

Abstract
High‐mobility group box‐1 (HMGB1) is a ubiquitous protein. While initially thought to be simply an architectural protein due to its DNA‐binding ability, evidence from the last decade suggests that HMGB1 is a key protein participating in the pathogenesis of acute liver injury and chronic liver disease. When it is passively released or actively secreted after injury, HMGB1 acts as a damage‐associated molecular pattern that communicates injury and inflammation to neighboring cells by the receptor for advanced glycation end products or toll‐like receptor 4, among others. In the setting of acute liver injury, HMGB1 participates in ischemia/reperfusion, sepsis, and drug‐induced liver injury. In the context of chronic liver disease, it has been implicated in alcoholic liver disease, liver fibrosis, nonalcoholic steatohepatitis, and hepatocellular carcinoma. Recently, specific posttranslational modifications have been identified that could condition the effects of the protein in the liver. Here, we provide a detailed review of how HMGB1 signaling participates in acute liver injury and chronic liver disease.
Abbreviations
- ALD
alcoholic liver disease
- APAP
acetaminophen
- DAMP
damage‐associated molecular pattern
- DILI
drug‐induced liver injury
- ERK
extracellular signal regulated kinase
- HCC
hepatocellular carcinoma
- HMGB1
high‐mobility group box‐1
- HSC
hepatic stellate cell
- I/R
ischemia/reperfusion
- IL
interleukin
- JNK
c‐Jun N‐terminal kinase
- LPS
lipopolysaccharide
- MyD88
myeloid differentiation primary response gene‐88
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- NFκB
nuclear factor kappa‐B
- NLS
nuclear localization signal
- PAMP
pathogen‐associated molecular pattern
- PI3K
phosphatidylinositol‐4,5‐bisphosphate 3‐kinase
- pMEK
phospho‐mitogen‐activated protein kinase kinase
- PTM
posttranslational modification
- RAGE
receptor for advanced glycation end products
- ROS
reactive oxygen species
- TLR
toll‐like receptor
- TNFα
tumor necrosis factor‐α
- WT
wild type
High‐mobility group (HMG) proteins were isolated in the early 1960s and were later characterized by their chemical and physical properties, receiving their name due to their fast electrophoretic mobility on polyacrylamide gels.1 They are among the most ubiquitous, abundant, and evolutionarily conserved proteins in eukaryotes. These and the other members of a larger HMG superfamily share a common structural HMG box (HMGB) motif that is a unique ~80 residue L‐shaped domain that mediates DNA binding.2
Native HMGB proteins have molecular weights of approximately 22‐25 kDa and are quite homologous within higher eukaryotic species (100% for human, mouse, and rat HMGB1 or HMGB3 and 99% for HMGB2).3 The expression of HMGB1 is ubiquitous, whereas HMGB2 is mainly expressed in lymphoid tissues, the gastrointestinal tract, and testis in adult animals. HMGB3 is present in embryos, lung, nasopharynx, bronchus, placenta, and hematopoietic stem cells, and HMGB4 is restricted to testis.4, 5, 6, 7, 8 Among the four HMGB proteins, HMGB1 is the most abundant nonhistone nuclear protein and is also cytoplasmically expressed as it shuttles back and forth from the nucleus to the cytoplasm.4, 9
HMGB1 has 215 residues and a predicted molecular mass of 24,894 Da in its native state. The protein is organized into two DNA‐binding domains (box A and box B, spanning nearly 75% of the protein) separated by a short linker sequence followed by a highly negatively charged C‐terminal domain with 30 glutamic and aspartic acid residues (Fig. 1). Key features in the secondary structure are four β‐strands and seven helical regions. The solution structure of the tandem HMGB domain has been resolved by nuclear magnetic resonance spectroscopy. Notably, the protein has three regions of interest in positions 80‐96 (lipopolysaccharide [LPS] binding10), 89‐108 (cytokine‐stimulating activity11), and 150‐183 (receptor for advanced glycation end products [RAGE] binding).
Figure 1.
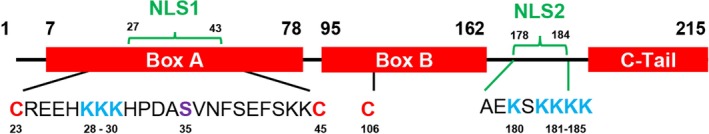
HMGB1 protein structure. HMGB1 has 215 amino acids and two NLS (NLS1 spanning from histidine 27 to lysine 43 and NLS2 spanning from alanine 178 to lysine 184). Cysteines that can undergo oxidation are written in red (C23 and C45 form a disulfide bond), lysines that can undergo acetylation are written in blue, and a serine that can undergo phosphorylation is written in purple. These posttranslational modifications have been found in liver disease.
In addition, HMGB1 has two nuclear localization signals (NLSs) in positions 27‐43 within box A (NLS1) and 178‐184 within box B (NLS2) (Fig. 1).
HMGB1 was originally discovered as a nuclear protein; however, when it is passively released or actively secreted after injury or cell stimulation, HMGB1 meets all the criteria of a damage‐associated molecular pattern (DAMP) and works as a necrosis signal for the immune system through cell‐surface receptors.12, 13, 14 In this latter role, HMGB1 acts as a proinflammatory cytokine that contributes to multiple injuries, including those from the liver, such as warm and cold ischemia/reperfusion (I/R) injury,15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27 sepsis,23, 28, 29, 30, 31, 32 acetaminophen (APAP) intoxication,23, 33, 34, 35, 36, 37, 38, 39, 40 alcoholic liver disease (ALD),41, 42, 43 fibrosis,44, 45, 46, 47, 48, 49 nonalcoholic steatohepatitis (NASH),50, 51, 52 and hepatocellular carcinoma (HCC).53, 54, 55, 56, 57, 58, 59, 60 By binding cell‐surface receptors on immune cells, HMGB1 activates intracellular signaling pathways that regulate immune cell function, including chemotaxis and cytokine production.37, 61, 62, 63 When HMGB1 targets hepatic stellate cells (HSCs), it induces a fibrogenic response.44, 49 Currently, less is known on how it signals in hepatocytes.
Our understanding of the role of HMGB1 has increasingly advanced during the last decade. Recent findings in molecular, structural, and functional analyses of HMGB1 have revealed that specific posttranslational modifications (PTMs) determine the effects of this powerful protein. Here, we aimed to provide an abridged review focusing on how HMGB1 signaling participates in acute and chronic liver disease.
Localization and Secretion
Under physiologic conditions, HMGB1 localizes to the nucleus due to its two NLSs; yet, its nuclear localization changes during development, aging, and injury.64 In the nucleus, HMGB1 binds double‐ or single‐stranded DNA and has affinity for four‐way junctions, bent DNA, and DNA adducts. The binding occurs in the minor groove of the DNA in a sequence‐independent manner that covers approximately 14 base pairs.2, 65 HMGB1 contributes to establishing the tridimensional structural framework and geometric requirements essential for the binding of some transactivators and therefore enables the formation of the pre‐initiation complex by recruiting various transcription factors.66, 67, 68, 69 HMGB1 induces DNA bending into a helical structure,70, 71, 72 facilitating gene expression or DNA replication by allowing the formation of large multiprotein complexes.2 The nuclear role of HMGB1 appears critical for survival as Hmgb1 –/– mice die within 24 hours after birth due to a deficit in the glucocorticoid receptor gene73; however, conditional ablation of Hmgb1 in specific cell subsets in the liver does not cause a deleterious phenotype.41, 74, 75 Moreover, under basal conditions, HMGB1 is not present or significantly reduced in the nucleus of some liver cells44, 49 looking otherwise healthy, suggesting that compensatory mechanisms may possibly exist; to date they are unknown.
Although HMGB1 constantly shuttles bidirectionally between the nucleus and the cytoplasm due to the action of importin and exportin in the nuclear pore complex, it preferentially accumulates in the nucleus during normal cellular homeostasis. However, acetylation of critical residues located in NLS1 (lysines 28‐30) and NLS2 (lysines 180, 182‐185) precludes HMGB1 from re‐entering the nucleus; as a result, it builds up in the cytoplasm and is eventually secreted to the extracellular space9, 76, 77 (Fig. 2).
Figure 2.
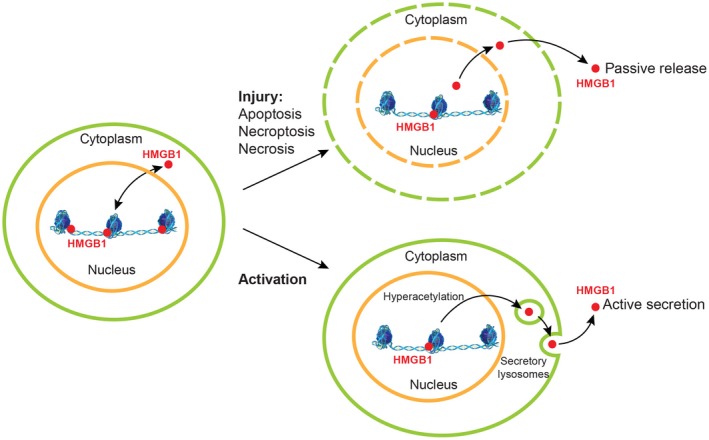
HMGB1 localization and secretion. During normal cellular homeostasis, HMGB1 preferentially accumulates in the nucleus due to its two NLSs; however, it can shuttle bidirectionally between the nucleus and the cytoplasm by the nuclear pore complex. Passive release of HMGB1 typically occurs in injured, apoptotic, necroptotic, or necrotic cells, where damage to the plasma and nuclear membranes occurs. Because HMGB1 lacks a hydrophobic secretion signal peptide, it is actively secreted by secretory lysosomes. Acetylation of critical residues located in both NLSs precludes HMGB1 from re‐entering the nucleus; as a result, it builds up in the cytoplasm and is eventually secreted to the extracellular space.
HMGB1 can be cleaved by thrombin and thrombomodulin between arginine 10 and glycine 11 and by caspase‐1 between aspartate 67 and lysine 68. It is unknown, however, if cleavage by caspase‐1 in this region localized within NLS1 also prevents the protein from re‐entering the nucleus or if the cleavage contributes to protein instability and degradation.
