κB-GSDMD axis and pyroptosis by LDH assay kit and Western blot. Then, we inhibited NF-κB activation with pyrrolidine dithiocarbamate (PDTC) and measured changes to GSDMD activity and pyroptosis by qRT-PCR, Western blot, and LDH assay kit.
Results
Suppression of oxidative stress by NAC reduced NF-κB and GSDMD activation and increased pyroptosis, characterized by LDH release and NLRP3 inflammasome activation in H9C2 cells under OGD. Moreover, inhibition of NF-κB activation reduced GSDMD transcription and activation and NLRP3 inflammasome-mediated pyroptosis of H9C2 cells under OGD.
Conclusions
We demonstrated that the NF-κB-GSDMD axis functioned as a bridge between oxidative stress and NLRP3 inflammasome-mediated cardiomyocyte pyroptosis. Our findings provide important insight into the mechanism of myocardial infarction-related ventricular remodeling.
MeSH Keywords: Inflammasomes, NF-kappa B, Oxidative Stress
Background
Myocardial infarction (MI) remains a leading cause of death worldwide [1]. Myocardial damage occurs during the onset of myocardial infarction, leading to ventricular remodeling, a series of changes in structure and function largely attributed to the loss of cardiomyocytes [2]. Currently, there is little breakthrough data characterizing cardiomyocyte loss induced by MI and the high rates of mortality and heart failure associated with MI [1]. Therefore, studying the mechanism of cardiomyocyte loss following MI will provide crucial insight for this field.
Pyroptosis, a recently identified form of programmed cell death, may be a potential mechanism of cardiomyocyte loss [3]. Initiation of pyroptosis by activation of caspases-1, -4, -5, and -11 can be triggered by various pathological stimuli, including MI [4,5], and features cell lysis and release of intracellular proinflammatory contents [6]. A recent study demonstrated that MI activated the multi-protein inflammasome complex, comprised of caspase-1, NLRP3, and ASC, and triggered pyroptosis in cardiomyocytes [5]. Although several studies have investigated the involvement of pyroptosis in cardiomyocyte loss caused by myocardial infarction, the exact regulatory mechanism remains elusive.
Oxidative stress occurs when the amount of oxide exemplified by reactive oxygen species (ROS) surpasses the antioxidant ability of antioxidant such as superoxide dismutase (SOD). ROS have been demonstrated to play a significant regulatory role in cardiomyocyte injury [7,8]. A recent study showed that inhibition of ROS generation suppressed pyroptosis of hematopoietic stem and progenitor cells [9]. However, the mechanistic relationship between oxidative stress and pyroptosis induced by MI is poorly understood.
It has been widely reported that NF-κB is a critical molecular switch for cellular response to oxidative stress [10]. NF-κB exists in the form of dimer and has been demonstrated to be involved in the development and progression of various diseases associated with inflammation, apoptosis, and proliferation, such as MI [11]. Therefore, we hypothesized that NF-κB may be a crucial effector molecule in the oxidative stress response of cardiomyocytes after MI.
A recent study showed that NF-κB was an essential transcription factor of gasdermin D (GSDMD) [12]. GSDMD was identified by 2 independent screening approaches as a key effector of pyroptosis [13]. Under normal cellular conditions, the C-terminus of GSDMD auto-inhibits the pore-forming activity of the N-terminus [14]. When extracellular signals associated with pyroptosis activate inflammasomes, such as NLRP3 inflammasomes, they subsequently cleave and activate caspases-1, -4, -5, and -11. Consequently, activated caspase-1 cleaves and separates the N- and C-terminals of GSDMD [14]. As a result, the N-terminal fragment of GSDMD forms nanoscopic pores in the cell membrane, leading to the release of proinflammatory materials and cell swelling [15,16]. Thus, we hypothesized that the NF-κB-GSDMD axis may function as a bridge between oxidative stress response and pyroptosis of cardiomyocytes. In the present study, we determined the relationship between oxidative stress and pyroptosis, and identified a potential mechanism through the NF-κB-GSDMD axis by which cardiomyocytes undergo pyroptosis following MI.
