

Abstract
Bone loss is common in advanced cirrhosis, although the precise mechanisms underlying bone loss in cirrhosis are unknown. We studied the profile and functionality of bone‐forming cells and bone‐building proteins in bone marrow (BM) of individuals with cirrhosis (n = 61) and individuals without cirrhosis as normal controls (n = 50). We also performed dual energy X‐ray absorptiometry for clinical correlation. BM mesenchymal cells (MSCs) were analyzed for colony‐forming units‐fibroblasts and their osteogenic (fibronectin‐1 [FN1], insulin‐like growth factor binding protein 3 [IGFBP3], collagen type 1 alpha 1 chain [COL1A1], runt‐related transcription factor 2 [RUNX2], and alkaline phosphatase, liver [ALPL]) and adipogenic ( adiponectin, C1Q, and collagen domain containing [ADIPOQ], peroxisome proliferator‐activated receptor gamma [PPARγ], and fatty acid binding protein 4 [FABP4]) potentials. Colony‐forming units‐fibroblasts were lower in patients with cirrhosis (P = 0.002) than in controls. Cirrhotic BM‐MSCs showed >2‐fold decrease in osteogenic markers. Compared to controls, patients with cirrhosis showed fewer osteocytes (P = 0.05), osteoblasts, chondroblasts, osteocalcin‐positive (osteocalcin+) area, clusters of differentiation (CD)169+ macrophages (P < 0.001, each), and nestin+ MSCs (P = 0.001); this was more apparent in Child‐Turcotte‐Pugh (CTP) class C than A (P < 0.001). Multivariate logistic regression showed low nestin+ MSCs (P = 0.004) as a predictor of bone loss. Bone‐resolving osteoclasts were comparable among CTP groups, but >2‐fold decreased anti‐osteoclastic and increased pro‐osteoclastic factors were noted in patients with CTP C compared to CTP A. Bone‐building proteins (osteocalcin [P = 0.008], osteonectin [P < 0.001], and bone morphogenic protein 2 [P = 0.001]) were decreased while anti‐bone repair factors (fibroblast growth factor 23 [P = 0.015] and dipeptidyl peptidase 4 [P < 0.001]) were increased in BM and peripheral blood; this was more apparent in advanced cirrhosis. The dual energy X‐ray absorptiometry scan T score significantly correlated with the population of osteoblasts, osteocytes, MSCs, and CD169+ macrophages. Conclusion: Osteoprogenitor cells are substantially reduced in patients with cirrhosis and more so in advanced disease. Additionally, increased anti‐bone repair proteins enhance the ineffective bone repair and development of osteoporosis in cirrhosis. Hepatology Communications 2018;0:0‐0)
Abbreviations
- +
positive
- BM
bone marrow
- BMP2
bone morphogenic protein 2
- CD
clusters of differentiation
- cDNA
complementary DNA
- CKD
chronic kidney disease
- CTP
Child‐Turcotte‐Pugh
- DEL1
developmental endothelial locus 1
- DEXA
dual energy X‐ray absorptiometry
- DPP4
dipeptidyl peptidase 4
- FGF23
fibroblast growth factor 23
- HSC
hematopoietic stem cell
- IGF1
insulin‐like growth factor 1
- IGFBP
insulin‐like growth factor binding protein
- IL
interleukin
- LFA1
lymphocyte function‐associated antigen 1
- LGR4
leucine‐rich repeat‐containing G‐protein coupled receptor 4
- MSC
mesenchymal stem cell
- NCPH
noncirrhotic portal hypertension
- NFATc1
nuclear factor of activated T cells, cytoplasmic 1
- PCR
polymerase chain reaction
- RANK
receptor activator of nuclear factor kappa B
- RANKL
receptor activator of nuclear factor kappa Β ligand
- RT
reverse transcription
- Runx
runt‐related transcription factor
- TNF‐α
tumor necrosis factor alpha
Liver cirrhosis is the end stage of chronic liver disease and often results in hepatic decompensation requiring liver transplantation. Metabolic dysfunction, portal hypertension, and proinflammatory and oxidative stresses predispose these patients to progressive organ dysfunction and failure.1 Bone loss in the form of osteopenia and osteoporosis are observed in 1%‐21% of patients with cirrhosis,2 with the risk of fracture in about 3%‐44% of these patients.3, 4, 5 According to a World Health Organization technical report,6 a dual energy x‐ray absorptiometry (DEXA) T value of <–2.5 is used to diagnose osteoporosis and values of –2.5 to –1 to diagnose osteopenia. Osteopenia is a common skeletal disorder seen in individuals with cirrhosis; although often asymptomatic, untreated osteopenia can result in fractures and impaired quality of life.7 Several mechanisms have been proposed for bone loss in cirrhosis, including low fibronectin, increased oncofetal fibronectin, low insulin‐like growth factor 1 (IGF1), increased proinflammatory cytokines (e.g., tumor necrosis factor α [TNF‐α]), interleukin‐6 (IL‐6), low level of sex hormones, altered vitamin D metabolism, high bilirubin, and corticosteroid therapy.1
Although bone is a rigid organ, it remains in dynamic balance throughout life that involves multiple bone marrow (BM) cell types.8 Bone formation by osteoblasts and resorption by osteoclasts is tightly controlled and is responsible for continuous bone remodeling and repair.8 Osteoclasts originate from hematopoietic stem cell (HSC) precursors along the myeloid differentiation lineage,9 whereas osteoblasts are short‐lived cells and are continuously derived from the differentiation of nestin‐positive (nestin+) mesenchymal stem cells (MSCs) of the BM.10
We recently reported that greater derangement of the hematopoietic niche occurs with increasing severity of cirrhosis. This includes dysfunction of HSCs and nestin+ MSCs, which contribute to hematologic and immunological derangements and reduced potential for hepatic regeneration.11 The present study was undertaken to assess the profile and osteogenic functionality of BM‐MSCs and status of bone‐forming cells in different stages of cirrhosis. We also studied the cellular and genetic basis of BM‐MSC dysfunction in individuals with cirrhosis.
Patients and Methods
Patients
We assumed that bone loss in patients with cirrhosis is around 20%,2 with alpha as 5% and permissible error as 10% (prevalence of bone loss lies between 10%‐30%). We therefore needed to enroll 61 patients. Considering the inadequate sample procurement rate to be nearly 20%, we decided to enroll 75 cases.
Study Group
We undertook a prospective study of BM aspirates and biopsies obtained from 61 patients with cirrhosis between January and December 2015 at the Institute of Liver and Biliary Sciences, New Delhi. The diagnosis of cirrhosis was made as described.12 BM biopsies were performed for clinical indications and diagnostic purposes in various Child‐Turcotte‐Pugh (CTP) groups; on occasion, biopsies were undertaken as part of a therapeutic trial for hepatic regeneration using granulocyte colony‐stimulating factor. After the diagnostic workup, leftover clinical material was used for this study. Written informed consent was obtained to use pathology data and the clinical profile, and the institutional scientific review board of the Institute of Liver and Biliary Sciences, New Delhi, India, approved the study. Patients with cirrhosis who had a history of chronic kidney disease (CKD), lymphoma, or malignancy or had received chemotherapy, immunosuppressants, drug intake, or primary BM neoplastic pathology were excluded. A BM biopsy of >1 cm × 2 mm (volume, >0.31 cm3) with three or more trabeculae and intertrabecular spaces was considered adequate for study purposes.
Disease Controls
BM samples from patients with CKD (n = 16) and noncirrhotic portal hypertension (NCPH; n = 16) were considered as disease controls to determine the effect of any chronic disease or portal hypertension on bone‐forming precursors.
Noncirrhotic Controls
Fifty individuals without cirrhosis who were >18 years of age and in whom BM was examined to rule out infective pathology but was found to be uninvolved were used as controls as described.11
Methods
MSC Isolation and Cell Culture
MSCs were isolated from freshly collected BM aspirates using the RosetteSep Human MSC Enrichment Kit (Stem Cell Technology) as per the manufacturer’s protocol. Briefly fresh BM was incubated with RosetteSep enrichment. After incubation samples were diluted with phosphate‐buffered saline containing 2% fetal bovine serum and 1 nM/mL ethylene diamine tetraacetic acid and centrifuged in a density gradient. Enriched cells from the density gradient medium:plasma interface were removed and washed once in dilution media and then resuspended in MesenCult MSC Culture Media (cat. no. 5402; Stem Cell Technology). Resuspended cells were plated on a tissue culture flask and incubated at 37°C in a CO2 incubator. For subculturing we used TrypLE Select 10× (cat. no. A12177‐01; Gibco)
Colony‐Forming Units‐Fibroblast Assay
To test the colony‐forming capacity of the MSCs, a total of 1 × 106 BM mononuclear cells were plated per 3.5 cm2 well area. Cells were incubated for 14 days in alpha minimal essential medium (low glucose with 10% fetal bovine serum) at 37C in 5% CO2. The cells were then fixed and stained with 0.1% toluidine blue in 1% paraformaldehyde (all reagents from Sigma‐ Aldrich, St. Louis, MO) to visualize the colonies. Stained colonies were manually counted. The assay for each sample was carried out in triplicate.