Because HMGB1 lacks a conventional hydrophobic secretion signal peptide,78 it is not secreted by conventional pathways but through passive and active mechanisms (Fig. 2). Passive release of HMGB1 typically occurs in injured, necroptotic, or necrotic cells where damage to the plasma and nuclear membranes occurs. In this setting, HMGB1 acts as an intracellular marker detected by the innate immune system that recognizes tissue damage and initiates reparative responses.12, 79 Both in vitro induction of apoptosis80 and injection of mice with the Fas agonistic Jo2 antibody81 induce HMGB1 release; yet, cells undergoing apoptosis induce negligible inflammation in the surrounding tissue82 as HMGB1 remains bound to DNA in apoptotic cells.12
Active secretion of HMGB1 by secretory lysosomes occurs, for example, in immune cells9, 13, 83, 84 where it acts as a proinflammatory cytokine during an immunologic challenge. Stimuli for secretion of HMGB1 in these cells include pathogen‐associated molecular patterns (PAMPs), cytokines, and oxidant stress, among others. Kupffer cells and monocyte‐derived macrophages release HMGB1 in response to LPS translocated from the gut lumen into the portal circulation,85 a key contributor to ALD and NASH, among others. Likewise, hepatocytes actively secrete HMGB1 in response to hypoxic conditions or oxidative stress induced by ischemia, pro‐oxidants, or alcohol consumption, and they appear to be the major cellular source of HMGB1 in the damaged liver.41, 86, 87, 88 Lastly, intestinal epithelial cells respond to local injury by secreting HMGB189 (Fig. 3).
Figure 3.
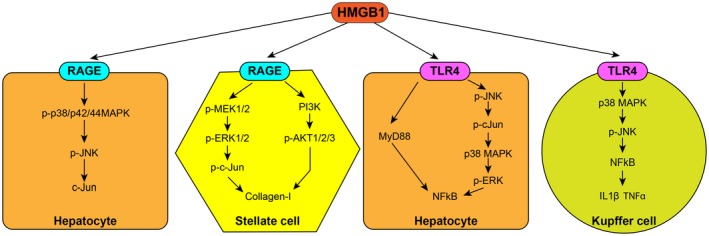
HMGB1 receptors and signaling. HMGB1 signals through RAGE and TLR4 in numerous cell types. In the hepatocyte, HMGB1 signals through RAGE, activating p‐p38/p42/44 MAPK, pJNK, and c‐Jun. In HSCs, HMGB1 can signal to RAGE to activate first pMEK1/2, pERK1/2, and p‐c‐Jun and second PI3K and pAKT1/2/3 to increase collagen‐I. HMGB1 activates TLR4 on hepatocytes to signal by MyD88‐dependent pathways or through pJNK, p‐c‐Jun, p38 MAPK, pERK to activate NFκB. In Kupffer cells, HMGB1 signals through TLR4, resulting in activation of p38 MAPK, pJNK, and NFκB, leading to induction of IL‐1β and TNFα. Abbreviations: MAPK, mitogen‐activated protein kinase; pMEK, phospho‐mitogen‐activated protein kinase kinase.
Receptors and Signaling
HMGB1 binds multiple receptors, including RAGE, toll‐like receptors (TLRs) 2/4/9, Mac‐1, syndecan‐1, phosphacan protein‐tyrosine phosphatase‐/β, and clusters of differentiation 24.11, 90, 91, 92, 93 Due to the dominant role of RAGE and TLR4 in liver disease, we focus on these two receptors.
RAGE
HMGB1 is a specific and saturable ligand that binds with high affinity to RAGE.94 The expression of RAGE is mediated, at least in part, through an autoregulatory loop whereby the interaction of HMGB1 with RAGE leads to nuclear factor κB (NFκB) and Sp1‐dependent transactivation of the advanced glycosylation end‐product receptor (AGER) gene.95, 96, 97 RAGE is expressed on hepatocytes, HSCs, sinusoidal endothelial cells, Kupffer cells, oval cells, and monocyte‐derived macrophages.44, 49, 59, 60, 98, 99, 100 RAGE activation leads to induction of NFκB‐dependent proinflammatory genes101 through a number of different pathways (Fig. 3): mitogen‐activated protein kinase, cell division control protein 42/Rac, phosphatidylinositol‐4,5‐bisphosphate 3‐kinase (PI3K)/pAkt, extracellular signal regulated kinase (ERK)/c‐Jun, and c‐Jun N‐terminal kinases (JNK), all inevitably leading to an inflammatory and fibrogenic signaling cascade.44, 102
RAGE mediates acute liver injury. In fact, during liver I/R, HMGB1 activates RAGE, leading to JNK and c‐Jun activation with subsequent ergosterol‐1 up‐regulation.103 In the context of APAP intoxication, HMGB1 acted through RAGE to trigger neutrophil recruitment during liver necrosis, which mediated subsequent liver damage.23 RAGE‐deficient neutrophils had 80% reduction in migration toward necrotic liver tissue. Furthermore, mice lacking Rage or Hmgb1 had reduced neutrophil recruitment, overall proinflammatory gene expression, and liver injury after APAP exposure. RAGE expressed on bone marrow‐derived cells was responsible for this effect as either global or bone marrow‐specific Rage null mice produced the same effect on neutrophil infiltration and liver injury.23 Of note, Tlr4 null mice had no difference in neutrophil migration or liver injury.
In the setting of chronic liver disease, our laboratory has shown an association between HMGB1 and RAGE signaling in ALD and fibrosis. Mice lacking Rage in myeloid cells were protected from alcohol‐induced steatosis, hepatocyte ballooning degeneration, inflammation, and liver injury (unpublished observations). Arriazu et al.44 demonstrated that in liver fibrosis, HMGB1 signals via RAGE through the PI3K‐pAkt1/2/3 pathway in HSCs. This was demonstrated by treating Rage‐ablated HSCs with recombinant HMGB1 and observing reduced collagen‐I expression along with blockade of PI3K and pAkt1/2/3 activation. Moreover, a neutralizing RAGE antibody prevented CCl4‐induced fibrosis in vivo. Further experiments from our laboratory have identified HMGB1 signaling by RAGE through phospho‐mitogen‐activated protein kinase kinase (pMEK) 1/2‐pERK1/2‐p‐c‐Jun in HSCs, which appears upstream of the PI3K‐pAkt1/2/3 pathway in regulating collagen‐I production.49 Lastly, in the setting of HCC, HMGB1 signaling by RAGE evoked NFκB activation in HCC cells.104 Rage –/– bone marrow‐derived macrophages had 70% lower tumor necrosis factor‐α (TNFα), interleukin (IL)‐1β, and IL‐6 production after stimulation with HMGB1 than cells expressing RAGE, indicating the importance of HMGB1 signaling by RAGE in these immune cells.
TLR4
TLRs recognize danger signals, such as PAMPs from bacteria or DAMPs, and subsequently activate the innate immune system. TLR4 is expressed on hepatocytes, Kupffer cells, HSCs, and sinusoidal endothelial cells.105, 106, 107 HMGB1 activation of TLR4 mainly occurs in myeloid cells,108, 109, 110 although the binding site in these and other cells has yet to be identified.
In Kupffer cells, HMGB1 signals through TLR4 to activate p38/pJNK and NFκB, which results in the production of the proinflammatory cytokines IL‐1β and TNFα.111 In experimental I/R models, HMGB1 signals via TLR4, phosphorylating JNK and activating NFκB.24 This will be discussed in detail later; however, other receptors have also been found to be involved.112, 113 In mouse models of nonalcoholic fatty liver disease (NAFLD), HMGB1 appears to signal through TLR4 and NFκB because primary hepatocytes treated with free fatty acids show HMGB1 activation of the TLR4/myeloid differentiation primary response gene‐88 (MyD88) pathway.114 Conversely, together with HMGB1 signaling through TLR4, some studies show that TLR4 signaling is likely involved in increasing HMGB1 expression in liver injury83, 115, 116, 117; nevertheless, additional studies are required to clarify this.
While, as described above, the literature points to a major role for HMGB1 signaling via RAGE, careful binding studies using surface plasmon resonance and/or nuclear magnetic resonance are needed to understand the impact of the HMGB1 PTMs on the binding. Should it be the case that one HMGB1 isoform binds RAGE faster than and/or dissociates slower to TLR4 or the reverse, this could represent a mechanism whereby these receptors regulate each other's signaling, a synergism or an alternative pathway that becomes activated (similar to an on/off system) when one receptor is saturated by its own ligand or an antagonist (e.g., LPS complex on TLR4). Future research should clarify this.
Posttranslational Modifications
HMGB1 can undergo multiple PTMs, including cysteine oxidation,41, 118, 119, 120, 121 lysine N‐acetylation,41, 122, 123, 124, 125, 126, 127, 128, 129, 130 serine phosphorylation,41, 125, 131, 132, 133, 134 lysine methylation,135, 136 serine adenosine diphosphate ribosylation,137 and asparagine, threonine, and lysine glycation.138 Prediction analysis suggests that HMGB1 may also undergo ubiquitination in lysines 146 and 147; however, this has not been experimentally confirmed. Some of these modifications influence DNA binding and stability, regulation of transcription, intracellular protein localization, secretion, cell motility, and the proinflammatory or profibrogenic effects.4 Of these, oxidation, acetylation, and phosphorylation appear to play a significant role in liver disease41, 44 (see modified residues in Fig. 1).
HMGB1 contains three conserved redox‐sensitive cysteines (23, 45, and 106), and their oxidation determines the bioactivity of extracellular HMGB1. Cells exclude HMGB1 from the nucleus by acetylating lysines, thereby neutralizing their basic charge and rendering them unable to function as NLSs. Although HMGB1 contains 43 lysines, only 21 can undergo acetylation, but only those in two key clusters appear to play a major role in preventing HMGB1 nuclear re‐entry due to acetylation, lysines 28‐30 and 43‐44 (representing a classic bipartite and functional NLS19, 139), and lysines 180 and 182‐185 within NLS2.9, 139 Histone N‐acetyltransferase can acetylate sites within HMGB1 that are structurally similar to those in histones, even though HMGB1 binds the enzyme with lower affinity.140 While a significant body of literature exists on HMGB1 oxidation, phosphorylation, and acetylation, the majority derives from in vitro experiments using non‐liver related cell lines; therefore, their relevance in acute and chronic liver disease needs to be demonstrated in vivo.
Acute Liver Injury
HMGB1 has been implicated in I/R injury,15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27 sepsis,23, 28, 29, 30, 31, 32 and APAP‐induced liver injury.23, 33, 34, 35, 36, 37, 38, 39, 40
I/R
In I/R injury, organ damage occurs due to the return of oxygen to a previously hypoxic tissue.141 Warm I/R occurs during liver resection, injury, or trauma as a result of low blood flow. Cold I/R occurs when a liver graft is preserved in the University of Wisconsin solution prior to liver transplant and reperfusion.
