

Abstract
We report on the presence of a rare nonsense mutation (rs149847328, p.Arg227Ter) in the glucokinase regulator (GCKR) gene in an adult patient with nonalcoholic fatty liver disease (NAFLD), morbid obesity, and type 2 diabetes; this patient developed a progressive histological form of the disease. Analysis of paired (5 years apart) liver biopsies (at baseline and follow‐up) showed progression of simple steatosis to severe nonalcoholic steatohepatitis and cirrhosis. Study design involved an initial exploration that consisted of deep sequencing of 14 chromosomal regions in 96 individuals (64 of whom were patients with NAFLD who were diagnosed by liver biopsy that showed the full spectrum of histological severity). We further performed a replication study to explore the presence of rs149847328 that included a sample of 517 unrelated individuals in a case‐control study (n = 390), including patients who were morbidly obese (n = 127). Exploration of sequence variation by next‐generation sequencing of exons, exon–intron boundaries, and 5′ and 3′ untranslated regions of 14 genomic loci that encode metabolic enzymes of the tricarboxylic acid cycle revealed the presence of heterozygosity for the p.Arg227Ter mutation, the frequency of which is 0.0003963 (4:10,000; Exome Aggregation Consortium database). GCKR protein expression was markedly decreased in the liver of the affected patient compared with patients with NAFLD who carry the wild‐type allele. Sequencing of the same 14 genomic loci in 95 individuals failed to reveal the rare mutation. The rarity of p.Arg227Ter was confirmed in a more extensive screening. Conclusion: While rare variants/mutations are difficult to detect in even reasonably large samples (frequency of the mutant allele of p.Arg227Ter was ~1:1,000 in our data set), the presence of this mutation should be suspected as potentially associated with NAFLD, particularly in young adults at the extreme of histological phenotypes. Hepatology Communications 2018;0:0‐0)
Abbreviations
- GCK
glucokinase
- GCKR
glucokinase regulator
- IDH
isocitrate dehydrogenase
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- NGS
next‐generation sequencing
- PNPLA3
patatin‐like phospholipase domain containing 3
- TCA
tricarboxylic acid
- TM6SF2
transmembrane 6 superfamily member 2
Nonalcoholic fatty liver disease (NAFLD) is a complex polygenic and multifactorial disease, the risk and course of which are determined by the interactions of a large, although not yet identified, collection of variants and environmental factors. Current knowledge indicates that genetic predisposition to NAFLD is partially explained by common (frequent) variants of genes involved in the regulation of metabolic pathways, including patatin‐like phospholipase domain containing 3 (PNPLA3), transmembrane 6 superfamily member 2 (TM6SF2), and glucokinase regulator (GCKR). Variants in these loci (rs738409,1 rs58542926,2 and rs7800943, 4 explain a modest fraction of the genetic variance in the manifestation of this disease. For example, summarized evidence suggests that the rs738409 variant is associated with a ~3.5‐fold risk of NAFLD and nonalcoholic steatohepatitis (NASH)1; carrying the T allele of rs58542926 imposes a ~2.1‐fold chance of having NAFLD2; and the rs780094 variant of GCKR is associated with a ~1.2‐fold higher risk of NAFLD.4 Variants in additional loci, including a missense (p.Gly17Glu, rs641738 C/T) variant in exon 1 of transmembrane channel‐like 4 (TMC4)/intergenic downstream of membrane bound O‐acyltransferase domain containing 7 (MBOAT7) has been associated with a modest risk of developing NAFLD and NASH.5 However, this association could not be replicated in other populations around the world.6, 7, 8, 9, 10 Finally, recent findings have shown that a splice variant (rs72613567:TA) in hydroxysteroid 17‐beta dehydrogenase 13 (HSD17B13) is associated with a reduced risk of NASH.11
There is also evidence on the putative role of rare variants in the susceptibility of NAFLD; for instance, a large study in a Japanese population suggests that ~1.79% of individuals carry rare variants potentially associated with NAFLD.12 Whole‐exome sequencing study on 6 Caucasian patients with extreme obesity (mean body mass index, 84) found two novel damaging mutations in Bardet‐Biedl syndrome 1 (BBS1) and a rare damaging mutation in PNPLA3.13
Whether rare variants are involved in the accelerated disease progression is not entirely known because of the lack of longitudinal studies that include patients with paired liver biopsies; however, significant effects and high penetrance would be anticipated if they were present. It is also uncertain whether the risk of NASH (the severe clinical form of NAFLD) or NASH‐cirrhosis could be associated with inherited mutations. While the latter appears highly unlikely, it would support the notion of NASH as a Mendelian disorder as well.