Material and Methods
Rat MI model
We modeled MI in Sprague-Dawley (SD) rats (8–12 weeks; 160–250 g) by ligating the left anterior descending (LAD) coronary artery, as previously described [17]. In brief, all rats were anesthetized by intraperitoneal injection of sodium pentobarbital (50 mg/kg; Merck, China) and mechanically ventilated with the HX-101E small-animal ventilator (Chengdu Thaimeng Software Ltd., Chengdu, China). The LAD coronary artery was identified through a left thoracotomy at the third and fourth intercostal space, and then MI was induced by ligating it with a size 6-0 polyester suture. Muscles and skin were sutured immediately. Successful coronary occlusion was confirmed by a pale area below the suture knots. Sham-treated animals underwent the same procedure, except the polyester suture was placed around the LAD coronary artery without being tied. This study was approved by the Institutional Animal Care and Use Committee of Guangdong Medical University in compliance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (NIH publication no. 85-23, revised 1996).
HE staining
Heart slices (4-μm) were obtained from paraffin-embedded tissues. The slices were stained with hematoxylin and eosin (HE; Beyotime, China) according to the manufacturer’s instructions. Images were photographed with a camera.
Transmission electron microscopy
After being fixed in glutaraldehyde, tissue samples were dehydrated, embedded, and stained with lead citrate and uranyl acetate. Samples were viewed by transmission electron microscopy (JEM-1200EXII, Japan).
H9C2 cell culture and oxygen-glucose deprivation (OGD)
The rat cardiomyocyte cell line, H9C2, was originally derived from normal rat heart tissue (ATCC, USA). Cells were cultured in low-glucose Dulbecco’s modified Eagle’s medium (HyClone, USA) supplemented with 10% fetal bovine serum (Gibco, USA), 100 U/mL penicillin G, and 100 mg/mL streptomycin (Invitrogen, Carlsbad, CA). H9C2 cells were maintained in a humidified incubator (Thermo Fisher Scientific, USA) with 74% N2, 5% CO2, and 21% O2 at 37°C. After synchronizing the cells by serum starvation, the OGD group was exposed to hypoxic conditions of 94% N2, 5% CO2, and 1% O2 at 37°C in glucose-free, serum-free DMEM. H9C2 cells cultured under normal conditions were used as the control group after cell cycle synchronization.
Lactate dehydrogenase (LDH) release
LDH is an indicator of cell membrane permeability, and we measured LDH release by use of the LDH assay kit (Beyotime, China). H9C2 cells were seeded in a 96-well plate. Following treatment, media from the cells was transferred to a new 96-well plate, incubated with LDH working reagent for 30 min, and measured according to the manufacturer’s instructions. In addition, serum samples were collected from animals and incubated with LDH working reagent for 30 min in a 96-well plate to measure LDH release. Results were obtained at 490 nm by a spectrophotometer (Epoch, USA).
Reactive oxygen species (ROS) activity
Intracellular generation of ROS was detected by dichloro-dihydro-luorescein diacetate (DCFH-DA) assay (Beyotime, China). H9C2 cells were incubated with DCFH-DA at 37°C for 30 min in the dark. Fluorescence was measured at 488 nm excitation and 525 nm emission using a fluorescence microplate reader (PerkinElmer, USA).
Superoxide dismutase (SOD) activity
SOD activity was evaluated using a kit according to the manufacturer’s instructions. Cells (1×107) were homogenized in 10 mM phosphate buffer solution (1 mL; pH 7.4). The homogenates were centrifuged at 12 000×g for 10 min at 4°C, and then the supernatant was used for SOD assay, performed using the WST-8 colorimetric method. SOD activity was measured at 450 nm using a spectrophotometer (Epoch, USA).
Western blot analysis
Protein samples were extracted from cultured cells or tissue using RIPA lysis buffer (Beyotime, China). Samples were centrifuged at 12 000×g at 4°C for 10 min, and then the supernatant was collected. Protein content was quantified via bicinchoninic acid (BCA) assay (Beyotime, China). Equal amounts of protein were separated on SDS–polyacrylamide gels (10%) and electro-transferred to polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA, USA). After blocking in 5% skim milk, membranes were incubated with primary antibodies, GAPDH (1: 1000; Cell Signaling Technology, USA), pro-caspase-1/cleaved caspase-1 (1: 100; Santa Cruz Biotechnology, USA), NLRP3 (1: 300; Abcam, UK), ASC (1: 350; Abcam, UK), NF-κB (1: 700; t-NF-κB p65, p-NF-κB p65, Abcam, UK), and GSDMD (1: 1000; Santa Cruz Biotechnology, USA) at 4°C overnight. After washing with TBS containing Tween-20, membranes were incubated with secondary antibody (1: 2000; Cell Signaling Technology, USA) for 1 h at room temperature. Western blots were developed using a chemiluminescent solution and quantified under Gel-Pro Analyzer 4.0 (Media Cybernetics, USA).
qRT-PCR analysis
Total RNA was isolated from H9C2 cells with NucleoZOL reagent (MACHEREY-NAGEL, Germany) according to the manufacturer’s instructions. RNA samples were quantified by spectrophotometer (Epoch, USA). Equal amounts of RNA were treated with TransScript All-in-One First-Strand cDNA Synthesis SuperMix Kit (TransGen Biotech, China) in a Cycler Thermal Cycler (Applied Biosystems, Life Technologies, Waltham, MA, USA). Reverse transcription polymerase chain reaction (RT-PCR) was performed with the primers listed in Table 1 (Sangon Biotechnology, Shanghai, China). mRNA levels of the target genes were quantified by q-PCR analysis using SYBR premix Ex Taq (Takara, Dalian, China), and constitutively expressed GAPDH gene was used as an internal control. qRT-PCR reactions were run using the LightCycler 480 II Real-Time PCR System (Roche Diagnostics, Germany), and data were collected and analyzed using LightCycler 480 software SW 1.5 (Roche Diagnostics).
Table 1.
Primer sequences for qRT-PCR.
| Gene | Primer sequences (5′ to 3′) | |
|---|---|---|
| Forward | Reward | |
| GSDMD | CCAGCATGGAAGCCTTAGAG | CAGAGTCGAGCACCAGACAC |
| GAPDH | CTCATGACCACAGTCCATGC | TTCAGCTCTGGGATGACCTT |
Statistical analysis
Data are presented as mean ± standard deviation (SD). Statistical analysis was performed with the statistical software GraphPad Prism 7 (GraphPad Software Inc. La Jolla, CA). Data following a normal distribution was evaluated via the unpaired t test and ANOVA with Bonferroni correction. Data that did not follow a normal distribution were analyzed via Mann-Whitney test and Kruskal-Wallis test. Values of p<0.05 were considered statistically significant. Levels of statistical significance are indicated in the figures.
Results
NLRP3 inflammasome-mediated pyroptosis contributed to cardiomyocyte loss following MI
To validate the role of NLRP3 inflammasome-mediated pyroptosis in cardiomyocyte loss, we established a MI model in SD rats. One week after surgery, the left ventricular wall of rats in the MI group was significantly thinner than that of rats in the sham-treated group (Figure 1A). Compared to the sham group, membrane pore formation significantly increased in the MI group (Figure 1B). Protein expression levels of caspase-1, cleaved caspase-1, NLRP3, and ASC (Figure 1C) and LDH release (Figure 1D) in serum were dramatically elevated in the MI group. These findings support the involvement of NLRP3 inflammasome-mediated pyroptosis in cardiomyocyte loss following MI.