Immunophenotyping MSCs
Screening of MSC+ and MSC‐negative markers was carried out by subjecting cells at passage 3 to flow cytometry analysis. We analyzed the expression levels of positive markers (clusters of differentiation (CD)105, CD73, CD90) and negative markers (CD34, CD45, and CD14).
Quantitative Reverse‐Transcription Polymerase Chain Reaction Analysis for Gene Expression
Total cellular RNA was isolated using an RNeasy Mini Kit (Qiagen, Venlo, Limburg, the Netherlands). RNA samples were treated with DNA‐free DNase I (Ambion, Life Technologies, Carlsbad, CA) according to the manufacturer’s instructions and were reverse transcribed into complementary DNA (cDNA) using a high‐capacity cDNA reverse‐transcription (RT) kit (Applied Biosystems, Life Technologies, Carlsbad, CA) as per the manufacturer’s instructions. cDNAs were further amplified using SYBR green quantitative polymerase chain reaction (PCR) master mix (Thermo Fischer Scientific), according to the manufacturer’s protocol, for 40 cycles on a ViiA 7 Real‐Time PCR machine (Applied Biosciences). Transcripts were amplified using gene‐specific primers (summarized in Supporting Table S1). A reverse‐transcriptase negative blank of each sample and a no template blank served as negative controls. Gene expression was normalized to the housekeeping gene 18S ribosomal RNA, which was used as an internal standard.
In Vitro Differentiation to Osteogenic and Adipogenic Lineage
Adipogenic differentiation was initiated in confluent cultures of MSCs using complete medium supplemented with 200 mM indomethacin, 0.5 mM 3‐isobutyl‐1‐methylxanthine, 10 mg/mL insulin, and 1 mM dexamethasone. After 18 days, adipogenic differentiation was detected by staining the lipid droplets with Oil Red O. Osteogenic differentiation was induced in confluent cultures of MSCs using complete medium supplemented with 0.1 mM dexamethasone, 10 mM beta‐glycerophosphate, and 0.2 mM ascorbic acid (all reagents from Sigma‐Aldrich).
BM Evaluation for Bone‐Forming Cells
Aspirates were analyzed for differential myelogram, erythroid precursors, megakaryocytes, and iron stores. BM biopsies were evaluated for cellularity (osteocytes, osteoblasts, fat spaces, and fibrosis). Chondroblasts were identified by alcian blue staining. Osteoclast progenitors and osteoclasts were assessed by tartrate‐resistant acid phosphatase staining. Twenty‐five consecutive high‐power fields were examined, and the average number/high‐power field was calculated.
BM Immunohistochemistry
HSCs were assessed by immunohistochemistry using CD34 (QBEnd/10; Biogenex, Fremont, CA) and CD117 (YR145; Biogenex). MSCs were assessed by the anti‐nestin antibody (N5413; Sigma‐Aldrich). Bone‐forming cells were identified by osteocalcin (OC4‐30; Novus Biologicals, Littleton, CO) and osteonectin (secreted protein acidic and rich in cysteine [SPARC], BM‐40; Novus Biologicals). Osteoprogenitor macrophages were determined by CD169 (HSn7D2; Novus Biologicals). Osteogenic precursors were identified by osterix (bs‐1110R; Bioss Inc., Woburn, MA). Runt‐related transcription factor (Runx)1 (4E7; LSBio, Seattle, WA) was used as an osteochrondroid differentiation marker. Perilipin (DK2069; Elabscience Biotechnology Inc., Houston, TX) was applied to identify adipogenic precursors. Positively stained cells were counted in a minimum of 25 high‐power fields (20×, area 1.3 mm2) by two pathologists (C.B., A.R.), and average numbers were calculated.
Image Analysis
Osteocalcin and osterix positivities are seen in the peritrabecular and intertrabecular areas, respectively, in a diffuse manner; hence, manual counting was not possible. We instead counted osteocalcin+ and osterix+ cells by capturing the entire biopsy in consecutive high‐resolution images at 20×. Osteocalcin‐ and osterix+ cell counting were done using Image J software (summarized in the Supporting Material).
Enzyme‐Linked Immunosorbent Assay
Enzyme‐linked immunosorbent assay for osteocalcin (ABIN368354; N‐MID, Aachen, Germany), osteonectin (E‐EL‐H1351; Elabscience), bone morphogenetic protein 2 (BMP2) (E13650313; Sincere Biotech, Beijing, China), fibroblast growth factor 23 (FGF23) (E13652282; Sincere Biotech), and dipeptidyl peptidase 4 (DPP4) (E‐EL‐H0058; Elabscience) was performed on BM plasma and peripheral blood plasma of cirrhosis and control cases.
Skeletal Survey
A baseline bone survey for bone loss was done by a DEXA scan of the lumbar spine and femoral neck in cases of cirrhosis and was classified as described.6
Statistical Analysis
Statistical analysis was carried out using Statistical Package for Social Sciences (SPSS, version 22.0; IBM Corp, Armonk, NY). Descriptive statistics were presented as proportions, mean ± SD/SEM, and median with interquartile range. Correlation was made by the Pearson/Spearman correlation coefficient test. Categorical data, i.e., sex, etiology, CTP score, were compared by chi‐square test. Student t test/Mann‐Whitney test was used to compare between two groups as appropriate (normal/non‐normal, respectively). One‐way analysis of variance and Kruskal‐Wallis test were applied followed by post‐hoc comparison using the Bonferroni method. To quantify the effect size in predictors of bone loss, univariate and multivariate logistic regression were applied with and without adjusting for confounders (age, sex, and vitamin D3). A diagnostic test was applied to determine the cut‐off value and area under the curve of selected variables. P < 0.05 was considered significant.
Results
Basic Attributes
A total of 203 BM examinations were performed from January 2015 to December 2015. Of these, 77 were from patients with cirrhosis, 62 from noncirrhotic controls, and the rest from patients with other diseases. BM of 16 patients with cirrhosis and 12 noncirrhotic controls were not included based on the exclusion criteria (Supporting Fig. S1). In the cirrhosis group, the mean Model for End‐Stage Liver Disease score was 15.74 ± 4.86, with 16.4% CTP A, 47.5% CTP B, and 36.1% CTP C stage. The clinical indications for the BM biopsy in cirrhosis were cytopenia (67.2%), pyrexia (13.1%), and evaluation for growth factor‐based therapy (19.1%) (Supporting Table S2). Serum calcium levels were comparable in the study and control groups, although patients with cirrhosis had lower vitamin D3 compared to controls (P = 0.007). The baseline parameters of cases with cirrhosis and noncirrhotic controls are summarized in Table 1.
Table 1.
Comparison of Basic Attributes in Study and Control Groups
| Parameters | Cirrhosis | Control | P Value |
|---|---|---|---|
| Number of cases (n) | 61 | 50 | |
| Age (years)* | 49.6 ± 11.6 | 44.66 ± 11.72 | 0.661 |
| Sex (M:F) | 48:13 | 30:10 | 0.44 |
| Etiology: alcohol/hepatitis B/hepatitis C/NASH/cryptogenic | 30/10/3/12/6 | ||
| Hemoglobin (g/dL)* | 10.97 ± 2.28 | 12.63 ± 1.78 | <0.001 |
| Total leucocyte count (mm3/dL)† | 5.35 (3.62‐9.12) × 103 | 7.55 (6‐8.55) × 103 | 0.005 |
| Platelet count (mm3/dL)† | 95 (60‐140) × 103 | 235 (175‐280) × 103 | <0.001 |
| Serum creatinine (mg/dL)† | 0.74 (0.61‐1.15) | 0.52 (0.45‐0.75) | <0.001 |
| Total bilirubin (mg/dL)† | 2.31 (1.42‐3.73) | 0.85 (0.7‐1) | <0.001 |
| Serum albumin (g/dL)* | 2.82 ± 0.66 | 4.99 ± 0.77 | <0.001 |
| International normalized ratio* | 1.41 ± 0.31 | 1.0 ± 0.07 | <0.001 |
| Serum calcium (mg/dL) | 8.35 ± 0.97 | 8.46 ± 0.71 | 0.542 |
| Vitamin D3 (ng/mL) | 20.7 (13.95‐28.47)‡ | 25 (23‐38)§ | 0.007 |
| MELD score† | 15.74 ± 4.86 | ||
| Child‐Turcotte‐Pugh Score; A/B/C (%) | 10 (16.4%)/29 (47.5%)/22 (36.9%) | ||
| Hepatic vein pressure gradient (mm of Hg*) | 16.92 ± 5.61 | ||
| Liver stiffness (Kpa)† | 40.2 ± 21.8 | ||
| Bone marrow parameters | |||
| Cellularity* | 49 ± 18.49 | 67 ± 17.32 | <0.001 |
| Myeloid: erythroid ratio* | 1.3 ± 0.55 | 1.4 ± 0.94 | 0.419 |
| Iron score (grade)* | 2.1 ± 1.16 | 2.8 ± 1.18 | 0.004 |
Mean ± SD.