Patients and Methods
Study Design and Patient Selection
Patients were included in the study if there was histopathologic evidence of NAFLD, either nonalcoholic fatty liver or NASH, based on a liver biopsy conducted within the study period. Exclusion criteria were secondary causes of steatosis, including alcohol abuse (≥30 g alcohol daily for men and ≥20 g for women), total parenteral nutrition, hepatitis B and hepatitis C virus infection, and the use of drugs known to precipitate steatosis. By using standard clinical and laboratory evaluation as well as liver biopsy features when applicable, autoimmune liver disease, metabolic liver disease, Wilson’s disease, and ‐1‐antitrypsin deficiency were likewise ruled out in all patients.
Healthy subjects were selected for inclusion into the control group if their age and sex matched those of the patients with NAFLD and, in addition to the standard health assessment, a careful ultrasonographic examination of the liver excluded fatty liver infiltration. The case and control participants were selected during the same study period from the same population of patients attending the Liver Unit, and all shared the same demographic characteristics.
The study involved an initial stage of exploration that consisted of deep sequencing of 14 chromosomal regions (Supporting Fig. S1) in 96 individuals, 64 of whom were patients with NAFLD diagnosed by liver biopsy and 32 were controls (Supporting Table S1). In addition, we performed a replication study to explore the presence of a rare variant in GCKR (rs149847328, p.Arg227Ter) that included a sample of 517 unrelated individuals. Patients and controls were selected from two different hospital‐based settings, including a cross‐sectional study of patients with NAFLD and metabolic syndrome (sample size n = 390) (Supporting Table S2) recruited in the Liver Unit, Hospital Abel Zubizarreta, Buenos Aires, Argentina, and an independent cohort of patients with morbid obesity who underwent bariatric surgery (sample size n = 127) (Supporting Table S3) in the Surgery Department, Hospital de Alta Complejidad en Red El Cruce, Florencio Varela, Argentina. In the population of patients with morbid obesity, control subjects were patients with obesity who also underwent bariatric surgery and had no features of NAFLD demonstrated in the liver biopsy.
All investigations performed followed the guidelines of the 1975 Declaration of Helsinki. Written consent from individuals was obtained by the procedures approved by the ethical committee of our institution (protocol numbers 104/HGAZ/09, 89/100, and 1204/2012).
Genetic Analysis
Genetic analyses were done on genomic DNA extracted from white blood cells.14 Next‐generation sequencing (NGS) was performed using the Ion Torrent Personal Genome Machine (Life Technologies, Carlsbad, CA) with 316 chips and barcoding for >60 times coverage.15 Library preparation for each sample was performed using the IT AmpliSeq 2.0 Beta kit following the manufacturer's instructions (Life Technologies). The data obtained from the Ion Torrent Personal Genome Machine were processed using the Ion Torrent Suite Software v. 4.2.1 (Life Technologies). Variants were annotated with the single‐nucleotide polymorphism database (https://www.ncbi.nlm.nih.gov/SNP/) identifications using SnpSift. Confirmation of the p.Arg227Ter mutation was performed using a TaqMan genotyping assay (C__33208488_10; Applied Biosystems, Foster City, CA).
Complete details of physical, anthropometric, and biochemical evaluation as well as NGS experiments, including variant calling, estimation of quality control, data analysis, and prediction of variant/mutation effect, are shown in the Supporting Information.
Results
Case Course and Series Description
We report on a 53‐year‐old male patient with NAFLD, arterial hypertension, morbid obesity, and type 2 diabetes and a family history of obesity and type 2 diabetes in first‐degree relatives (both parents, two siblings, and two daughters) who developed a progressive histological form of the disease (NASH‐cirrhosis). Since the age of 48 (at the time of the first clinical consultation), the patient gained weight and presented persistently elevated aminotransferase levels (Fig. 1A). Secondary causes of steatosis, including alcohol abuse, viral hepatitis, and drug‐induced liver injury, were excluded; autoimmune liver disease, metabolic liver disease, Wilson’s disease, and ‐1‐antitrypsin deficiency were also ruled out. Analysis of paired liver biopsies (performed at baseline and follow‐up, 5 years apart) showed liver fibrosis progression from stage F0 to cirrhosis (Fig. 1B,C). The patient was found to be heterozygous CG for PNPLA3 (738409 variant) but homozygous CC for TM6SF2 (rs58542926 variant), the latter being a protective condition. In addition, the patient was heterozygous for the common variant located in GCKR rs1260326 (c.1337C>T; p.P446L), which has been reproducibly associated with metabolic traits, including NAFLD.16
Figure 1.