Figure 1.

NLRP3 inflammasome-mediated pyroptosis contributed to cardiomyocyte loss following MI. SD rats of sham group or MI group were sacrificed 7 d after surgery. (A) Representative images of HE staining of rat heart in sham and MI groups (n=3). (B) Representative electron micrographs (8,000×) of myocardial tissue. Yellow arrowheads indicate membrane pores (n=3). (C) Relative protein levels of caspase-1, cleaved caspase-1, NLRP3, and ASC in sham and MI groups (n=6). (D) LDH levels in serum (n=6). Values are presented as means ±SD. * p<0.05, ** p<0.01 relative to MI group.
Oxidative stress induced NLRP3 inflammasome-mediated pyroptosis of H9C2 cells
To determine the effect of oxidative stress on NLRP3 inflammasome-mediated cardiomyocyte pyroptosis, we developed an ischemic model using H9C2 cells with OGD for 24 h, 36 h, and 48 h. NLRP3 inflammasome-mediated pyroptosis was characterized by significantly increased expression of caspase-1 and cleaved caspase-1 and LDH release. Expression levels of caspase-1 and cleaved caspase-1 were dramatically elevated in the OGD group at 36 h (Figure 2A). Due to serum starvation, protein levels of caspase-1 and cleaved caspase-1 in the control and OGD groups at 48 h were also higher than those in the control group at 36 h (Figure 2A). LDH release was markedly higher in the OGD group at 24 h, 36 h, and 48 h compared to the control group (Figure 2B). We analyzed cardiomyocyte pyroptosis at 36 h due to the stability of the ischemic model. Protein levels of NLRP3 and ASC significantly increased in the OGD group at 36 h (Figure 2C). These findings indicate that NLRP3 inflammasome-mediated pyroptosis was involved in cardiomyocyte loss following MI. OGD conditions elevated ROS activity while reducing SOD content in the OGD group at 36 h compared to the control group (Figure 2D, 2E), showing a relationship between oxidative stress and NLRP3-mediated cardiomyocyte pyroptosis.
Figure 2.

Oxidative stress induced NLRP3 inflammasome-mediated pyroptosis of H9C2 cells. H9C2 cells were cultured under normal conditions or OGD for 24 h, 36 h, and 48 h. (A) Relative protein levels of caspase-1 and cleaved caspase-1 in control and OGD groups at 24 h, 36 h, and 48 h (n=3). (B) LDH levels in media in control and OGD groups at 24 h, 36 h, and 48 h (n=3). (C) Relative protein levels of NLRP3 and ASC in control and OGD groups at 36 h (n=3). (D) ROS levels in control and OGD groups at 36 h. (E) SOD content in control and OGD groups at 36 h (n=3). Values are means ±SD. * p<0.05, ** p<0.01, *** p<0.001 relative to control group.
Suppression of oxidative stress alleviated OGD-induced pyroptosis in H9C2 cells and reduced NF-κB and GSDMD activity
To characterize the relationship between oxidative stress and NLRP3-mediated pyroptosis, we suppressed levels of oxidative stress using the antioxidant N-acetyl cysteine (NAC). Results showed that NAC significantly reduced ROS at concentrations of 50 μM and 100 μM and increased SOD content at a concentration of 50 μM (Figure 3A, 3B). Therefore, we selected 50 μM as the optimal concentration of NAC to suppress oxidative stress in subsequent experiments. Suppression of oxidative stress significantly inhibited LDH release and downregulated expression of caspase-1, cleaved caspase-1, NLRP3, and ASC (Figure 3C, 3D). Furthermore, protein levels of phosphorylated NF-κB and GSDMD were significantly upregulated by OGD, and this upregulation was reversed by addition of 50 μM NAC (Figure 3D).
Figure 3.

