

Abstract
Short-form ATP phosphoribosyltransferase (ATPPRT) is a hetero-octameric allosteric enzyme comprising four catalytic subunits (HisGS) and four regulatory subunits (HisZ). ATPPRT catalyzes the Mg2+-dependent condensation of ATP and 5-phospho-α-d-ribosyl-1-pyrophosphate (PRPP) to generate N1-(5-phospho-β-d-ribosyl)-ATP (PRATP) and pyrophosphate, the first reaction of histidine biosynthesis. While HisGS is catalytically active on its own, its activity is allosterically enhanced by HisZ in the absence of histidine. In the presence of histidine, HisZ mediates allosteric inhibition of ATPPRT. Here, initial velocity patterns, isothermal titration calorimetry, and differential scanning fluorimetry establish a distinct kinetic mechanism for ATPPRT where PRPP is the first substrate to bind. AMP is an inhibitor of HisGS, but steady-state kinetics and 31P NMR spectroscopy demonstrate that ADP is an alternative substrate. Replacement of Mg2+ by Mn2+ enhances catalysis by HisGS but not by the holoenzyme, suggesting different rate-limiting steps for nonactivated and activated enzyme forms. Density functional theory calculations posit an SN2-like transition state stabilized by two equivalents of the metal ion. Natural bond orbital charge analysis points to Mn2+ increasing HisGS reaction rate via more efficient charge stabilization at the transition state. High solvent viscosity increases HisGS’s catalytic rate, but decreases the hetero-octamer’s, indicating that chemistry and product release are rate-limiting for HisGS and ATPPRT, respectively. This is confirmed by pre-steady-state kinetics, with a burst in product formation observed with the hetero-octamer but not with HisGS. These results are consistent with an activation mechanism whereby HisZ binding leads to a more active conformation of HisGS, accelerating chemistry beyond the product release rate.
Allosteric control of catalysis is a widespread strategy evolved in biosynthetic pathways.1−4 The modulation of biochemical pathways for synthetic biology applications often requires overcoming or manipulating allosteric regulation.5,6 Furthermore, allosteric sites provide a more selective avenue for drug design in comparison with active sites, which tend to be more conserved.3,7,8 Accordingly, the elucidation of allosteric mechanisms in multiprotein enzymatic complexes paves the way for future therapeutic and biotechnological applications.
The allosteric enzyme adenosine 5′-triphosphate phosphoribosyltransferase (ATPPRT) (EC 2.4.2.17), responsible for the first and flux-controlling step in histidine biosynthesis,9 is a potential drug target in some pathogenic organisms,8,10−12 the focus of synthetic biology endeavors to harness the histidine biosynthetic pathway for histidine production in bacteria,6,13,14 and a model system for the study of allosteric regulation of catalysis.3,8,15,16
ATPPRT catalyzes the Mg2+-dependent and reversible nucleophilic substitution of adenosine 5′-triphosphate (ATP) N1 on 5-phospho-α-d-ribosyl-1-pyrophosphate (PRPP) C1 to generate N1-(5-phospho-β-d-ribosyl)-ATP (PRATP) and inorganic pyrophosphate (PPi) (Scheme 1),9 with the chemical equilibrium highly displaced toward reactants.17 The metabolic status of the cell regulates ATPPRT activity via allosteric inhibition by histidine9,18 and orthosteric inhibition by adenosine 5′-monophosphate (AMP).19 Intriguingly, orthosteric inhibition by adenosine 5′-diphosphate (ADP) is also reported.19
Scheme 1. ATPPRT-Catalyzed Nucleophilic Substitution Reaction.

The hisG gene encodes two forms of ATPPRT. Most histidine-synthesizing organisms possess a long-form of the protein, HisGL,16,20 a homohexamer with each subunit consisting of two N-terminal catalytic domains and a C-terminal allosteric domain responsible for histidine inhibition.11 HisGL ATPPRTs operate by a steady-state ordered kinetic mechanism where ATP is the first substrate to bind to, and PRATP the last product to dissociate from, the enzyme.21,22
Archaea and some eubacteria have instead a short-form of the protein, HisGS,23,24 a homodimer with each subunit comprising two catalytic domains homologous to HisGL’s, but lacking the C-terminal allosteric domain.20,25 Thus, HisGS is catalytically active on its own but insensitive to inhibition by histidine.20,26 HisGS binds HisZ, the product of the hisZ gene, a catalytically inactive paralogue of histidyl-tRNA synthetase,23 forming the hetero-octameric ATPPRT holoenzyme, where two HisGS dimers flank a HisZ tetramer.24,26,27 HisZ has two distinct allosteric functions: in the absence of histidine, it activates catalysis by HisGS, and in the presence of histidine, it binds the final product of the pathway and mediates allosteric inhibition of HisGS.20,24,26,28 The kinetic mechanism of HisGS ATPPRTs has not been investigated, but recent crystal structures suggest that the order of substrate binding may be different from HisGL’s.29 Moreover, little is known about the kinetics of allosteric activation.
We recently reported several crystal structures of the psychrophilic bacterium Psychrobacter arcticus dimeric HisGS (PaHisGS) and hetero-octameric ATPPRT holoenzyme (PaATPPRT),26,29 from which an activation mechanism was inferred that involves tightening of the PaHisGS dimer in the hetero-octamer when both substrates are bound (Figure 1), which facilitates leaving group stabilization at the transition state.29 Here we employ initial rate studies, isothermal titration calorimetry (ITC), differential scanning fluorimetry (DSF), 31P nuclear magnetic resonance (31P NMR), liquid chromatography–mass spectrometry (LC-MS), density functional theory, solvent viscosity effects, and pre-steady-state kinetics to unveil a distinct kinetic mechanism for PaATPPRT, the role of ADP as a substrate instead of an inhibitor, the basis for charge-stabilization at the transition state, and a shift in the rate-liming step upon allosteric activation of the enzyme.
Figure 1.
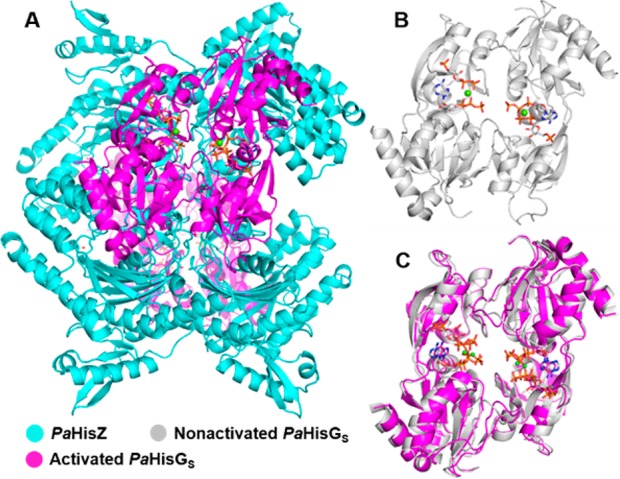
PaHisGS and PaATPPRT quaternary structures and allosteric activation. (A) PaATPPRT hetero-octamer, where catalysis is enhanced. The second PaHisGS homodimer is behind the PaHisZ tetramer. (B) Nonactivated PaHisGS homodimer and (C) overlay of activated and nonactivated PaHisGS dimers. In all structures, PaHisGS is bound to PRPP, ATP, and Mg2+.29
Materials and Methods
Materials
ATP, PRPP, PPi, AMP, ADP, MgCl2, MnCl2, D2O (99.9 atom % deuterium), tricine, dithiothreitol (DTT), and glycerol were purchased from Sigma-Aldrich. All other chemicals were purchased from readily available commercial sources, and all chemicals were used without further purification. PaHisGS, PaHisZ, Mycobacterium tuberculosis pyrophosphatase (MtPPase), and tobacco etch virus protease were obtained as previously published.26 PRATP was produced as previously described.29
PaHisGS and PaATPPRT Activity Assay
All assays were performed under initial rate conditions in the forward direction at 20 °C as previously described26 by monitoring the increase in absorbance at 290 nm due to formation of PRATP (ε290 nm = 3600 M–1 cm–1)30 in 1 cm path length quartz cuvettes (Hellma) in a Shimadzu UV-2600 spectrophotometer. Unless stated otherwise, for PaHisGS activity, PaHisGS concentration was 2.2 μM, and for PaATPPRT activity, PaHisGS and PaHisZ concentrations were 0.38 and 15 μM, respectively. Reactions were started by addition of PRPP. Control reactions lacked either ATP, PRPP, PaHisGS, or PaHisZ. In all kinetic experiments under the various different conditions described below, controls were carried out to ensure that the rate did not depend on MtPPase. Kinetic measurements were performed at least in duplicates unless stated otherwise.
PaATPPRT Equilibrium Dissociation Constant (KD) in Glycerol
Initial velocities were measured in the presence of 5.6 mM ATP, 2 mM PRPP, 0.38 μM PaHisGS, and varying concentrations of PaHisZ (0.9–8.5 μM) in 0%, (0.5–16.3 μM) in 18% and 27% glycerol (v/v). PaHisZ-PaHisGSKD values were obtained by fitting initial rate data to a kinetic equation (vide infra) as previously reported.26
PaATPPRT and PaHisGS Saturation Kinetics with ATP and PRPP
PaATPPRT initial rates were measured at saturating concentrations of one substrate and varying concentrations of the other, either ATP (0.4–5.6 mM) or PRPP (0.1–2.0 mM). Initial rates for PaHisGS were determined at saturating concentrations of one substrate and varying concentrations of the other, either ATP (either 0.4–2.8 or 0.4–5.6 mM) or PRPP (0.1–2.0 mM).
PaATPPRT and PaHisGS Saturation Kinetics with MnCl2
PaATPPRT initial rates were measured at saturating concentrations of one substrate and varying concentrations of the other, either ATP (0.1–1.4 mM) or PRPP (0.1–2.0 mM), while initial rates for PaHisGS (1.1 μM) were determined at saturating concentrations of one substrate and varying concentrations of the other, either ATP (0.1–1.4 mM) or PRPP (0.05–2.0 mM), in the presence of 15 mM MnCl2 instead of MgCl2.
Analysis of PaHisGS Reaction with MnCl2 by LC-MS
Reaction mixtures (500 μL) contained 100 mM tricine pH 8.5, 100 mM KCl, 4 mM DTT, 15 mM MnCl2, 19.7 μM MtPPase, 1.4 mM ATP, 2.0 mM PRPP, and 10.3 μM PaHisGS. Reactions were incubated for 1 h at 20 °C, after which proteins were removed by passage through 10000 MWCO Vivaspin centrifugal concentrators. Reactions were run in duplicate, and control reactions lacked PaHisGS. LC-MS analysis of the protein-free reaction mixtures was performed on an EC250/4.6 Nucleodur 100–10 C18 ec HPLC column (10 μm × 4.6 mm × 250 mm) (Macherey-Nagel) in a 1260 infinity HPLC system coupled to a G6130B Single Quadrupole mass spectrometer (Agilent Technologies). Separation of PRATP and ATP was carried out in (A) 50 mM triethylamine-acetic acid pH 7.4 and (B) methanol as a mobile phase in the following sequence: 0–3 min 100% A, 3–3.1 min 90% A and 10% B, 3.1–12 min 80% A and 20% B at a flow rate of 1 mL min–1, with UV absorbance monitored at 260 and 290 nm. Electrospray ionization-mass spectrometry (ESI-MS) data were acquired in negative mode with a capillary voltage of 4500 V.
PaATPPRT and PaHisGS Saturation Kinetics in Glycerol
PaATPPRT and PaHisGS initial rates were measured at saturating concentrations of one substrate and varying concentrations of the other, either ATP (0.4–5.6 mM) or PRPP (0.1–2.0 mM), in the presence of 0%, 18%, and 27% glycerol (v/v).
PaHisGS Inhibition by AMP
The half-maximal inhibitory concentration of AMP was determined by measuring initial rates for PaHisGS (4.5 μM) in the presence of 5.6 mM ATP, 2 mM PRPP, and varying concentrations of AMP (0–0.8 mM). The inhibition mechanism was investigated by measuring initial rates for PaHisGS (4.5 μM) at saturating concentrations of one substrate and varying concentrations of the other, either ATP (0.4–5.6 mM) with different concentrations of AMP (0–0.1 mM) or PRPP (0.1–2.0 mM) with different concentrations of AMP (0–0.05 mM). PaHisGS concentration was more than 4-fold higher than the lowest AMP concentration used, and pseudo-first-order approximation was assumed.
PaHisGS Saturation Kinetics with ADP and PRPP
Initial rates for PaHisGS were determined at saturating concentrations of one substrate and varying concentrations of the other, either ADP (0.4–5.6 mM) or PRPP (0.1–2.0 mM).
Comparison of PaHisGS Reactions with ADP and ATP by 31P NMR Spectroscopy
Analysis of PaHisGS reactions by 31P NMR spectroscopy was carried out as previously described,26 except that PaHisGS concentration was 10.3 μM and ADP replaced ATP in half of the reactions. All reactions were run in duplicate, and control reactions lacked PaHisGS.
PaATPPRT and PaHisGS Initial Velocity Patterns
Initial rates for PaATPPRT were measured in the presence of varying ATP (0.4–5.6 mM) and PRPP (0.1–2.0 mM), with 1 μM PaHisGS and 20 μM PaHisZ. Initial rates for PaHisGS were determined in the presence of varying ATP (0.2–2.8 mM) and PRPP (0.1–2.0 mM). Measurements were performed in quadruplicates.
PaHisGS Binding by ITC
ITC measurements were carried out at 20 °C in a MicroCal PEAQ-ITC calorimeter (Malvern Instruments). Protein and ligand were solubilized in the same ATPPRT assay buffer. After a small injection of 0.4 μL, 18 successive injections of 2 μL of ligand (either 0.8 mM PRPP or 10 mM ATP) were made into 300 μL of 50 μM PaHisGS, with 150-s intervals between successive injections and a reference power of 10 μcal s–1. Heat of dilution for each experiment was measured by titrating ligand into assay buffer, and subtracted from the corresponding binding curve. All measurements were performed in duplicate. Data for PRPP binding were fitted to a single-site binding model as implemented in the PEAQ-ITC analysis software (Malvern Instruments).
PaHisGS Thermal Denaturation by DSF
DSF measurements (λex = 490 nm, λem 610 nm) were performed in 96-well plates on a Stratagene Mx3005p instrument. Thermal denaturation assays (50 μL) for 7.5 μM PaHisGS were measured in the presence and absence of ligands (6 mM ATP, 2 mM PRPP, 208 μM PRATP, 3.6 mM PPi), with or without 22% glycerol (v/v) (apoenzyme) in 100 mM tricine, 100 mM KCl, 4 mM DTT and 15 mM MgCl2 pH 8.5. The assay for apoenzyme was also performed in 10 mM KH2PO4, 10 mM KF pH 8.0. Sypro Orange (5×) (Invitrogen) was added to all wells. Thermal denaturation curves were recorded over a temperature range from 25–93 °C with 1 °C min–1 increments. Control curves lacked enzyme and were subtracted from curves containing enzyme. All measurements were carried out in triplicate.
Density Functional Theory Calculations
Theoretical structures were derived from B3LYP calculations using a 6-31G* basis set with a Lanl2DZ basis set on Mg2+ and Mn2+ and a Lanl2DZ pseudopotential added to Mg2+ and Mn2+ as implemented in Gaussian 09.31 A model system was chosen by including all residues within 5 Å of ADP and PRPP in the crystallographic dimer of the PaHisGS-PRPP-Mg-ADP complex crystal structure29 and by flipping the adenine ring from its crystal structure orientation to bring N1 in proximity to PRPP. The system was further paired down to include only functional groups, metal ions and water molecules within the 5-Å cutoff that were essential for stabilization of the transition structure. In addition to the divalent metal found in the crystal structure, a second divalent metal had to be included in the system for a transition structure to be located. Initial searches exploring structures with fixed distances along the reaction coordinate were located by performing an optimization of an input structure with the key bond-forming or bond-breaking distances held constant, and frequency calculations resulted in only one imaginary frequency along the reaction coordinate. Final transition structures for the system complexed with either Mg2+ or Mn2+ were located as stationary points with no geometrical constrains and exhibit only one imaginary frequency along the reaction coordinate. Coordinates for all optimized structures are available in the Supporting Information.
Pre-Steady-State Kinetics
Approach to steady-state in PaHisGS and PaATPPRT reactions was investigated under multiple-turnover conditions by monitoring the increase in absorbance at 290 nm upon PRATP formation at 20 °C in an Applied Photophysics SX-20 stopped-flow spectrophotometer outfitted with a 5 μL mixing cell (0.5 cm path length and 0.9 ms dead-time). Each syringe contained 100 mM tricine pH 8.5, 100 mM KCl, 4 mM DTT, 15 mM MgCl2, and 20 μM MtPPase. In addition, one syringe carried 40 μM PaHisGS (with or without 100 μM PaHisZ) and 4 mM PRPP, while the other carried 11.2 mM ATP. Reaction was triggered by rapidly mixing 55 μL from each syringe. Absorbance increase with PaHisGS was monitored in a linear-time base for 5 s with 5000 data points collected, and with PaATPPRT, in a split-time base for 2 s, with 4000 data points collected in the first 0.2 s and 4000 in the following 1.8 s. At least 8 traces were acquired for each enzyme, and controls lacked PRPP.
Data Analysis of Kinetics and Thermal Denaturation
Kinetic and DSF data were analyzed by the nonlinear regression function of SigmaPlot 13 (SPSS Inc.). Data points and error bars in graphs are represented as mean ± standard error, and kinetic and equilibrium constants are presented as mean ± fitting error. Initial rate data with varying concentrations of PaHisZ were fitted to eq 1. The concentration of PaATPPRT at any concentration of PaHisGS and PaHisZ was calculated according to eq 2. Substrate saturation data were fitted to eq 3. Inhibition data at fixed substrate concentrations were fitted to eq 4, and competitive inhibition data were fitted to eq 5. Initial velocity patterns were fitted to eq 6, and pre-steady-state kinetics under multiple-turnover conditions was fitted to eq 7. In eqs 1–7, v is the initial rate, Vmax is the maximal velocity, G is the concentration of PaHisGS, Z is the concentration of PaHisZ, KD is the equilibrium dissociation constant, PaATPPRT is the concentration of PaHisGS-PaHisZ complex, S is the concentration of the varying substrate, kcat is the steady-state turnover number, KM is the apparent Michaelis constant, ET is total enzyme concentration, vi is the initial rate in the presence of inhibitor, IC50 is the half-maximal inhibitory concentration, Ki is the inhibitor dissociation constant, A and B are the first and second substrates to bind to the enzyme, respectively, Ka and Kb are their respective Michaelis constants, Kia is the apparent dissociation constant for the complex between enzyme and substrate A when the concentration of B approaches zero, t is time, P(t) is product concentration at time t, A0 is the amplitude of the burst phase, and kburst is the first-order rate constant of product formation in the burst phase. DSF thermal denaturation data were fitted to eq 8,32 where FU is fraction unfolded, T is the temperature in °C, Tm is the melting temperature, c is the slope of the transition region, and LL and UL are folded and unfolded baselines, respectively.
| 1 |
| 2 |
| 3 |
| 4 |
| 5 |
| 6 |
| 7 |
| 8 |
Results and Discussion
PaHisGS and PaATPPRT Kinetic Mechanism
A steady-state ordered kinetic mechanism in which ATP is the first substrate to bind to the enzyme, and PRATP is the last product to dissociate from it, has long been demonstrated for HisGL ATPPRTs.21,22 This mechanism has been supported by several structures of Campylobacter jejuni and M. tuberculosis ATPPRT-ATP binary complexes,10,16 and by the recent structure of the C. jejuni ATPPRT catalytic core in complex with PRPP, where despite being able to bind to the free enzyme, PRPP drifts into the ATP binding site, which would lead to a dead-end complex.33 The kinetic mechanism of HisGS ATPPRTs, on the other hand, has not been explored. We recently published the crystal structures of PaHisGS and PaATPPRT in binary complexes with PRPP and PRATP, and in ternary complexes with PRPP-ATP, but were unable to obtain structures of enzyme-ATP binary complexes, suggesting a reverse order of substrate binding in comparison with HisGL ATPPRTs.29
To test this hypothesis, the kinetic mechanism of PaHisGS and PaATPPRT was investigated. Intersecting patterns of double-reciprocal plots with both ATP and PRPP in initial velocity studies were determined for PaATPPRT (Figure 2A,B) and PaHisGS (Figure 2C,D), indicating a ternary complex is formed in a sequential mechanism. The double-reciprocal plots intersecting to the left of the y-axes rule out a rapid equilibrium ordered mechanism.34 Fitting the data to eq 6 (Figure 2E,F) yielded steady-state kinetic parameters summarized in Table S1.
Figure 2.
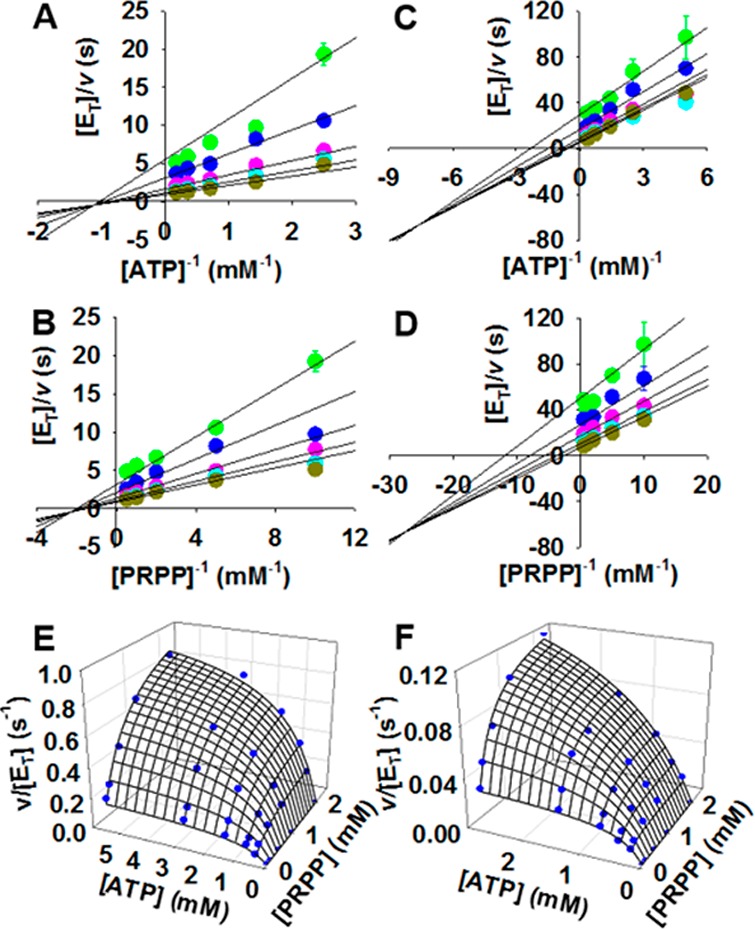
Initial velocity patterns for PaATPPRT and PaHisGS. Intersecting double-reciprocal plots for PaATPPRT with (A) ATP and (B) PRPP as varying substrates and for PaHisGS with (C) ATP and (D) PRPP as varying substrates. Each color represents a different fixed concentration of the cosubstrate. Data points are mean ± SE. Three-dimensional plot of (E) PaATPPRT and (F) PaHisGS initial rate data, where lines are data fitting to eq 6.
Binding studies were performed with PaHisGS to elucidate the substrate binding order. Binding of PRPP to PaHisGS was detected by ITC (Figure S1), and fitting the data from two independent experiments to a single-site binding model (stoichiometry of 1:1 and no cooperativity) resulted in KD's of 15.4 ± 0.2 and 8.3 ± 0.1 μM (one from each experiment, yielding a mean ± SE of 12 ± 2 μM). ATP binding to PaHisGS, on the other hand, could not be detected, as no signal was observed beyond heat of dilution (Figure S2). This corroborates the hypothesis that PRPP can bind to the free enzyme, while ATP cannot.
To confirm and expand these results, PaHisGS thermal denaturation curves in the presence and absence of substrates and products were determined by DSF (Figure 3), and data fitting to eq 8 produced Tm’s shown in Table S2. PRPP and PRATP increased PaHisGSTm by 6 and 5 °C, respectively, indicating that these molecules can bind to the free enzyme. Conversely, ATP and PPi did not alter PaHisGSTm. The latter observation alone does not necessarily rule out the possibility that ATP and PPi can bind to the free enzyme, but the integration of crystallography,29 initial velocity patterns, ITC, and DSF data supports a steady-state ordered mechanism where PRPP is the first substrate to bind to PaHisGS and PRATP is the last product to dissociate from it. The strong parallels in corresponding binding modes seen in the PaHisGS and PaATPPRT crystal structures29 suggest that PaATPPRT follows the same mechanism. Moreover, given the conservation of PRPP position in PaATPPRT29 and Lactococcus lactis ATPPRT binary complexes,27 this mechanism may be valid for other HisGS ATPPRTs.
Figure 3.
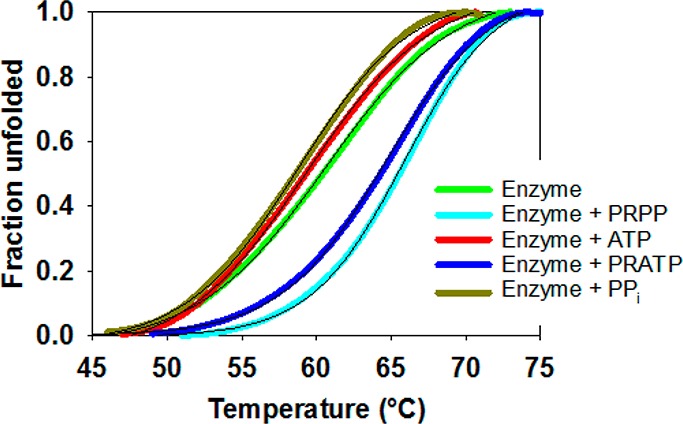
DSF-based thermal denaturation of PaHisGS apoenzyme and in the presence of substrates and products. Thin black lines are data fitting to eq 8.
AMP Is an Inhibitor of PaHisGS
AMP is a competitive inhibitor of HisGL ATPPRTs against both substrates,16,19 which is explained structurally by the simultaneous partial occupation of the PRPP and ATP binding sites by AMP’s phosphoribosyl and adenine moieties, respectively.11,16,35 AMP is also a competitive inhibitor against PRPP in L. lactis ATPPRT,28 and the recent crystal structure of the PaHisGS-AMP complex shows a similar binding mode as in HisGL ATPPRTs.11,16,29,35 AMP inhibits PaHisGS with an IC50 of 79 ± 6 μM (Figure S3A), and inhibition is competitive against both PRPP and ATP, with Ki’s of 25 ± 5 and 52 ± 8 μM, respectively (Figure S3B,C). These values are on average ca. 7- and 10-fold lower than those for HisGL ATPPRTs,16,19 and over 27-fold lower than that for L. lactis ATPPRT,28 suggesting PaHisGS activity is more stringently regulated by this metabolite.
ADP Is a Substrate for PaHisGS
ADP has been shown to be an inhibitor of HisGL ATPPRTs.19 However, crystal structures of PaHisGS and PaATPPRT in complex with PRPP-ADP reveal that ADP binds in the same manner as ATP.29 In order to evaluate the ability of PaHisGS to use ADP as a substrate, we compared the reactions with ADP and ATP by 31P NMR spectroscopy (Figure S4). The spectra of reactions containing ADP (Figure S4A) and ATP (Figure S4C) are similar except for the peak at −19.2 to −19.4 corresponding to the γ-PO42– phosphorus of ATP and PRATP, since this group is absent in ADP and N1-(5-phospho-β-d-ribosyl)-ADP (PRADP). Spectra for both reactions differ from the controls lacking PaHisGS (Figure S4B,D). The characteristic peak at ca. 3.3 ppm corresponding to the phosphorus in the N1-5-phospho-β-d-ribose moiety of the product26 is present in the reaction spectra with ADP (Figure S4A, inset) and ATP (Figure S4C, inset), and absent in the controls (Figure S4B,D, insets), establishing that ADP can replace ATP as a substrate for PaHisGS.
For a quantitative comparison of the reactions with ATP and ADP, steady-state kinetic analysis of the reaction with either substrate was carried out (Figure 4) and kinetic parameters are summarized in Table S3. Values of kcat are the same within error with either ATP or ADP as a substrate, indicating that once saturated PaHisGS turns over ATP and ADP just as effectively. The main difference is in the KM for ADP, which is over 3-fold that for ATP, suggesting some small loss of affinity for the steady-state with ADP.
Figure 4.
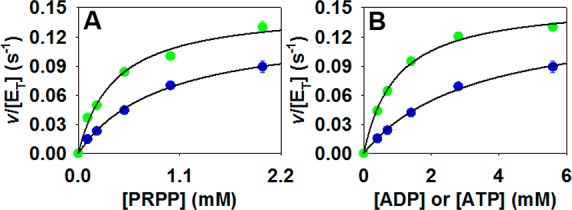
PaHisGS substrate saturation curves with either ATP (green) or ADP (blue) as a substrate. (A) Varying PRPP concentration with saturating concentration of the nucleotide. (B) Varying the nucleotide concentration with saturating concentration of PRPP. Data points are mean ± SE, and lines are data fitting to eq 3.
PaHisGS and PaATPPRT Kinetics with Mn2+
Replacement of Mg2+ by Mn2+ is a common strategy in enzymology,36 having been employed to uncover rate-limiting steps in reactions involving stabilization of phosphate groups.37 HisGL ATPPRTs have been reported to have their activities either unaltered or decreased by changing the divalent metal in the reaction from the physiological Mg2+ to Mn2+, but no mechanistic inference has been drawn.17,38 To evaluate the effect of Mn2+ on a HisGS enzyme, saturation curves for PaHisGS and PaATPPRT with either divalent metal were determined (Figure 5), and kinetic constants are displayed in Table S4. Mn2+ led to 2.6- and 11-fold increases in PaHisGSkcat and kcat/KMATP, respectively, as compared with Mg2+. The change in kcat/KMATP was driven in large part by a reduction in KMATP. LC-MS analysis of the PaHisGS reaction with Mn2+ confirmed the same product, PRATP, was being formed (Figure S5).
Figure 5.
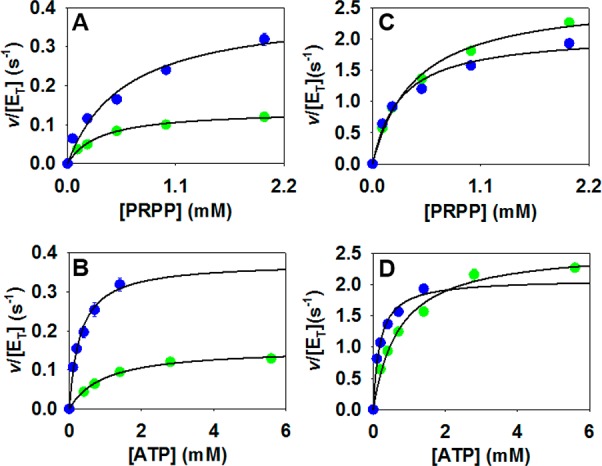
Steady-state kinetics with Mg2+ (green) and Mn2+ (blue). Saturation curves varying either PRPP or ATP concentration with saturating concentration of the cosubstrate for (A and B) PaHisGS and (C and D) PaATPPRT. Data points are mean ± SE, and lines are data fitting to eq 3.
In contrast to PaHisGS, steady-state constants for PaATPPRT were unchanged by Mn2+, except for a 2.9-fold increase in kcat/KMATP owing to a reduction in KMATP. These results raise the possibility that PaHisGS and PaATPPRT reactions have distinct rate-limiting steps. Crystal structures of PaHisGS and PaATPPRT with various ligands do not depict any specific interaction between Mg2+ and the enzyme,29 raising the possibility the off-rates of products from the active site might not be affected by the nature of the metal ion. It is possible, nonetheless, that a solution metal stabilizes charges of either PRATP or PPi concomitantly with product release from the enzyme. The metal ion seen in the structures acts as a Lewis acid to stabilize negative charges in the Michaelis complex upon binding of ATP, with a putative second metal ion likely present at the transition state to facilitate departure of the PPi leaving group.29 Thus, one might expect Mn2+ to increase the reaction rate if rate-limiting steps are located between ATP binding to the enzyme-PRPP complex and product formation, since a stronger Lewis acid would facilitate catalysis by stabilizing charges more efficiently. The kinetic constants affected would be kcat and kcat/KMATP, which is exactly what is observed with PaHisGS. If, on the other hand, ternary complex formation played a minor role in limiting the reaction rate, chemistry was fast, and product release was the slowest step, a modest increase in kcat/KMATP only would be expected, which is the case with PaATPPRT.
A Transition-State Hypothesis for the PaHisGS Reaction
An SN1-like, DN*AN‡39,40 transition-state structure has recently been proposed for C. jejuni and M. tuberculosis (HisGL) and L. lactis (HisGS) ATPPRT-catalyzed reaction based on kinetic isotope effects and computational chemistry, using a simplified model of the reaction for density functional theory calculations.10 Having established that PaHisGS utilizes ADP as a substrate with a similar kcat as it does ATP (Table S3), the crystal structure of the PaHisGS-PRPP-Mg-ADP Michaelis complex29 served as a starting point for density functional theory calculations in order to find a theoretical transition state for the reaction that includes not only the full substrates but also several active-site residue side-chain surrogates and water molecules essential to stabilize the system, with either Mg2+ or Mn2+ as the metal ion (Figure 6). Transition structures were located as stationary points (i.e., without any constraints on distances or dihedral angles) and possess only one imaginary frequency reflecting vibration along the N1–C1–O1 axis. Inclusion of a second equivalent of the divalent metal ion to stabilize the departing PPi leaving group was essential to locate transition structures, lending support to a recent proposal based on the crystal structures of PaHisGS and PaATPPRT Michaelis complexes29 and the transition structures of other phosphoribosyltransferases.41
Figure 6.

Transition-state model for the PaHisGS-catalyzed reaction. (A) Transition structure with magnesium, (B) transition structure with manganese, and (C) overlay of the transition structures with magnesium and manganese. Substrates are represented as stick models, side-chain mimics as wireframe, and metal ions and water oxygens as spheres. Carbon is in either cyan or yellow for substrates and in gray for side-chain mimics, with oxygen in red, nitrogen in blue, phosphorus in orange, magnesium in green, and manganese in purple. Hydrogens are omitted for simplicity. Partial bonds and metal ion coordination bonds are represented by dashed lines. Distances are shown for the N1–C1 and the C1–O1 bonds. Key residue side-chain mimics are labeled, and the prime denotes a residue of the adjacent subunit in the PaHisGS dimer.29
The optimized structures indicate an SN2-like, almost synchronous ANDN transition state is possible for the PaHisGS-catalyzed reaction with either Mg2+ (Figure 6A) or Mn2+ (Figure 6B) as a Lewis acid. The 6-NH2 group of ADP is protonated in all transition structures and is likely to lose a proton to form the 6-NH group of PRADP only after the nucleophilic substitution is complete, as recently hypothesized.29 Nucleophilic attack occurs from the charge-neutral resonance structure of adenine in which N1 has transiently a negative charge due to electron donation from N6. This natural resonance structure represents 6.64% of the distribution of adenine resonances.42 The ANDN transition state located here contrasts with the DN*AN‡ one proposed for HisGL and HisGS ATPPRT reaction.10 This would mean that different orthologues of ATPPRT catalyze the same reaction via different transition states, which is not uncommon in ribosyl-transfer reactions. For instance, distinct transition-state models based on kinetic isotope effects and density functional theory have been suggested for bovine and human purine nucleoside phosphorylases,43,44 and for wild-type and mutant human purine nucleoside phosphorylases.43,45 Kinetic isotope effect measurements for PaHisGS could test the ANDN transition-state hypothesis put forth in this work.
Overlay of the transition structures with Mg2+ and Mn2+ demonstrates an almost identical arrangement (Figure 6C), indicating transition-state geometry cannot explain the discrimination in PaHisGS reactivity between the metal ions. Natural bond orbital (NBO) analysis of the transition structures, however, revealed significant differences in charge distribution in the metal ions and the PPi at the transition state depending on which metal is included (Table S5). Most atoms have very similar charges in the two transition structures, except for the metal ions and PPi oxygens. The average charge of the two magnesium ions at the transition state is 1.439, over 2-fold higher than the average charge of the manganese ions, 0.649. This is due to more efficient attenuation of the negative charge of the PPi leaving group by Mn2+ through d-orbital bonding to coordinating oxygens, as shown by orbital population analysis. As compared with Mg2+, therefore, Mn2+ improves catalysis in the PaHisGS reaction by more effectively stabilizing the negatively charged leaving group at the transition state.
Solvent Viscosity Effects on PaHisGS and PaATPPRT Kinetics
In order to probe further the distinct rate-limiting steps governing PaHisGS and PaATPPRT catalyses, the effect of solvent viscosity on reaction rates was evaluated (Figure 7), and the data are summarized in Tables S6 and S7. Increasing solvent viscosity by increasing glycerol concentration46 slows down diffusional steps such as substrate binding and release and product release, and values of kinetic constants will be reduced if such steps are rate-limiting.47−49PaHisGS rate constants did not decrease with increasing glycerol concentration (Table S6), consistent with diffusional steps not contributing to limit the reaction rate. Instead, as shown in Figure 7A and Table S6, glycerol led to an increase in PaHisGSkcat and kcat/KMPRPP of up to 2.7- and 2.4-fold, respectively, while kcat/KMATP was only marginally affected. Inverse solvent viscosity effects generally suggest that a more active dynamic conformation of the enzyme or the Michaelis complex is favored at high viscosity.47−49 To rule out the possibility that glycerol might be affecting the overall stability of the enzyme, a thermal denaturation curve was determined by DSF in 22% glycerol (Figure S6), and no difference in Tm was observed in comparison with that determined without glycerol (Table S2). Crystal structures of PaHisGS apoenzyme and PaHisGS-PRPP-ATP were also obtained with and without soaking crystals in glycerol, and no electron density for glycerol was visualized in any of the structures.29 This suggests that glycerol is acting as part of bulk solvent, not as a ligand, but with the caveat that crystal lattice might have prevented binding.
Figure 7.
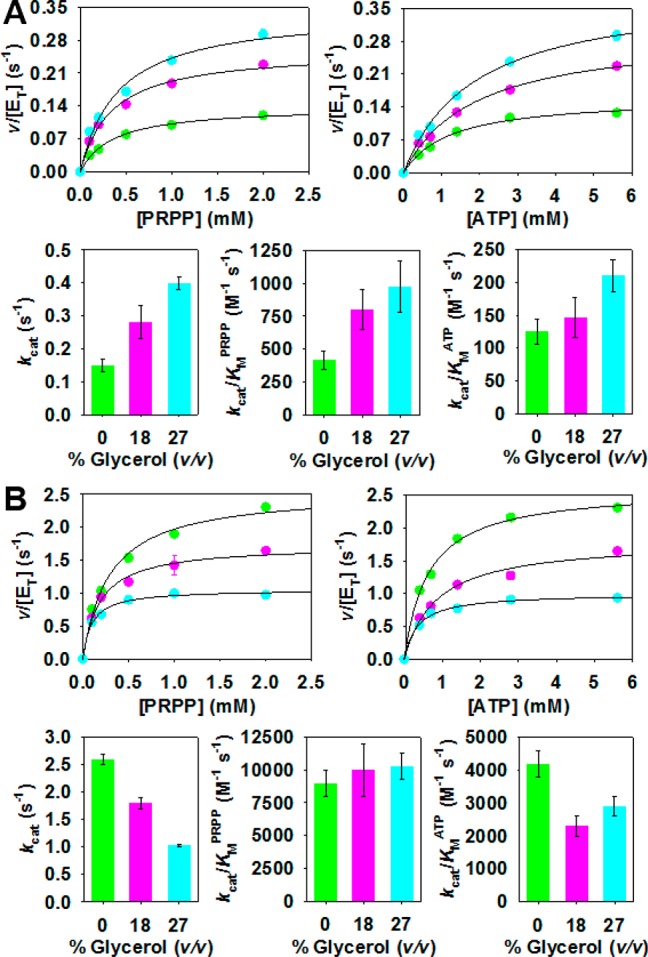
Solvent viscosity effects on steady-state kinetics determined at 0% (green), 18% (pink), and 27% (cyan) glycerol (v/v). (A) PaHisGS saturation curves (top) and steady-state constants (bottom) dependence on glycerol concentration. (B) PaATPPRT saturation curves (top) and steady-state constants (bottom) dependence on glycerol concentration. Data represent either mean ± SE (scatter plots) or value ± fitting error (bar plots). Lines are data fitting to eq 3.
To assess the effect of solvent viscosity on PaATPPRT, first the KD for the PaHisGS-PaHisZ complex had to be measured in glycerol (Figure S7), and data fitting to eq 1 yielded KD’s of 1.3 ± 0.1, 1.1 ± 0.2, and 0.5 ± 0.1 μM in 0%, 18%, and 27% glycerol, respectively. Knowledge of the KD’s allowed calculation, using eq 2, of PaATPPRT concentrations at different glycerol concentrations for measurement of kcat. In contrast to the effect on PaHisGS, increasing solvent viscosity resulted in a decrease of up to 2.5-fold in PaATPPRT kcat, with negligible effects on kcat/KM for either substrate, as shown in Figure 7B and Table S7. This points to product dissociation from PaATPPRT as the rate-limiting step in the reaction, as is the case with HisGL ATPPRTs.8,50
Burst in Product Formation by PaATPPRT
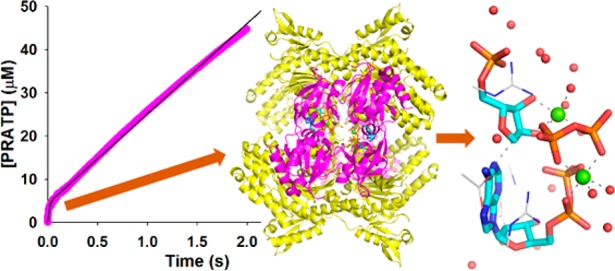
To glean additional support for distinct rate-limiting steps controlling nonactivated and activated PaHisGS reactions, product formation time courses were monitored under pre-steady-state conditions for PaHisGS and PaATPPRT (Figure 8). PRATP formation with PaHisGS varies linearly with time with a steady-state rate constant of 0.091 ± 0.001 s–1, in reasonable agreement with kcat (Tables S3, S4, S6). This rules out a slow step after formation of enzyme-bound products51 and suggests interconversion between ternary complexes (k5 + k6 in Scheme 2) is rate-limiting.
Figure 8.
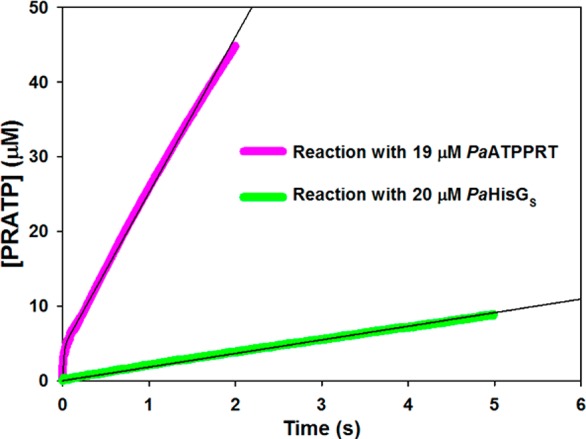
Pre-steady-state kinetics with PaATPPRT and PaHisGS, with a burst in product formation observed with the former but not the latter. Black lines are data fitting to eq 7 for PaATPPRT and a linear regression for PaHisGS.
Scheme 2. Interpretation of the Pre-Steady-State of PaHisGS and PaATPPRT Reactions.

On the other hand, a burst in PRATP formation precedes the steady-state with PaATPPRT, and data fitting to eq 7 yielded a kburst of 80 ± 1 s–1, a steady-state rate constant of 1.11 ± 0.01 s–1, and an A0 of 4.3 μM. This is consistent with a step after chemistry, likely product release (k′7 in Scheme 2), limiting the reaction rate,51,52 in agreement with the conclusion drawn from solvent viscosity effects. M. tuberculosis HisGL ATPPRT also displays a burst in product formation with a kburst of 0.67 s–1 at 25 °C.8 Thus, PaATPPRT kburst at 20 °C is over 119-fold higher than M. tuberculosis HisGL ATPPRT’s at 25 °C, which may be a feature of HisGS ATPPRTs and/or a consequence of PaATPPRT being psychrophilic.26
The amplitude of the burst phase (A0) generally reflects the concentration of the Michaelis complex, which at saturating substrate concentrations could be as high as the concentration of enzyme.53 The A0 of 4.3 μM is over 4.4-fold lower than the concentration of PaATPPRT used in the experiment (19 μM). Two main reasons may account, separately or in combination, for this result without invoking the unlikely scenario where ca. 75% of enzyme molecules are inactive. First, the enzyme might not be fully saturated by one or both substrates, which would also explain the steady-state rate constant being slightly smaller than the kcat values extrapolated from substrate saturation curves. This may be the case with the PaHisGS steady-state rate constant as well. Second, chemical reversibility decreases A0. Both kburst and A0 are dependent on all rate constants depicted in Scheme 2, the forward and reverse rate constants for interconversion between enzyme-bound substrates and products, k5 and k6, respectively, and the net rate constant for release of products from the enzyme, k′7, according to eqs 9 and 10.51
| 9 |
| 10 |
Upon inspection of eqs 9 and 10, one must conclude that under the most favorable conditions, full expression of A0 can only occur when chemistry is irreversible (k6 = 0) and much faster than product release (k5 ≫ k′7). Internal reversibility, described by the magnitude of k6, will increase kburst while decreasing A0. Equilibrium in the ATPPRT reaction strongly favors the reactants,17 making it possible for the crystal structure of the PaATPPRT-PRPP-ATP ternary-complex to be attained with wild-type enzyme.29 Hence, k6 is likely to be much larger than k5, making k6 the main contributor to kburst and significantly reducing A0 from its theoretical upper limit of 19 μM. Relative contributions of k5 and k6 to kburst and A0 notwithstanding, it is clear that activation of PaHisGS by PaHisZ switches the rate-limiting step of the reaction from interconversion between the ternary complexes to product release.
PaHisZ-Induced Shift in the Rate-Limiting Step
The results presented here demonstrate that two long-established mechanistic features of HisGL ATPPRTs, namely, ATP as the first substrate to bind to the enzyme and ADP as an inhibitor,16,19,21,22,33 do not apply to PaHisGS, and possibly other HisGS ATPPRTs. Providing functional data to support hypotheses proposed based on extensive crystallography work on PaHisGS and PaATPPRT,29PaHisGS is shown to be able to replace ATP for ADP as a substrate and to operate by a steady-state ordered mechanism where PRPP is the first substrate to bind to the enzyme (Scheme 3). PaHisGSkcat increases when Mn2+ replaces Mg2+, which can be accounted for owing to more efficient charge stabilization by Mn2+ upon leaving group departure at the transition state. The observation that PaATPPRT steady-state kinetics is unaltered with Mn2+ raises the possibility of kcat’s for the activated and nonactivated enzyme forms reporting on distinct steps. This is confirmed by solvent viscosity effects on steady-state parameters and by pre-steady-state kinetics under multiple-turnover conditions, which indicate that interconversion between PaHisGS–PRPP-ATP and PaHisGS–PRATP-PPi complexes limits the reaction rate for the nonactivated enzyme, likely with a significant contribution from chemistry given the effect of Mn2+. However, allosteric activation by PaHisZ accelerates this interconversion well beyond the steady-state rate, which now reflects the off-rate of either PPi from the PaATPPRT–PRATP-PPi ternary complex or PRATP from the PaATPPRT–PRATP binary complex (Scheme 3). This provides fundamental insight into the allosteric regulation of a complex multiprotein enzyme.
Scheme 3. Kinetic Mechanism and Rate-Limiting Steps of PaHisGS (top) and PaATPPRT (bottom) Reactions and the Corresponding Crystal Structures26,29 .

The second PaHisGS homodimers lie behind the PaHisZ tetramers.
Acknowledgments
This work was supported by a Wellcome Trust Institutional Strategic Support Fund to the University of St Andrews and the Biotechnology and Biological Sciences Research Council (BBSRC) [grant no. BB/M010996/1] via an EASTBIO Doctoral Training Partnership studentship to G.F. The computational work used the Extreme Science and Engineering Discovery Environment (XSEDE) resource comet at the SDSC through allocation CHE150007, supported by the National Science Foundation [grant no. ACI-1548562]. R.S. was the recipient of an Erasmus Undergraduate Fellowship. The authors thank Dr. Eoin R. Gould for his assistance with 31P NMR experiments.
Glossary
Abbreviations
- ATP
adenosine 5′-triphosphate
- AMP
adenosine 5′-monophosphate
- ADP
adenosine 5′-diphosphate
- ATPPRT
ATP phosphoribosyltransferase
- PRPP
5-phospho-α-d-ribosyl-1-pyrophosphate
- PRATP
N1-(5-phospho-β-d-ribosyl)-ATP
- PPi
inorganic pyrophosphate
- DTT
dithiothreitol
- ITC
isothermal titration calorimetry
- DSF
differential scanning fluorimetry
- LC-MS
liquid chromatography–mass spectrometry
- PaATPPRT
P. arcticus ATPPRT
- PaHisGS
P. arcticus HisGS
- PaHisZ
P. arcticus HisZ
- 31P NMR
31P nuclear magnetic resonance
- MtPPase
Mycobacterium tuberculosis inorganic pyrophosphatase
- MWCO
molecular weight cut off
- ESI-MS
electrospray ionization mass spectrometry
- KD
equilibrium dissociation constant
- EcPRPPS
E. coli PRPP synthetase
- PRADP
N1-(5-phospho-β-d-ribosyl)-ADP
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.8b00559.
ITC curves, PaHisGS inhibition by AMP, spectra, DSF-based thermal denaturation of PaHisGS, determination of equilibrium dissociation, PaATPPRT and PaHisGS steady-state parameters from initial velocity patterns, PaHisGSTm’s by DSF in the presence and absence of ligands, steady-state kinetic constants, effect of Mn2+ on PaATPPRT and PaHisGS steady-state kinetic parameters, NBO charge distribution, solvent viscosity effects, and coordinates for all transition structures (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Barends T. R.; Dunn M. F.; Schlichting I. (2008) Tryptophan synthase, an allosteric molecular factory. Curr. Opin. Chem. Biol. 12, 593–600. 10.1016/j.cbpa.2008.07.011. [DOI] [PubMed] [Google Scholar]
- Fan Y.; Cross P. J.; Jameson G. B.; Parker E. J. (2018) Exploring modular allostery via interchangeable regulatory domains. Proc. Natl. Acad. Sci. U. S. A. 115, 3006–3011. 10.1073/pnas.1717621115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisco J. P.; de Chiara C.; Pacholarz K. J.; Garza-Garcia A.; Ogrodowicz R. W.; Walker P. A.; Barran P. E.; Smerdon S. J.; de Carvalho L. P. S. (2017) Uncoupling conformational states from activity in an allosteric enzyme. Nat. Commun. 8, 203. 10.1038/s41467-017-00224-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Carvalho L. P.; Argyrou A.; Blanchard J. S. (2005) Slow-onset feedback inhibition: Inhibition of mycobacterium tuberculosis alpha-isopropylmalate synthase by l-leucine. J. Am. Chem. Soc. 127, 10004–10005. 10.1021/ja052513h. [DOI] [PubMed] [Google Scholar]
- Buller A. R.; Brinkmann-Chen S.; Romney D. K.; Herger M.; Murciano-Calles J.; Arnold F. H. (2015) Directed evolution of the tryptophan synthase beta-subunit for stand-alone function recapitulates allosteric activation. Proc. Natl. Acad. Sci. U. S. A. 112, 14599–14604. 10.1073/pnas.1516401112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schendzielorz G.; Dippong M.; Grunberger A.; Kohlheyer D.; Yoshida A.; Binder S.; Nishiyama C.; Nishiyama M.; Bott M.; Eggeling L. (2014) Taking control over control: Use of product sensing in single cells to remove flux control at key enzymes in biosynthesis pathways. ACS Synth. Biol. 3, 21–29. 10.1021/sb400059y. [DOI] [PubMed] [Google Scholar]
- Cramer J. T.; Führing J. I.; Baruch P.; Brütting C.; Knölker H.-J.; Gerardy-Schahn R.; Fedorov R. (2018) Decoding allosteric networks in biocatalysts: Rational approach to therapies and biotechnologies. ACS Catal. 8, 2683–2692. 10.1021/acscatal.7b03714. [DOI] [Google Scholar]
- Pedreno S.; Pisco J. P.; Larrouy-Maumus G.; Kelly G.; de Carvalho L. P. (2012) Mechanism of feedback allosteric inhibition of atp phosphoribosyltransferase. Biochemistry 51, 8027–8038. 10.1021/bi300808b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ames B. N.; Martin R. G.; Garry B. J. (1961) The first step of histidine biosynthesis. J. Biol. Chem. 236, 2019–2026. [PubMed] [Google Scholar]
- Moggre G. J.; Poulin M. B.; Tyler P. C.; Schramm V. L.; Parker E. J. (2017) Transition state analysis of adenosine triphosphate phosphoribosyltransferase. ACS Chem. Biol. 12, 2662–2670. 10.1021/acschembio.7b00484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y.; Sharma V.; Sacchettini J. C. (2003) Crystal structure of atp phosphoribosyltransferase from mycobacterium tuberculosis. J. Biol. Chem. 278, 8333–8339. 10.1074/jbc.M212124200. [DOI] [PubMed] [Google Scholar]
- Cho Y.; Ioerger T. R.; Sacchettini J. C. (2008) Discovery of novel nitrobenzothiazole inhibitors for mycobacterium tuberculosis atp phosphoribosyl transferase (hisg) through virtual screening. J. Med. Chem. 51, 5984–5992. 10.1021/jm800328v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulis-Horn R. K.; Persicke M.; Kalinowski J. (2015) Corynebacterium glutamicum atp-phosphoribosyl transferases suitable for l-histidine production–strategies for the elimination of feedback inhibition. J. Biotechnol. 206, 26–37. 10.1016/j.jbiotec.2015.04.001. [DOI] [PubMed] [Google Scholar]
- Kulis-Horn R. K.; Persicke M.; Kalinowski J. (2014) Histidine biosynthesis, its regulation and biotechnological application in corynebacterium glutamicum. Microb. Biotechnol. 7, 5–25. 10.1111/1751-7915.12055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacholarz K. J.; Burnley R. J.; Jowitt T. A.; Ordsmith V.; Pisco J. P.; Porrini M.; Larrouy-Maumus G.; Garlish R. A.; Taylor R. J.; de Carvalho L. P. S.; Barran P. E. (2017) Hybrid mass spectrometry approaches to determine how l-histidine feedback regulates the enzyzme mtatp-phosphoribosyltransferase. Structure 25, 730–738. 10.1016/j.str.2017.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittelstadt G.; Moggre G. J.; Panjikar S.; Nazmi A. R.; Parker E. J. (2016) Campylobacter jejuni adenosine triphosphate phosphoribosyltransferase is an active hexamer that is allosterically controlled by the twisting of a regulatory tail. Protein Sci. 25, 1492–1506. 10.1002/pro.2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell R. M.; Koshland D. E. (1971) Allosteric properties of the first enzyme of the histidine operon. Bioorg. Chem. 1, 409–423. 10.1016/0045-2068(71)90043-5. [DOI] [Google Scholar]
- Martin R. G. (1963) The first enzyme in histidine biosynthesis: The nature of feedback inhibition by histidine. J. Biol. Chem. 238, 257–268. [Google Scholar]
- Morton D. P.; Parsons S. M. (1977) Inhibition of atp phosphoribosyltransferase by amp and adp in the absence and presence of histidine. Arch. Biochem. Biophys. 181, 643–648. 10.1016/0003-9861(77)90270-3. [DOI] [PubMed] [Google Scholar]
- Livingstone E. K.; Mittelstadt G.; Given F. M.; Parker E. J. (2016) Independent catalysis of the short form hisg from lactococcus lactis. FEBS Lett. 590, 2603–2610. 10.1002/1873-3468.12277. [DOI] [PubMed] [Google Scholar]
- Morton D. P.; Parsons S. M. (1976) Biosynthetic direction substrate kinetics and product inhibition studies on the first enzyme of histidine biosynthesis, adenosine triphosphate phosphoribosyltransferase. Arch. Biochem. Biophys. 175, 677–686. 10.1016/0003-9861(76)90559-2. [DOI] [PubMed] [Google Scholar]
- Kleeman J. E.; Parsons S. M. (1976) Reverse direction substrate kinetics and inhibition studies on the first enzyme of histidine biosynthesis, adenosine triphosphate phosphoribosyltransferase. Arch. Biochem. Biophys. 175, 687–693. 10.1016/0003-9861(76)90560-9. [DOI] [PubMed] [Google Scholar]
- Sissler M.; Delorme C.; Bond J.; Ehrlich S. D.; Renault P.; Francklyn C. (1999) An aminoacyl-trna synthetase paralog with a catalytic role in histidine biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 96, 8985–8990. 10.1073/pnas.96.16.8985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega M. C.; Zou P.; Fernandez F. J.; Murphy G. E.; Sterner R.; Popov A.; Wilmanns M. (2005) Regulation of the hetero-octameric atp phosphoribosyl transferase complex from thermotoga maritima by a trna synthetase-like subunit. Mol. Microbiol. 55, 675–686. 10.1111/j.1365-2958.2004.04422.x. [DOI] [PubMed] [Google Scholar]
- Bovee M. L.; Champagne K. S.; Demeler B.; Francklyn C. S. (2002) The quaternary structure of the hisz-hisg n-1-(5′-phosphoribosyl)-atp transferase from lactococcus lactis. Biochemistry 41, 11838–11846. 10.1021/bi020243z. [DOI] [PubMed] [Google Scholar]
- Stroek R.; Ge Y.; Talbot P. D.; Glok M. K.; Bernas K. E.; Thomson C. M.; Gould E. R.; Alphey M. S.; Liu H.; Florence G. J.; Naismith J. H.; da Silva R. G. (2017) Kinetics and structure of a cold-adapted hetero-octameric atp phosphoribosyltransferase. Biochemistry 56, 793–803. 10.1021/acs.biochem.6b01138. [DOI] [PubMed] [Google Scholar]
- Champagne K. S.; Sissler M.; Larrabee Y.; Doublie S.; Francklyn C. S. (2005) Activation of the hetero-octameric atp phosphoribosyl transferase through subunit interface rearrangement by a trna synthetase paralog. J. Biol. Chem. 280, 34096–34104. 10.1074/jbc.M505041200. [DOI] [PubMed] [Google Scholar]
- Champagne K. S.; Piscitelli E.; Francklyn C. S. (2006) Substrate recognition by the hetero-octameric atp phosphoribosyltransferase from lactococcus lactis. Biochemistry 45, 14933–14943. 10.1021/bi061802v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alphey M. S.; Fisher G.; Ge Y.; Gould E. R.; Machado T. G.; Liu H.; Florence G. J.; Naismith J. H.; da Silva R. G. (2018) Catalytic and anticatalytic snapshots of a short-form atp phosphoribosyltransferase. ACS Catal. 8, 5601–5610. 10.1021/acscatal.8b00867. [DOI] [Google Scholar]
- Smith D. W.; Ames B. N. (1965) Phosphoribosyladenosine monophosphate, an intermediate in histidine biosynthesis. J. Biol. Chem. 240, 3056–3063. [PubMed] [Google Scholar]
- Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Mennucci B., Petersson G. A., Nakatsuji H., Caricato M., Li X., Hratchian H. P., Izmaylov A. F., Bloino J., Zheng G., Sonnenberg J. L., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Montgomery J. A. Jr., Peralta J. E., Ogliaro F., Bearpark M. J., Heyd J. J., Brothers E. N., Kudin K. N., Staroverov V. N., Kobayashi R., Normand J., Raghavachari K., Rendell A. P., Burant J. C., Iyengar S. S., Tomasi J., Cossi M., Rega N., Millam J. M., Klene M., Knox J. E., Cross J. B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R. E., Yazyev O., Austin A. J., Cammi R., Pomelli C., Ochterski J. W., Martin R. L., Morokuma K., Zakrzewski V. G., Voth G. A., Salvador P., Dannenberg J. J., Dapprich S., Daniels A. D., Farkas O., Foresman J. B., Ortiz J. V., Cioslowski J., and Fox D. J. (2009), Gaussian 09, Gaussian Inc., Wallingford, CT.
- Niesen F. H.; Berglund H.; Vedadi M. (2007) The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2, 2212–2221. 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- Mittelstadt G.; Jiao W.; Livingstone E. K.; Moggre G. J.; Nazmi A. R.; Parker E. J. (2018) A dimeric catalytic core relates the short and long forms of atp-phosphoribosyltransferase. Biochem. J. 475, 247–260. 10.1042/BCJ20170762. [DOI] [PubMed] [Google Scholar]
- Cleland W. W. (1967) Enzyme kinetics. Annu. Rev. Biochem. 36, 77–112. 10.1146/annurev.bi.36.070167.000453. [DOI] [PubMed] [Google Scholar]
- Lohkamp B.; McDermott G.; Campbell S. A.; Coggins J. R.; Lapthorn A. J. (2004) The structure of escherichia coli atp-phosphoribosyltransferase: Identification of substrate binding sites and mode of amp inhibition. J. Mol. Biol. 336, 131–144. 10.1016/j.jmb.2003.12.020. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Cole P. A. (2014) Catalytic mechanisms and regulation of protein kinases. Methods Enzymol. 548, 1–21. 10.1016/B978-0-12-397918-6.00001-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace M. R.; Walsh C. T.; Cole P. A. (1997) Divalent ion effects and insights into the catalytic mechanism of protein tyrosine kinase csk. Biochemistry 36, 1874–1881. 10.1021/bi962138t. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Shang X.; Deng A.; Chai X.; Lai S.; Zhang G.; Wen T. (2012) Genetic and biochemical characterization of corynebacterium glutamicum atp phosphoribosyltransferase and its three mutants resistant to feedback inhibition by histidine. Biochimie 94, 829–838. 10.1016/j.biochi.2011.11.015. [DOI] [PubMed] [Google Scholar]
- Guthrie R. D.; Jencks W. P. (1989) Iupac recommendations for the representation of reaction mechanisms. Acc. Chem. Res. 22, 343–349. 10.1021/ar00166a001. [DOI] [Google Scholar]
- In IUPAC recommendation for reaction mechanism nomenclature (see ref (39)), ANDN describes an associative nucleophilic substitution reaction mechanism where the electrophile is partially bonded to both incoming nucleophile and departing leaving group at the transition state. DN*AN‡ describes a dissociative nucleophilic substitution reaction mechanism where the leaving group departs to form an intermediate, and the highest-energy transition state is the one for subsequent nucleophilic attack to the intermediate.
- Burgos E. S.; Vetticatt M. J.; Schramm V. L. (2013) Recycling nicotinamide. The transition-state structure of human nicotinamide phosphoribosyltransferase. J. Am. Chem. Soc. 135, 3485–3493. 10.1021/ja310180c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun G.; Nicklaus M. C. (2007) Natural resonance structures and aromaticity of the nucleobases. Theor. Chem. Acc. 117, 323–332. 10.1007/s00214-006-0154-9. [DOI] [Google Scholar]
- Lewandowicz A.; Schramm V. L. (2004) Transition state analysis for human and plasmodium falciparum purine nucleoside phosphorylases. Biochemistry 43, 1458–1468. 10.1021/bi0359123. [DOI] [PubMed] [Google Scholar]
- Kline P. C.; Schramm V. L. (1993) Purine nucleoside phosphorylase. Catalytic mechanism and transition-state analysis of the arsenolysis reaction. Biochemistry 32, 13212–13219. 10.1021/bi00211a033. [DOI] [PubMed] [Google Scholar]
- Silva R. G.; Hirschi J. S.; Ghanem M.; Murkin A. S.; Schramm V. L. (2011) Arsenate and phosphate as nucleophiles at the transition states of human purine nucleoside phosphorylase. Biochemistry 50, 2701–2709. 10.1021/bi200279s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- sheely M. L. (1932) Glycerol viscosity tables. Ind. Eng. Chem. 24, 1060–1064. 10.1021/ie50273a022. [DOI] [Google Scholar]
- Karsten W. E.; Lai C.-J.; Cook P. F. (1995) Inverse solvent isotope effects in the nad-malic enzyme reaction are the result of the viscosity difference between d2o and h2o: Implications for solvent isotope effect studies. J. Am. Chem. Soc. 117, 5914–5918. 10.1021/ja00127a002. [DOI] [Google Scholar]
- Lin Y.; West A. H.; Cook P. F. (2008) Potassium is an activator of homoisocitrate dehydrogenase from saccharomyces cerevisiae. Biochemistry 47, 10809–10815. 10.1021/bi801370h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.; Volkman J.; Nicholas K. M.; Yamamoto T.; Eguchi T.; Nimmo S. L.; West A. H.; Cook P. F. (2008) Chemical mechanism of homoisocitrate dehydrogenase from saccharomyces cerevisiae. Biochemistry 47, 4169–4180. 10.1021/bi702361j. [DOI] [PubMed] [Google Scholar]
- Goitein R. K.; Chelsky D.; Parsons S. M. (1978) Primary 14c and alpha secondary 3h substrate kinetic isotope effects for some phosphoribosyltransferases. J. Biol. Chem. 253, 2963–2971. [PubMed] [Google Scholar]
- Johnson K. A. (1992) 1 transient-state kinetic analysis of enzyme reaction pathways. In The enzymes (Sigman D. S., Ed.), pp 1–61, Academic Press. [Google Scholar]
- Johnson K. A. (1995) Rapid quench kinetic analysis of polymerases, adenosinetriphosphatases, and enzyme intermediates. Methods Enzymol. 249, 38–61. 10.1016/0076-6879(95)49030-2. [DOI] [PubMed] [Google Scholar]
- Hartley B. S.; Kilby B. A. (1954) The reaction of p-nitrophenyl esters with chymotrypsin and insulin. Biochem. J. 56, 288–297. 10.1042/bj0560288. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.