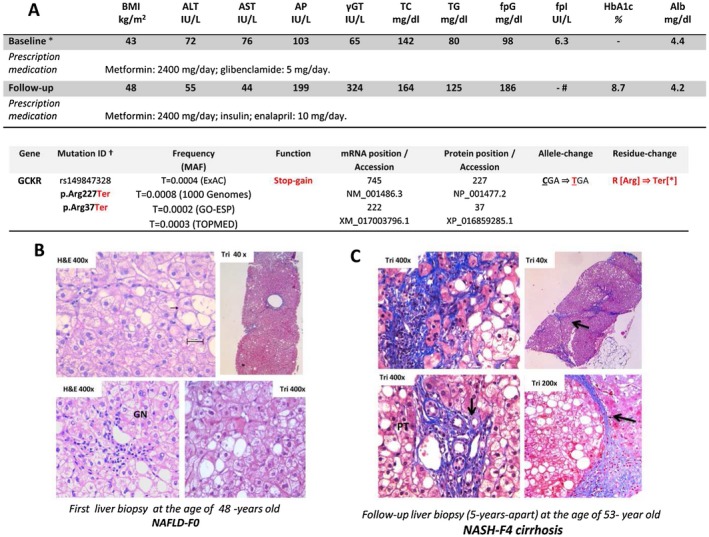
Clinical and histological evaluation of the patient carrying a rare mutation in the GCKR gene. Features (progressive NASH and difficult to manage glycemic control) show severity and extent of organ involvement. (A) Patient’s laboratory and clinical features. *First diagnosis of NAFLD and follow‐up were 5 years apart. Serum ALT and AST (normal levels, <37 and <34 IU/L, respectively), GT (normal, <64 IU/L), AP (normal, <98 IU/L); #means that the patient was under insulin treatment. Results are expressed as means. Further laboratory features were unremarkable, including hepatitis B and C, ceruloplasmin, alpha‐1‐antitrypsin, and iron studies and antinuclear, smooth muscle, and anti‐mitochondrial antibodies. Liver ultrasound showed normal liver parenchyma and biliary tree. Prescription medications that the patient received during the time interval of paired‐biopsy evaluation were dietary, oral hypoglycemic medication, or insulin treatment for diabetes. Accession numbers refer to the National Center for Biotechnology Information Reference Sequence identification. Mutation ID + include p.Arg227Ter (forward strand of the reference sequence genome assembly) and p.Arg37Ter (forward strand of the curated reference sequence records, which are created by a process that includes automated computational methods). (B,C) Histological examination of paired liver biopsies at (B) baseline and (C) follow‐up; the second follow‐up biopsy specimen was fragmented (five fragments were considered for histological assessment). Liver parenchyma shows steatosis, ballooned‐enlarged hepatocytes, lobular inflammation, and glycogenated nuclei. Paired histological evaluation shows the progression of fibrosis score from F0 to F4; histological features were assessed according to the system developed by Kleiner et al.27 (C) Illustrated are nodular formation (arrow), fibrous septa‐entrapping hepatocytes, portal tracts with inflammatory infiltrate, microcirculatory remodeling (arrows indicate microvessels), deposition of collagen in perisinusoidal space of Disse, and pericellular fibrosis (Masson’s trichrome); nuclear features of hepatocyte regeneration (anisonucleosis/nuclear duplication) are also present. Histological specimens were assessed by a LEICA DM 2000 (Leica, Germany) trinocular microscope equipped with a high‐definition camera (Leica MC190 HD); all images were recorded using the Leica Application Suite software. Abbreviations: γGT, gamma‐glutamyltransferase; alb, albumin; ALT, alanine aminotransferase; AP, alkaline phosphatase; AST, aspartate aminotransferase; BMI, body mass index; ExAC, Exome Aggregation Consortium; fpG, fasting plasma glucose; fpI, fasting plasma insulin; GN, glycogenated nuclei; GO‐ESP, National Heart, Lung, and Blood Institute GO Exome Sequencing Project; H&E, hematoxylin and eosin stain; HbA1c, hemoglobin A1c; MAF, minor allele frequency; mRNA, messenger RNA; NA, not applicable; PT, portal tract; TC, total cholesterol; Ter, premature stop codon; TG, triglycerides; TOPMED, Trans‐Omics for Precision Medicine; TRI, Masson’s trichrome stain.