Suppression of oxidative stress reduced OGD-induced pyroptosis of H9C2 cells and NF-κB and GSDMD activity. H9C2 cells were cultured under OGD for 36 h and treated with or without NAC (25 μM, 50 μM, and 100 μM). (A) ROS levels in control, OGD, and NAC-treated (25 μM, 50 μM, and 100 μM) groups (n=6). (B) SOD content in control, OGD, and NAC-treated (25 μM, 50 μM, and 100 μM) groups (n=3). (C) LDH levels of media in control, OGD, and NAC-treated (50 μM) groups (n=6). (D) Relative protein levels of caspase-1, cleaved caspase-1, NLRP3, ASC, GSDMD, cleaved GSDMD, p-NF-κB p65, and NF-κB p65 in control, OGD, and NAC-treated (50 μM) groups (n=3). Values are means ±SD. * p<0.05, ** p<0.01, *** p<0.001 relative to OGD.
Inhibition of NF-κB reduced oxidative stress-mediated pyroptosis via GSDMD
To establish a connection between NF-κB and NLRP3 inflammasome-mediated pyroptosis, we inhibited NF-κB using pyrrolidine dithiocarbamate (PDTC). Addition of PDTC reduced phosphorylation levels of NF-κB at concentrations of 25 μM and 50 μM (Figure 4A). We used 25 μM PDTC in subsequent experiments. As depicted in Fig. 4B, the mRNA level of GSDMD was significantly enhanced by OGD, which was drastically reversed by addition of 25 μM PDTC (Figure 4B). In addition, inhibition of NF-κB significantly decreased protein expression of GSDMD, caspase-1, cleaved caspase-1, and NLRP3, and ASC and LDH release (Figure 4C, 4D).
Figure 4.

Inhibition of NF-κB reduced oxidative stress-mediated pyroptosis via GSDMD. H9C2 cells were cultured under OGD for 36 h and treated with or without PDTC (10 μM, 25 μM, and 50 μM). (A) Relative protein levels of p-NF-κB p65 and NF-κB p65 in control, OGD, and PDTC-treated (10 μM, 25 μM, and 50 μM) groups (n=3). (B) Relative mRNA levels of GSDMD in control, OGD, and PDTC-treated (25 μM) groups (n=3). (C) LDH levels in media in control, OGD, and PDTC-treated (25 μM) groups (n=3). (D) Relative protein levels of caspase-1, cleaved caspase-1, NLRP3, ASC, GSDMD, and cleaved GSDMD in control, OGD, and PDTC-treated (25 μM) groups (n=3). Values are means ±SD. * p<0.05, ** p<0.01, *** p<0.001 relative to OGD.
Discussions
Targeting the reduction of cardiomyocyte loss may provide critical direction in research on MI therapy. Pyroptosis was identified as an alternative form of programmed cell death, which may be detrimental to cardiomyocyte loss [5,18]. Therefore, it is important to characterize the mechanisms underlying cardiomyocyte pyroptosis induced by MI. In our study, we found that oxidative stress triggered the NF-κB-GSDMD signaling axis, which functioned as a crucial pathway in regulating cardiomyocyte pyroptosis. Ultimately, our observations provide new understanding and insight for the improvement of ventricular remodeling following MI.
The inflammasome complex comprises various components, including NLRP3 and ASC, and cleaves and activates caspase-1, initiating pyroptosis when MI occurs [19,20]. A previous study demonstrated that mice with caspase-1 deficiency exhibited less fibrosis and lower mortality rate than wild-type mice for 9 days after surgery [18]. Thus, NLRP3 inflammasome-mediated pyroptosis is critical in the process of cardiomyocyte loss induced by MI. Oxidative stress resulting from MI leads to excessive ROS accumulation in the heart, leading to myocardial injury [21,22], suggesting that oxidative stress also plays a role in ventricular remodeling following MI. Recent studies focusing on age-related degenerative diseases reported that oxidative stress activated NLRP3 inflammasome-mediated pyroptosis [23]. Chen et al. demonstrated that the addition of antioxidants significantly reduced activation of NLRP3 inflammasome and subsequently decreased pyroptosis induced by cadmium [24]. As a result, we hypothesized that oxidative stress plays a key role in cardiomyocyte pyroptosis. Our data show that inhibition of oxidative stress suppressed pyroptosis of H9C2 cells.