Warm I/R
Tsung et al.24 identified HMGB1 as an early mediator of inflammatory organ damage, where levels increased within 1 hour and up to 24 hours after I/R injury in mice. Inhibition of HMGB1 with a neutralizing antibody decreased liver damage through phosphorylation of JNK and increased NFκB binding. TLR4 was identified as being responsible for the effect of HMGB1 because global Tlr4 –/– mice showed lessened I/R injury.24 Other studies support that an HMGB1 antibody protects the liver from I/R injury.25 HMGB1 was released from necrotic cells within the first 12 hours of injury and during the inflammatory phase in the subsequent 12 hours.25, 26 Further work revealed that HMGB1 was actively released from hypoxic hepatocytes through regulation by reactive oxygen species (ROS) that required TLR4 signaling; this was confirmed in Tlr4 null hepatocytes under hypoxia as they released less ROS and HMGB1.88 Furthermore, global Tlr4–/– mice subjected to I/R had less liver injury and inflammatory cytokine production that could not be further reduced by the antioxidant N‐acetylcysteine. This suggests that hepatocytes release ROS during I/R and HMGB1 is regulated by TLR4 signaling, which leads to liver injury and inflammation.88 However, TLR4 is not the only receptor involved in HMGB1‐mediated I/R injury as both TLR9 and RAGE inhibition protect from I/R injury in mice.112, 113
Numerous studies show that blocking TLR4, either ablating it in hepatocytes or using an inhibitor, attenuated liver injury, inflammatory pathways, and HMGB1 serum levels in I/R injury mouse models.20, 142 The TLR4, JNK, and p38 signaling pathway was activated during hepatocyte hypoxia in which JNK inhibition decreased HMGB1 release. Taken together, this indicates that TLR4 mediated proinflammatory signaling and that HMGB1 release from hepatocytes plays a harmful role in I/R injury.142 Ablation of Hmgb1 in hepatocytes reduced the number of infiltrating neutrophils and proinflammatory gene expression following I/R and ameliorated liver injury at later time points.23
However, HMGB1 plays a more complex role than first anticipated and could potentially be protective to hepatocytes rather than deleterious.143 In certain studies, mice with Hmgb1 deleted from hepatocytes had significantly greater hepatocellular injury after I/R and greater DNA damage, contrasting its well‐known role in promoting inflammation and tissue damage. Huang et al.143 observed increased liver damage plus serum TNFα and IL‐6, and 1 hour after I/R, pJNK, p38, pERK, and NFκB were increased. This correlated with more recruitment of immune cells to the liver and enhanced chemokine expression, indicating a greater inflammatory response when Hmgb1 is ablated from hepatocytes. Lack of HMGB1 increased poly adenosine diphosphate‐ribose polymerase‐1 activation, depleting nicotinamide adenine dinucleotide+ and adenosine triphosphate stores, and resulting in increased mitochondrial injury, oxidative stress in the hepatocytes, and subsequent cell death. Further evidence shows that pretreatment of mice with HMGB1 before I/R protects against injury. Cytokine levels were decreased and NFκB DNA binding reduced in the pretreated group.144 Higher hepatic IL‐1 receptor‐associated kinase‐M levels, a negative regulator of TLR signaling, reflected the reduced IL‐1 receptor‐associated kinase‐1 levels observed. This protection by HMGB1 was blocked in Tlr4 null mice; hence, HMGB1 plays a protective role during I/R through down‐regulation of TLR4 signaling.144
Additional support for the role of HMGB1 in warm I/R comes from the plethora of compounds targeting HMGB1 signaling that have been found to successfully ameliorate injury.32, 145, 146, 147, 148, 149, 150 Thus, the role of hepatic HMGB1 in warm I/R is still contradictory. To clarify whether it amplifies or ameliorates injury or has no involvement whatsoever, as one study claims,74 more research is essential.
Cold I/R
Research into the role of cold I/R has been less extensive, but there is evidence to suggest a role for HMGB1. Serum HMGB1 increased in serum of liver transplant recipients after graft reperfusion for only 10 minutes, with continued release of HMGB1 as well as up‐regulation in the hepatocytes.27 This increase in HMGB1 secretion and expression correlated with alanine aminotransferase activity; nonetheless, HMGB1 levels decreased 1‐2 hours after surgery. Remarkably, HMGB1 levels did not correlate with TNFα or IL‐6 and neutrophils had low HMGB1 expression. It is still unknown if hepatocytes, macrophages, HSCs, neutrophils, or sinusoidal endothelial cells were the key source of HMGB1; the exact mechanism for the role of HMGB1 has yet to be determined. These results do, however, highlight HMGB1 as a potential biomarker of liver injury after transplant.
SEPSIS
HMGB1 has been identified as a mediator of systemic inflammation leading to multiorgan dysfunction in sepsis. In 2003, a role for HMGB1 in the pathogenesis of sepsis‐induced multiple organ failure was first described.31 In 2009, Nagano et al.30 showed that recombinant soluble thrombomodulin decreased HMGB1, improving acute liver injury and survival rates in experimental endotoxemia. Later studies by Ye et al.29 showed that human hematopoietic cells are required to induce sepsis‐induced mortality following cecal ligation and puncture in severely immune‐deficient, nonobese, diabetic, NOD scid gamma ice and that small interfering RNA treatment to inhibit HMGB1 release by human macrophages and dendritic cells dramatically reduced sepsis‐induced mortality. Work from Wang et al.28 demonstrated that glycyrrhizin is a potential agent for the treatment of sepsis as it reduces the serum level and gene expression of HMGB1 and other proinflammatory cytokines. However, more recent studies indicate that in LPS‐induced shock, HMGB1 ablation did not ameliorate inflammation or lethality, despite efficient reduction of HMGB1 serum levels.23 Hence, additional work is needed to further clarify the role of HMGB1 in sepsis.
DRUG‐INDUCED LIVER INJURY
Drug‐induced liver injury (DILI) is the most common cause of acute liver failure in the United States.40 Although a plethora of drugs cause severe liver damage as a side effect, APAP is by far the prominent culprit and a significant cause of mortality. As HMGB1 is known to be released from necrotic cells12 and activated immune cells,9 it is a likely candidate involved in DILI.
APAP
Antoine et al.151 found that HMGB1 is released into the serum of mice following an APAP overdose prior to alanine aminotransferase and correlated with the pathology scores of liver injury,151 suggesting that HMGB1 could be a serum biomarker of DILI. Moreover, neutralization of HMGB1 was therapeutic by reducing the inflammatory reaction initiated by APAP,118 providing further evidence of the protein's role in DILI. Later, this theory was solidified clinically as concentrations of total and acetylated HMGB1 were found to increase 10‐fold to 180‐fold, respectively, in the serum from patients with liver injury after an APAP overdose152; this correlated with worse prognosis due to more severe liver injury, failure, or death.152
Huebener et al.23 later showed that mice lacking Hmgb1 in hepatocytes were protected from a lethal dose of APAP, with 100% of mice surviving compared to only 22% of wild‐type (WT) mice. HMGB1 promoted neutrophil migration toward the necrotic tissue, which was blocked in mice lacking Hmgb1 in hepatocytes. When neutrophil function was inactivated, these mice were protected from APAP‐induced injury.23
Studies from Lundback et al.153 revealed that a humanized HMGB1 antibody significantly attenuated APAP‐induced liver injury and inflammation in mice, with higher efficacy than treatment with N‐acetylcysteine, supporting the idea that HMGB1 could act as a therapeutic target to treat patients with DILI. Moreover, pretreatment or posttreatment with the HMGB1 inhibitor glycyrrhizin, also with antioxidant properties, markedly reduced hepatic inflammation, neutrophil recruitment, and injury caused by APAP in mouse models.23, 154 Despite the overwhelming evidence that HMGB1 is implicated in APAP‐induced liver injury, the mechanism by which HMGB1 mediates injury, the receptors involved, and signaling pathways activated remain largely unknown.
Other Hepatotoxins
There is less in‐depth research on the role of HMGB1 in DILI caused by other drugs. Mice dosed with flucloxacillin had elevated HMGB1 and other TLR4 ligands in serum, correlating with liver injury and inflammatory markers.155 D‐galactosamine increases HMGB1 release into plasma, correlating with a decrease in nuclear expression in rat liver156; and, an HMGB1 neutralizing antibody suppressed plasma HMGB1 and inflammatory cytokines and improved liver injury and survival.156 Methimazole increased plasma HMGB1 3‐6 hours after exposure157; this was ameliorated when Kupffer cell activation was blocked, suggesting that HMGB1 may be involved in mediating DILI by Kupffer cells. Rats treated with diclofenac had increased HMGB1 in plasma,158 and sulfamethoxazole and flucloxacillin stimulated HMGB1 release from human hepatocytes along with markers of cell death and inflammation.159 Moreover, disulfide HMGB1 stimulated cytokine release from dendritic cells, indicating that it may be involved in stimulating an immune response to the drugs. There is, however, a lack of data from human patients on the role of HMGB1 in DILI caused by drugs other than APAP; hence, more clinical evidence is needed.
Chronic Liver Disease
ALD
Ge et al.41 demonstrated that liver biopsies from patients with ALD showed a robust increase in HMGB1 expression and translocation that correlated with disease stage compared to healthy explants. This was also accompanied by enhanced HMGB1 levels in the serum compared to healthy individuals. Similar findings were observed in chronic ethanol‐fed WT mice. Using primary cell culture, our laboratory validated the ability of hepatocytes from ethanol‐fed mice to translocate HMGB1 from the nucleus to the cytoplasm and ultimately secrete a large concentration of HMGB1 that was far superior to that from Kupffer cells. Of note, HMGB1 secretion was time and dose dependent and responsive to pro‐oxidants and blocked by antioxidants. We further demonstrated that selective ablation of Hmgb1in hepatocytes protected from alcohol‐induced liver injury in mice due to the increase in key enzymes involved in fatty acid β‐oxidation. Likewise, there was a more efficient low‐density lipoprotein plus very low‐density lipoprotein export from the liver into the circulation that largely prevented steatosis and ultimately liver injury.41
In subsequent studies, we detected native and posttranslationally modified HMGB1 in humans and in mice with ALD. Specifically, in liver and in serum from control mice and in serum from healthy volunteers, the lysine residues within NLS1 and NLS2 were nonacetylated and all cysteine residues were reduced. However, in livers from ethanol‐fed mice, in addition to all thiol/nonacetylated isoforms of HMGB1, we observed acetylated NLS1 and NLS2, a unique phosphorylation site in serine 35, and an increase in oxidation of HMGB1 to the disulfide isoform. In serum from ethanol‐fed mice and from patients with ALD, there was disulfide‐bonded hyperacetylated‐HMGB1, disulfide‐bonded nonacetylated‐HMGB1, and phosphorylated HMGB1 in serine 35. Hepatocytes appeared to be a major source of all these HMGB1 isoforms. Thus, hepatocyte HMGB1 participates in the pathogenesis of ALD and undergoes PTMs that could condition its toxic effects. Kupffer cells, however, produced mostly the all thiol/nonacetylated and acetylated versions of the protein.41 Current studies from our laboratory using genetic approaches are focusing on trying to understand the specific role of each of these PTMs and the contribution of HMGB1 acetylation in Kupffer cells to the pathogenesis of ALD.