We thus decided to search for rare variant(s) or mutation(s) that explain the severe clinical histological phenotype; we reasoned that the observed metabolic derangement could be driven by genetic diversity in metabolic pathways. Hence, we proceeded with the exploration of sequence variation by NGS of exons, exon–intron boundaries, and 5′ and 3′ untranslated regions of 14 genomic loci that encode enzymes involved in the regulation of metabolic processes, including the tricarboxylic acid (TCA) cycle, cellular respiration, and components of the mitochondrial matrix that control glutamine and glutamate metabolism. The last group included glutamate dehydrogenase 1 and 2 (GLUD1 and GLUD2), glutamate‐ammonia ligase (GLUL), glutaminase‐2 (GLS2), and glutaminase (GLS). The list of explored loci also included isocitrate dehydrogenases (IDH1, IDH2, IDH3A, and IDH3B) that have a central role in intermediary metabolism and energy production, as well as GCKR. Other TCA‐cycle genes that we included in this study were aconitase‐2 (ACO2), involved in the second step of the TCA cycle, 2‐oxoglutarate‐dehydrogenase complex (DLST), and malate and oxoglutarate dehydrogenases (MDH1 and OGDH).
Our findings revealed heterozygosity for a rare nonsense (rs149847328 C/T, p.Arg227Ter) mutation in the GCKR gene, the frequency of which is 0.0003963 in the Exome Aggregation Consortium database (4:10,000; 48 of 121,126 alleles). The GCKR mutation was further confirmed using a TaqMan genotyping assay (C__33208488_10; Applied Biosystems).
The substitution of a C with a T converts the arginine codon to a premature stop codon (Fig. 1A), resulting in the protein transcribed from the mutant messenger RNA being incomplete (Fig. 2A,B). Previous in vitro mutagenesis experiments confirmed that p.Arg227Ter truncates the protein length by about two thirds.17
Figure 2.
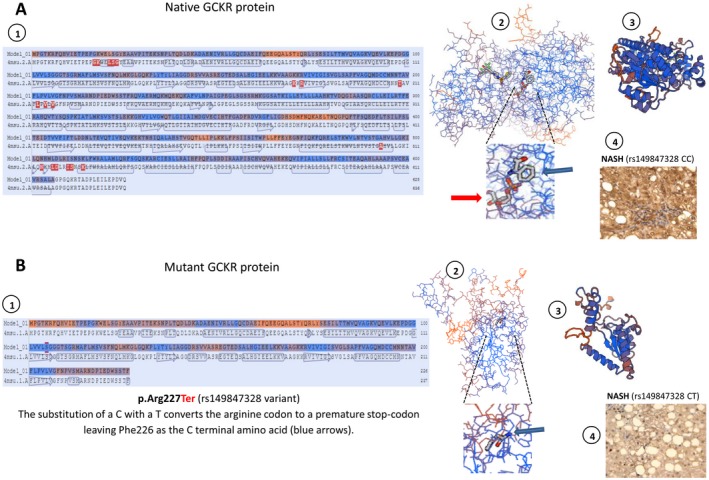
Protein structure modeling of the p.Arg227Ter mutation in the GCKR gene. Prediction was built using the SWISS‐MODEL re(https://swissmodel.expasy.org/). (A) Representation of native GCKR protein and (B) mutant protein (stop gained) showing structural changes as the result of a premature stop codon in residue 227. A1 and B1 show amino acid sequence alignment of native and mutant protein, respectively. A2 and B2 show the backbone protein structure; insets highlight the effect of the p.Arg227Ter mutation that is responsible for a loss in the binding of a sugar (D‐sorbitol‐6‐phosphate 9, red arrows); its binding sites involve several amino acids downstream of Phe226 that are then lost in the mutated protein (B2). A3 and B3 show a three‐dimensional ribbon diagram of wild‐type and mutant protein conformational structures, respectively; p.Arg227Ter truncates the protein length by about two thirds. A4 and B4 show representative protein expression patterns of GCKR in the liver of a patient with NASH who carries two copies of the wild‐type (rs149847328 CC) and one copy of the mutant (rs149847328 CT) allele, respectively. A4 shows positive staining in more than 50% of cells; B4 shows positive staining in less than 20% of cells. Liver protein expression of GCKR was evaluated using immunohistochemistry; immunostaining for GCKR was performed on a subsample of six liver specimens previously included in paraffin (four samples of patients with NASH and similar phenotypic and clinical features of the affected patient and two liver specimens of the affected patient, including baseline and follow‐up liver biopsies). Immunoreactivity was examined using light microscopy of liver sections; original magnification ×400.