NF-κB has been shown to be essential in the process of ventricular remodeling following MI. Some studies have identified NF-κB as a potential target for treatment of MI by using mice with a targeted deletion of the NF-κB gene [25]. Furthermore, NF-κB is a key regulator of both pyroptosis and cellular response to oxidative stress [26]. A recent report revealed that lymphoid-derived cells triggered pyroptosis in HIV-infected CD4 T cells that were naturally pyroptosis-resistant, partly due to NF-κB activation [27]. Inhibition of NF-κB transcription reduced the activation of NLRP3 inflammasome and restricted proinflammatory responses [28]. Therefore, we postulated that the NF-κB-GSDMD axis functions as a bridge between oxidative stress and cardiomyocyte pyroptosis. As expected, suppressing NF-κB activation led to decreased transcription of GSDMD and alleviated pyroptosis of H9C2 cells, supporting our hypothesis that the NF-κB-GSDMD signaling axis was the mechanistic connection between oxidative stress and pyroptosis induced by MI (Figure 5).
Figure 5.
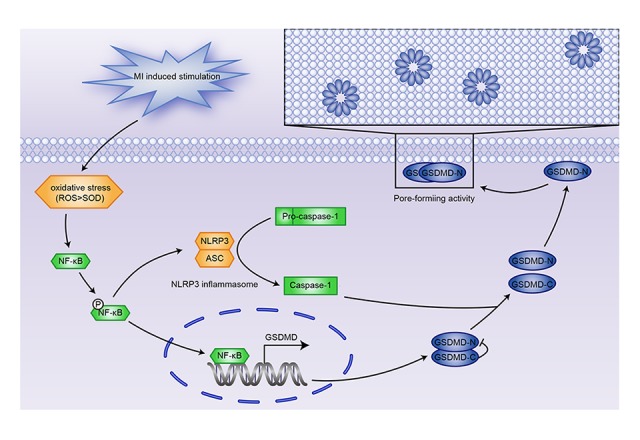
Oxidative stress induces NLRP3 inflammasome-mediated pyroptosis in MI via the NF-κB-GSDMD signaling axis.
There were several limitations to our study. We did not identify whether NF-κB directly activated transcription of GSDMD to induce cardiomyocyte pyroptosis. This could be accomplished by luciferase assay and chromatin immunoprecipitation (ChIP) analysis. Furthermore, the effect of NF-κB inhibition on GSDMD activity and pyroptosis must be further validated. NF-κB gene knockdown or knockout may be more effective than inhibition by PDTC. Knockdown of NF-κB and overexpression of GSDMD in cardiomyocytes in vitro and in vivo would validate the regulatory role of NF-κB on GSDMD-dependent cardiomyocyte pyroptosis. Finally, we conducted numerous cell experiments to demonstrate the vital role of the NF-κB-GSDMD axis in cardiomyocyte pyroptosis, but further investigation using animal models is necessary. For example, targeted nano-delivery of NF-κB inhibitor to cardiomyocytes in rats following MI, as tracked by MRI, would provide valuable data on the importance of the NF-κB-GSDMD axis during pyroptosis in vivo [29,30].
We demonstrated that inhibition of NF-κB mitigated pyroptosis induced by oxidative stress. However, clinical trials of targeted therapy of NF-κB identified many adverse effects, primarily caused by the lack of cell and tissue specificity of the chemotherapeutics [11,31]. Therefore, we must focus on enhancing the specificity of currently available drugs or identify novel targets to improve MI treatment. Our results show that GSDMD, which plays a crucial role in inducing pyroptosis, may be a potential candidate for the development of novel targeted therapies.