FIBROSIS
Liver fibrosis is a frequent life‐threatening complication of most chronic liver diseases. Our laboratory observed that liver HMGB1 protein expression correlated with fibrosis stage in patients with chronic hepatitis C virus infection and primary biliary cirrhosis.49 Serum levels of HMGB1 were also increased in these patients compared to healthy controls, suggesting protein secretion. These findings were equally replicated in four mouse models of liver fibrosis due to DILI caused by chronic CCl4 injections or thioacetamide administration, cholestasis triggered by common bile duct ligation, and NASH induced by feeding a methionine‐ and choline‐deficient diet. Overall, these data suggested that HMGB1 has a key role in liver fibrosis.49
Studies initiated by Arriazu et al.44 demonstrated that, while HSC secretion of HMGB1 is minimal, in response to a noxious stimuli, HSCs acetylated HMGB1 in vitro, which contributed to collagen‐I deposition. In a follow‐up study, Ge et al.49 showed that HMGB1 also stimulated HSC migration in vitro and in vivo, a critical event driving the progression of liver fibrosis. Importantly, the authors demonstrated that ablation of Hmgb1 in HSCs in vivo reduced fibrosis about 25%. This finding unlocked the possibility that additional cellular sources of HMGB1 may have a more meaningful impact in liver fibrosis.
The role of HMGB1 was later proven as neutralization of HMGB1 protected liver fibrosis whereas injection of HMGB1 promoted liver fibrosis. The authors observed that HMGB1 was up‐regulated and secreted mostly by hepatocytes and also Kupffer cells along with infiltrating macrophages following chronic CCl4 treatment. Moreover, Hmgb1 ablation in hepatocytes or myeloid cells was also partially protective; more notably, ablation in both reduced fibrosis about 75% compared to WT mice, thus indicating a stronger contribution of HMGB1 from these two cell types to liver fibrosis.49
In line with the in vivo data, coculture with hepatocytes or Kupffer cells from CCl4‐injected WT mice up‐regulated collagen‐I production by HSCs; however, coculture with hepatocytes from CCl4‐injected conditional Hmgb1 knockout mice or with Kupffer cells from CCl4‐injected conditional Hmgb1knockout mice partially blunted this effect. Overall, the data suggested that signaling from hepatocytes and Kupffer cells by HMGB1 was driving collagen‐I production by HSCs.49
Our laboratory also investigated whether induction of HMGB1 could participate in the pathogenesis of liver fibrosis by RAGE cell‐specific signaling mechanisms.44, 49 Notably, while Rage ablation in hepatocytes and myeloid cells did not cause significant protection, Rage ablation in HSCs and RAGE neutralization almost fully prevented liver fibrosis. Mechanistically, we then demonstrated that HMGB1 signaled through RAGE to up‐regulate collagen‐I expression by activating the pMEK 1/2/pERK1/2/p‐c‐Jun signaling pathway upstream of PI3K/pAkt, thereby contributing to scarring.49
While two studies59, 60 did not find a role for HMGB1 in liver fibrosis, work from Kao et al.160 found that HMGB1 up‐regulated α‐smooth muscle actin expression and suppressed the activity of the collagen‐degrading matrix metalloproteinases 2, leading to extracellular matrix accumulation. Likewise, it was shown that an Hmgb1 small interfering RNA inhibited the synthesis of α‐smooth muscle actin and collagen in rat fibrosis.161 Although the role of HMGB1 in fibrosis progression appears clear, the contribution of the various HMGB1 isoforms as well as their potential role in fibrosis regression and the receptor involved during the resolution phase remain open questions and may clarify some aspects of the role of HMGB1 in liver fibrosis.
NASH
NAFLD is the most common cause of chronic liver disease in developed countries and particularly in the United States due to the major consumption of junk food.162 In 2018, work from Chen et al.51 proved that HMGB1 expression was detected in high‐fat diet‐induced NAFLD in mice and that liver injury was mitigated by an HMGB1 neutralizing antibody. In this study, HMGB1 expression was regulated by both the JNK1/2‐activating transcription factor 2 axis and the microRNA‐200 family. Li et al.114 later established that HMGB1 release from hepatocytes in response to free fatty acid infusion was vital for TLR4/MyD88 activation and cytokine expression both in vitro and in vivo. They then treated mice with a neutralizing antibody to HMGB1 that protected against free fatty acid‐induced TNFα and IL‐6 production. They concluded that TLR4/MyD88 signaling in liver parenchymal cells played a pivotal role during the early progression of NAFLD in which free HMGB1 served as a ligand for TLR4 activation.114
Chandrashekaran et al.163 identified HMGB1 as a key mediator of intestinal inflammation in NAFLD that is RAGE and redox signaling dependent and promotes ectopic intestinal inflammation. These studies point to HMGB1 as an important mediator of liver injury in the early stages of NAFLD. Nonetheless, the key receptor participating in the later stages of the disease as well as during the resolution phase and the HMGB1 isoforms involved remain elusive. Likewise, the contribution of HMGB1 from intestinal origin to the overall pathogenesis of NASH is unknown.
HCC
HMGB1 induces chronic inflammation and extracellular matrix accumulation, creating a prime microenvironment for the development of HHC. In 2008, serum HMGB1 levels were found to be strongly increased in patients with HCC and correlated with clinicopathologic features, such as with α‐fetoprotein levels and tumor size,164 and Masuda et al.56 established that serum HMGB1 predicted worse clinical prognosis in patients with HCC.
Likewise, Dong et al.165 found that HMGB1 was highly expressed in human liver cancer compared with normal tissue and was positively associated with pathologic grade and distant metastases of liver cancer. They further demonstrated that knockdown of Hmgb1 down‐regulated the expression of pAKT, Ki67, and matrix metalloproteinases 2; inhibited the proliferation and metastatic potential of SMMC‐7721 cells; induced cell cycle arrest and apoptosis; and slowed the growth of xenograft tumors in mice. Thus, this study suggested that HMGB1 may be involved in liver cancer development and progression, representing a potential therapeutic target for this aggressive malignancy.
Later, Chen et al.53 showed that HMGB1 plays a role in the regulation of the Hippo pathway during liver tumorigenesis as binding of HMGB1 to GA‐binding protein α promoted the expression of Yes‐associated protein (the downstream effector of the Hippo pathway) to induce liver tumors in mice. Additionally, HMGB1 depletion in hepatocytes blocked diethylnitrosamine‐induced liver cancer initiation and short hairpin RNA‐mediated gene silencing of Hmgb1 inhibited HCC cell proliferation.
Two of our studies showed that Hmgb1ablation prevents ductular reaction, a major mechanism contributing to portal fibrosis.44, 49 Recently, Kambu et al.60 reported that HMGB1 release is a critical mechanism in hepatic pathogenesis in autophagy‐deficient conditions and leads to ductular reaction, hepatic progenitor cell expansion, and tumor development. A similar conclusion was put forward by Hernandez et al.,59 pointing to HMGB1 as linking hepatocyte death to ductular reaction, progenitor signature, and hepatocarcinogenesis in chronic liver disease.
Regarding the receptors involved, Yaser et al.166 showed that RAGE is overexpressed in primary HCC compared to adjacent nontumor liver samples and demonstrated that it regulates cellular proliferation in HCC due to HMGB1 binding. The Tsung laboratory showed that in hypoxic HCC cells, HMGB1 activates TLR4‐ and RAGE‐signaling pathways to induce caspase‐1 activation with the ensuing production of multiple inflammatory mediators, which in turn promote cancer invasion and metastasis167; yet, the authors did not discuss the potential effect of caspase‐1 cleavage of HMGB1. In a follow‐up publication, they established that tumors lacking HMGB1 had a significant reduction in mitochondrial biogenesis and increased mitochondrial dysfunction. In these studies, they showed that in hypoxic conditions HMGB1 that translocated from the nucleus to the cytoplasm interacted with TLR9, activated p38, and phosphorylated peroxisome proliferator‐activated receptor gamma coactivator 1α with subsequent up‐regulation of mitochondrial biogenesis.57
An area that remains to be investigated is the likely possibility that specific HMGB1 isoforms may have a dominant role in the onset and progression of HCC. Likewise, the signaling cascades that they could activate and whether they drive the phenotype of cancer stem cells remain elusive.
Therapeutic Implications
HMGB1 orchestrates the regulation of both inflammation and wound healing.44, 64, 116, 168, 169 There is significant evidence that HMGB1 plays a role in the initiation, exacerbation, or prolongation of acute liver injury and chronic liver disease, although the receptors involved may vary among the clinical conditions and their activation is likely regulated by the presence of various HMGB1 isoforms or even HMGB1 protein complexes, some of which may have significant immune effects. HMGB1 has promise as a target for treatment as well as potential to be used as a biomarker. Nevertheless, additional research into the molecular mechanisms underlying its effects, the overall contribution of the PTMs, and if one isoform dominates or regulates the others is essential in order to elucidate the importance of HMGB1 in liver disease and to design drugs or develop antibodies that specifically target each HMGB1 isoform. It is likely that the isoforms play different roles in liver disease, perhaps by binding with different affinity to key receptors.
Supported by American Association for the Study of Liver Diseases Pinnacle Research Award (G3156 to X.G.), Chicago Biomedical Consortium (PDR‐088 to H.G. and N.N.), National Institute of Diabetes and Digestive and Kidney Diseases Public Health Service grant R01 DK111677 (to N.N.) and National Institute on Alcohol Abuse and Alcoholism Public Health Service grant R01 AA021887 (to N.N.).
Potential conflict of interest
Nothing to report.
References
- 1. Johns E. The Chromosomal Proteins. Elsevier Science; 2012. [Google Scholar]
- 2. Bustin M, Reeves R. High‐mobility‐group chromosomal proteins: architectural components that facilitate chromatin function. Prog Nucleic Acid Res Mol Biol 1996;54:35‐100. [DOI] [PubMed] [Google Scholar]
- 3. Bustin M, Lehn DA, Landsman D. Structural features of the HMG chromosomal proteins and their genes. Biochim Biophys Acta 1990;1049:231‐243. [DOI] [PubMed] [Google Scholar]
- 4. Stros M. HMGB proteins: interactions with DNA and chromatin. Biochim Biophys Acta 2010;1799:101‐113. [DOI] [PubMed] [Google Scholar]
- 5. Yang H, Tracey KJ. Targeting HMGB1 in inflammation. Biochim Biophys Acta 2010;1799:149‐156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ronfani L, Ferraguti M, Croci L, Ovitt CE, Scholer HR, Consalez GG, et al. Reduced fertility and spermatogenesis defects in mice lacking chromosomal protein Hmgb2. Development 2001;128:1265‐1273. [DOI] [PubMed] [Google Scholar]
- 7. Bustin M. Regulation of DNA‐dependent activities by the functional motifs of the high‐mobility‐group chromosomal proteins. Mol Cell Biol 1999;19:5237‐5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vaccari T, Beltrame M, Ferrari S, Bianchi ME. Hmg4, a new member of the Hmg1/2 gene family. Genomics 1998;49:247‐252. [DOI] [PubMed] [Google Scholar]
- 9. Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, et al. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J 2003;22:5551‐5560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Youn JH, Kwak MS, Wu J, Kim ES, Ji Y, Min HJ, et al. Identification of lipopolysaccharide‐binding peptide regions within HMGB1 and their effects on subclinical endotoxemia in a mouse model. Eur J Immunol 2011;41:2753‐2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li J, Kokkola R, Tabibzadeh S, Yang R, Ochani M, Qiang X, et al. Structural basis for the proinflammatory cytokine activity of high mobility group box 1. Mol Med 2003;9:37‐45. [PMC free article] [PubMed] [Google Scholar]
- 12. Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002;418:191‐195. [DOI] [PubMed] [Google Scholar]
- 13. Gardella S, Andrei C, Ferrera D, Lotti LV, Torrisi MR, Bianchi ME, et al. The nuclear protein HMGB1 is secreted by monocytes via a non‐classical, vesicle‐mediated secretory pathway. EMBO Rep 2002;3:995‐1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Degryse B, Bonaldi T, Scaffidi P, Muller S, Resnati M, Sanvito F, et al. The high mobility group (HMG) boxes of the nuclear protein HMG1 induce chemotaxis and cytoskeleton reorganization in rat smooth muscle cells. J Cell Biol 2001;152:1197‐1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gendy AM, Abdallah DM, El‐Abhar HS. The potential curative effect of rebamipide in hepatic ischemia/reperfusion injury. Naunyn Schmiedebergs Arch Pharmacol 2017;390:691‐700. [DOI] [PubMed] [Google Scholar]
- 16. Kim MS, Lee S, Jung N, Lee K, Choi J, Kim SH, et al. The vitamin D analogue paricalcitol attenuates hepatic ischemia/reperfusion injury through down‐regulation of Toll‐like receptor 4 signaling in rats. Arch Med Sci 2017;13:459‐469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li X, Wu Y, Zhang W, Gong J, Cheng Y. Pre‐conditioning with tanshinone IIA attenuates the ischemia/reperfusion injury caused by liver grafts via regulation of HMGB1 in rat Kupffer cells. Biomed Pharmacother 2017;89:1392‐1400. [DOI] [PubMed] [Google Scholar]
- 18. Sun J, Guo E, Yang J, Yang Y, Liu S, Hu J, et al. Carbon monoxide ameliorates hepatic ischemia/reperfusion injury via sirtuin 1‐mediated deacetylation of high‐mobility group box 1 in rats. Liver Transpl 2017;23:510‐526. [DOI] [PubMed] [Google Scholar]
- 19. Yamamoto T, Tajima Y. HMGB1 is a promising therapeutic target for acute liver failure. Expert Rev Gastroenterol Hepatol 2017;11:673‐682. [DOI] [PubMed] [Google Scholar]
- 20. Yokoi T, Yokoyama Y, Kokuryo T, Yamaguchi J, Nagino M. Inhibition of Toll‐like receptor 4 ameliorates experimental postischemic injury in the cholestatic liver through inhibition of high‐mobility group box protein b1 (HMGB1) signaling. Surgery 2018;163:270‐276. [DOI] [PubMed] [Google Scholar]
- 21. Zhang S, Wotzkow C, Bongoni AK, Shaw‐Boden J, Siegrist M, Taddeo A, et al. Role of the plasma cascade systems in ischemia/reperfusion injury of bone. Bone 2017;97:278‐286. [DOI] [PubMed] [Google Scholar]
- 22. Zhang T, Wei W, Dirsch O, Kruger T, Kan C, Xie C, et al. Identification of proteins interacting with cytoplasmic high‐mobility group box 1 during the hepatocellular response to ischemia reperfusion injury. Int J Mol Sci 2017;18pii:E167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huebener P, Pradere JP, Hernandez C, Gwak GY, Caviglia JM, Mu X, et al. The HMGB1/RAGE axis triggers neutrophil‐mediated injury amplification following necrosis. J Clin Invest 2015;125:539‐550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia‐reperfusion. J Exp Med 2005;201:1135‐1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Watanabe T, Kubota S, Nagaya M, Ozaki S, Nagafuchi H, Akashi K, et al. The role of HMGB‐1 on the development of necrosis during hepatic ischemia and hepatic ischemia/reperfusion injury in mice. J Surg Res 2005;124:59‐66. [DOI] [PubMed] [Google Scholar]
- 26. Yang M, Antoine DJ, Weemhoff JL, Jenkins RE, Farhood A, Park BK, et al. Biomarkers distinguish apoptotic and necrotic cell death during hepatic ischemia/reperfusion injury in mice. Liver Transpl 2014;20:1372‐1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ilmakunnas M, Tukiainen EM, Rouhiainen A, Rauvala H, Arola J, Nordin A, et al. High mobility group box 1 protein as a marker of hepatocellular injury in human liver transplantation. Liver Transpl 2008;14:1517‐1525. [DOI] [PubMed] [Google Scholar]
- 28. Wang W, Zhao F, Fang Y, Li X, Shen L, Cao T, et al. Glycyrrhizin protects against porcine endotoxemia through modulation of systemic inflammatory response. Crit Care 2013;17:R44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ye C, Choi JG, Abraham S, Wu H, Diaz D, Terreros D, et al. Human macrophage and dendritic cell‐specific silencing of high‐mobility group protein B1 ameliorates sepsis in a humanized mouse model. Proc Natl Acad Sci U S A 2012;109:21052‐21057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nagato M, Okamoto K, Abe Y, Higure A, Yamaguchi K. Recombinant human soluble thrombomodulin decreases the plasma high‐mobility group box‐1 protein levels, whereas improving the acute liver injury and survival rates in experimental endotoxemia. Crit Care Med 2009;37:2181‐2186. [DOI] [PubMed] [Google Scholar]
- 31. Zhang LT, Yao YM, Lu JQ, Dong N, Yu Y, Yan XJ, et al. The potential role of high mobility group‐1 protein in the pathogenesis of sepsis‐induced multiple organ dysfunction syndrome in rats. Zhonghua Wai Ke Za Zhi 2003;41:303‐306. [PubMed] [Google Scholar]
- 32. McDonald KA, Huang H, Tohme S, Loughran P, Ferrero K, Billiar T, et al. Toll‐like receptor 4 (TLR4) antagonist eritoran tetrasodium attenuates liver ischemia and reperfusion injury through inhibition of high‐mobility group box protein B1 (HMGB1) signaling. Mol Med 2015;20:639‐648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brillant N, Elmasry M, Burton NC, Rodriguez JM, Sharkey JW, Fenwick S, et al. Dynamic and accurate assessment of acetaminophen‐induced hepatotoxicity by integrated photoacoustic imaging and mechanistic biomarkers in vivo. Toxicol Appl Pharmacol 2017;332:64‐74. [DOI] [PubMed] [Google Scholar]
- 34. Horiuchi T, Sakata N, Narumi Y, Kimura T, Hayashi T, Nagano K, et al. Metformin directly binds the alarmin HMGB1 and inhibits its proinflammatory activity. J Biol Chem 2017;292:8436‐8446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jiang L, Ke M, Yue S, Xiao W, Yan Y, Deng X, et al. Blockade of Notch signaling promotes acetaminophen‐induced liver injury. Immunol Res 2017;65:739‐749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Weemhoff JL, Woolbright BL, Jenkins RE, McGill MR, Sharpe MR, Olson JC, et al. Plasma biomarkers to study mechanisms of liver injury in patients with hypoxic hepatitis. Liver Int 2017;37:377‐384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yang G, Zhang L, Ma L, Jiang R, Kuang G, Li K, et al. Glycyrrhetinic acid prevents acetaminophen‐induced acute liver injury via the inhibition of CYP2E1 expression and HMGB1‐TLR4 signal activation in mice. Int Immunopharmacol 2017;50:186‐193. [DOI] [PubMed] [Google Scholar]
- 38. Yang H, Wang H, Wang Y, Addorisio M, Li J, Postiglione MJ, et al. The haptoglobin beta subunit sequesters HMGB1 toxicity in sterile and infectious inflammation. J Intern Med 2017;282:76‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zheng L, Yin L, Xu L, Qi Y, Li H, Xu Y, et al. Protective effect of dioscin against thioacetamide‐induced acute liver injury via FXR/AMPK signaling pathway in vivo. Biomed Pharmacother 2018;97:481‐488. [DOI] [PubMed] [Google Scholar]
- 40. Lee WM. Acute liver failure. Semin Respir Crit Care Med 2012;33:36‐45. [DOI] [PubMed] [Google Scholar]
- 41. Ge X, Antoine DJ, Lu Y, Arriazu E, Leung TM, Klepper AL, et al. High mobility group box‐1 (HMGB1) participates in the pathogenesis of alcoholic liver disease (ALD). J Biol Chem 2014;289:22672‐22691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Seo YS, Kwon JH, Yaqoob U, Yang L, De Assuncao TM, Simonetto DA, et al. HMGB1 recruits hepatic stellate cells and liver endothelial cells to sites of ethanol‐induced parenchymal cell injury. Am J Physiol Gastrointest Liver Physiol 2013;305:G838‐G848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang X, Bu HF, Zhong W, Asai A, Zhou Z, Tan XD. MFG‐E8 and HMGB1 are involved in the mechanism underlying alcohol‐induced impairment of macrophage efferocytosis. Mol Med 2013;19:170‐182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Arriazu E, Ge X, Leung TM, Magdaleno F, Lopategi A, Lu Y, et al. Signalling via the osteopontin and high mobility group box‐1 axis drives the fibrogenic response to liver injury. Gut 2017;66:1123‐1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bai L, Jin W, Kong M, Zhang X, Zheng S, Chen Y, et al. Injury resistance in the setting of liver fibrosis is accompanied by the inhibition of high‐mobility group box‐1 translocation and release. Dig Dis 2018;36:167‐176. [DOI] [PubMed] [Google Scholar]
- 46. Hu YB, Hu DP, Fu RQ. Correlation between high mobility group box‐1 protein and chronic hepatitis B infection with severe hepatitis B and acute‐on‐chronic liver failure: a meta‐analysis. Minerva Med 2017;108:268‐276. [DOI] [PubMed] [Google Scholar]
- 47. Inkaya AC, Demir NA, Kolgelier S, Sumer S, Demir LS, Ural O, et al. Is serum high‐mobility group box 1 (HMGB‐1) level correlated with liver fibrosis in chronic hepatitis B? Medicine (Baltimore) 2017;96:e7547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Khanjarsim V, Karimi J, Khodadadi I, Mohammadalipour A, Goodarzi MT, Solgi G, et al. Ameliorative effects of nilotinib on CCl4 induced liver fibrosis via attenuation of RAGE/HMGB1 gene expression and oxidative stress in rat. Chonnam Med J 2017;53:118‐126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ge X, Arriazu E, Magdaleno F, Antoine DJ, Dela Cruz R, Theise N, et al. High mobility group box‐1 drives fibrosis progression signaling via the receptor for advanced glycation end‐products in mice. Hepatology 2018;. 10.1002/hep.30093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Afrin R, Arumugam S, Rahman A, Wahed MI, Karuppagounder V, Harima M, et al. Curcumin ameliorates liver damage and progression of NASH in NASH‐HCC mouse model possibly by modulating HMGB1‐NF‐kappaB translocation. Int Immunopharmacol 2017;44:174‐182. [DOI] [PubMed] [Google Scholar]
- 51. Chen X, Ling Y, Wei Y, Tang J, Ren Y, Zhang B, et al. Dual regulation of HMGB1 by combined JNK1/2‐ATF2 axis with miR‐200 family in nonalcoholic steatohepatitis in mice. FASEB J 2018;32:2722‐2734. [DOI] [PubMed] [Google Scholar]
- 52. Gan LT, Van Rooyen DM, Koina ME, McCuskey RS, Teoh NC, Farrell GC. Hepatocyte free cholesterol lipotoxicity results from JNK1‐mediated mitochondrial injury and is HMGB1 and TLR4‐dependent. J Hepatol 2014;61:1376‐1384. [DOI] [PubMed] [Google Scholar]
- 53. Chen R, Zhu S, Fan XG, Wang H, Lotze MT, Zeh HJ 3rd, et al. High mobility group protein B1 controls liver cancer initiation through yes‐associated protein ‐dependent aerobic glycolysis. Hepatology 2018;67:1823‐1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li S, Huang Y, Huang Y, Fu Y, Tang D, Kang R, et al. The long non‐coding RNA TP73‐AS1 modulates HCC cell proliferation through miR‐200a‐dependent HMGB1/RAGE regulation. J Exp Clin Cancer Res 2017;36:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lv G, Wu M, Wang M, Jiang X, Du J, Zhang K, et al. miR‐320a regulates high mobility group box 1 expression and inhibits invasion and metastasis in hepatocellular carcinoma. Liver Int 2017;37:1354‐1364. [DOI] [PubMed] [Google Scholar]
- 56. Masuda K, Ono A, Aikata H, Kawaoka T, Nelson Hayes C, Teraoka Y et al. Serum HMGB1 concentrations at 4 weeks is a useful predictor of extreme poor prognosis for advanced hepatocellular carcinoma treated with sorafenib and hepatic arterial infusion chemotherapy. J Gastroenterol 2018;53:107‐118. Erratum. In: J Gastroenterol 2018;53:161‐162. [DOI] [PubMed] [Google Scholar]
- 57. Tohme S, Yazdani HO, Liu Y, Loughran P, van der Windt DJ, Huang H, et al. Hypoxia mediates mitochondrial biogenesis in hepatocellular carcinoma to promote tumor growth through HMGB1 and TLR9 interaction. Hepatology 2017;66:182‐197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Xiao Y, Sun L, Fu Y, Huang Y, Zhou R, Hu X, et al. High mobility group box 1 promotes sorafenib resistance in HepG2 cells and in vivo. BMC Cancer 2017;17:857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hernandez C, Huebener P, Pradere JP, Antoine DJ, Friedman RA, Schwabe RF. HMGB1 links chronic liver injury to progenitor responses and hepatocarcinogenesis. J Clin Invest 2018;128:2436‐2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Khambu B, Huda N, Chen X, Antoine DJ, Li Y, Dai G, et al. HMGB1 promotes ductular reaction and tumorigenesis in autophagy‐deficient livers. J Clin Invest 2018;128:2419‐2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jhun J, Lee S, Kim H, Her YM, Byun JK, Kim EK, et al. HMGB1/RAGE induces IL‐17 expression to exaggerate inflammation in peripheral blood cells of hepatitis B patients. J Transl Med 2015;13:310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Su S, Wu J, Gong T, He K, Feng C, Zhang M, et al. Inhibition of high mobility group box 1‐toll‐like receptor‐4 signaling by glycyrrhizin contributes to the attenuation of cold ischemic injury of liver in a rat model. Transplant Proc 2016;48:191‐198. [DOI] [PubMed] [Google Scholar]
- 63. Wan Z, Zhang X, Peng A, He M, Lei Z, Wang Y. TLR4‐HMGB1 signaling pathway affects the inflammatory reaction of autoimmune myositis by regulating MHC‐I. Int Immunopharmacol 2016;41:74‐81. [DOI] [PubMed] [Google Scholar]
- 64. Muller S, Ronfani L, Bianchi ME. Regulated expression and subcellular localization of HMGB1, a chromatin protein with a cytokine function. J Intern Med 2004;255:332‐343. [DOI] [PubMed] [Google Scholar]
- 65. Butler AP, Mardian JK, Olins DE. Nonhistone chromosomal protein HMG 1 interactions with DNA. Fluorescence and thermal denaturation studies. J Biol Chem 1985;260:10613‐10620. [PubMed] [Google Scholar]
- 66. Chau KY, Lam HY, Lee KL. Estrogen treatment induces elevated expression of HMG1 in MCF‐7 cells. Exp Cell Res 1998;241:269‐272. [DOI] [PubMed] [Google Scholar]
- 67. Verrier CS, Roodi N, Yee CJ, Bailey LR, Jensen RA, Bustin M, et al. High‐mobility group (HMG) protein HMG‐1 and TATA‐binding protein‐associated factor TAF(II)30 affect estrogen receptor‐mediated transcriptional activation. Mol Endocrinol 1997;11:1009‐1019. [DOI] [PubMed] [Google Scholar]
- 68. Zappavigna V, Falciola L, Helmer‐Citterich M, Mavilio F, Bianchi ME. HMG1 interacts with HOX proteins and enhances their DNA binding and transcriptional activation. EMBO J 1996;15:4981‐4991. [PMC free article] [PubMed] [Google Scholar]
- 69. Sutrias‐Grau M, Bianchi ME, Bernues J. High mobility group protein 1 interacts specifically with the core domain of human TATA box‐binding protein and interferes with transcription factor IIB within the pre‐initiation complex. J Biol Chem 1999;274:1628‐1634. [DOI] [PubMed] [Google Scholar]
- 70. Onate SA, Prendergast P, Wagner JP, Nissen M, Reeves R, Pettijohn DE, et al. The DNA‐bending protein HMG‐1 enhances progesterone receptor binding to its target DNA sequences. Mol Cell Biol 1994;14:3376‐3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sheflin LG, Fucile NW, Spaulding SW. The specific interactions of HMG 1 and 2 with negatively supercoiled DNA are modulated by their acidic C‐terminal domains and involve cysteine residues in their HMG 1/2 boxes. Biochemistry 1993;32:3238‐3248. [DOI] [PubMed] [Google Scholar]
- 72. Grosschedl R, Giese K, Pagel J. HMG domain proteins: architectural elements in the assembly of nucleoprotein structures. Trends Genet 1994;10:94‐100. [DOI] [PubMed] [Google Scholar]
- 73. Calogero S, Grassi F, Aguzzi A, Voigtlander T, Ferrier P, Ferrari S, et al. The lack of chromosomal protein Hmg1 does not disrupt cell growth but causes lethal hypoglycaemia in newborn mice. Nat Genet 1999;22:276‐280. [DOI] [PubMed] [Google Scholar]
- 74. Yanai H, Matsuda A, An J, Koshiba R, Nishio J, Negishi H, et al. Conditional ablation of HMGB1 in mice reveals its protective function against endotoxemia and bacterial infection. Proc Natl Acad Sci U S A 2013;110:20699‐20704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Huebener P, Gwak GY, Pradere JP, Quinzii CM, Friedman R, Lin CS, et al. High‐mobility group box 1 is dispensable for autophagy, mitochondrial quality control, and organ function in vivo. Cell Metab 2014;19:539‐547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundback P, et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature 2012;488:670‐674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lu B, Antoine DJ, Kwan K, Lundback P, Wahamaa H, Schierbeck H, et al. JAK/STAT1 signaling promotes HMGB1 hyperacetylation and nuclear translocation. Proc Natl Acad Sci U S A 2014;111:3068‐3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gorr SU, Darling DS. An N‐terminal hydrophobic peak is the sorting signal of regulated secretory proteins. FEBS Lett 1995;361:8‐12. [DOI] [PubMed] [Google Scholar]
- 79. Dumitriu IE, Baruah P, Manfredi AA, Bianchi ME, Rovere‐Querini P. HMGB1: guiding immunity from within. Trends Immunol 2005;26:381‐387. [DOI] [PubMed] [Google Scholar]
- 80. Bell CW, Jiang W, Reich CF 3rd, Pisetsky DS. The extracellular release of HMGB1 during apoptotic cell death. Am J Physiol Cell Physiol 2006;291:C1318‐C1325. [DOI] [PubMed] [Google Scholar]
- 81. Jiang G, Wang Y, Yun J, Hajrasouliha AR, Zhao Y, Sun D, et al. HMGB1 release triggered by the interaction of live retinal cells and uveitogenic T cells is Fas/FasL activation‐dependent. J Neuroinflammation.2015;12:179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol 2002;2:965‐975. [DOI] [PubMed] [Google Scholar]
- 83. Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, et al. HMG‐1 as a late mediator of endotoxin lethality in mice. Science 1999;285:248‐251. [DOI] [PubMed] [Google Scholar]
- 84. Abraham E, Arcaroli J, Carmody A, Wang H, Tracey KJ. HMG‐1 as a mediator of acute lung inflammation. J Immunol 2000;165:2950‐2954. [DOI] [PubMed] [Google Scholar]
- 85. Chen G, Li J, Ochani M, Rendon‐Mitchell B, Qiang X, Susarla S, et al. Bacterial endotoxin stimulates macrophages to release HMGB1 partly through CD14‐ and TNF‐dependent mechanisms. J Leukoc Biol 2004;76:994‐1001. [DOI] [PubMed] [Google Scholar]