As expected, GCKR protein expression was markedly decreased in the liver of the affected patient compared with patients with NAFLD carrying the wild‐type allele (Fig. 2A,B).
In addition, we deep sequenced the same chromosomal regions in 95 individuals, of whom 63 were patients with NAFLD diagnosed by liver biopsy and 32 were controls, and we failed to reveal the rare mutation. A more extensive screening by a TaqMan assay was carried out in a cross‐sectional case‐control study involving a sample of 517 individuals, including patients who were morbidly obese. The results confirmed the rarity of p.Arg227Ter because neither the controls nor the patients were found to carry the mutation.
Discussion
Common variants in the GCKR gene are associated with alterations in metabolic traits, including NAFLD16, 18 and plasma triglyceride levels.19 The gene product is a regulatory protein that inhibits glucokinase (GCK, hexokinase 4) in the liver and pancreatic islet cells by binding noncovalently to form an inactive complex with the enzyme. The affinity of GCKR for GCK is modulated by fructose metabolites; GCKR with bound fructose 6‐phosphate has increased affinity for GCK, while GCKR with bound fructose 1‐phosphate has strongly decreased affinity for GCK and does not inhibit GCK activity.20 The GCKR‐p.Arg227Ter variant abolishes two fructose 1‐phosphate sites (positions 348 and 514); likewise, mutant protein lacks domains that are essential for interaction with GCK (Fig. 2B).
GCKR is considered a susceptibility gene for a form of maturity‐onset diabetes of the young (https://www.omim.org/entry/600842) and familial clustering of type 2 diabetes.21, 22 In addition, cosegregation analysis of obesity and the GCKR‐p.Arg227Ter mutation has been elegantly assessed by Veiga‐da‐Cunha and coworkers,17 who initially described the presence of the stop codon in five of six members of an obese family. While it was suggested that p.Arg227Ter does not necessarily predispose to obesity,17 it was shown that GCK activity is decreased by about 50% in subjects with obesity and diabetes who carry the mutation.23
The patient we present here not only carries the p.Arg227Ter mutation but also developed a complex phenotype clustering morbid obesity, type 2 diabetes, and NASH, the later condition associated with impaired hepatic glucose metabolic machinery rendering the organ prone to rapidly progressive severe damage.
To our knowledge, this is the first report on the role of a rare GCKR mutation in the susceptibility of the NASH phenotype; heterozygosity for the GCKR mutation may explain onset of the disease in adulthood. In addition, due to the critical role of GCKR in modulating hepatic glucose and lipid metabolism, it is expected that mutations in this gene may have a significant impact in the phenotype even in the heterozygote state.24 This observation is supported by phenotypic characterization of GCKR‐knockout mice that showed that both heterozygote (+/–) and homozygote (–/–) mice present a substantial decrease in hepatic GCK expression and enzymatic activity when compared with wild‐type mice and fail in a glucose tolerance test to sustain appropriate glucose clearance.23, 25
Some limitations of our study should be highlighted; specifically, the patient’s phenotype cannot be fully ascribed to the presence of the p.Arg227Ter mutation because other variants/mutations not tested in this study might be involved in the course of the disease. Furthermore, we cannot rule out that other nongenetic factors might have modulated the accelerated course of the disease, including poor glucose control or the presence of morbid obesity. It is known that adiposity increases the genetic risk of NAFLD at multiple loci that predispose to the disease.26 Finally, it would have been particularly informative having information on the p.Arg227Ter mutation in family members of the proband; unfortunately, family members opted not to take part in the genetic testing. Therefore, phasing with other variants was not possible.