Conclusions
In our study, we demonstrated that the NF-κB-GSDMD axis acts as a bridge between oxidative stress and pyroptosis. Our results provide a new perspective for understanding ventricular remodeling cause by MI.
Acknowledgements
We thank Dr. Yuan He and Dr. Wei Lei of the Affiliated Hospital of Guangdong Medical University for providing technical guidance.
Footnotes
Conflict of interest
None.
Source of support: Departmental sources
References
- 1.Benjamin EJ, Blaha MJ, Chiuve SE, et al. Heart disease and stroke statistics – 2017 update: A report from the American Heart Association. Circulation. 2017;135(10):e146–603. doi: 10.1161/CIR.0000000000000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling – concepts and clinical implications: A consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J Am Coll Cardiol. 2000;35(3):569–82. doi: 10.1016/s0735-1097(99)00630-0. [DOI] [PubMed] [Google Scholar]
- 3.Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol. 2001;9(3):113–14. doi: 10.1016/s0966-842x(00)01936-3. [DOI] [PubMed] [Google Scholar]
- 4.Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7(2):99–109. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mezzaroma E, Toldo S, Farkas D, et al. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc Natl Acad Sci USA. 2011;108(49):19725–30. doi: 10.1073/pnas.1108586108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: Mechanistic description of dead and dying eukaryotic cells. Infect Immun. 2005;73(4):1907–16. doi: 10.1128/IAI.73.4.1907-1916.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aikawa R, Komuro I, Yamazaki T, et al. Oxidative stress activates extracellular signal-regulated kinases through Src and Ras in cultured cardiac myocytes of neonatal rats. J Clin Invest. 1997;100(7):1813–21. doi: 10.1172/JCI119709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alfadda AA, Sallam RM. Reactive oxygen species in health and disease. J Biomed Biotechnol. 2012;2012 doi: 10.1155/2012/936486. 936486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Basiorka AA, McGraw KL, Eksioglu EA, et al. The NLRP3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood. 2016;128(25):2960–75. doi: 10.1182/blood-2016-07-730556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sies H, Berndt C, Jones DP. Oxidative stress. Annu Rev Biochem. 2017;86:715–48. doi: 10.1146/annurev-biochem-061516-045037. [DOI] [PubMed] [Google Scholar]
- 11.Gordon JW, Shaw JA, Kirshenbaum LA. Multiple facets of NF-kappaB in the heart: To be or not to NF-kappaB. Circ Res. 2011;108(9):1122–32. doi: 10.1161/CIRCRESAHA.110.226928. [DOI] [PubMed] [Google Scholar]
- 12.Liu Z, Gan L, Xu Y, et al. Melatonin alleviates inflammasome-induced pyroptosis through inhibiting NF-kappaB/GSDMD signal in mice adipose tissue. J Pineal Res. 2017;63(1) doi: 10.1111/jpi.12414. [DOI] [PubMed] [Google Scholar]
- 13.Man SM, Kanneganti TD. Gasdermin D: The long-awaited executioner of pyroptosis. Cell Res. 2015;25(11):1183–84. doi: 10.1038/cr.2015.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi J, Zhao Y, Wang K, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526(7575):660–65. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 15.Ding J, Wang K, Liu W, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535(7610):111–16. doi: 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
- 16.Sborgi L, Ruhl S, Mulvihill E, et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 2016;35(16):1766–78. doi: 10.15252/embj.201694696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Selye H, Bajusz E, Grasso S, Mendell P. Simple techniques for the surgical occlusion of coronary vessels in the rat. Angiology. 1960;11:398–407. doi: 10.1177/000331976001100505. [DOI] [PubMed] [Google Scholar]
- 18.Frantz S, Ducharme A, Sawyer D, et al. Targeted deletion of caspase-1 reduces early mortality and left ventricular dilatation following myocardial infarction. J Mol Cell Cardiol. 2003;35(6):685–94. doi: 10.1016/s0022-2828(03)00113-5. [DOI] [PubMed] [Google Scholar]
- 19.He Y, Hara H, Nunez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. 2016;41(12):1012–21. doi: 10.1016/j.tibs.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Toldo S, Mezzaroma E, Mauro AG, et al. The inflammasome in myocardial injury and cardiac remodeling. Antioxid Redox Signal. 2015;22(13):1146–61. doi: 10.1089/ars.2014.5989. [DOI] [PubMed] [Google Scholar]
- 21.Neri M, Fineschi V, Di Paolo M, et al. Cardiac oxidative stress and inflammatory cytokines response after myocardial infarction. Curr Vasc Pharmacol. 2015;13(1):26–36. doi: 10.2174/15701611113119990003. [DOI] [PubMed] [Google Scholar]
- 22.Sun Y. Oxidative stress and cardiac repair/remodeling following infarction. Am J Med Sci. 2007;334(3):197–205. doi: 10.1097/MAJ.0b013e318157388f. [DOI] [PubMed] [Google Scholar]
- 23.Salminen A, Ojala J, Kaarniranta K, Kauppinen A. Mitochondrial dysfunction and oxidative stress activate inflammasomes: Impact on the aging process and age-related diseases. Cell Mol Life Sci. 2012;69(18):2999–3013. doi: 10.1007/s00018-012-0962-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen H, Lu Y, Cao Z, et al. Cadmium induces NLRP3 inflammasome-dependent pyroptosis in vascular endothelial cells. Toxicol Lett. 2016;246:7–16. doi: 10.1016/j.toxlet.2016.01.014. [DOI] [PubMed] [Google Scholar]
- 25.Timmers L, van Keulen JK, Hoefer IE, et al. Targeted deletion of nuclear factor kappaB p50 enhances cardiac remodeling and dysfunction following myocardial infarction. Circ Res. 2009;104(5):699–706. doi: 10.1161/CIRCRESAHA.108.189746. [DOI] [PubMed] [Google Scholar]
- 26.Nakajima S, Kitamura M. Bidirectional regulation of NF-kappaB by reactive oxygen species: A role of unfolded protein response. Free Radic Biol Med. 2013;65:162–74. doi: 10.1016/j.freeradbiomed.2013.06.020. [DOI] [PubMed] [Google Scholar]
- 27.Munoz-Arias I, Doitsh G, Yang Z, et al. Blood-derived CD4 T cells naturally resist pyroptosis during abortive HIV-1 infection. Cell Host Microbe. 2015;18(4):463–70. doi: 10.1016/j.chom.2015.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garcia JA, Volt H, Venegas C, et al. Disruption of the NF-kappaB/NLRP3 connection by melatonin requires retinoid-related orphan receptor-alpha and blocks the septic response in mice. FASEB J. 2015;29(9):3863–75. doi: 10.1096/fj.15-273656. [DOI] [PubMed] [Google Scholar]
- 29.Liang R, Xie J, Li J, et al. Liposomes-coated gold nanocages with antigens and adjuvants targeted delivery to dendritic cells for enhancing antitumor immune response. Biomaterials. 2017;149:41–50. doi: 10.1016/j.biomaterials.2017.09.029. [DOI] [PubMed] [Google Scholar]
- 30.Harel-Adar T, Ben MT, Amsalem Y, et al. Modulation of cardiac macrophages by phosphatidylserine-presenting liposomes improves infarct repair. Proc Natl Acad Sci. 2011;108(5):1827–32. doi: 10.1073/pnas.1015623108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Durand JK, Baldwin AS. Targeting IKK and NF-kappaB for therapy. Adv Protein Chem Struct Biol. 2017;107:77–115. doi: 10.1016/bs.apcsb.2016.11.006. [DOI] [PubMed] [Google Scholar]