Median (interquartile range).
n = 34.
n = 47.
Abbreviation: MELD, Model for End‐Stage Liver Disease.
Cellular and Osteogenic Functional Loss of BM MSCs in Cirrhosis
MSCs support HSCs and also serve as the progenitors of bone cells. To confirm the loss of BM‐MSCs in cirrhosis we analyzed the number of colony‐forming units‐fibroblasts in total BM mononuclear cells in cirrhotic (n = 20) and control BM (n = 10). We found a significant decrease (P = 0.002) in the number of colony‐forming units‐fibroblast colonies per million BM mononuclear cells in individuals with cirrhosis (3.5 ± 0.65) compared to controls (12.16 ± 1.16) (Fig. 1A). This indicates a loss of self‐renewing potential of MSCs in patients with cirrhosis. Quantitative RT‐PCR analysis of isolated MSCs in response to in vitro osteogenic and adipogenic differentiation showed more than a 2‐fold decrease in osteogenic gene (fibronectin‐1 [FN1], insulin‐like growth factor binding protein 3 [IGFBP3], collagen type I alpha 1 chain [COL1A1], RUNX2, and alkaline phosphatase, liver [ALPL]) and increase in adipogenic gene (adiponectin, C1Q, and collagen domain containing [ADIPOQ], peroxisome proliferator‐activated receptor gamma [PPARγ], and fatty acid binding protein 4 [FABP4]) expression in cirrhotic BM‐MSCs compared to controls (Fig. 1B). This was further confirmed for osterix, Runx1, and perilipin by BM immunohistochemistry, which showed fewer osterix+ and Runx1+ and more perilipin+ cells in cirrhotic BM than in control BM (Fig. 1C). These data suggest that the number of BM nestin+ MSCs decline in cirrhosis and that the potential of these cells toward the osteogenic lineage also decreases.
Figure 1.
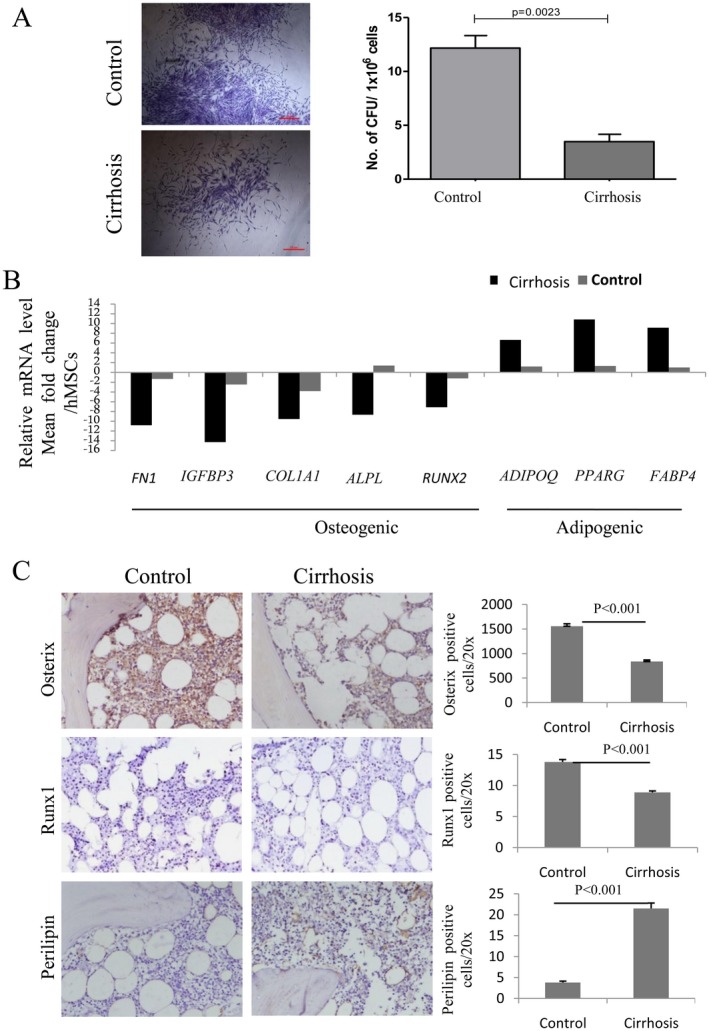
Cirrhosis versus control cases. (A) Individual colonies captured at 4× magnification for control and cirrhosis BM cells (left) and bar graph (right) showing average number of CFU‐F colonies (mean ± SE) generated per million BM mononuclear cells in control and cirrhotic BM. (B) Relative mRNA level of indicated osteogenic and adipogenic marker genes in cirrhosis with respect to control BM‐derived MSC after in vitro osteogenic and adipogenic differentiation. (C) Representative images of osterix‐, Runx1‐, and perilipin‐stained sections of control and cirrhosis BM (left); bar graph (right) showing number of osteogenic cells and adipogenic cells (mean ± SE) in cirrhosis and control BM. All immunohistochemistry images ×200; area 1.3 mm2. Abbreviations: ADIPOQ, adiponectin, C1Q, and collagen domain containing; ALPL, alkaline phosphatase, liver; CFU‐F, colony‐forming units‐fibroblasts; COL1A1, collagen type I alpha 1 chain; FABP4, fatty acid binding protein 4; FN1, fibronectin‐1; hMSC, human MSC; mRNA, messenger RNA; PPARγ, peroxisome proliferator activated receptor gamma.
Loss of Bone‐Forming Cells in Cirrhosis
MSC‐derived osteoblasts and chondroblasts secrete osteocalcin and osteonectin proteins that play a role in bone metabolic regulation.13 CD169+ macrophages are responsible for the retention of hematopoietic stem and progenitor cells in the MSC niche.14 We quantified these cells in the BM biopsies of patients with cirrhosis and controls. Nestin+ MSCs in the BM positively correlated with the osteoblasts (r = 0.816; P < 0.001), chondroblasts (r = 0.469; P < 0.001), and osteocalcin+ area (r = 0.417; P = 0.011). Bone‐forming cells (osteoblasts [P < 0.001], osteocytes [P = 0.05], chondroblasts [P < 0.001]) (Fig. 2A,I‐III); osteocalcin+ cells (P < 0.001), osteocalcin+ area (P < 0.001), and osteonectin+ cells (P = 0.001) (Fig. 2B,I‐III); nestin+ MSCs (P = 0.001) (Fig. 2C); and CD169 macrophages (P < 0.001) (Fig. 2D) were found to be significantly reduced in patients with cirrhosis compared to controls.
Figure 2.
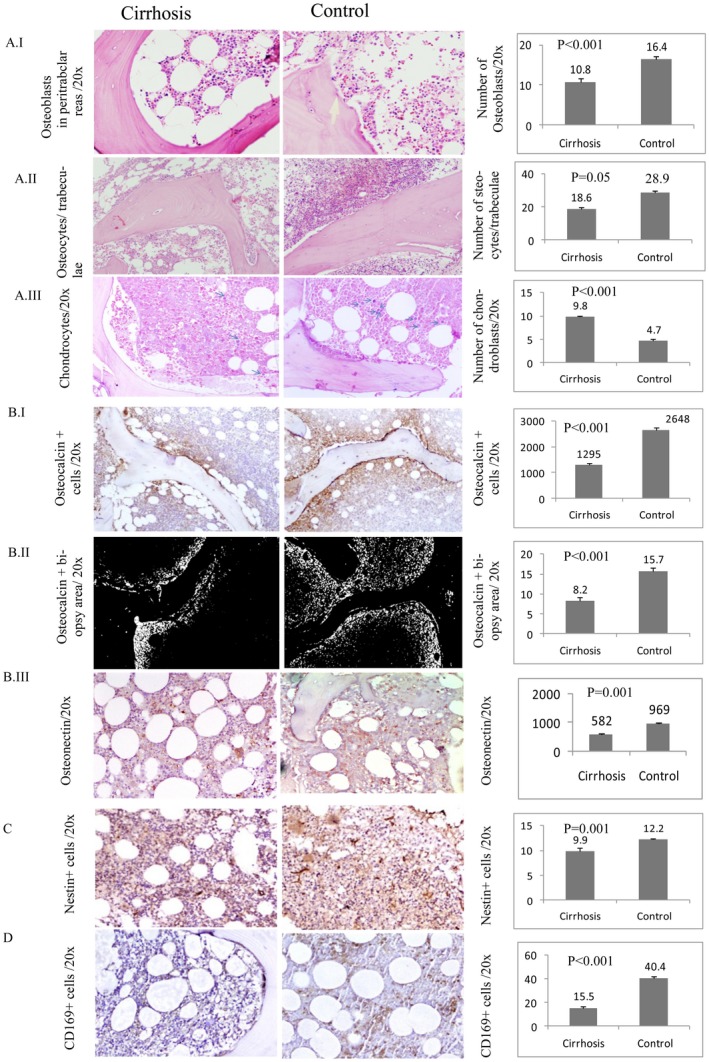
Images of BM biopsy in Cirrhosis and Control cases. (A) Panel I. Representative Images of cirrhosis and control BM (left) showing the osteoblast cells and bar graph (right) showing number of osteoblast (mean ± SE) in cirrhosis and control BM (H&E). Panel II. Cirrhosis and control BM (left) showing the osteocytes and bar graph (right) showing number of osteocytes (mean ± SE) in cirrhosis and control BM (H&E). Panel III. Alcian blue‐stained chondroblasts in cirrhosis and control BM biopsy section (left) and bar graph (right) showing number of chondroblasts (mean ± SE) in cirrhosis and control BM. (B) Panel I. Representing osteocalcin+ cells (mean ± SE) in cirrhosis and control BM biopsy section (left) and bar graph (right) showing number of osteocalcin+ cells in cirrhosis and control BM. Panel II. Osteocalcin+ area (mean ± SE) in cirrhosis and control BM biopsy section (left) and bar graph (right) showing percentage osteocalcin+ area in cirrhosis and control BM. Panel III. Showing osteonectin+ cells (mean ± SE) in cirrhosis and control BM biopsy section (left) and bar graph (right) showing osteonectin in cirrhosis and control BM (C) Nestin+ BM MSCs in cirrhosis and control BM biopsy section (left) and bar graph (right) showing number of these cells in cirrhosis and control BM. (D) CD169+ BM macrophage (mean ± SE) in cirrhosis and control BM biopsy section (left) and bar graph (right) showing number of CD169+ cells in cirrhosis and control BM. (All image magnification ×200, area 1.3 mm2). Abbreviations: BM, bone marrow; H&E, hematoxylin and eosin.
No Effect of Etiology, Isolated Portal Hypertension, and Chronicity on Bone‐Forming Cells
We investigated the influence of the etiology of cirrhosis on the cellular profiles in BM. No significant difference was noted in patients with different etiologies (alcohol [30 patients], viral [13 patients], nonalcoholic steatohepatitis [13 patients], and five others) on the osteoblasts (P = 0.357), osteocytes (P = 0.345), chondroblasts (P = 0.478), osteocalcin+ cells (P = 0.577), osteocalcin+ area (P = 0.441), nestin+ cells (P = 0.991), and CD169+ niche‐protecting macrophages (P = 0.621).
Moreover, to discern the effect of chronicity and portal hypertension on bone‐forming cells, we studied these cells in the BM biopsies of patients with CKD (n = 16) and NCPH (n = 16). These cell populations were comparable to controls in NCPH biopsies (Supporting Fig. S2). Similarly, these were akin to the controls in CKD (Supporting Fig. S2), except for significantly lower osteocalcin+ cells (P < 0.001). Patients with cirrhosis compared to patients with NCPH and those with CKD had fewer osteoblasts (P < 0.001 and P = 0.041, respectively), osteocytes (P < 0.001 and P = 0.027), chondroblasts (P = 004 and P < 0.001), osteocalcin+ area (P < 0.001 and P < 0.001), osteonectin+ cells (P < 0.001 and P < 0.001), nestin+ cells (P = 0.002, P = 0.03), and CD169+ cells (P < 0.001 and P < 0.001). Patients with cirrhosis also showed lower osteocalcin+ cells (P < 0.001) than those with NCPH, but no difference was noted with CKD cases (P = 0.126). These results indicate that the uniquely altered cirrhotic microenvironment induces adverse effects on bone‐forming cells rather than portal hypertension alone.
Osteoprogenitor Cell Reduction with Progression of Cirrhosis
We determined the bone‐forming cells in various cirrhosis groups according to the CTP score. In individuals with cirrhosis, progressive reduction of osteoblasts, osteocytes, osteocalcin+ cells, osteocalcin+ BM area, osteonectin+ cells, nestin+ MSCs, and CD169+ niche macrophages was found with advancing stage of cirrhosis (Table 2). Bone‐forming cells in the CTP A group were comparable to controls (Supporting Table S3), and these were significantly lower in CTP B and C (Table 2).
Table 2.
Bone‐Forming Cells in Different Child‐Turcotte‐Pugh (CTP) Groups
| Bone‐Forming Cells |
CTP A (10) Median (Interquartile Range) |
CTP B (29) Median (Interquartile Range) |
CTP C (22) Median (Interquartile Range) |
P Value CTP A Versus CTP B |
P Value CTP A Versus CTP C |
P Value CTP B Versus CTP C |
|---|---|---|---|---|---|---|
| Osteoblasts/peritrabecular area | 18 (14‐19) | 13 (7‐16) | 5 (4‐8.25) | 0.137 | 0.001 | <0.001 |
| Osteocytes/trabeculae | 28 (15‐35) | 20 (16‐25) | 12.5 (10‐15) | 0.403 | 0.002 | 0.001 |
| Chondrocytes/20× | 8.5 (8‐9.75) | 5 (4‐7) | 2 (1.75‐3) | 0.001 | <0.001 | <0.001 |
| Osteocalcin+ cells/20× | 1,201 (1,093‐1,684) | 1,309 (937‐2,105) | 877 (639‐1,306) | 0.753 | 0.024 | 0.012 |
| Osteocalcin+ BM biopsy area, % | 8.37 (4.46‐14.71) | 7.59 (3.36‐13.67) | 3.4 (2.9‐7.3) | 0.750 | 0.002 | 0.049 |
| Osteonectin+ cells/20× | 972 (892.5‐1,226) | 652 (542‐786) | 322 (200‐432.25) | 0.004 | <0.001 | <0.001 |
| Nestin+ cells/20× | 16 (15‐16) | 12 (9‐13) | 6 (4‐8) | 0.003 | <0.001 | <0.001 |
| CD169+ cells/20× | 26(17‐29) | 17 (13‐20) | 8.5 (6.75‐14.25) | 0.150 | <0.001 | <0.001 |
Osteoclastogenic Effector Transcription Factor Alterations are Discernible with Progression of Cirrhosis
It is difficult to quantify osteoclasts in the BM biopsy; hence, we studied the osteoclasts in BM aspirates. Mononucleated osteoclast progenitor cells (4.5 ± 1.61 versus 4.7 ± 1.74; P = 0.553) (Fig. 3A,B) and multinucleated osteoclasts (1.8 ± 1.2 versus 1.68 ± 1.25; P = 0.598) (Fig. 3C,D) were comparable in individuals with cirrhosis and controls. No significant difference was noted in osteoclast progenitors and osteoclast numbers in different CTP groups (Fig. 3B,D). However, activated forms of osteoclasts with cytoplasmic podia were more common in CTP B and C than in CTP A cases and controls (Fig. 3C). To discern activated status of osteoclasts in advanced cirrhosis, we performed a BM biopsy quantitative RT‐PCR analysis for the genes associated with osteoclast homeostasis. This showed a decrease in the expression of the anti‐osteoclastic factors14, 15 developmental endothelial locus 1 (DEL1), nuclear factor of activated T‐cells, cytoplasmic 1 (NFATc1), lymphocyte function‐associated antigen 1 (LFA1), and leucine‐rich repeat‐containing G‐protein coupled receptor 4 (LGR4) and an increase in the pro‐osteoclastic factors15, 16 receptor activator of nuclear factor kappa B (RANK) and IGFBP1 in CTP C compared to CTP A (Fig. 3E). These data suggest that there is increased osteoclastogenic activity with increasing severity of cirrhosis.
Figure 3.
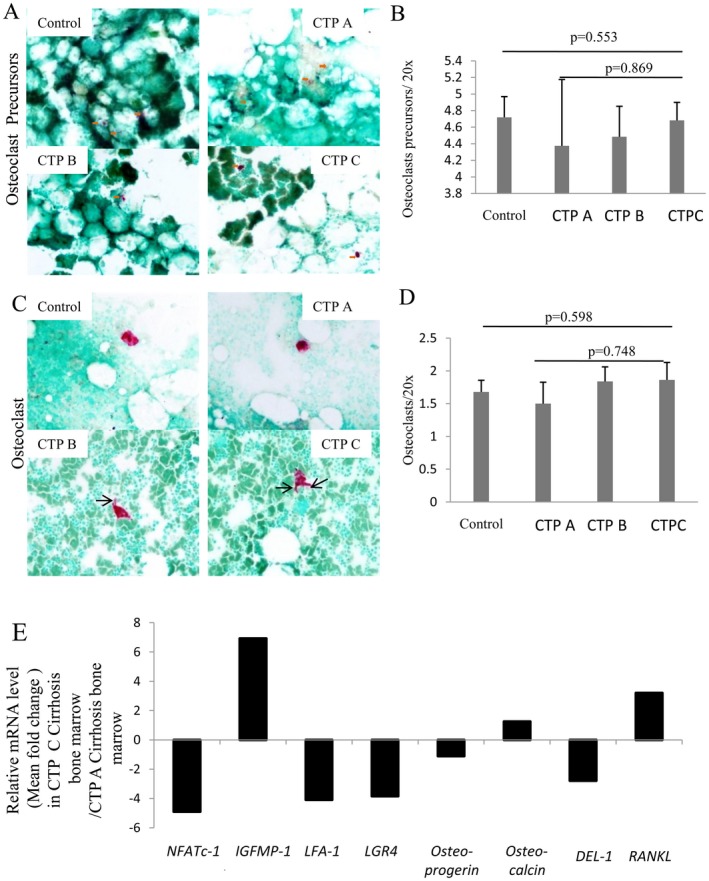
Osteoclasts and osteoclast precursors. (A) Representative photomicrographs showing mononucleated osteoclastic progenitor cells (red, with orange arrows) in control and in cirrhosis subgroups CTP A, B, and C (TRAP staining, magnification ×200). (B) Bar graph showing mean ± SE of osteoclastic progenitors in control and cirrhosis subgroups CTP A, B, and C. (C) Multinucleated osteoclasts (red) in control and in cirrhosis subgroups CTP A, B, and C (TRAP staining, magnification ×200). Osteoclast in CTP C shows cytoplasmic podia, indicating the activated osteoclast form (black arrow). Activation is less in CTP B, and there is no activated form in CTP A and control cases. (D) Bar graph showing mean ± SE of osteoclasts in control and in CTP A, B, and C. (E) Relative mRNA level of indicated osteoclast effecter genes in BM tissue of CTP C with respect to CTP A patients with cirrhosis. Abbreviations: CTP, Child‐Turcotte‐Pugh; IGFBP‐1, insulin like growth factor protein‐1; mRNA, messenger RNA; TRAP, tartrate‐resistant acid phosphatase.
Bone‐Forming Proteins Decrease with Advancement of Cirrhosis
Osteoblast‐derived osteocalcin, osteonectin, and FGF23 have opposing effects for bone matrix formation.13, 17 BM stromal cells secrete BMP2, which enhances the osteoblast differentiation from MSCs and osteoblast functions.18 Accumulated adipocytic cells in BM lead to generation of DPP4, which impairs bone regeneration.19 We observed that osteocalcin (P = 0.008 and P = 0.011), osteonectin (P < 0.001 and P < 0.001), and BMP2 (P = 0.001 and P = 0.001) were significantly decreased whereas FGF23 (P = 0.015 and P = 0.028) and DPP4 (P < 0.001 and P = 0.004) were increased in the BM and peripheral blood plasma, respectively; this was more apparent with increasing severity of liver cirrhosis (Fig. 4).
Figure 4.
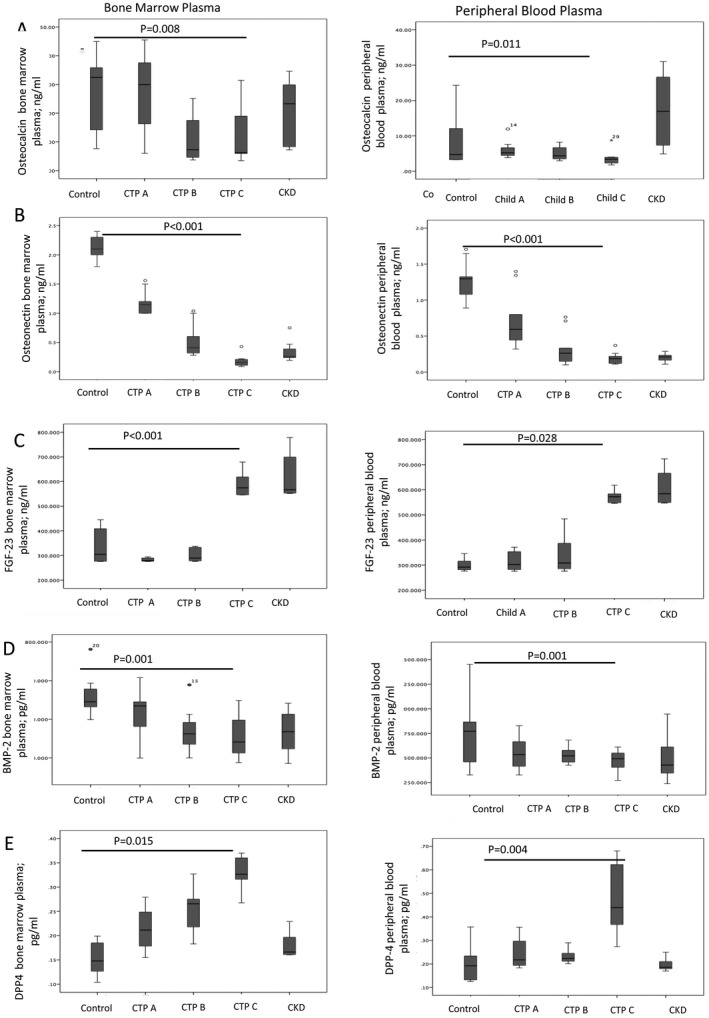
Graphs showing BM plasma (left) and peripheral blood plasma (right) of the same patients displaying levels of bone‐building proteins. (A) Osteocalcin, (B) osteonectin, (C) bone morphogenic protein, and (D) anti‐bone building proteins FGF23 and (E) DPP4 in control and patients with cirrhosis with Child‐Turcotte‐Pugh (CTP) A, B, or C { median (interquartile range) }.
Osteoprogenitor Cells Correlate with Bone Density Parameters in Patients with Cirrhosis
Osteopenia and osteoporosis were significantly more common in CTP B and CTP C than in CTP A cirrhosis (CTP A/B/C normal bone density, 7/10/1; osteopenia, 1/12/4; and osteoporosis, 2/7/17; P = 0.001). To investigate the clinical significance of loss of bone‐forming cells, we correlated the whole body T score with the population of these cells in the BM. The T score correlated with osteoblasts (r = 0.667; P < 0.001), osteocytes (r = 0.670; P < 0.001), nestin+ MSCs (r = 0.622; P < 0.001), osteocalcin+ area (r = 0.419; P = 0.01), and CD169+ macrophages (r = 0.525; P = 0.009). The osteoprogenitors were significantly lower in cirrhotic cases with osteoporosis and osteopenia (Fig. 5) compared to cirrhotic cases without bone loss. No specific predilection of osteoporosis was seen among the various etiologies of cirrhosis (alcohol/viral/nonalcoholic steatohepatitis + cryptogenic; normal bone density, 7/3/2; osteopenia, 13/4/8; osteoporosis, 10/5/8; P = 0.793).
Figure 5.
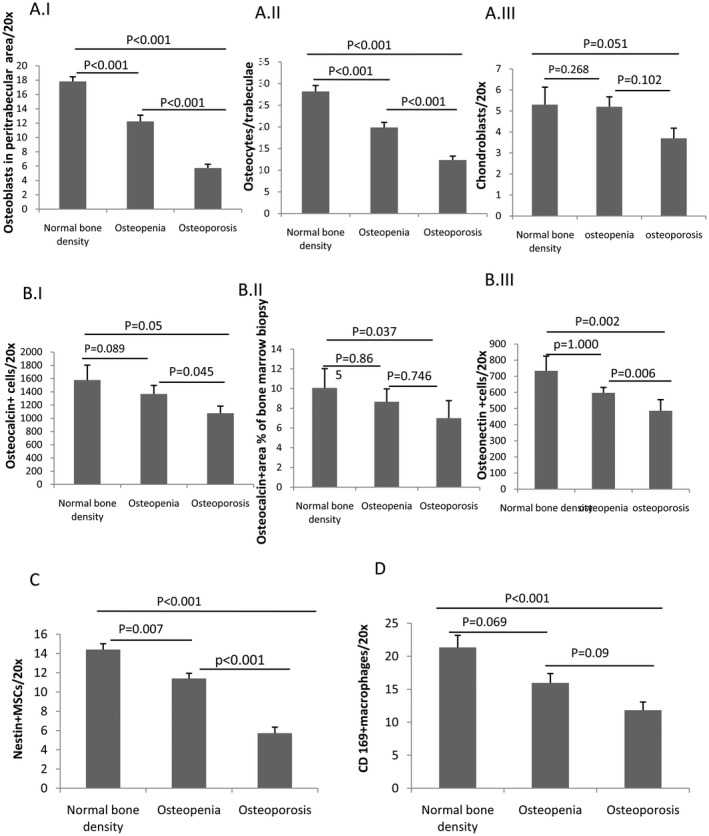
Distribution graphs. (A) Panel I. Osteoblasts. Panel II. Osteocytes. Panel III. Chondroblasts. (B) Panel I. Osteocalcin+ cells. Panel II. Percentage osteocalcin+ area. Panel III. Osteonectin+ cells. (C) Nestin+ MSCs. (D) CD169+ BM macrophages (all expressed in mean ± SE) in BM sections of patients with cirrhosis with normal bone density, osteopenia, and osteoporosis; evaluated by T score on bone densitometry scan.
Univariate and multivariate logistic regression analyses after adjusting for vitamin D, age, sex, liver disease severity score, Model for End‐Stage Liver Disease, and bone‐forming cells (nestin+, osteoblasts, and osteocytes) were found to be significant factors associated with bone loss in patients with cirrhosis. Multivariate logistic regression analysis was done for all those parameters that had P < 0.2 (Table 3).
Table 3.
Univariate and Multivariate Analysis for Predictors Associated With Cirrhosis‐Related Osteoporosis
| Unadjusted Univariate Analysis | Adjusted for Age, Sex, VitD | ||||||
|---|---|---|---|---|---|---|---|
| Attributes | Bone Loss (n = 26) | No Bone Loss (n = 35) | P Value | OR (95% CI) | P Value | OR (95% CI) | |
| Age (year)* | 53.42 ± 10.98 | 49.77 ± 10.71 | 0.184 | 1.03 (0.98‐1.88) | |||
| Sex | |||||||
| Male | 18 | 30 | 0.127 | 0.375 (0.10‐1.32) | |||
| Female | 8 | 5 | 1 | ||||
| Etiology | |||||||
| Nonalcoholic | 14 | 17 | 1 | ||||
| Alcoholic | 12 | 18 | 0.681 | 0.81(0.29‐2.24) | 0.927 | 1.07 (0.23‐5.02) | |
| Child‐Turcotte‐Pugh stage | |||||||
| A | 2 | 08 | 1 | ||||
| B | 7 | 22 | 0.821 | 1.23 (0.21‐7.27) | 0.935 | 1.12 (0.066‐12.15) | |
| C | 17 | 05 | 0.047 | 6.43(1.03‐40.27) | 0.635 | 1.8 (1.26‐25.67) | |
| Model for End‐Stage Liver Disease* | 18.38 ± 4.66 | 13.77 ± 4.04 | 0.001 | 1.26 (1.11‐1.45) | 0.050 | 1.24 (1.02‐1.54) | |
| Vitamin D3 (ng/mL)† | 17.75 (12.6‐24.8); n = 14 | 22.75 (15.23‐34.78); n = 20 | 0.45 | 0.98 (0.94‐1.027) | |||
| Serum calcium (mg/dL)* | 8.49 ± 1.24 | 8.26 ± 0.75 | 0.415 | 1.29 (0.70‐2.39) | 0.341 | 1.4 (0.68‐2.88) | |
| Serum phosphorus (mg/dL)* | 3.11 ± 0.85 | 3.03 ± 0.88 | 0.773 | 1.11(0.54‐2.30) | 0.252 | 0.48 (0.14‐1.59) | |
| Serum bilirubin (mg/dL)† | 2.30 (1.36‐3.73) | 2.15 (1.5‐3.76) | 0.201 | 1.08 (0.96‐1.23) | 0.15 | 0.86 (0.57‐1.5) | |
| Osteoblasts / paratrabecular area* | 6.1 ± 2.75 | 14.26 ± 4.6 | <0.001 | 0.66 (0.55‐0.79) | 0.02 | 0.57 (0.36‐0.92) | |
| Osteocytes / trabeculae* | 12.89 ± 4.19 | 22.83 ± 6.88 | <0.001 | 0.72 (0.61‐0.85) | 0.01 | 0.68(0.50‐0.92) | |
| Osteocalcin area (%)* | 4.19 ± 3.80 | 6.3 ± 4.45 | 0.422 | 0.97 (0.90‐1.04) | 0.78 | 1.01 (0.92‐1.11) | |
| Nestin+ cells/20×* | 6.16 ± 3.12 | 12.61 ± 2.75 | <0.001 | 0.52 (0.38‐0.71) | 0.006 | 0.45 (0.25‐0.80) | |
| CD169/20×* | 12.26 ± 6.00 | 18.00 ± 7.32 | 0.004 | 0.881 (0.81‐0.96) | 0.11 | 0.89 (0.78‐1.03) | |
Mean ± SD.
Median (interquartile range).
Nestin loss was found to be a significant predictor of osteoporosis (P = 0.004; odds ratio, 0.468; 95% confidence interval, 0.282‐0.781) with overall predictive value of 81.6%. With area under the receiver operating characteristic analysis, the cut‐off value of 9.5 nestin+ cells (P < 0.001) was identified for cirrhosis‐associated osteoporosis with area under the curve of 93.5% (95% confidence interval, 87.2%‐99.8%) with 83.9% sensitivity and 80% specificity (Supporting Fig. S3).
Clinical follow‐up was available in 55 of 61 (90.1%) patients; 3 patients had a traumatic fracture (1 vertebral and 2 hip), 1 had probable skull lesions, and 11 had complaints of bone pain (7 vertebral region and 4 hip).
Discussion
To our knowledge, this is the first human study that has examined the status of bone‐forming cells in cirrhosis. The results of this novel study show that MSCs and their progeny cells, which are responsible for bone formation, are significantly reduced in the BM of patients with cirrhosis. This change was more evident in individuals with advanced cirrhosis, irrespective of etiology, portal hypertension, and chronicity of disease. These data add a new dimension to the spectrum of systemic changes due to cirrhosis of the liver, particularly because the functional mass of parenchymal and nonparenchymal hepatic cells in progressive cirrhosis is substantially reduced, which in turn can affect the skeletal system.
The results show that in patients with cirrhosis there is a loss of bone‐forming cells (bone‐building proteins, bone loss‐preventing transcription factors) and an increased level of factors associated with increased osteoclastic activity and bone loss. These changes were more severe with advancing stages of cirrhosis (Fig. 6). The bone‐forming cell population in the BM of patients with cirrhosis also correlated with the T score obtained by the DEXA scan.
Figure 6.
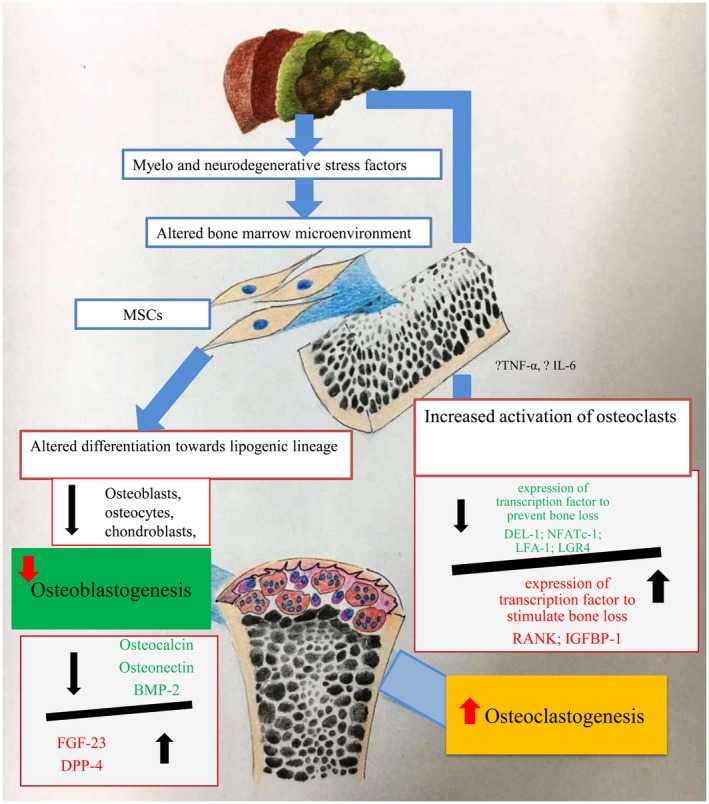
Concept diagram showing the altered status of bone‐forming cells in an advanced stage of cirrhosis.
Bone mass is determined by two factors: peak bone mass accumulated throughout growth and continuous consolidation and resorption of the bone thereafter.20 Most of our knowledge has been attained from studies on primary biliary cirrhosis.21, 22 It is known that bone fractures are more frequent in patients with cirrhosis than in the general population, even after adjusting for the confounding variables, such as sex, presence of cholestasis, and high alcohol consumption.23 The results of our study, which demonstrate CTP score rather than the etiology of cirrhosis as the main factor contributing to bone loss, are supported by others,24 and this has been described both in cholestatic and noncholestatic chronic liver diseases.24
Chronic exposure of BM‐MSCs to inflammatory cytokines (such as TNF‐α, interferon‐gamma, IL‐β), increased oxidative stress,11 and other harmful biological toxins produced in the process of chronic liver injury25 might be responsible for their deterioration and altered differentiation toward the adipogenic lineage26, 27 of BM‐MSCs in patients with advanced cirrhosis. Such compromised MSCs are unable to give rise to functionally active osteoblasts; hence, their products, such as osteocalcin and osteonectin, also decrease in cirrhosis. It has been shown that osteocalcin levels inversely correlate with liver disease severity.28
Bone loss in cirrhosis is not uniform and is common in advanced cirrhosis; this has been reported in various studies19, 29, 30 and can be explained by the fact that bone‐forming cells progressively decrease with severity of cirrhosis. Our group has provided an explanation for the reduction of bone stem cells and niche cells due to increased proinflammatory cytokines11 and probably due to increased oxidative stress. Both are markedly increased in advanced cirrhosis compared to early cirrhotic stages.31
A low turnover from decreased bone‐forming cells and matrix‐related proteins as well as a high turnover of pro‐osteoclastic activity contribute to bone loss in cirrhosis. Bone remodeling and homeostasis are complex processes, and several transcription factors are involved in the process of fine‐tuning the osteoclastic functions. In this study, we demonstrated that there is decreased expression of DEL1, NFATc1, LFA1, and LGR4 and increased expression of RANK and IGFBP1 in CTP C compared to CTP A cirrhotic BM tissue. DEL1 is an endothelial cell‐secreted protein that regulates LFA1 integrin‐dependent leukocyte recruitment and inflammation. DEL1 is also expressed by osteoclasts and regulates NFATc1‐mediated osteoclastogenesis and excessive bone resorption.15, 16 RANK and LGR4 are expressed on osteoclasts and are the receptors for RANK ligand (RANKL; tumor necrosis factor ligand superfamily member 11). LGR4 competes with RANK for binding with RANKL and inhibits the canonical pathway that is responsible for osteoclast activation and differentiation.16 Thus, there is a trend of up‐regulation of osteoclastogenic activity and down‐regulation of anti‐bone resorption functions in advanced individuals with cirrhosis.
This observation led us to further explore the pathogenesis of these altered pro‐osteoclastic and anti‐osteoclastic factors in advanced cirrhosis. Activation of inflammatory cells in patients with cirrhosis induces proinflammatory cytokine production, such as TNF‐α, IL‐1, IL‐13, IL‐6, IL‐7, IL‐11, IL‐15, and IL‐17.28, 32, 33 These inflammatory mediators have been described to be elevated in advanced cirrhosis and promote osteoclast precursor activation or RANKL production by osteoblasts, which are associated with bone resorption.28 Another study identified an increased level of liver‐derived factor IGFBP1 as an osteoclast differentiation factor in chronic liver disease that was proposed to induce increased bone resorption in cirrhotic cases.34 Osteoprotegerin is synthesized by liver and is considered to be anti‐osteoclastic, and a lower level of osteoprotegerin in cirrhosis promotes bone loss.35 An experimental study by Nussler et al.36 highlighted that cirrhotic livers produce cytokines and growth factors that may inhibit the function of osteoblasts by TGF‐β while favoring osteoclastogenesis.
We also investigated any confounding effect of portal hypertension or chronicity of a disease process in the genesis of altered bone cell milieu seen in patients with cirrhosis. Similar to results in our previous study,11 we observed near normal BM cells in patients with NCPH and thus no adverse effect of isolated portal hypertension on bone‐forming cells. The effect of CKD on bone‐forming cells revealed significantly lower osteocalcin+ cells but comparable nestin+ mesenchymal cells, osteoblasts, and osteocytes to controls. Of the 16 patients with CKD, 8 had high turnover bone disease, 2 had osteomalacia due to poor bone mineralization and excessive osteoid formation37 (Supporting Fig. S4), and 6 were on dialysis and were found to have adynamic bone disease (Supporting Fig. S4). Studies have shown uremia‐induced parathyroid hormone resistance as a cause of low osteocalcin production in these patients, leading to low bone turnover in adynamic bone disease.37, 38 Overall, our study highlights that ostodystrophy seen in patients with CKD is different and is not related to direct reduction of osteoprogenitor cells as noted in advanced cirrhosis.
Vitamin D deficiency per se is not considered to be implicated in the development of hepatic ostodystrophy; rather it is reduced organ sensitivity to vitamin D due to genetic pleomorphism of vitamin D receptor, e.g., in primary biliary cirrhosis, that contributes to hepatic ostodystrophy.39 Vitamin D levels were not found to be associated with bone density in cirrhotic animal models or in patients with cirrhosis.36
A limitation of our study is that only a subset of patients with cirrhosis was analyzed where BM biopsies were indicated. Because the profile of patients advised for BM biopsy (and thus included in this study) could be more serious when compared with those not advised for biopsy, potential selection bias could limit the generalizability of our study results to all individuals with cirrhosis. However, the distribution of patients included in our study according to clinical indication for BM biopsy (viz., cytopenia, pyrexia of unknown origin, and regenerative therapy) across the CTP A, B, and C categories were similar. Hence, the results obtained across these categories of patients (Supporting Table S1) are authentic and generalizable beyond the study participants.
In conclusion, bone‐forming cells decrease in cirrhosis, and this is more evident in advanced cirrhosis. The decline of osteoprogenitors correlates with bone density parameters. Ineffective repair, regeneration, and remodeling of bone in advanced cirrhosis occur due to suboptimal levels of osteoprogenitors and increased pro‐osteoclastic factors. Cellular therapy and modulation of pro‐osteoblastic factors could be explored as therapeutic options in cirrhosis.
Supporting information
Acknowledgment
We thank Mr. Amit, Ms. Shagufta, Ms. Rekha, Ms. Meenakshi, Mr. Parveen, Mr. Amit Rathore, and Mr. Mukesh for their support in laboratory experiments.
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1234/full.
References
- 1. Møller S, Henriksen JH, Bendtsen F. Extrahepatic complications to cirrhosis and portal hypertension: haemodynamic and homeostatic aspects. World J Gastroenterol 2014;20:15499‐15517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nakchbandi IA. Osteoporosis and fractures in liver disease: relevance, pathogenesis and therapeutic implications. World J Gastroenterol 2014;20:9427‐9438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Leslie WD, Bernstein CN. Leboff MS; American Gastroenterological Association Clinical Practice Commitee. AGA technical review on osteoporosis in hepatic disorders. Gastroenterology 2003;125:941‐966. [DOI] [PubMed] [Google Scholar]
- 4. Nakchbandi IA, van der Merwe SW. Current understanding of osteoporosis associated with liver disease. Nat Rev Gastroenterol Hepatol 2009;6:660‐670. [DOI] [PubMed] [Google Scholar]
- 5. Blachier M, Leleu H, Peck‐Radosavljevic M, Valla DC, Roudot‐Thoraval F. The burden of liver disease in Europe: a review of available epidemiological data. J Hepatol 2013;58:593‐608. [DOI] [PubMed] [Google Scholar]
- 6. WHO Scientific Group on the Prevention and Management of Osteoporosis . Prevention and management of osteoporosis: report of a WHO Scientific Group. WHO Technical Report Series, No. 921. Geneva, Switzerland: World Health Organization; 2003. [Google Scholar]
- 7. Yadav A, Carey EJ. Osteoporosis in chronic liver disease. Nutr Clin Pract 2013;28:52‐64. [DOI] [PubMed] [Google Scholar]
- 8. Chen Q, Shou P, Zheng C, Jiang M, Cao G, Yang Q, et al. Fate decision of mesenchymal stem cells: adipocytes or osteoblasts? Cell Death Differ 2016;23:1128‐1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Teitelbaum SL. Bone resorption by osteoclasts. Science 2000;289:1504‐1508. [DOI] [PubMed] [Google Scholar]
- 10. Horwitz EM, LeBlanc K, Dominici M, Mueller I, Slaper‐Cortenbach I, Marini FC, et al. International Society for Cellular Therapy. Clarification of the nomenclature for MSC: the International Society for Cellular Therapy position statement. Cytotherapy 2005;7:393‐395. [DOI] [PubMed] [Google Scholar]
- 11. Bihari C, Anand L, Rooge S, Kumar D, Saxena P, Shubham S, et al. Bone marrow stem cells and their niche components are adversely affected in advanced cirrhosis of the liver. Hepatology 2016;64:1273‐1288. [DOI] [PubMed] [Google Scholar]
- 12. D’Amico G, Garcia‐Tsao G, Cales P, Escorsell A, Nevens F, Cestari R,et al. Diagnosis of portal hyper‐tension: how and when In: de Franchis R, ed. Portal Hypertension III: Proceedings of the Third Baverno International Consensus Workshop on Definitions, Methodology and Therapeutic Strategies. Oxford: Blackwell Science Ltd.; 2001:36‐64. [Google Scholar]
- 13. Lee NK, Sowa H, Hinoi E, Ferron M, Ahn JD, Confavreux C, et al. Endocrine regulation of energy metabolism by the skeleton. Cell 2007;130:456‐469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chow A, Lucas D, Hidalgo A, Méndez‐Ferrer S, Hashimoto D, Scheiermann C, et al. Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J Exp Med 2011;208:261‐271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shin J, Maekawa T, Abe T, Hajishengallis E, Hosur K, Pyaram K, et al. DEL‐1 restrains osteoclastogenesis and inhibits inflammatory bone loss in nonhuman primates. Sci Transl Med 2015;7:307ra155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Luo J, Yang Z, Ma Y, Yue Z, Lin H, Qu G, et al. LGR4 is a receptor for RANKL and negatively regulates osteoclast differentiation and bone resorption. Nat Med 2016;22:539‐546. [DOI] [PubMed] [Google Scholar]
- 17. Wang H, Yoshiko Y, Yamamoto R, Minamizaki T, Kozai K, Tanne K, et al. Overexpression of fibroblast growth factor 23 suppresses osteoblast differentiation and matrix mineralization in vitro. J Bone Miner Res 2008;23:939‐948. [DOI] [PubMed] [Google Scholar]
- 18. Marupanthorn K, Tantrawatpan C, Kheolamai P, Tantikanlayaporn D, Manochantr S. Bone morphogenetic protein‐2 enhances the osteogenic differentiation capacity of mesenchymal stromal cells derived from human bone marrow and umbilical cord. Int J Mol Med 2017;39:654‐662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ambrosi TH, Scialdone A, Graja A, Gohlke S, Jank AM, Bocian C, et al. Adipocyte accumulation in the bone marrow during obesity and aging impairs stem cell‐based hematopoietic and bone regeneration. Cell Stem Cell 2017;20:771‐784.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hajiabbasi A, Shafaghi A, Fayazi HS, Shenavar Masooleh I, Hedayati Emami MH, Ghavidel Parsa P, et al. The factors affecting bone density in cirrhosis. Hepat Mon 2015;15:e26871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Luxon BA. Bone disorders in chronic liver diseases. Curr Gastroenterol Rep 2011;13:40‐48. [DOI] [PubMed] [Google Scholar]
- 22. Guañabens N, Parés A. Liver and bone. Arch Biochem Biophys 2010;503:84‐94. [DOI] [PubMed] [Google Scholar]
- 23. Gao R, Gao F, Li G, Hao JY. Health‐related quality of life in Chinese patients with chronic liver disease. Gastroenterol Res Pract 2012;516140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Goel V, Kar P. Hepatic osteodystrophy. Trop Gastroenterol 2010;31:82‐86. [PubMed] [Google Scholar]
- 25. Clària J, Stauber RE, Coenraad MJ, Moreau R, Jalan R, Pavesi M, et al. CANONIC Study Investigators of the EASL‐CLIF Consortium and the Europe Foundation for the Study of Chronic Liver Failure (EF‐CLIF). Systemic inflammation in decompensated cirrhosis: characterization and role in acute‐on‐chronic liver failure. Hepatology 2016;64:1249‐1264. [DOI] [PubMed] [Google Scholar]
- 26. Atashi F, Modarressi A, Pepper MS. The role of reactive oxygen species in mesenchymal stem cell adipogenic and osteogenic differentiation: a review. Stem Cells Dev 2015;24:1150‐1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tormos KV, Anso E, Hamanaka RB, Eisenbart J, Joseph J, Kalyanaraman B, et al. Mitochondrial complex III ROS regulate adipocyte differentiation. Cell Metab 2011;14:537‐544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Guarino M, Loperto I, Camera S, Cossiga V, Di Somma C, Colao A, et al. Osteoporosis across chronic liver disease. Osteoporos Int 2016;27:1967‐1977. [DOI] [PubMed] [Google Scholar]
- 29. Arase Y, Suzuki F, Suzuki Y, Akuta N, Kobayashi M, Sezaki H, et al. Virus clearance reduces bone fracture in postmenopausal women with osteoporosis and chronic liver disease caused by hepatitis C virus. J Med Virol 2010;82:390‐395. [DOI] [PubMed] [Google Scholar]
- 30. Karoli Y, Karoli R, Fatima J, Manhar M. Study of hepatic osteodystrophy in patients with chronic liver disease. J Clin Diagn Res 2016;10:OC31‐OC34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Albillos A, Lario M, Alvarez‐Mon M. Cirrhosis‐associated immune dysfunction: distinctive features and clinical relevance. J Hepatol 2014;61:1385‐1396. [DOI] [PubMed] [Google Scholar]
- 32. Patel N, Muñoz SJ. Bone disease in cirrhosis. Clin Liver Dis 2015;6:96‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Handzlik‐Orlik G, Holecki M, Wilczyński K, Duława J. Osteoporosis in liver disease: pathogenesis and management. Ther Adv Endocrinol Metab 2016;7:128‐135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang X, Wei W, Krzeszinski JY, Wang Y, Wan Y. A liver‐bone endocrine relay by IGFBP1 promotes osteoclastogenesis and mediates FGF21‐induced bone resorption. Cell Metab 2015;22:811‐824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Moschen AR, Kaser A, Stadlmann S, Millonig G, Kaser S, Mühllechner P, et al. The RANKL/OPG system and bone mineral density inpatients with chronic liver disease. J Hepatol 2005;43:973‐983. [DOI] [PubMed] [Google Scholar]
- 36. Nussler AK, Wildemann B, Freude T, Litzka C, Soldo P, Friess H, et al. Chronic CCl4 intoxication causes liver and bone damage similar to the human pathology of hepatic osteodystrophy: a mouse model to analyse the liver‐bone axis. Arch Toxicol 2014;88:997‐1006. [DOI] [PubMed] [Google Scholar]
- 37. Brandenburg VM, Floege J. Adynamic bone disease‐bone and beyond. NDT Plus 2008;1:135‐147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Coen G. Adynamic bone disease: an update and overview. J Nephrol 2005;18:117‐122. [PubMed] [Google Scholar]
- 39. Rouillard S, Lane NE. Hepatic osteodystrophy. Hepatology 2001;33:301‐307. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials