

Abstract
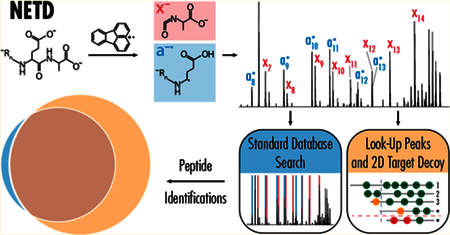
Negative electron-transfer dissociation (NETD) has emerged as a premier tool for peptide anion analysis, offering access to acidic post-translational modifications and regions of the proteome that are intractable with traditional positive-mode approaches. Whole-proteome scale characterization is now possible with NETD, but proper informatic tools are needed to capitalize on advances in instrumentation. Currently only one database search algorithm (OMSSA) can process NETD data. Here we implement NETD search capabilities into the Byonic platform to improve the sensitivity of negative-mode data analyses, and we benchmark these improvements using 90 min LC−MS/MS analyses of tryptic peptides from human embryonic stem cells. With this new algorithm for searching NETD data, we improved the number of successfully identified spectra by as much as 80% and identified 8665 unique peptides, 24639 peptide spectral matches, and 1338 proteins in activated-ion NETD analyses, more than doubling identifications from previous negative-mode characterizations of the human proteome. Furthermore, we reanalyzed our recently published large-scale, multienzyme negative-mode yeast proteome data, improving peptide and peptide spectral match identifications and considerably increasing protein sequence coverage. In all, we show that new informatics tools, in combination with recent advances in data acquisition, can significantly improve proteome characterization in negative-mode approaches
Keywords: negative mode, peptide anions, negative electron-transfer dissociation, activated ion, search algorithm, two-dimensional target decoy, multiple proteases
INTRODUCTION
The emergence of dissociation methods suitable for reprodu-cible, sequence-informative fragmentation of peptide anions has recently enabled high-throughput proteomics in the negative mode.1–3 Nearly all proteomic analysis has hitherto been done in the positive mode and consequently preferentially detects basic peptides and proteins. Complementary proteome analysis in the negative mode holds strong potential to offer a unique view of acidic species that may be discriminated against using the standard methodology. Negative electron-transfer dissoci-ation (NETD) is a primary method for peptide anion dissociation, mitigating the challenges seen with collision-based dissociation of deprotonated peptides.3–8 By leveraging electron rearrangements driven by oxidation of peptide anions with reagent cations, NETD generates α•- and x-type product ions through cleavage of the C−Cα backbone bond to consistently yield the sequence information necessary for shotgun proteomic experiments.9
NETD enabled the first description of large-scale proteomic studies on peptide anions in 2012, but low NETD dissociation efficiencies and poor chromatographic reproducibility with high-pH solvents remained major challenges to routine use.10,11 We recently addressed these shortcomings by (1) modifying a dual-cell linear ion trap−Orbitrap hybrid mass spectrometry (MS) system to perform activated-ion NETD (AI-NETD) for improved precursor-to-product ion conversion and (2) devoting significant effort to investigating five different high-pH buffer systems.3 Ultimately these improvements facilitated the largest scale negative-mode proteomic study to date, characterizing more than 1100 proteins in yeast with single-shot analyses and more than 3700 yeast proteins using an offine fractionation approach. With a platform suitable for whole-proteome-scale characterization of peptide anions, the stage is now set to pursue high-throughput characterization of acidic peptides, post-translational modifications (PTMs), and other biomolecules that are recalcitrant to positive-mode methods.12–22
The aforementioned advances in instrumentation, however, are not yet paired with modern informatic tools. Specifically, shotgun proteome analysis, whether in the positive or negative mode, relies on search algorithms for automated spectral interpretation and peptide identification. While dozens of algorithms have been developed for the interpretation of positive-mode tandem mass spectra, only one search algorithm, the Open Mass Spectrometry Search Algorithm (OMSSA), is capable of searching NETD and AI-NETD spectra.10,23 (We do note that MassMatrix was recently modified to handle negative-mode ultraviolet photodissociation (UVPD) spectra, and this algorithm might also be adapted to support NETD spectral analysis.2) Further compounding this issue is that OMSSA development ceased several years ago. So, in a very real sense, there exists no modern and regularly maintained software algorithm to interpret NETD spectra. We conclude that for negative-mode proteome analysis to reach its full potential considerable effort to develop appropriate data analysis tools must be exerted.
Toward this overall goal, we describe here the modification of the Byonic search algorithm to accommodate NETD and AI-NETD spectra. Byonic is a modern and fully functional proteomics software suite that incorporates advanced database searching approaches and postsearch data visualization.24 As a hybrid between pure database search approaches (such as SEQUEST25 and OMSSA) and De Novo sequencing programs, Byonic employs a lookup-peak strategy for improved sensitivity, and a scoring algorithm for tandem MS (MS/MS) spectra takes account of both predicted and observed peak intensities as well as accuracy of mass matches.26 Byonic also includes a 2D target-decoy strategy that estimates and controls peptide spectral match (PSM) and protein false discovery rates (FDRs) simultaneously, further improving PSM sensitivity for high-scoring proteins.27 After adapting Byonic for NETD tandem mass spectra, we characterize the algorithm’s perform-ance. Beyond improving peptide and protein identifications over OMSSA, we demonstrate that the combination of AI-NETD fragmentation and Byonic data processing enables the largest negative-mode analysis of the human proteome to date, doubling protein identifications from previous work.2 We also report a reanalysis of our published large-scale AI-NETD data set from yeast, including analyses from five proteases,3 where Byonic significantly improves peptide identifications and protein sequence coverage.
EXPERIMENTAL SECTION
Sample Preparation
Human embryonic stem cells (hESC, line H1) were prepared as described in Phanstiel et al.28 Cells were resuspended in lysis buffer (50 mM Tris, pH 8; 8 M urea; 75 mM sodium chloride; 10 mM sodium butyrate; protease and phosphatase inhibitor tablets (Roche Diagnostics, Indianapolis, IN)) and were lysed by glass bead milling (Retsch, Haan, Germany). Lysate protein concentration was measured via BCA (Thermo Pierce, Rockford, IL). Proteins were reduced with 5 mM dithiothreitol (45 min at 58 °C), free cysteines were alkylated with 15 mM iodoacetamide (in the dark for 30 min), and alkylation was quenched with 5 mM DTT. A 1 mg protein aliquot was digested overnight at room temperature in 1.5 M urea with trypsin (Promega, Madison, WI) added at a 1:50 (w/w) enzyme to protein ratio. A second trypsin addition was performed in the morning at a 1:100 (w/w) enzyme to protein ratio for 1 h. The digestion was quenched by the addition of TFA and desalted over tC18 Sep-Pak cartridges (Waters, Milford, MA). Yeast samples were prepared as previously described.3
LC−MS/MS
All data collection was performed on a modified dual-cell linear ion trap-Orbitrap hybrid MS system (Thermo Fisher Scientific, San Jose, CA) outfitted with a multipurpose dissociation cell (MDC) and 10.6 μm continuous wave CO2 laser (Synrad, Mukilteo, WA), as previously described.3,29–31 Mobile phase A consisted of 5 mM piperidine in water and mobile phase B was 5 mM piperidine in 85% acetonitrile, 15% water. A nano-electrospray tip was laser pulled (Sutter Instrument Company, Novato, CA) and packed with 3.5 μm diameter, 130 Å pore size ethylene-bridged hybrid C18 particles (Waters, Milford, MA) to a length of 30−35 cm. Reversed-phase columns were packed in-house using 75 μm inner diameter, 360 μm outer diameter bare fused silica capillary and were installed on a nano-ACQUITY UltraPerformance LC (Waters) using a stainless-steel ultrahigh pressure union formatted for 360 μm outer diameter columns (IDEX). One microgram of human peptides was loaded onto the column in 95% A for 10 min, and the elution gradient increased linearly from 5 to 30% B over 70 min, followed by an increase to 70% B at 76 min and a wash at 70% B for 4 min (all at 400 nL per min). The column was then re-equilibrated at 5% B for 10 min. Eluting peptide anions were converted to gas-phase ions by electrospray ionization at −1.5 kV with respect to ground, and the inlet capillary temperature was held at 300 °C. Survey scans of peptide precursors were collected with an automatic gain control (AGC) target value of 1 000 000 over the 300−1250 Th range, followed by data-dependent NETD MS/MS scans of the 10 most intense precursors with AGC target value of 100 000. Maximum injection times were 200 ms for both full and tandem MS scans. Precursors with charge states equal to one or unassigned were rejected. Precursors were isolated using a ±0.9 Th isolation window, and an exclusion window of 10 ppm was constructed around the monoisotopic peak of each selected precursor for 45 s. Resolving powers of 60 000 and 15 000 at 400 m/z were used for survey scans and MS/MS scans in the Orbitrap, respectively. Reagent accumulation times were set to 20 ms, and reaction times were 40 ms for z = −2, 30 ms for z = −3, 20 ms for z = −4, 15 ms for z = −5, and 10 ms for z = −6 and higher charge states. The laser was triggered using instrument firmware and modification to instrument code in conjunction with a gated laser controller for AI-NETD reactions. The laser irradiated the trapping volume of the MDC during the entirety of the NETD reaction at 70% total output. Three triplicate injections (1 μg peptides each) of the same complex mixture of hESC tryptic peptides were collected for both NETD and AI-NETD analyses.
Data Analysis
OMSSA was previously modified to accommodate NETD spectra of anionic peptides, allowing for α•- and x-type productions.10 Prior to the OMSSA search, a ±3 Da window around the unreacted precursor and the window 55 Da below and 5 Da above the charge-reduced precursor peaks were cleaned from the spectra. This was done because inclusion of unreacted precursor peaks, charge-reduced precursor peaks, and neutral loss peaks from the charge-reduced precursor ions are known to drastically hurt scoring in OMSSA and other search algorithms.32,33 A multi-isotope search using three isotopes with a mass tolerance of ±40 ppm was used for precursors, and a monoisotopic mass tolerance of ±0.01 Da was used for product ions (α•- and x-type). Data processing was performed using COMPASS software designed for OMSSA outputs.34 Byonic was modified for NETD and AI-NETD spectra as described below. No spectral cleaning was performed prior to analysis with the Byonic pipeline. A precursor mass tolerance was set to ±10 ppm with a Precursor off by x setting of “too high (narrow)” and a product ion tolerance of ±20 ppm, and peptide spectral score cuts were set to automatic. For both OMSSA and Byonic, oxidation of methionine and pyro-glu from N-terminal glutamine and glutamate were specified as variable modifications, while carbamidomethylation of cysteine was a set as a fixed modification. For all enzymes, three missed cleavages were allowed with the following specificity: trypsin (RK|), lysC (K|), GluC (DE|), chymotrypsin (FWYL|), and AspN (|D). For human data for both search algorithms, PSMs were made against the UniProt human database (canonical plus isoforms) downloaded on November 6, 2015, which was concatenated with a reversed sequence version of the forward database. Yeast data were searched similarly with both OMSSA and Byonic using the UniProt yeast database downloaded on September 29, 2014. For OMSSA, unique peptides were filtered to a 1% FDR using both e-value and precursor mass accuracy, and proteins were also filtered to 1% FDR. For Byonic, protein-oblivious FDR estimation reports 1% FDR at the unique peptide level, while protein-aware reports FDR on the PSMs from proteins ranked above the top decoy protein, using the 2D target decoy approach available in Byonic.27 When pooling spectra from multiple nLC−MS/MS analyses, FDR was calculated for the aggregate set of data rather than calculating a separate false discovery rate for each run prior to combining results.
RESULTS AND DISCUSSION
With the ultimate goal to improve large-scale negative-mode proteome analysis by the development of postacquisition informatics tools, we modified the Byonic algorithm to analyze NETD and AI-NETD spectra. To guide this modification process we utilized a data set comprising 11 433 AI-NETD MS/MS spectra from the analysis of yeast tryptic peptides, although no formal training of predicted peak weights for scoring NETD spectra was performed. From these experiments we made several spectral processing decisions. First, we employed a simple scorer for singly charged α•- and x-type product ions. Following successful spectral annotation, we expanded the scorer to include multiply charged fragments and isotope peaks from the higher-mass fragments. When analyzing high-resolution MS/MS spectra, we allowed the algorithm to score each production’s isotope series. For low-resolution MS/ MS data, we considered only the monoisotopic m/z peak for product ions up to 1200 Da, switching to an approximation of the average isotopic mass for larger ones.
In Byonic each ion predicted and observed adds to the score; each ion predicted and not observed subtracts from the score. The amount added depends on the predicted intensity, observed intensity, and closeness of the m/z match (where a peak within 0.01 of the predicted m/z scores better than a peak within 0.02). The amount subtracted depends only on the predicted intensity. Intensities are not raw intensities but depend instead on the rank of the peak (tallest, second tallest, and so forth). For NETD spectra, the algorithm predicted roughly equal intensities for all α•- and x-type product ions. On the basis of the yeast data set, we adjusted scoring for doubly deprotonated precursors to put the remaining charge preferentially on the more acidic half. With this, the algorithm predicts low intensities for product ions with less than half the peptide’s total acidity (number of carboxyl groups) for z = −2 precursors. The predicted intensities can have a rank of none, weak, or strong. The smaller or less acidic half of a z = −2 precursor is predicted to be weak, while all other scored peaks are predicted to be strong. Finally, we introduced scoring of NETD spectral features apart from product ion series, including residual precursor ions, charge-reduced precursor ions, and neutral loss of CO2 from charge-reduced precursors. We elected not to remove m/z peaks from spectra prior to scoring. Inclusion of nonsequence-informative m/z peaks characteristic of ion−ion reactions can significantly hurt scoring in many search algorithms. Byonic’s use of rank, rather than raw, intensity limits the influence of a small number of intense peaks, such as unreacted precursors and side-chain neutral losses, making it well-suited to handle NETD and AI-NETD spectra without spectral preprocessing.11,33,35 With these requisite modifications outlined, we anticipate a straightforward recipe for the adoption of other search algorithms to accommodate NETD spectra.
Following these modifications, we characterized the perform-ance of Byonic for both NETD and AI-NETD spectra using technical triplicate 90 min LC−MS/MS analyses (i.e., three injections of the same digest) of tryptic peptides from human embryonic stem cells (hESCs). The average number of NETD and AI-NETD MS/MS spectra acquired per analysis was 13 963 and 13 753, respectively. We also compared Byonic’s performance to OMSSA, focusing on identifications of unique peptides, PSMs, and proteins as benchmarks. Note that the 2D target decoy approach (2D-FDR) employed by Byonic can contribute to significant gains for unique peptides and PSMs27 but does not necessarily reflect improvements in the spectral scoring algorithm. To decouple the improvements from the search algorithm and the gains from the 2D-FDR method, we report two results from Byonic. The protein-oblivious results provide a more straightforward comparison to OMSSA, where spectral hits are filtered to 1% FDR at the unique peptide level. The protein-aware results represent standard Byonic output and use the 2D-FDR approach to rerank PSMs based on high-scoring protein identities.
To compare the sensitivity and specificity of the different search algorithms, we generated a receiver operating character-istic (ROC) curve for batched AI-NETD LC−MS/MS analyses where the number of unique peptides identified are plotted against percent FDR (Figure 1A). These data reveal that operation of Byonic in either protein-oblivious or protein-aware mode results in more unique peptide identifications than OMSSA (1% FDR). The protein-aware option adds an additional 1139 identifications, while the protein-oblivious iteration provides a modest boost of 349 additional peptides (15 and 5% boosts, respectively). We conclude that the standalone Byonic scoring algorithm is superior to OMSSA for NETD spectral analysis, but only moderately so; however, the combination of the improved scoring and the 2D-FDR calculation provides the more substantial improvement.
Figure 1.
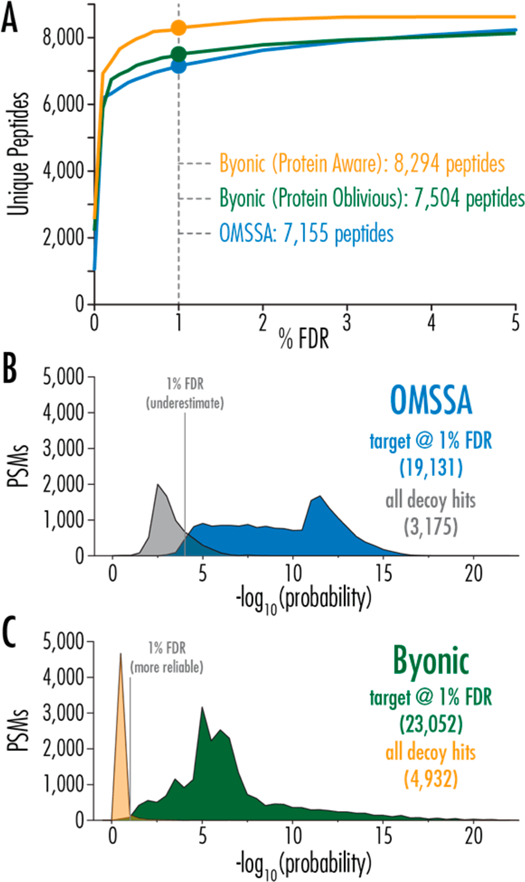
(A) Receiver−operator characteristic curve for AI-NETD data demonstrates the improved sensitivity of the Byonic scoring algorithm. OMSSA (blue) is compared with Byonic with both the protein-oblivious (standard 1D FDR, green) and the protein-aware (advanced 2D FDR, gold) results shown. For each curve, unique peptides are reported for the given percent FDR at the peptide level. (B,C) Comparison of the probability distributions of target and decoy hits for OMSSA and Byonic, respectively. For both plots, all decoy hits (not filtered) from the searches are shown (gray and gold, respectively), while the PSM scores graphed (blue and green, respectively) are filtered based on 1% FDR at the peptide level, suggesting that OMSSA’s FDR estimate may be an underestimate.
To further compare the spectral scoring algorithms, we plotted the probabilities scores (adjusted p values)36 of PSMs returned from OMSSA (Figure 1B) and Byonic (protein-oblivious) (Figure 1C), which were filtered to 1% FDR at the peptide level (i.e., the PSM hits corresponding to the unique peptide identifications shown in the blue and green lines in panel A). The adjusted p values for all decoy hits (unfiltered) are also included, showcasing the differences in scoring for incorrect hits between the two algorithms. The improved sensitivity of Byonic is represented by the narrower decoy distribution, allowing inclusion of more confidently matched spectra in the target (PSM) distribution. This is likely due to use of robust statistics (peak ranks rather than presence/ absence or raw intensities) and soft decision boundaries (for example, a penalty rather than a hard threshold for mass errors) used by Byonic but not OMSSA.27,37
Figure 2A compares the average number of PSMs identified by OMSSA and Byonic algorithms in triplicate NETD and AI-NETD analyses, where the error bars indicate one standard deviation. Figure 2B,C shows the overlap in unique peptides from OMSSA and Byonic (protein-aware) for NETD and AI-NETD, respectively. The high degree of overlap in peptide identifications between the two search engines, rather than simply identifying disparate populations of peptides from the same population of spectra, lends further confidence to the additional peptide identifications using the modified Byonic algorithm for NETD and AI-NETD spectra. For the few peptides that were identified by OMSSA but not Byonic, the majority of identifications come from low quality spectra (Supplemental Figure 1), which likely did not pass Byonic’s scoring/FDR thresholds. These may also be a result of the slight FDR underestimate seen with OMSSA in Figure 1. Most importantly, however, >95% of peptides sequenced with OMSSA were also identified with Byonic for both NETD and AI-NETD.
Figure 2.
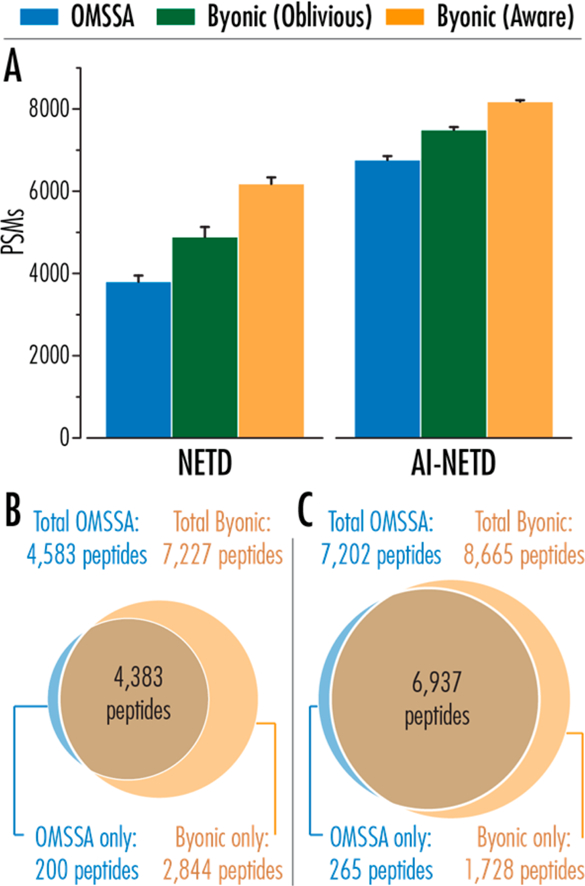
(A) Numbers of peptide spectral matches from NETD and AI-NETD data are improved by both the protein-oblivious and protein-aware FDR calculations. Results are an average from three replicate LC−MS/MS analyses and error bars show one standard deviation. (B,C) Overlap in identified peptides between OMSSA and Byonic for batched triplicate analyses of NETD and AI-NETD, respectively.
To understand the parameters that contributed to improve-ments in identifications with Byonic, we next investigated the performance of each algorithm as a function of precursor ion charge state and m/z (Figure 3). Here performance is described in terms of MS/MS success rate (%). For comparing charge states, success rate is defined as the number of identified spectra (i.e., PSMs) at a given charge state divided by the total number of MS/MS scans of precursors with that charge state. Similarly, success rate when comparing precursor ion m/z is defined as the number of PSMs accepted at 1% FDR in a given 50 Th bin divided by the number of total MS/MS scans of precursor ions with m/z in that bin. Both the protein-oblivious and protein-aware Byonic search strategies improve success rates over OMSSA at all precursor charge states for NETD and AI-NETD. The Byonic scoring algorithm alone (i.e., protein-oblivious strategy) provided the largest improvements in identification of precursor ions with high charge states (z = −4 and higher), while protein-aware algorithm afforded the substantial gain in identifications of z = −2 precursor ions (Figure 3A). The protein-oblivious algorithm performs similarly to OMSSA for NETD MS/MS success rate for the lowest m/z precursors but continues to improve success rates over OMSSA as precursor m/z increases (Figure 3B). In contrast, the protein-aware algorithm provides consistent boost in NETD MS/MS success rate over OMSSA across the m/z range. In all, the Byonic scoring algorithm (i.e., protein-oblivious) improves precursor identification success rates over OMSSA for all precursor charge states across the m/z range, while the protein-aware algorithm contributes varying degrees of improvement in addition to the protein-oblivious strategy depending on precursor m/z and charge. Figure 3 also highlights the superior performance of AI-NETD over NETD for all precursors, especially low-charge density (z = −2) and high m/z precursor ions.38–40
Figure 3.
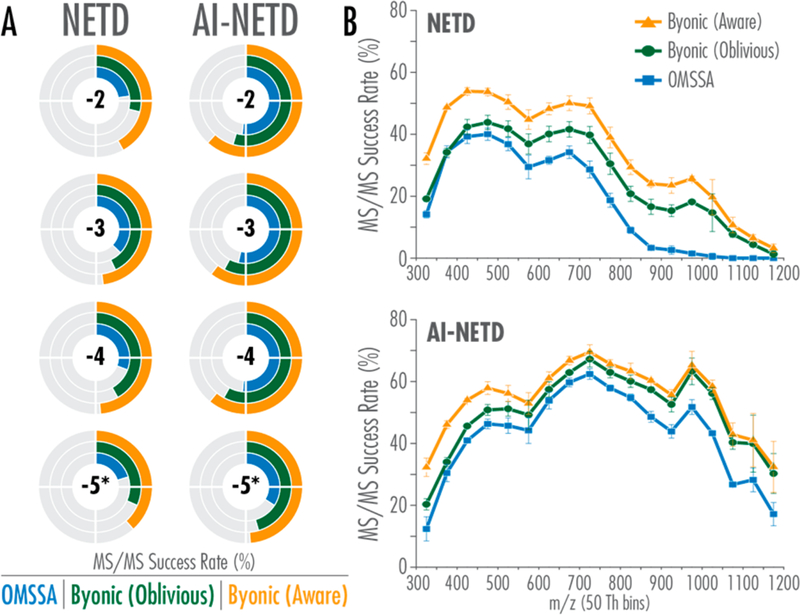
(A) Both protein-oblivious (green) and protein-aware (gold) FDR calculations improve the MS/MS success rate over OMSSA (blue) at all charge states for both NETD and AI-NETD. Reported success rates are average values for three triplicate injections. For each graph, a full circle would indicate a success rate of 100%. * includes z = −5 and all higher charge states. (B) MS/MS success rates in NETD and AI-NETD are improved across the m/z range as well. Plotted values are an average of three triplicate injections, and error bars show one standard deviation.
For all of the metrics in Figures 2 and 3, the newly modified search algorithm offers a more considerable improvement for NETD analyses than for AI-NETD. This result reiterates the differences in scoring approaches between Byonic and OMSSA and confirms previously observed characteristics of NETD and AI-NETD spectra.3,41,42 The reliance of OMSSA on spectra with higher numbers of matching fragment ions gives AI-NETD a distinct advantage in OMSSA workflows because it improves dissociation efficiency and generates more product ions than NETD, especially for z = −2 precursor ions. Thus, the more sophisticated scoring and 2D-FDR approaches of Byonic have more to offer to the (generally) lower quality NETD spectra because they can salvage informative spectra even when only a modest number of matching fragment ions are present, as shown in Supplemental Figure 2. This is especially evident in Figure 3 and Supplemental Figure 2, where Byonic (protein-aware) nearly doubles the NETD MS/MS success rate for z = −2 precursors and significantly improves success rates at all m/z values for NETD. Note, however, that even with these improvements, NETD cannot match the efficacy of AI-NETD fragmentation.
The improvements in PSM and peptide identification we obtained with our modified Byonic search algorithm translate to improved protein characterization. Figure 4A presents the average number of protein identifications from the triplicate injections of hESC tryptic peptides with both NETD and AI-NETD, again with error bars indicating one standard deviation. Only protein-aware results are shown, as the protein grouping and scoring algorithm requires the 2D-FDR component. Figure 4B displays the overlap in protein identifications between the two search algorithms from the batched AI-NETD analyses, where nearly 98% of proteins identified by OMSSA are also identified by Byonic. Of the 73 proteins that are only identified by OMSSA, most of them can be explained by the “OMSSA Only” peptides discussed above (Supplemental Figure 3C). The remaining proteins are likely identified differently in OMSSA than Byonic because protein grouping is a complex process that relies on several facets to distinguish non-subsumable protein groups from each other based on evidence at the peptide level.43,44 Beyond the difference in algorithms used, the difference in peptide identifications (i.e., the more unique peptides identified with Byonic) can contribute to differences of protein grouping, making it a difficult incongruency to address. We hypothesize that the more sophisticated protein grouping performed by Byonic27 is more parsimonious and conservative than the grouping done with OMSSA results. Even so, the substantial majority of protein identifications from both search engines overlapped. A greater number of PSMs are identified per protein in the Byonic analysis than the OMSSA analysis (Figure 4C), reflecting the improved number of total PSMs observed with Byonic. There are a few artifacts in this comparison from the protein grouping differences, but the general trend clearly shows more PSMs per protein with Byonic, offering improved protein characterization and confidence in its identification. Similar results were observed for NETD as well (Supplemental Figure 3).
Figure 4.

(A) Byonic (gold) identifies more protein groups than OMSSA (blue) for both NETD and AI-NETD analyses. Results are an average of triplicate injections, and error bars show one standard deviation. (B) Overlap in identified proteins from batched triplicate AI-NETD analyses using each search algorithm. (C) For the protein groups identified by both programs, the number of PSMs per protein group is plotted with Byonic on the y axis and OMSSA on the x axis (for those with 120 or fewer PSMs per protein). The inset histogram shows the distribution of ΔPSMs, which is the difference of PSMs identified with Byonic minus PSMs identified with OMSSA, for all 1167 protein groups shared between the two algorithms. A positive number indicates more PSMs for a given protein group with Byonic.
In total, our modified Byonic search algorithm facilitated the identification of 18 837 PSMs (7277 unique) and 1089 proteins from hESCs using NETD. AI-NETD further improved upon these results, producing 24 639 PSMs (8665 unique) and 1338 protein identifications. For NETD and AI-NETD our new search algorithm provided 81.1 and 28.7% increases in PSM identifications over OMSSA, respectively (see Supplemental Figure 4 for full set of comparisons). Beyond introducing an improved and more accessible tool for automated NETD spectral interpretation, this study is the first to report analysis of human peptides with NETD and AI-NETD and represents the most thorough negative-mode characterization of the human proteome to date.
Multiple-Protease Data Re-Analysis
The gains in peptide and protein identifications above motivated us to re-examine the large-scale AI-NETD yeast (Saccharomyces cerevisiae) data set from our previous work.3 That data, representing over 83% of the yeast proteome and the largest negative-mode proteomics data set available, includes single-shot experiments (90 min LC−MS/MS analyses) and deep-sequencing analyses (10 fractions from offine low pH RP fractionation with each fraction being a 90 min LC-MS/MS acquisition) for five different proteases (trypsin, LysC, chymotrypsin, GluC, and AspN). Figure 5 summarizes the performance of the modified Byonic search algorithm (protein-aware and protein-oblivious) compared with the previous OMSSA analysis for all of these data.
Figure 5.
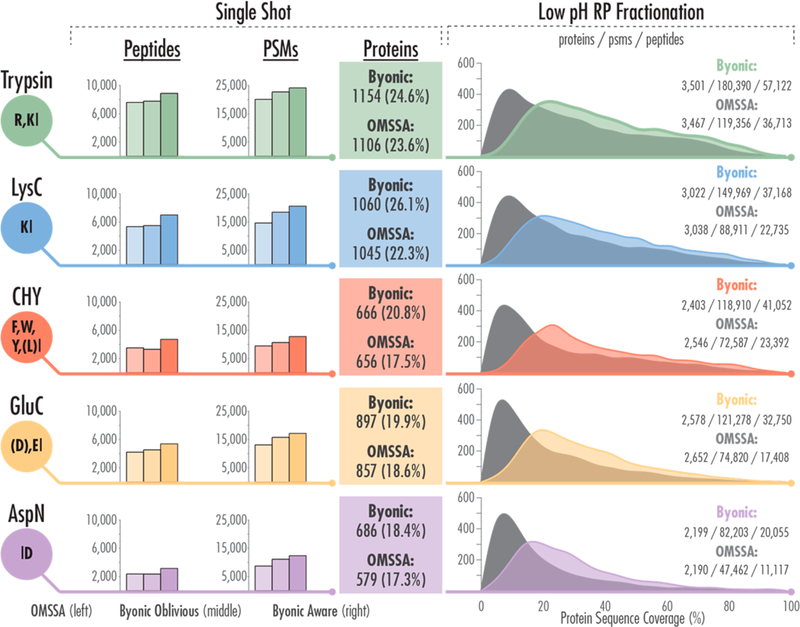
We compared our new informatics pipeline to previously reported AI-NETD data, which included MS/MS spectra of peptides generated from multiple proteases (trypsin, LysC, chymotrypsin [CHY], GluC, and AspN). Each color shows results from a different protease. Data from single-shot analyses are given on the left side of the figure, showing bar charts for peptides and PSMs, with the left most bar in each triad being OMSSA, the middle being Byonic with protein-oblivious FDR, and the rightmost bar being Byonic with protein-aware FDR for each. Protein identifications from the single shot work are given in the colored boxes with average percent protein sequence coverage shown in parentheses. The deep-sequencing (fractionated) results are shown on the right of the Figure with histograms depicting percent protein sequence coverage for proteins identified from Byonic (color) and OMSSA (gray). Numbers of protein identifications, PSMs, and unique peptide identifications are also given as indicated.
Overall, Byonic outperformed OMSSA for data from each protease, especially using the 2D-FDR approach. The number of PSMs identified with both protein-oblivious and protein-aware algorithms is greater than with OMSSA for all five proteases. Additionally, the protein-aware approach not only increased the number of proteins identified for each protease in the single-shot experiments but also increased the average protein sequence coverage (colored text boxes in middle of Figure 5). The number of peptide identified from each protease relative to the other proteases remained similar between the two search algorithms, with the exception, with the exception of AspN and chymotrypsin. With OMSSA, more PSMs and proteins were identified from the chymotrypsin data than the AspN data, whereas Byonic favored AspN slightly for both metrics.
The slight decreases in unique peptide identifications seen with the protein-oblivious algorithm for the chymotrypsin and AspN single-shot experiments (left bar graphs in Figure 5) may reflect bias in using a tryptic peptide data set to develop NETD/AI-NETD capabilities of Byonic; this indicates a metric where the scoring algorithm may be improved in future iterations. A similar trend is also evident in the deep-sequencing data, where Byonic identified 0.5−5.6% fewer proteins than OMSSA for lysC, chymotrypsin, and gluC data (but marginally outperformed OMSSA for AspN and trypsin). Although the protein identifications are roughly equivalent or slightly fewer with Byonic compared with OMSSA, Byonic greatly increased the number of unique peptide and PSM identifications, as reflected in the increased protein sequence coverage for each deep-sequencing data set (histograms to the right in Figure 5). This shows that the modified Byonic algorithm is not only is fully capable of processing large negative-mode proteomic data from multiple proteases but also can significantly increase the information gleaned for each protein identification and increase the confidence in its characterization over the current OMSSA approach.
CONCLUSIONS
We have demonstrated a substantial improvement in NETD and AI-NETD data analysis using a modified search algorithm that was originally designed for positive mode use, providing evidence that further gains in negative-mode proteomics can be accomplished through continued development of informatic tools. The Byonic features that proved the most critical for the improvements described in this work include the (1) rank-based scoring that does not necessitate spectral preprocessing, (2) the use of m/z errors in the scoring algorithm, (3) the NETD fragment ion predicted intensities based on longer or more acidic sequences of precursor peptides, and (4) the 2D-FDR protein-aware calculations. Approximately half of the sensitivity gain is accounted for with the improved scoring (items 1−3) and the other half is from 2D-FDR.
While this is a step forward in providing necessary improvements over current informatics tools, there is still room for improvement to design an even more tailored algorithm to better process NETD data. These improvements will hinge on the continued collection of large NETD data sets, from which patterns of NETD can be deduced and incorporated into spectral scoring. Future studies will seek to improve scoring algorithms based on fragment ion abundances, favored cleavages, hydrogen atom rearrangements, and other spectral features, which are now commonly known for positive-mode fragmentation methods but remain to be elucidated for NETD and AI-NETD. Experiments exploring the benefits NETD and AI-NETD in combination with this new search algorithm for PTM analyses are currently underway as well. On the whole, this work represents a crucial step forward in the informatics tools available for analysis of large-scale negative-mode proteomic data, which has been made possible by recent advances in instrumentation.3,9–11 High-throughput NETD and AI-NETD experiments can now be analyzed within the framework of a full-featured, modern proteomics search engine, complete with sophisticated target decoy approaches, auto-mated spectral annotation, and other visualization tools. This advance makes data analysis more robust and streamlined for proteomic users and offers a route to accessible informatics tools for negative-mode proteomic experiments.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge support from Thermo Fisher Scientific and NIH grants R35 GM118110 and R21 GM118341. N.M.R. was funded through an NSF Graduate Research Fellowship (DGE-1256259). We also thank Alicia Richards and the Jamie Thomson lab for help preparing the hESC sample.
Footnotes
Notes
The authors declare no competing financial interest.
All raw files for hESC tryptic peptide analyses with NETD and AI-NETD are available on Chorus (Project ID 1042, Experiment ID 2225). Supporting Information, also available on Chorus, includes peptide spectral matches and protein identifications from hESC samples with OMSSA and Byonic for NETD and AI-NETD data. Also included are identifications from Byonic from the reanalysis of the multiple protease data from yeast. Supporting documents include: NETD Byonic Supporting Information.pdf, NETD hESC PSMs.xlsx, NETD hESC Proteins.xlsx, AI-NETD hESC PSMs.xlsx, AI-NETD hESC Proteins.xlsx, Byonic_AI-NETD_Yeast_MultiProtease_-SingleShot.xlsx, and Byonic_AI-NETD_Yeast_MultiProtease_-Fractionated.xlsx.
REFERENCES
- (1).Brodbelt JS Ion Activation Methods for Peptides and Proteins. Anal. Chem 2016, 88 (1), 30–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Madsen JA; Xu H; Robinson MR; Horton AP; Shaw JB; Giles DK; Kaoud TS; Dalby KN; Trent MS; Brodbelt JS High-throughput database search and large-scale negative polarity liquid chromatography-tandem mass spectrometry with ultraviolet photodissociation for complex proteomic samples. Mol. Cell. Proteomics 2013, 12 (9), 2604–2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Riley NM; Rush MJP; Rose CM; Richards AL; Kwiecien NW; Bailey DJ; Hebert AS; Westphall MS; Coon JJ The Negative Mode Proteome with Activated Ion Negative Electron Transfer Dissociation. Mol. Cell. Proteomics 2015, 14, mcp.M115.049726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Brinkworth CS; Dua S; McAnoy AM; Bowie JH Negative ion fragmentations of deprotonated peptides: backbone cleavages directed through both Asp and Glu. Rapid Commun. Mass Spectrom 2001, 15 (20), 1965–1973. [DOI] [PubMed] [Google Scholar]
- (5).Steinborner ST; Bowie JH A comparison of the positive- and negative-ion mass spectra of bio-active peptides from the dorsal secretion of the Australian red tree frog, Litoria rubella. Rapid Commun. Mass Spectrom 1996, 10 (10), 1243–1247. [DOI] [PubMed] [Google Scholar]
- (6).Steinborner ST; Bowie JH The Negative Ion Mass Spectra of [MH]− Ions Derived From Caeridin and Dynastin Peptides. Internal Backbone Cleavages Directed Through Asp and Asn Residues. Rapid Commun. Mass Spectrom 1997, 11 (No. 11), 253–258. [Google Scholar]
- (7).Bowie JH; Brinkworth CS; Dua S Collision-induced fragmentations of the (M-H)- parent anions of underivatized peptides: an aid to structure determination and some unusual negative ion cleavages. Mass Spectrom. Rev 2002, 21 (2), 87–107. [DOI] [PubMed] [Google Scholar]
- (8).Huzarska M; Ugalde I; Kaplan DA; Hartmer R; Easterling ML; Polfer NC Negative electron transfer dissociation of deprotonated phosphopeptide anions: choice of radical cation reagent and competition between electron and proton transfer. Anal. Chem 2010, 82 (7), 2873–2878. [DOI] [PubMed] [Google Scholar]
- (9).Coon JJ; Shabanowitz J; Hunt DF; Syka JEP Electron transfer dissociation of peptide anions. J. Am. Soc. Mass Spectrom 2005, 16 (6), 880–882. [DOI] [PubMed] [Google Scholar]
- (10).McAlister GC; Russell JD; Rumachik NG; Hebert AS; Syka JEP; Geer LY; Westphall MS; Pagliarini DJ; Coon JJ Analysis of the acidic proteome with negative electron-transfer dissociation mass spectrometry. Anal. Chem 2012, 84 (6), 2875–2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Rumachik NG; McAlister GC; Russell JD; Bailey DJ; Wenger CD; Coon JJ Characterizing peptide neutral losses induced by negative electron-transfer dissociation (NETD). J. Am. Soc. Mass Spectrom 2012, 23 (4), 718–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Robinson MR; Moore KL; Brodbelt JS Direct identification of tyrosine sulfation by using ultraviolet photo-dissociation mass spectrometry. J. Am. Soc. Mass Spectrom 2014, 25 (8), 1461–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Flora JW; Muddiman DC Selective, Sensitive, and Rapid Phosphopeptide Identification in Enzymatic Digests Using ESI-FTICR-MS with Infrared Multiphoton Dissociation. Anal. Chem 2001, 73 (14), 3305–3311. [DOI] [PubMed] [Google Scholar]
- (14).Kjeldsen F; Horning OB; Jensen SS; Giessing AM; Jensen ON Towards liquid chromatography time-scale peptide sequencing and characterization of post-translational modifications in the negative-ion mode using electron detachment dissociation tandem mass spectrometry. J. Am. Soc. Mass Spectrom 2008, 19 (8), 1156–1162. [DOI] [PubMed] [Google Scholar]
- (15).Hersberger KE; Hakansson K Characterization of O-sulfopeptides by negative ion mode tandem mass spectrometry: superior performance of negative ion electron capture dissociation. Anal. Chem 2012, 84 (15), 6370–6377. [DOI] [PubMed] [Google Scholar]
- (16).Nwosu CC; Strum JS; An HJ; Lebrilla CB Enhanced detection and identification of glycopeptides in negative ion mode mass spectrometry. Anal. Chem 2010, 82 (23), 9654–9662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Madsen JA; Ko BJ; Xu H; Iwashkiw JA; Robotham SA; Shaw JB; Feldman MF; Brodbelt JS Concurrent automated sequencing of the glycan and peptide portions of O-linked glycopeptide anions by ultraviolet photodissociation mass spectrom-etry. Anal. Chem 2013, 85 (19), 9253–9261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Huang Y; Yu X; Mao Y; Costello CE; Zaia J; Lin C De novo sequencing of heparan sulfate oligosaccharides by electron-activated dissociation. Anal. Chem 2013, 85 (24), 11979–11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Wolff JJ; Leach FE 3rd; Laremore TN; Kaplan DA; Easterling ML; Linhardt RJ; Amster IJ Negative electron transfer dissociation of glycosaminoglycans. Anal. Chem 2010, 82 (9), 3460–3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Leach FE 3rd; Wolff JJ; Xiao Z; Ly M; Laremore TN; Arungundram S; Al-Mafraji K; Venot A; Boons GJ; Linhardt RJ; et al. Negative electron transfer dissociation Fourier transform mass spectrometry of glycosaminoglycan carbohydrates. Eur. Mass Spectrom 2011, 17 (2), 167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Smith SA; Kalcic CL; Safran KA; Stemmer PM; Dantus M; Reid GE Enhanced characterization of singly protonated phosphopeptide ions by femtosecond laser-induced ionization/dissociation tandem mass spectrometry (fs-LID-MS/MS). J. Am. Soc. Mass Spectrom 2010, 21 (12), 2031–2040. [DOI] [PubMed] [Google Scholar]
- (22).Antoine R; Lemoine J; Dugourd P Electron photodetach-ment dissociation for structural characterization of synthetic and bio-polymer anions. Mass Spectrom. Rev 2014, 33 (6), 501–522. [DOI] [PubMed] [Google Scholar]
- (23).Geer LY; Markey SP; Kowalak JA; Wagner L; Xu M; Maynard DM; Yang X; Shi W; Bryant SH Open mass spectrometry search algorithm. J. Proteome Res 2004, 3 (5), 958–964. [DOI] [PubMed] [Google Scholar]
- (24).Bern M; Kil YJ; Becker C Byonic: advanced peptide and protein identification software. Curr. Protoc. Bioinformatics 2012, Chapter 13, Unit13.20. 10.1002/0471250953.bi1320s40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Eng JK; McCormack AL; Yates JR An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom 1994, 5 (11), 976–989. [DOI] [PubMed] [Google Scholar]
- (26).Bern M; Cai Y; Goldberg D Lookup peaks: a hybrid of de novo sequencing and database search for protein identification by tandem mass spectrometry. Anal. Chem 2007, 79 (4), 1393–1400. [DOI] [PubMed] [Google Scholar]
- (27).Bern MW; Kil YJ Two-dimensional target decoy strategy for shotgun proteomics. J. Proteome Res 2011, 10 (12), 5296–5301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Phanstiel DH; Brumbaugh J; Wenger CD; Tian S; Probasco MD; Bailey DJ; Swaney DL; Tervo MA; Bolin JM; Ruotti V; et al. Proteomic and phosphoproteomic comparison of human ES and iPS cells. Nat. Methods 2011, 8 (10), 821–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Rose CM; Russell JD; Ledvina AR; McAlister GC; Westphall MS; Griep-Raming J; Schwartz JC; Coon JJ; Syka JEP Multipurpose dissociation cell for enhanced ETD of intact protein species. J. Am. Soc. Mass Spectrom 2013, 24 (6), 816–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Ledvina AR; Rose CM; McAlister GC; Syka JEP; Westphall MS; Griep-Raming J; Schwartz JC; Coon JJ Activated ion ETD performed in a modified collision cell on a hybrid QLT-Oribtrap mass spectrometer. J. Am. Soc. Mass Spectrom 2013, 24 (11), 1623–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Riley NM; Westphall MS; Coon JJ Activated Ion Electron Transfer Dissociation for Improved Fragmentation of Intact Proteins. Anal. Chem 2015, 87 (14), 7109–7116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Good DM; Wenger CD; McAlister GC; Bai DL; Hunt DF; Coon JJ Post-acquisition ETD spectral processing for increased peptide identifications. J. Am. Soc. Mass Spectrom 2009, 20 (8), 1435–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Good DM; Wenger CD; Coon JJ The effect of interfering ions on search algorithm performance for electron-transfer dissociation data. Proteomics 2010, 10 (1), 164–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Wenger CD; Phanstiel DH; Lee MV; Bailey DJ; Coon JJ COMPASS: a suite of pre- and post-search proteomics software tools for OMSSA. Proteomics 2011, 11 (6), 1064–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Good DM; Wenger CD; McAlister GC; Bai DL; Hunt DF; Coon JJ Post-acquisition ETD spectral processing for increased peptide identifications. J. Am. Soc. Mass Spectrom 2009, 20 (8), 1435–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Kall L; Storey JD; MacCoss MJ; Noble WS Assigning significance to peptides identified by tandem mass spectrometry using decoy databases. J. Proteome Res 2008, 7 (1), 29–34. [DOI] [PubMed] [Google Scholar]
- (37).Bern M; Goldberg D Improved ranking functions for protein and modification-site identifications. J. Comput. Biol 2008, 15 (7), 705–719. [DOI] [PubMed] [Google Scholar]
- (38).Good DM; Wirtala M; McAlister GC; Coon JJ Performance characteristics of electron transfer dissociation mass spectrometry. Mol. Cell. Proteomics 2007, 6 (11), 1942–1951. [DOI] [PubMed] [Google Scholar]
- (39).Swaney DL; McAlister GC; Wirtala M; Schwartz JC; Syka JEP; Coon JJ Supplemental activation method for high-efficiency electron-transfer dissociation of doubly protonated peptide precursors. Anal. Chem 2007, 79 (2), 477–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Ledvina AR; McAlister GC; Gardner MW; Smith SI; Madsen JA; Schwartz JC; Stafford GC Jr.; Syka JE; Brodbelt JS; Coon JJ Infrared photoactivation reduces peptide folding and hydrogen-atom migration following ETD tandem mass spectrometry. Angew. Chem., Int. Ed 2009, 48 (45), 8526–8528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Shaw JB; Madsen J. a; Xu H; Brodbelt JS Systematic comparison of ultraviolet photodissociation and electron transfer dissociation for peptide anion characterization. J. Am. Soc. Mass Spectrom 2012, 23 (10), 1707–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Shaw JB; Kaplan D. a; Brodbelt JS Activated ion negative electron transfer dissociation of multiply charged peptide anions. Anal. Chem 2013, 85 (9), 4721–4728. [DOI] [PubMed] [Google Scholar]
- (43).Serang O; Noble WA review of statistical methods for protein identification using tandem mass spectrometry. Stat. Interface 2012, 5 (1), 3–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Nesvizhskii AI; Aebersold R Interpretation of shotgun proteomic data: the protein inference problem. Mol. Cell. Proteomics 2005, 4 (10), 1419–1440. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.