In conclusion, while rare variants/mutations are difficult to detect in even reasonably large samples, the presence of this mutation should be suspected as potentially associated with NAFLD, particularly in young adults at the extreme of histological phenotypes.
Supporting information
Author names in bold designate shared co‐first authorship.
Funding
Supported in part by the National Agency for Scientific and Technological Promotion (ANPCyT) (grants PID‐C2012‐0061 to CJP, PICT 2014‐0432 to SS, PICT 2014‐1816 to CJP, and PICT 2015‐0551 to SS) and by the The National Scientific and Technical Research Council (S.S. and C.J.P.)
Potential conflict of interest: Nothing to report.
Contributor Information
Carlos J. Pirola, Email: pirola.carlos@lanari.fmed.uba.ar.
Silvia Sookoian, Email: sookoian.silvia@lanari.fmed.uba.ar.
References
- 1. Sookoian S, Pirola CJ. Meta‐analysis of the influence of I148M variant of patatin‐like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology 2011;53:1883‐1894. [DOI] [PubMed] [Google Scholar]
- 2. Pirola CJ, Sookoian S. The dual and opposite role of the TM6SF2‐rs58542926 variant in protecting against cardiovascular disease and conferring risk for nonalcoholic fatty liver: a meta‐analysis. Hepatology 2015;62:1742‐1756. [DOI] [PubMed] [Google Scholar]
- 3. Palmer ND, Musani SK, Yerges‐Armstrong LM, Feitosa MF, Bielak LF, Hernaez R, et al. Characterization of European ancestry nonalcoholic fatty liver disease‐associated variants in individuals of African and Hispanic descent. Hepatology 2013;58:966‐975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zain SM, Mohamed Z, Mohamed R. Common variant in the glucokinase regulatory gene rs780094 and risk of nonalcoholic fatty liver disease: a meta‐analysis. J Gastroenterol Hepatol 2015;30:21‐27. [DOI] [PubMed] [Google Scholar]
- 5. Mancina RM, Dongiovanni P, Petta S, Pingitore P, Meroni M, Rametta R, et al. The MBOAT7‐TMC4 variant rs641738 increases risk of nonalcoholic fatty liver disease in individuals of European descent. Gastroenterology 2016;150:1219‐1230.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sookoian S, Flichman D, Garaycoechea ME, Gazzi C, Martino JS, Castano GO, et al. Lack of evidence supporting a role of TMC4‐rs641738 missense variant‐MBOAT7‐ intergenic downstream variant‐in the susceptibility to nonalcoholic fatty liver disease. Sci Rep 2018;8:5097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kawaguchi T, Shima T, Mizuno M, Mitsumoto Y, Umemura A, Kanbara Y, et al. Risk estimation model for nonalcoholic fatty liver disease in the Japanese using multiple genetic markers. PLoS One 2018;13:e0185490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Koo BK, Joo SK, Kim D, Bae JM, Park JH, Kim JH, et al. Additive effects of PNPLA3 and TM6SF2 on the histological severity of non‐alcoholic fatty liver disease. J Gastroenterol Hepatol 2018;33:1277‐1285. [DOI] [PubMed] [Google Scholar]
- 9. Lin YC, Chang PF, Chang MH, Ni YH. Genetic determinants of hepatic steatosis and serum cytokeratin‐18 fragment levels in Taiwanese children. LiverInt 2018;38:1300‐1307. [DOI] [PubMed] [Google Scholar]
- 10. Krawczyk M, Rau M, Schattenberg JM, Bantel H, Pathil A, Demir M, et al. NAFLD Clinical Study Group. Combined effects of the PNPLA3 rs738409, TM6SF2 rs58542926, and MBOAT7 rs641738 variants on NAFLD severity: a multicenter biopsy‐based study. J Lipid Res 2017;58:247‐255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Abul‐Husn NS, Cheng X, Li AH, Xin Y, Schurmann C, Stevis P, et al. A protein‐truncating HSD17B13 variant and protection from chronic liver disease. N Engl J Med 2018. Mar 22;378:1096‐1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boonvisut S, Yoshida K, Nakayama K, Watanabe K, Miyashita H, Iwamoto S. Identification of deleterious rare variants in MTTP, PNPLA3, and TM6SF2 in Japanese males and association studies with NAFLD. Lipids Health Dis 