- 86. Evankovich J, Cho SW, Zhang R, Cardinal J, Dhupar R, Zhang L, et al. High mobility group box 1 release from hepatocytes during ischemia and reperfusion injury is mediated by decreased histone deacetylase activity. J Biol Chem 2010;285:39888‐39897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Sun Q, Loughran P, Shapiro R, Shrivastava IH, Antoine DJ, Li T, et al. Redox‐dependent regulation of hepatocyte absent in melanoma 2 inflammasome activation in sterile liver injury in mice. Hepatology 2017;65:253‐268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Tsung A, Klune JR, Zhang X, Jeyabalan G, Cao Z, Peng X, et al. HMGB1 release induced by liver ischemia involves Toll‐like receptor 4 dependent reactive oxygen species production and calcium‐mediated signaling. J Exp Med 2007;204:2913‐2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Liu S, Stolz DB, Sappington PL, Macias CA, Killeen ME, Tenhunen JJ, et al. HMGB1 is secreted by immunostimulated enterocytes and contributes to cytomix‐induced hyperpermeability of Caco‐2 monolayers. Am J Physiol Cell Physiol 2006;290:C990‐C999. [DOI] [PubMed] [Google Scholar]
- 90. Bianchi ME, Manfredi AA. High‐mobility group box 1 (HMGB1) protein at the crossroads between innate and adaptive immunity. Immunol Rev 2007;220:35‐46. [DOI] [PubMed] [Google Scholar]
- 91. Yu M, Wang H, Ding A, Golenbock DT, Latz E, Czura CJ, et al. HMGB1 signals through toll‐like receptor (TLR) 4 and TLR2. Shock 2006;26:174‐179. [DOI] [PubMed] [Google Scholar]
- 92. Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, et al. Toll‐like receptor 9‐dependent activation by DNA‐containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol 2007;8:487‐496. [DOI] [PubMed] [Google Scholar]
- 93. Chen GY, Tang J, Zheng P, Liu Y. CD24 and Siglec‐10 selectively repress tissue damage‐induced immune responses. Science 2009;323:1722‐1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Hori O, Brett J, Slattery T, Cao R, Zhang J, Chen JX, et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co‐expression of rage and amphoterin in the developing nervous system. J Biol Chem 1995;270:25752‐25761. [DOI] [PubMed] [Google Scholar]
- 95. Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, et al. RAGE and amyloid‐beta peptide neurotoxicity in Alzheimer's disease. Nature 1996;382:685‐691. [DOI] [PubMed] [Google Scholar]
- 96. Li J, Schmidt AM. Characterization and functional analysis of the promoter of RAGE, the receptor for advanced glycation end products. J Biol Chem 1997;272:16498‐16506. [DOI] [PubMed] [Google Scholar]
- 97. Li J, Qu X, Schmidt AM. Sp1‐binding elements in the promoter of RAGE are essential for amphoterin‐mediated gene expression in cultured neuroblastoma cells. J Biol Chem 1998;273:30870‐30878. [DOI] [PubMed] [Google Scholar]
- 98. Yamagishi S, Matsui T. Role of receptor for advanced glycation end products (RAGE) in liver disease. Eur J Med Res 2015;20:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Lohwasser C, Neureiter D, Popov Y, Bauer M, Schuppan D. Role of the receptor for advanced glycation end products in hepatic fibrosis. World J Gastroenterol 2009;15:5789‐5798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Zen K, Chen CX, Chen YT, Wilton R, Liu Y. Receptor for advanced glycation endproducts mediates neutrophil migration across intestinal epithelium. J Immunol 2007;178:2483‐2490. [DOI] [PubMed] [Google Scholar]
- 101. Xie J, Mendez JD, Mendez‐Valenzuela V, Aguilar‐Hernandez MM. Cellular signalling of the receptor for advanced glycation end products (RAGE). CellSignal 2013;25:2185‐2197. [DOI] [PubMed] [Google Scholar]
- 102. Huttunen HJ, Fages C, Rauvala H. Receptor for advanced glycation end products (RAGE)‐mediated neurite outgrowth and activation of NF‐kappaB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J Biol Chem 1999;274:19919‐19924. [DOI] [PubMed] [Google Scholar]
- 103. Zeng S, Feirt N, Goldstein M, Guarrera J, Ippagunta N, Ekong U, et al. Blockade of receptor for advanced glycation end product (RAGE) attenuates ischemia and reperfusion injury to the liver in mice. Hepatology 2004;39:422‐432. [DOI] [PubMed] [Google Scholar]
- 104. Gong W, Wang ZY, Chen GX, Liu YQ, Gu XY, Liu WW. Invasion potential of H22 hepatocarcinoma cells is increased by HMGB1‐induced tumor NF‐kappaB signaling via initiation of HSP70. Oncol Rep 2013;30:1249‐1256. [DOI] [PubMed] [Google Scholar]
- 105. Bigorgne AE, Crispe IN TLRs in hepatic cellular crosstalk. Gastroenterol Res Pract 2010;2010.pii:618260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Knolle PA, Limmer A. Neighborhood politics: the immunoregulatory function of organ‐resident liver endothelial cells. Trends Immunol 2001;22:432‐437. [DOI] [PubMed] [Google Scholar]
- 107. Schwabe RF, Seki E, Brenner DA. Toll‐like receptor signaling in the liver. Gastroenterology 2006;130:1886‐1900. [DOI] [PubMed] [Google Scholar]
- 108. Seong SY, Matzinger P. Hydrophobicity: an ancient damage‐associated molecular pattern that initiates innate immune responses. Nat Rev Immunol 2004;4:469‐478. [DOI] [PubMed] [Google Scholar]
- 109. Palumbo R, Sampaolesi M, De Marchis F, Tonlorenzi R, Colombetti S, Mondino A, et al. Extracellular HMGB1, a signal of tissue damage, induces mesoangioblast migration and proliferation. J Cell Biol 2004;164:441‐449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Treutiger CJ, Mullins GE, Johansson AS, Rouhiainen A, Rauvala HM, Erlandsson‐Harris H, et al. High mobility group 1 B‐box mediates activation of human endothelium. J Intern Med 2003;254:375‐385. [DOI] [PubMed] [Google Scholar]
- 111. Chen XL, Sun L, Guo F, Wang F, Liu S, Liang X, et al. High‐mobility group box‐1 induces proinflammatory cytokines production of Kupffer cells through TLRs‐dependent signaling pathway after burn injury. PLoS One 2012;7:e50668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Zeng S, Dun H, Ippagunta N, Rosario R, Zhang QY, Lefkowitch J, et al. Receptor for advanced glycation end product (RAGE)‐dependent modulation of early growth response‐1 in hepatic ischemia/reperfusion injury. J Hepatol 2009;50:929‐936. [DOI] [PubMed] [Google Scholar]
- 113. Bamboat ZM, Balachandran VP, Ocuin LM, Obaid H, Plitas G, DeMatteo RP. Toll‐like receptor 9 inhibition confers protection from liver ischemia‐reperfusion injury. Hepatology 2010;51:621‐632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Li L, Chen L, Hu L, Liu Y, Sun HY, Tang J, et al. Nuclear factor high‐mobility group box1 mediating the activation of Toll‐like receptor 4 signaling in hepatocytes in the early stage of nonalcoholic fatty liver disease in mice. Hepatology 2011;54:1620‐1630. [DOI] [PubMed] [Google Scholar]
- 115. Ding N, Zhang Y, Loughran PA, Wang Q, Tsung A, Billiar TR. TIFA upregulation after hypoxia‐reoxygenation is TLR4‐ and MyD88‐dependent and associated with HMGB1 upregulation and release. Free Radic Biol Med 2013;63:361‐367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Lotze MT, Tracey KJ. High‐mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol 2005;5:331‐342. [DOI] [PubMed] [Google Scholar]
- 117. Andersson U, Wang H, Palmblad K, Aveberger AC, Bloom O, Erlandsson‐Harris H, et al. High mobility group 1 protein (HMG‐1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med 2000;192:565‐570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Antoine DJ, Williams DP, Kipar A, Laverty H, Park BK. Diet restriction inhibits apoptosis and HMGB1 oxidation and promotes inflammatory cell recruitment during acetaminophen hepatotoxicity. Mol Med 2010;16:479‐490. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 119. Liu A, Fang H, Dirsch O, Jin H, Dahmen U. Oxidation of HMGB1 causes attenuation of its pro‐inflammatory activity and occurs during liver ischemia and reperfusion. PLoS One 2012;7:e35379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Urbonaviciute V, Meister S, Furnrohr BG, Frey B, Guckel E, Schett G, et al. Oxidation of the alarmin high‐mobility group box 1 protein (HMGB1) during apoptosis. Autoimmunity 2009;42:305‐307. [DOI] [PubMed] [Google Scholar]
- 121. Zandarashvili L, Sahu D, Lee K, Lee YS, Singh P, Rajarathnam K, et al. Real‐time kinetics of high‐mobility group box 1 (HMGB1) oxidation in extracellular fluids studied by in situ protein NMR spectroscopy. J Biol Chem 2013;288:11621‐11627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Carneiro VC, de Moraes Maciel R, de Abreu da Silva IC, da Costa RF, Paiva CN, Bozza MT, et al. The extracellular release of Schistosoma mansoni HMGB1 nuclear protein is mediated by acetylation. Biochem Biophys Res Commun 2009;390:1245‐1249. [DOI] [PubMed] [Google Scholar]
- 123. Elenkov I, Pelovsky P, Ugrinova I, Takahashi M, Pasheva E. The DNA binding and bending activities of truncated tail‐less HMGB1 protein are differentially affected by Lys‐2 and Lys‐81 residues and their acetylation. Int J Biol Sci 2011;7:691‐699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Kim YM, Park EJ, Kim JH, Park SW, Kim HJ, Chang KC. Ethyl pyruvate inhibits the acetylation and release of HMGB1 via effects on SIRT1/STAT signaling in LPS‐activated RAW264.7 cells and peritoneal macrophages. Int Immunopharmacol 2016;41:98‐105. [DOI] [PubMed] [Google Scholar]
- 125. Pelovsky P, Pashev IG, Pasheva E. Interplay between in vitro acetylation and phosphorylation of tailless HMGB1 protein. Biochem Biophys Res Commun 2009;380:138‐142. [DOI] [PubMed] [Google Scholar]
- 126. Qi Z, Zhang Y, Qi S, Ling L, Gui L, Yan L, et al. Salidroside inhibits HMGB1 acetylation and release through upregulation of SirT1 during inflammation. Oxid Med Cell Longev 2017;2017:9821543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Topalova D, Ugrinova I, Pashev IG, Pasheva EA. HMGB1 protein inhibits DNA replication in vitro: a role of the acetylation and the acidic tail. Int J Biochem Cell Biol 2008;40:1536‐1542. [DOI] [PubMed] [Google Scholar]
- 128. Ugrinova I, Mitkova E, Moskalenko C, Pashev I, Pasheva E. DNA bending versus DNA end joining activity of HMGB1 protein is modulated in vitro by acetylation. Biochemistry 2007;46:2111‐2117. [DOI] [PubMed] [Google Scholar]
- 129. Ugrinova I, Pashev IG, Pasheva EA. Post‐synthetic acetylation of HMGB1 protein modulates its interactions with supercoiled DNA. Mol Biol Rep 2009;36:1399‐1404. [DOI] [PubMed] [Google Scholar]
- 130. Yang Z, Li L, Chen L, Yuan W, Dong L, Zhang Y, et al. PARP‐1 mediates LPS‐induced HMGB1 release by macrophages through regulation of HMGB1 acetylation. J Immunol 2014;193:6114‐6123. [DOI] [PubMed] [Google Scholar]
- 131. Kim SW, Jin Y, Shin JH, Kim ID, Lee HK, Park S, et al. Glycyrrhizic acid affords robust neuroprotection in the postischemic brain via anti‐inflammatory effect by inhibiting HMGB1 phosphorylation and secretion. Neurobiol Dis 2012;46:147‐156. [DOI] [PubMed] [Google Scholar]
- 132. Shin JH, Kim ID, Kim SW, Lee HK, Jin Y, Park JH, et al. Ethyl pyruvate inhibits HMGB1 phosphorylation and release by chelating calcium. Mol Med 2015;20:649‐657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Shin JH, Lee HK, Lee HB, Jin Y, Lee JK. Ethyl pyruvate inhibits HMGB1 phosphorylation and secretion in activated microglia and in the postischemic brain. Neurosci Lett 2014;558:159‐163. [DOI] [PubMed] [Google Scholar]
- 134. Youn JH, Shin JS. Nucleocytoplasmic shuttling of HMGB1 is regulated by phosphorylation that redirects it toward secretion. J Immunol 2006;177:7889‐7897. [DOI] [PubMed] [Google Scholar]
- 135. Ito I, Fukazawa J, Yoshida M. Post‐translational methylation of high mobility group box 1 (HMGB1) causes its cytoplasmic localization in neutrophils. J Biol Chem 2007;282:16336‐16344. [DOI] [PubMed] [Google Scholar]
- 136. Yang Y, Huang JQ, Zhang X, Shen LF. MiR‐129‐2 functions as a tumor suppressor in glioma cells by targeting HMGB1 and is down‐regulated by DNA methylation. Mol Cell Biochem 2015;404:229‐239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Yang M, Liu L, Xie M, Sun X, Yu Y, Kang R, et al. Poly‐ADP‐ribosylation of HMGB1 regulates TNFSF10/TRAIL resistance through autophagy. Autophagy 2015;11:214‐224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Kim YH, Kwak MS, Park JB, Lee SA, Choi JE, Cho HS, et al. N‐linked glycosylation plays a crucial role in the secretion of HMGB1. J Cell Sci 2016;129:29‐38. [DOI] [PubMed] [Google Scholar]
- 139. Cokol M, Nair R, Rost B. Finding nuclear localization signals. EMBO Rep 2000;1:411‐415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Wong LC, Sharpe DJ, Wong SS. High‐mobility group and other nonhistone substrates for nuclear histone N‐acetyltransferase. Biochem Genet 1991;29:461‐475. [DOI] [PubMed] [Google Scholar]
- 141. Shibayama Y, Asaka S, Nishijima A. Mechanism of liver injury following ischemia. Exp Mol Pathol 1991;55:251‐260. [DOI] [PubMed] [Google Scholar]
- 142. Nace GW, Huang H, Klune JR, Eid RE, Rosborough BR, Korff S, et al. Cellular‐specific role of toll‐like receptor 4 in hepatic ischemia‐reperfusion injury in mice. Hepatology 2013;58:374‐387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Huang H, Nace GW, McDonald K‐A, Tai S, Klune JR, Rosborough BR, et al. Hepatocyte specific high‐mobility group box 1 deletion worsens the injury in liver ischemia/reperfusion: a role for intracellular high‐mobility group box 1 in cellular protection. Hepatology 2014;59:1984‐1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Izuishi K, Tsung A, Jeyabalan G, Critchlow ND, Li J, Tracey KJ, et al. Cutting edge: high‐mobility group box 1 preconditioning protects against liver ischemia‐reperfusion injury. J Immunol 2006;176:7154‐7158. [DOI] [PubMed] [Google Scholar]
- 145. Cardinal J, Pan P, Dhupar R, Ross M, Nakao A, Lotze M, et al. Cisplatin prevents high mobility group box 1 release and is protective in a murine model of hepatic ischemia/reperfusion injury. Hepatology 2009;50:565‐574. [DOI] [PubMed] [Google Scholar]
- 146. Ogiku M, Kono H, Hara M, Tsuchiya M, Fujii H. Glycyrrhizin prevents liver injury by inhibition of high‐mobility group box 1 production by Kupffer cells after ischemia‐reperfusion in rats. J Pharmacol Exp Ther 2011;339:93‐98. [DOI] [PubMed] [Google Scholar]
- 147. Li F, Chen Z, Pan Q, Fu S, Lin F, Ren H, et al. The protective effect of PNU‐282987, a selective alpha7 nicotinic acetylcholine receptor agonist, on the hepatic ischemia‐reperfusion injury is associated with the inhibition of high‐mobility group box 1 protein expression and nuclear factor kappaB activation in mice. Shock 2013;39:197‐203. [DOI] [PubMed] [Google Scholar]
- 148. Liu A, Fang H, Yang Y, Sun J, Fan H, Liu S, et al. The fibrin‐derived peptide bbeta15‐42 attenuates liver damage in a rat model of liver ischemia/reperfusion injury. Shock 2013;39:397‐403. [DOI] [PubMed] [Google Scholar]
- 149. Oishi K, Hagiwara S, Koga S, Kawabe S, Uno T, Iwasaka H, et al. The vitamin E derivative, EPC‐K1, suppresses inflammation during hepatic ischemia‐reperfusion injury and exerts hepatoprotective effects in rats. J Surg Res 2012;176:164‐170. [DOI] [PubMed] [Google Scholar]
- 150. Kang JW, Koh EJ, Lee SM. Melatonin protects liver against ischemia and reperfusion injury through inhibition of toll‐like receptor signaling pathway. J Pineal Res 2011;50:403‐411. [DOI] [PubMed] [Google Scholar]
- 151. Antoine DJ, Williams DP, Kipar A, Jenkins RE, Regan SL, Sathish JG, et al. High‐mobility group box‐1 protein and keratin‐18, circulating serum proteins informative of acetaminophen‐induced necrosis and apoptosis in vivo. Toxicol Sci 2009;112:521‐531. [DOI] [PubMed] [Google Scholar]
- 152. Antoine DJ, Jenkins RE, Dear JW, Williams DP, McGill MR, Sharpe MR, et al. Molecular forms of HMGB1 and keratin‐18 as mechanistic biomarkers for mode of cell death and prognosis during clinical acetaminophen hepatotoxicity. J Hepatol 2012;56:1070‐1079. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 153. Lundback P, Lea JD, Sowinska A, Ottosson L, Furst CM, Steen J, et al. A novel high mobility group box 1 neutralizing chimeric antibody attenuates drug‐induced liver injury and postinjury inflammation in mice. Hepatology 2016;64:1699‐1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Wang X, Sun R, Wei H, Tian Z. High‐mobility group box 1 (HMGB1)‐Toll‐like receptor (TLR)4‐interleukin (IL)‐23‐IL‐17A axis in drug‐induced damage‐associated lethal hepatitis: interaction of gammadelta T cells with macrophages. Hepatology 2013;57:373‐384. [DOI] [PubMed] [Google Scholar]
- 155. Takai S, Higuchi S, Yano A, Tsuneyama K, Fukami T, Nakajima M, et al. Involvement of immune‐ and inflammatory‐related factors in flucloxacillin‐induced liver injury in mice. J Appl Toxicol 2015;35:142‐151. [DOI] [PubMed] [Google Scholar]
- 156. Takano K, Shinoda M, Tanabe M, Miyasho T, Yamada S, Ono S, et al. Protective effect of high‐mobility group box 1 blockade on acute liver failure in rats. Shock 2010;34:573‐579. [DOI] [PubMed] [Google Scholar]
- 157. Akai S, Uematsu Y, Tsuneyama K, Oda S, Yokoi T. Kupffer cell‐mediated exacerbation of methimazole‐induced acute liver injury in rats. J Appl Toxicol 2016;36:702‐715. [DOI] [PubMed] [Google Scholar]
- 158. Ramm S, Mally A. Role of drug‐independent stress factors in liver injury associated with diclofenac intake. Toxicology 2013;312:83‐96. Erratum. In: Toxicology 2014;316:71‐74. [DOI] [PubMed] [Google Scholar]
- 159. Ogese MO, Faulkner L, Jenkins RE, French NS, Copple IM, Antoine DJ, et al. Characterization of drug‐specific signaling between primary human hepatocytes and immune cells. Toxicol Sci 2017;158:76‐89. [DOI] [PubMed] [Google Scholar]
- 160. Kao YH, Jawan B, Goto S, Hung CT, Lin YC, Nakano T, et al. High‐mobility group box 1 protein activates hepatic stellate cells in vitro. Transplant Proc 2008;40:2704‐2705. [DOI] [PubMed] [Google Scholar]
- 161. Ge WS, Wu JX, Fan JG, Wang YJ, Chen YW. Inhibition of high‐mobility group box 1 expression by siRNA in rat hepatic stellate cells. World J Gastroenterol 2011;17:4090‐4098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Shaker M, Tabbaa A, Albeldawi M, Alkhouri N. Liver transplantation for nonalcoholic fatty liver disease: new challenges and new opportunities. World J Gastroenterol 2014;20:5320‐5330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Chandrashekaran V, Seth RK, Dattaroy D, Alhasson F, Ziolenka J, Carson J, et al. HMGB1‐RAGE pathway drives peroxynitrite signaling‐induced IBD‐like inflammation in murine nonalcoholic fatty liver disease. Redox Biol 2017;13:8‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Cheng BQ, Jia CQ, Liu CT, Lu XF, Zhong N, Zhang ZL, et al. Serum high mobility group box chromosomal protein 1 is associated with clinicopathologic features in patients with hepatocellular carcinoma. Dig Liver Dis 2008;40:446‐452. [DOI] [PubMed] [Google Scholar]
- 165. Dong YD, Cui L, Peng CH, Cheng DF, Han BS, Huang F. Expression and clinical significance of HMGB1 in human liver cancer: knockdown inhibits tumor growth and metastasis in vitro and in vivo. Oncol Rep 2013;29:87‐94. [DOI] [PubMed] [Google Scholar]
- 166. Yaser AM, Huang Y, Zhou RR, Hu GS, Xiao MF, Huang ZB, et al. The role of receptor for advanced glycation end products (RAGE) in the proliferation of hepatocellular carcinoma. Int J Mol Sci 2012;13:5982‐5997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Yan W, Chang Y, Liang X, Cardinal JS, Huang H, Thorne SH, et al. High‐mobility group box 1 activates caspase‐1 and promotes hepatocellular carcinoma invasiveness and metastases. Hepatology 2012;55:1863‐1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Ulloa L, Tracey KJ. The "cytokine profile": a code for sepsis. Trends Mol Med 2005;11:56‐63. [DOI] [PubMed] [Google Scholar]
- 169. Ulloa L, Batliwalla FM, Andersson U, Gregersen PK, Tracey KJ. High mobility group box chromosomal protein 1 as a nuclear protein, cytokine, and potential therapeutic target in arthritis. Arthritis Rheum 2003;48:876‐881. [DOI] [PubMed] [Google Scholar]