2017;16:183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gerhard GS, Chu X, Wood GC, Gerhard GM, Benotti P, Petrick AT, et al. Next‐generation sequence analysis of genes associated with obesity and nonalcoholic fatty liver disease‐related cirrhosis in extreme obesity. Hum Hered 2013;75:144‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sookoian S, Castano GO, Burgueno AL, Gianotti TF, Rosselli MS, Pirola CJ. A nonsynonymous gene variant in the adiponutrin gene is associated with nonalcoholic fatty liver disease severity. J Lipid Res 2009;50:2111‐2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sookoian S, Castano GO, Scian R, Fernández Gianotti T, Dopazo H, Rohr C, et al. Serum aminotransferases in nonalcoholic fatty liver disease are a signature of liver metabolic perturbations at the amino acid and Krebs cycle level. Am J Clin Nutr 2016;103:422‐434. [DOI] [PubMed] [Google Scholar]
- 16. Speliotes EK, Yerges‐Armstrong LM, Wu J, Hernaez R, Kim LJ, Palmer CD, et al. NASH CRN; GIANT Consortium; MAGIC Investigators; GOLD Consortium. Genome‐wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet 2011;7:e1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Veiga‐da‐Cunha M, Delplanque J, Gillain A, Bonthron DT, Boutin P, Van Schaftingen E, et al. Mutations in the glucokinase regulatory protein gene in 2p23 in obese French caucasians. Diabetologia 2003;46:704‐711. [DOI] [PubMed] [Google Scholar]
- 18. Santoro N, Zhang CK, Zhao H, Pakstis AJ, Kim G, Kursawe R, et al. Variant in the glucokinase regulatory protein (GCKR) gene is associated with fatty liver in obese children and adolescents. Hepatology 2012;55:781‐789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Diabetes Genetics Initiative of Broad Institute of Harvard and MIT , Lund University , and Novartis Institutes of BioMedical Research , Saxena R, Voight BF, Lyssenko V, Burtt NP, deBakker PI, Chen H, et al. Genome‐wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 2007;316:1331‐1336. [DOI] [PubMed] [Google Scholar]
- 20. Choi JM, Seo MH, Kyeong HH, Kim E, Kim HS. Molecular basis for the role of glucokinase regulatory protein as the allosteric switch for glucokinase. Proc Natl Acad Sci U S A 2013;110:10171‐10176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tanaka D, Nagashima K, Sasaki M, Yamada C, Funakoshi S, Akitomo K, et al. GCKR mutations in Japanese families with clustered type 2 diabetes. Mol Genet Metab 2011;102:453‐460. [DOI] [PubMed] [Google Scholar]
- 22. Iwata M, Maeda S, Kamura Y, Takano A, Kato H, Murakami S, et al. Genetic risk score constructed using 14 susceptibility alleles for type 2 diabetes is associated with the early onset of diabetes and may predict the future requirement of insulin injections among Japanese individuals. Diabetes Care 2012;35:1763‐1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Caro JF, Triester S, Patel VK, Tapscott EB, Frazier NL, Dohm GL. Liver glucokinase: decreased activity in patients with type II diabetes. Horm Metab Res 1995;27:19‐22. [DOI] [PubMed] [Google Scholar]
- 24. de la Iglesia N, Mukhtar M, Seoane J, Guinovart JJ, Agius L. The role of the regulatory protein of glucokinase in the glucose sensory mechanism of the hepatocyte. J Biol Chem 2000;275:10597‐10603. [DOI] [PubMed] [Google Scholar]
- 25. Grimsby J, Coffey JW, Dvorozniak MT, Magram J, Li G, Matschinsky FM, et al. Characterization of glucokinase regulatory protein‐deficient mice. J Biol Chem 2000;275:7826‐7831. [DOI] [PubMed] [Google Scholar]
- 26. Stender S, Smagris E, Lauridsen BK, Kofoed KF, Nordestgaard BG, Tybjaerg‐Hansen A, et al. Relationship between genetic variation at PPP1R3B and levels of liver glycogen and triglyceride. Hepatology 2018;67:2182‐2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Nonalcoholic Steatohepatitis Clinical Research Network. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005;41:1313‐1321. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials