

Abstract
Non-homologous end joining (NHEJ) is the primary pathway of DNA double-strand break repair in vertebrate cells, yet it remains unclear how NHEJ factors assemble a synaptic complex that bridges DNA ends. To address the role of XRCC4-like factor (XLF) in synaptic complex assembly, we employed single-molecule fluorescence imaging in Xenopus laevis egg extract, a system that efficiently joins DNA ends. We find that a single XLF dimer binds to DNA substrates just prior to formation of a ligation-competent synaptic complex between DNA ends. The interaction of both globular head domains of the XLF dimer with XRCC4 is required for efficient formation of this synaptic complex. In contrast to a model in which filaments of XLF and XRCC4 bridge DNA ends, our results indicate that binding of a single XLF dimer facilitates the assembly of a stoichiometrically well-defined synaptic complex.
Introduction
Canonical non-homologous end joining (NHEJ) is the major pathway of DNA double-strand break (DSB) repair in vertebrate cells. Successful re-joining of broken DNA ends requires that they be held together in a manner that permits their processing and ligation. DNA ends are first bound by the Ku heterodimer, composed of Ku70 and Ku80, which fits over the DNA end like a ring1. Ku recruits the DNA-dependent protein kinase catalytic subunit (DNA-PKcs), whose kinase activity is required for efficient end joining2–4. Ends are ultimately ligated by DNA ligase 4 (LIG4), which resides in a complex with XRCC45,6. A homolog of XRCC4, named XRCC4-like factor (XLF) or Cernunnos, was identified as an XRCC4-interacting protein whose disruption in humans results in immunodeficiency, developmental defects, and cellular radiosensitivity7,8. Despite the identification of these core components of the NHEJ machinery, the nature of the synaptic complex that aligns broken DNA ends, in particular the role of XLF and XRCC4:LIG4, remains poorly defined.
Structural characterization of XLF and XRCC4 reveals that both proteins form symmetric homodimers that interact through their globular head domains9–14, and mutations in XLF or XRCC4 that disrupt this interaction impair NHEJ in cell culture-based assays15–17. Two possible roles of the XLF-XRCC4 interaction in NHEJ have been proposed: First, XLF may directly promote catalysis by XRCC4:LIG4. XLF stimulates XRCC4:LIG4 activity in vitro, especially on non-cohesive and mismatched ends17–21. XLF likewise increases the rate of LIG4 autoadenylation, the initial step in the LIG4 catalytic cycle22. The second proposal is that alternating filaments of XLF and XRCC4 bridge broken DNA ends. Filaments of purified XLF and XRCC4 are seen in crystal structures and electron micrographs12–14,23–25, and a mixture of XLF and XRCC4 bridges DNA molecules in bulk and single-molecule assays16,17,25,26. Additionally, super-resolution imaging of fixed cells stained with anti-XLF or anti-XRCC4 antibodies revealed elongated nuclear foci proposed to be XLF-XRCC4 filaments27. While these results are suggestive, it has not been demonstrated that XLF-XRCC4 filaments are necessary for physiological end joining.
To study the role of XLF-XRCC4 interaction in NHEJ, we employed Xenopus laevis egg extract, which performs efficient NHEJ dependent on the core pathway components Ku, DNA-PKcs, XLF, XRCC4, and LIG428–38. We previously combined this cell-free system with a single-molecule Förster resonance energy transfer (smFRET) assay that allowed us to visualize synapsis of single pairs of DNA ends during NHEJ37,38. These experiments revealed a stepwise transition from an initial, “long-range” synaptic complex (LR-complex), in which DNA ends are physically tethered but not held closely together, to a “short-range” synaptic complex (SR-complex) in which DNA ends are closely aligned for ligation. The SR-complex either ligates DNA ends or dissociates, requiring another round of SR-complex formation prior to eventual ligation. Formation of the LR-complex requires Ku and DNA-PKcs, consistent with previously reported dimerization and DNA end-bridging activities of the DNA-PK holoenzyme39–42. Transition from the LR-complex to the SR-complex requires DNA-PK catalytic activity, XLF, and XRCC4:LIG4, but not LIG4 catalytic activity. A non-catalytic role of LIG4 in the stable bridging of DNA ends is consistent with previous results from pull-down experiments in mammalian cell-free extracts43. A subsequent study in a reconstituted mixture of human NHEJ proteins observed a transition from a transient synaptic complex dependent on Ku and DNA-PKcs to a more stable complex dependent on XLF and XRCC4:LIG444. These observations closely parallel our results in Xenopus egg extract, suggesting that the mechanisms of synaptic complex assembly are conserved between frogs and humans.
Here, we investigated the role of the XLF-XRCC4 interaction in synaptic complex formation. Using X. laevis egg extract, we show that XLF-XRCC4 interaction is required for formation of the SR-complex. In three-color single-molecule imaging experiments, we visualized binding of fluorescently labeled XLF protein to individual DNA substrates undergoing synapsis. Strikingly, formation of the SR-complex involves association of only a single XLF dimer with DNA ends. Experiments with a synthetic XLF tandem dimer imply that XLF nonetheless interacts with XRCC4 through both globular head domains of the XLF dimer. These results support a model in which XLF binds with defined stoichiometry to the synaptic complex, facilitating a structural rearrangement that closely aligns DNA ends.
Results
XLF-XRCC4 interaction is required for end joining in Xenopus egg extract
To explore the function of the XLF-XRCC4 interaction during physiological end joining, we first tested whether Xenopus laevis XLF and XRCC4 interact. To this end, we used bio-layer interferometery (BLI) to measure association of X. laevis XLF with immobilized X. laevis XRCC4 (unless otherwise specified, “XLF” and “XRCC4” will refer to the X. laevis proteins). Full-length XLF bound XRCC4 and only partially dissociated when washed with buffer, suggesting irreversible aggregation on the surface (Supplementary Fig. 1A). Similar aggregation was previously observed in surface plasmon resonance (SPR) experiments with human XLF and XRCC410. However, a truncated version of XLF lacking the unstructured C-terminal region (XLF1−226, corresponding to human XLF1−224 [ref. 13]) bound XRCC4 and completely dissociated when washed with buffer (Fig. 1A and Supplementary Fig. 1B). X. laevis XLF1−226 and XRCC4 interact with slightly higher affinity than was reported for the human XLF and XRCC4 (apparent Kd of 0.28 ± 0.03 μM, compared to 1.2 μM to 7.8 μM for hXLF-hXRCC4; see, however, Fig. 5C below and Supplementary Table 1) 10,13,45. Thus, XLF-XRCC4 interaction is conserved between Xenopus and humans. Dynamic light scattering (DLS) revealed that XRCC4 and XLF formed large complexes when mixed, suggesting assembly of filaments (Supplementary Fig. 1C). Also, similar to human XLF and XRCC4, a mixture of XLF and XRCC4, but neither protein alone, bridged DNA in pull-down experiments (Supplementary Fig. 1D). Thus, XLF-XRCC4 interaction, in vitro filament formation, and DNA bridging are conserved between X. laevis and humans.
Figure 1: XLF-XRCC4 interaction is required for NHEJ in Xenopus egg extract.
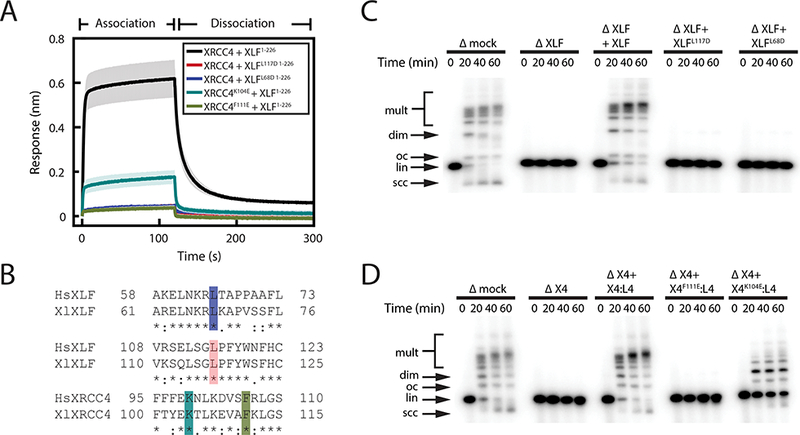
(A) Bio-layer interferometery (BLI) measurements of interaction between point mutants of XRCC4 and C-terminally truncated XLF (XLF1−226). Traces represent the average and the shading around each trace represents the standard deviation of three replicates.
(B) Alignment of human (Hs) and Xenopus laevis (Xl) XLF and XRCC4 around residues required for the XLF-XRCC4 interaction: XlXLF L68 (HsXLF L65, blue)13, XlXLF L117 (HsXLF L115, red)13, XlXRCC4 K104 (HsXRCC4 K99, cyan)11,16, and XlXRCC4 F111 (HsXRCC4 F106, chartreuse)13.
(C-D) Bulk NHEJ assay in egg extract immunodepleted of XLF (C) or XRCC4 (D) and supplemented with purified recombinant protein. The molar ratio of recombinant protein:DNA ends for XLF and XRCC4:LIG4 in the rescue experiments shown in C and D is 500:1 and 50:1, respectively. Δmock, immunodepletion with nonspecific rabbit IgG; ΔX4, XRCC4 depletion, ΔXLF, XLF depletion; X4:L4, recombinant XRCC4:LIG4 complex. DNA species: scc, supercoiled closed-circular; lin, linear; oc, open-circular; dim, dimer; mult, multimer. Uncropped images of C and D are in Supplementary Data Set 1.
Figure 5: Characterization of human XLFL115A and Xenopus XLFL117A.
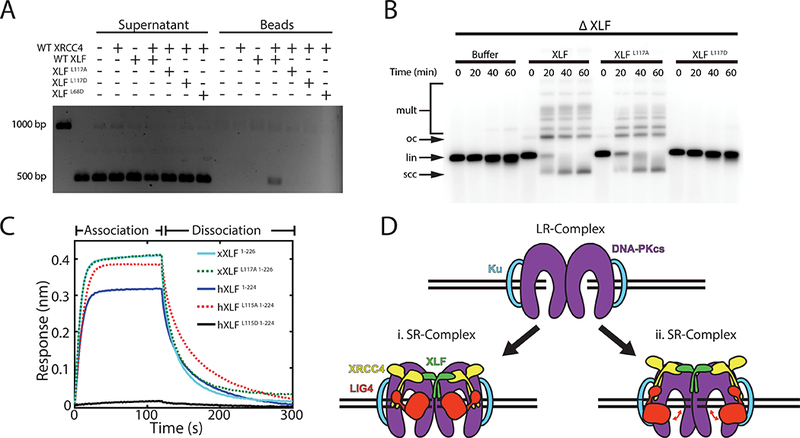
(A) XLF-XRCC4 DNA pull-down assay. Beads coated with a biotinylated 1000 bp DNA fragment were incubated with a soluble 500 bp DNA fragment and the indicated proteins. Deproteinized supernatant and bead-bound fractions were separated on a 1x TBE agarose gel. 1000 and 500 bp DNA fragments alone were loaded in the first two lanes.
(B)) Bulk NHEJ assay in egg extract immunodepleted of XLF (ΔXLF) and supplemented with purified recombinant protein. lin, linear substrate; oc, open-circular products; scc, supercoiled closed-circular products; mult, dimeric and higher-order multimeric products. Uncropped images of A and B are in Supplementary Data Set 1.
(C)) Bio-layer interferometry of XRCC4 binding by wild-type and L115A/L117A mutant human and Xenopus XLF. Response curves represent the average of two experimental replicates for each condition. XLF variants were tested at a concentration of 250 nM, with the exception of hXLF1−224,L115D, which was tested at 2 μM. Fitted Kon and Koff values are listed in Supplementary Table 1.
(D) Model of the role of XLF in short-range synaptic complex assembly. Binding of both head domains of XLF to XRCC4:LIG4 is required for SR-complex formation, which may involve (i) direct engagement of both DNA ends by the XLF:XRCC4:LIG4 ternary complex, or (ii) a conformational change in the DNA-PK holoenzyme that is facilitated allosterically by binding of the XLF:XRCC4:LIG4 ternary complex. In the latter model, conformational flexibility in LIG4 (red double arrows) permits binding and unbinding of the LIG4 catalytic domain to DNA ends.
Mutations of human XLF and XRCC4 that disrupt the interaction between the two proteins impair NHEJ in cell-based assays13,15–17. We tested the effect of generating the corresponding mutations in the X. laevis proteins (see Fig. 1B). Interaction between XLF and XRCC4 was abolished by the XLF L68D and L117D mutations and by the XRCC4 F111E mutation (Fig. 1A-B). The XRCC4 K104E mutation weakened but did not eliminate interaction with XLF (Fig. 1A), reflecting a higher dissociation rate (Supplementary Fig. 1E-F). The denaturation temperatures of all mutants, measured by differential scanning fluorimetry (DSF), were within 1.45°C of the wild-type protein, arguing that the defect is not due to large-scale misfolding (Supplementary Fig. 1G). Mutations that disrupted XLF-XRCC4 interaction also disrupted formation of large XLF-XRCC4 complexes in DLS experiments and abolished DNA bridging in pull-down experiments (Supplementary Fig. 1C-D).
We next tested whether interaction-deficient XLF and XRCC4 mutants support cell-free NHEJ. Egg extract was immunodepleted of XLF or XRCC4, which abolished end joining of linear DNA substrates (Fig. 1C-D; ref. 38). This effect was fully rescued by addition of recombinant XLF to XLF-depleted extract (Fig. 1C) or by addition of recombinant XRCC4:LIG4 to XRCC4-depleted extract (Fig. 1D; note that depletion of XRCC4 co-depletes LIG4 [see ref. 38]). In contrast, XLFL117D and XLFL68D did not rescue end joining in XLF-depleted egg extract (Fig. 1C), and XRCC4F111E:LIG4 did not rescue end joining in XRCC4-depleted extract (Fig. 1D). XRCC4K104E:LIG4 partially rescued end joining in XRCC4-depleted extract (Fig. 1D), consistent with the residual low-affinity interaction between XRCC4K104E and XLF (Fig. 1A and Supplementary Fig. 1E-F). Taken together, these results argue that, as in mammals, the interaction between XLF and XRCC4 is required for end joining in frog egg extract.
XLF-XRCC4 interaction is required for short-range synapsis of DNA ends
We previously described two related single-molecule fluorescence assays for monitoring synaptic complex formation in egg extract38. In the “intermolecular” assay, a surface-tethered DNA duplex labeled with Cy3 is exposed to extract containing a DNA duplex labeled with Cy5. Co-localization of Cy3 and Cy5 without FRET reports on the formation of the “long-range” synaptic complex (LR-complex), whereas the appearance of FRET signals the formation of the short-range synaptic complex (SR-complex), in which DNA ends are closely aligned and poised for ligation. To increase the efficiency of SR-complex formation, we also employed an “intramolecular” assay in which a 2-kilobasepair DNA substrate labeled 7 nucleotides from one end with Cy3 and 7 nucleotides from the other end with Cy5 is tethered to a glass coverslip via an internal biotin (Fig. 2A). FRET between Cy3 and Cy5, detected by TIRF microscopy, indicates assembly of the SR-complex. Because the Cy3 and Cy5 labels on the intramolecular substrate always co-localize, this assay cannot detect LR-complex assembly.
Figure 2: XLF-XRCC4 interaction is required for SR-complex formation.
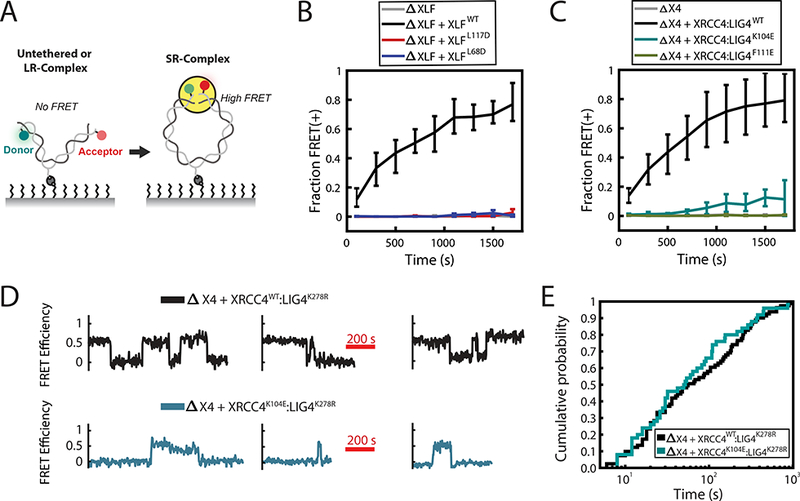
(A) Schematic of intramolecular single-molecule FRET reporter for monitoring short-range synaptic complex (SR-complex) formation38. DNA is tethered internally via a biotin-streptavidin interaction and labeled 7 bp from each end with Cy3 (Donor) and Cy5 (Acceptor) fluorophores. No FRET is detected when DNA ends are untethered or in the long-range synaptic complex (LR-complex). Close alignment of DNA ends within the SR-complex is indicated by energy transfer between Cy3 and Cy5. The data shown in B and C were collected using a 0.5 s exposure time, alternating between two frames of Cy3 excitation and one frame of Cy5 excitation. The data shown in D and E were collected using a 1 s exposure with a 1 s delay between exposures, alternating between four frames of Cy3 excitation and one frame of Cy5 excitation.
(B-C) Kinetics of SR-complex formation in extract depleted of XLF or XRCC4 and supplemented with purified recombinant protein, as in Fig. 1C-D. The mean fraction of FRET-positive (SR-complex or ligated) substrates is plotted as a function of time after extract addition. Error bars represent the minimum and maximum values obtained from multiple experimental replicates. See Supplementary Table 2 for sample sizes.
(D) Sample smFRET trajectories showing SR-complex formation and dissociation in XRCC4-immunodepleted extract supplemented with either XRCC4WT:LIG4K278R or XRCC4K104E:LIG4K278R. Catalytically inactive LIG4K278R was used here and in panel E to prevent ligation of DNA ends.
(E) Cumulative distribution functions of SR-complex lifetimes. The difference between the two conditions was not statistically significant (p = 0.45, log-rank test). See Supplementary Table 3 for sample sizes.
Given our previous demonstration that XLF and XRCC4:LIG4 are dispensable for LR-complex formation38, we employed the intramolecular assay to address whether the XLF-XRCC4 interaction is required for SR-complex formation. For these experiments, many fields of view were imaged sequentially, and the fraction of DNA substrates with FRET efficiency > 0.25 was plotted as a function of time after extract addition (Figure 2B and C). As shown previously38, depletion of XLF or XRCC4 abolished SR-complex formation, and these defects were rescued by purified recombinant XLF or XRCC4:LIG4, respectively (Fig. 2B-C). By contrast, interaction-deficient XLFL68D, XLFL117D, and XRCC4F111E:LIG4 failed to support SR-complex formation (Fig. 2B and 2C), while XRCC4K104E:LIG4 exhibited low activity, consistent with its residual binding to XLF (Fig. 2C). Thus, a physical interaction between XLF and XRCC4 is required for the close alignment of DNA ends within the short-range synaptic complex.
The above results do not distinguish whether XLF-XRCC4 interaction is required for the formation or maintenance of the SR-complex. To address this question, we performed long time-course imaging experiments to compare the kinetics of SR-complex formation and dissociation in the presence of wild-type XRCC4 and the hypomorphic K104E mutant. To prevent ligation of DNA substrates in these experiments, extract immunodepleted of XRCC4 was supplemented with XRCC4 WT or K104E in complex with catalytically inactive LIG4K278R. The SR-complex formed at a 5-fold slower rate in the presence of XRCC4K104E compared to XRCC4WT (2.7 × 10−4 ± 0.5 × 10−4 s−1, standard error of the mean, compared to 1.4 × 10−3 ± 0.3 × 10−3 s−1, S.E.M.), implying that the XLF-XRCC4 interaction is required for efficient establishment of the SR-complex. Once formed, however, the SR-complex had an equivalent lifetime in the presence of XRCC4K104E (129 ± 27 s) and XRCC4WT (153 ± 23 s) (Fig. 2D-E), suggesting that the stability of the assembled SR-complex becomes independent of XRCC4-XLF interaction. In both cases, the lifetime distribution of the SR-complex is significantly non-exponential (p < 0.001, Lilliefors test46), suggesting that SR-complex dissociation is not governed by a single rate constant.
The short-range synaptic complex contains a single XLF dimer
A model in which end synapsis involves the formation of XLF-XRCC4 filaments predicts binding of numerous XLF dimers to each pair of DNA ends. To determine the stoichiometry of XLF within the synaptic complex, we employed three-color imaging of Cy3/Cy5-labeled DNA ends and Alexa Fluor 488-labeled, Halo-tagged XLF (AF488-XLF). AF488-XLF rescued end joining in XLF-immunodepleted extract, demonstrating that XLF labeled in this manner is functional (Supplementary Fig. 2A). Three-color imaging allowed us to quantify binding of AF488-XLF to DNA substrates while also monitoring FRET between Cy3- and Cy5-labeled DNA ends (Fig. 3A). Biotinylated AF488-Halo tag deposited on the coverslip surface was used as an internal standard to determine the intensity of single AF488 fluorophores in egg extract (see Methods).
Figure 3: Three-color imaging of XLF binding and SR-complex formation.
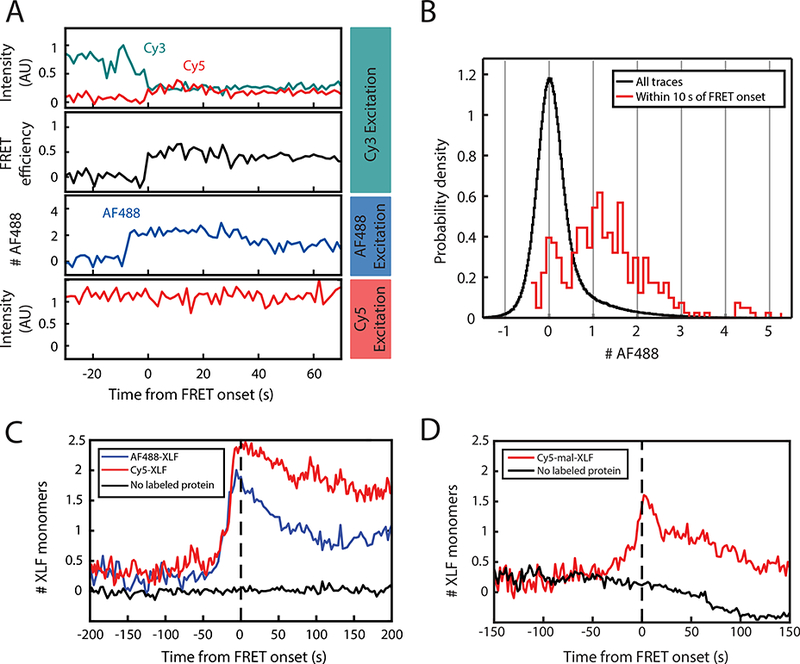
(A) 3-color single-molecule imaging of FRET between Cy3 and Cy5-labeled DNA ends, along with binding of Halo-tagged XLF protein labeled with Alexa Fluor 488 (AF488-XLF). Top panel: Intensity in the Cy3 (cyan) and Cy5 (red) channels with excitation of Cy3. Second panel: Calculated Cy3-Cy5 FRET efficiency. Third panel: AF488 intensity, expressed as a multiple of the intensity of single-AF488 reference spots. Bottom panel: Cy5 intensity with direct excitation. See Supplementary Fig. 2F for additional traces.
(B) Histogram of AF488 intensity, expressed as a multiple of the average intensity of single-AF488 reference spots. The black curve includes all substrates, while the red curve includes only the subset of frames within 10 s of the transition to high FRET.
(C) Average of AF488-XLF and Cy5-XLF stoichiometry for traces aligned at the time of FRET onset. AF488 intensity, normalized to the intensity of single-AF488 reference spots, was divided by the protein labeling fraction to give XLF protein stoichiometry. Cy5 intensity, normalized to single-Cy5 intensity (see Supplementary Fig. 3A), was divided by protein labeled fraction after subtracting 1 to account for the single Cy5 label on the DNA. Black curve shows data from a mock AF488 imaging experiment without AF488-XLF protein.
(D) Average stoichiometry of XLF nonspecifically labeled on cysteines with Cy5-maleimide, aligned at the time of FRET onset. Cy5-mal-XLF stoichiometry was calculated as for Cy5-XLF in panel (C). Black curve shows data from a control experiment in the absence of Cy5-mal-XLF. The decay of the black curve below zero results from photobleaching of the Cy5 label on DNA.
At any instant, most DNA substrates exhibited no AF488 signal (Fig. 3B, black curve). Given that AF488-XLF monomer was 68% labeled based on UV-Vis absorbance measurements (see Methods), this result implies low average occupancy of XLF on DNA substrates, and it is inconsistent with widespread formation of XLF-XRCC4 filaments under these conditions. We next focused on substrates that undergo a transition to the SR-complex, as indicated by the appearance of FRET between Cy3 and Cy5. Strikingly, SR-complex formation was typically preceded by step-like AF488-XLF binding events (Fig. 3A and Supplementary Fig. 2F). The number of AF488 fluorophores present within 10 s of SR-complex formation, determined by comparison with the internal single-AF488 standard, was mostly between 0 and 2 (Fig. 3B, red curve), consistent with binding of a single, incompletely labeled dimer of XLF. To test this interpretation, we fit the distribution of fluorophore number in the AF488 frame just prior to SR-complex formation to a binomial distribution with n = 2 (Supplementary Fig. 2E). The fraction of labeled XLF obtained from the fit (0.60; bootstrap 95% C.I., 0.46–0.71) is in good agreement with the value of 0.68 estimated from absorbance measurements. An ensemble average of XLF stoichiometry, aligned at the time of SR-complex formation and corrected for the labeled fraction, showed a peak occupancy of ~2 XLF monomers (i.e., 1 XLF dimer) several seconds prior to SR-complex formation (Fig. 3C, blue curve). No such peak was observed in the absence of labeled protein (Fig. 3C, black curve), ruling out a photophysical artifact. Similar results were obtained when Halo-XLF was labeled with Cy5 (Cy5-XLF; Fig. 3C, red curve, and Supplementary Fig. 3A-C). Remarkably, despite the ability of Halo-XLF to support NHEJ with similar efficiency as untagged XLF (Supplementary Fig. 2A), a mixture of Halo-XLF and XRCC4 did not bridge DNA in bulk pull-down experiments (Supplementary Fig. 1H), indicating that the DNA bridging activity detected by this pull-down assay is not essential for NHEJ. To ensure that the observed low stoichiometry of XLF in single-molecule experiments was not due to the inability of Halo-XLF to form DNA-bridging filaments with XRCC4, we labeled untagged XLF on endogenous cysteines with Cy5-maleimide. Cy5-maleimide-labeled XLF (Cy5-mal-XLF) rescued end joining in XLF-depleted extract (Supplementary Fig. 3D), and unlike Halo-XLF, it assembled DNA-bridging complexes with XRCC4 in bulk pull-down experiments (Supplementary Fig. 3E). Nonetheless, Cy5-mal-XLF still bound with an average stoichiometry consistent with 1 dimer per DNA substrate at the time of SR-complex formation (Fig. 3D). These results imply that SR-complex formation involves binding of a single XLF dimer.
End synapsis requires interaction of both XLF head domains with XRCC4
Although our data indicate that extensive XLF-XRCC4 filaments are not required for SR-complex formation, the possibility remained that both subunits of the XLF dimer must interact with XRCC4. To address this point, we generated an XLF tandem dimer construct (tdXLF; Fig. 4A) in which the number of XRCC4 interaction sites could be precisely defined. tdXLF had the same molecular weight as the wild-type XLF dimer, as measured by size-exclusion chromatography with multi-angle light scattering (SEC-MALS; Supplementary Fig. 4A-B). End joining in XLF-immunodepleted egg extract was rescued by tdXLF, demonstrating that this construct functions in NHEJ (Fig. 4B-C).
Figure 4: End joining in the presence of a synthetic tandem dimer of XLF.
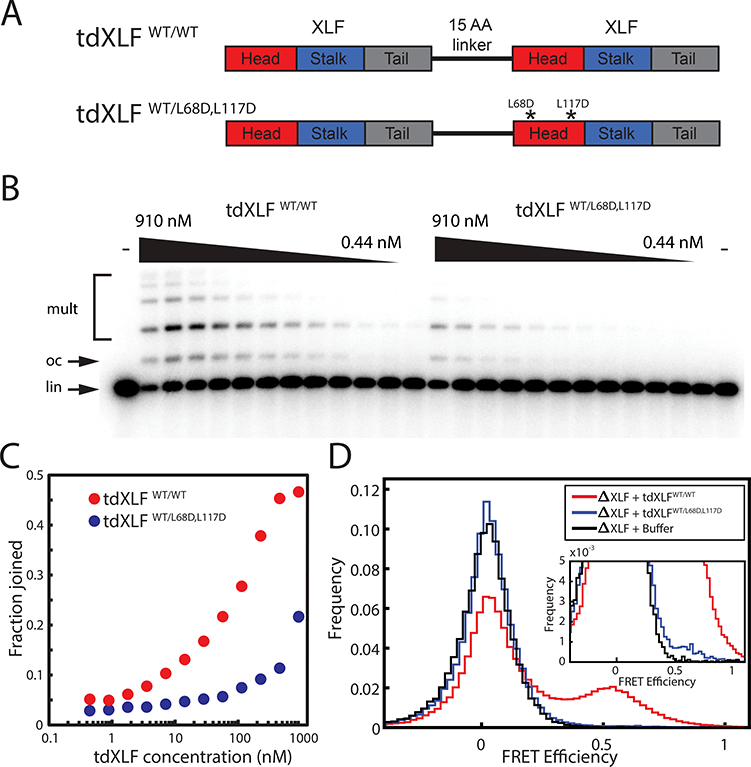
(A) Schematic of XLF tandem dimer (tdXLF) construct. Two XLF coding sequences are concatenated by a flexible 15-amino acid-long linker. Asterisks denote point mutations.
(B) Dose-dependence of end joining as a function of tdXLF concentration for WT/WT and WT/L68D,L117D constructs. Black wedges represent 2-fold serial dilution series from 910 nM to 0.44 nM. Control reactions not supplemented with tdXLF are labeled “-”. lin, linear DNA substrate; oc, open-circular product; mult, dimeric and multimeric products. An uncropped image of B is in Supplementary Data Set 1.
(C) Quantification of product formation in the gel in panel (B). Fraction joined was quantified as background-subtracted intensity of product bands divided by total background-subtracted intensity of substrate and product bands.
(D) Histogram of FRET efficiency from tdXLF rescue of smFRET intramolecular circularization experiments, accumulated over 20 min.
To test whether interaction with XRCC4 on both sides of the XLF dimer is required for end joining, XLF-depleted extract was supplemented with tdXLFWT/WT or tdXLFWT/L68D,L117D, in which the second XLF sequence in the tandem dimer was mutated to prevent interaction with XRCC4 (Fig. 4A). While tdXLFWT/L68D,L117D partially rescued end joining, its activity was impaired ~8 to 16-fold relative to tdXLFWT/WT—more so than would be expected from a 50% reduction in the effective concentration of wild-type XLF head domains (Fig. 4B-C). Equivalent results were obtained for the tdXLFWT/L117D single mutant (not shown). Importantly, tdXLFWT/L68D,L117D exhibited a 15-fold reduction in SR-complex formation rate (1.9×10−4 ± 1.1×10−4 s−1) relative to tdXLFWT/WT (2.8×10−3 ± 1.1×10−3 s−1) in intramolecular smFRET circularization assays (Fig. 4D). However, a low level of SR-complex formation and end joining still occurred (Fig. 4B-D). Given the presence of two interaction-blocking mutations, it is unlikely that this residual activity of tdXLFWT/L68D,L117D reflects binding of the mutant head domain to XRCC4. Thus, binding of XRCC4 to both head domains of XLF is required to form the SR-complex with normal efficiency, while binding of a single XLF head domain to XRCC4 permits SR-complex formation at a substantially reduced rate.
As an independent means of examining the requirement for XRCC4-XLF interaction through both XLF head domains, we generated synthetic XLF heterodimers by co-expression of XLF monomers with different affinity tags, followed by tandem affinity purification (see Methods). Exchange of monomers between purified XLF dimers is negligible (Supplementary Fig. 4C-D). Consistent with results for tdXLF, XLF heterodimers with one wild-type subunit and one L117D mutant subunit were severely deficient in end joining compared to WT:WT heterodimers (Supplementary Fig. 4E-F). Taken together, these results indicate that efficient SR-complex formation and end joining require that both head domains of a single XLF monomer interact with XRCC4.
NHEJ requires XLF-XRCC4 binding but not DNA bridging by filaments
As noted above, N-terminally Halo-tagged XLF supports end joining despite failing to bridge DNA in a pulldown assay (Supplementary Fig. 1H and 2A). The behavior of this fusion protein is reminiscent of the L115A mutant of human XLF, which fails to bridge DNA in vitro while rescuing the DNA damage sensitivity of various XLF-deficient cell lines17,47. To investigate further whether the in vitro DNA bridging activity of XLF is separable from its function in end joining, we examined the corresponding L117A mutant in X. laevis XLF. Similar to hXLFL115A, XLFL117A failed to bridge DNA in pulldown experiments with XRCC4 (Fig. 5A). However, XLFL117A supported robust end joining in XLF-immunodepleted egg extract (Fig. 5B). Previous results from a native PAGE binding assay suggested that hXLFL115A is deficient in interacting with XRCC411. In contrast, our quantitative BLI measurements reveal that both hXLFL115A and Xenopus XLFL117A interact with XRCC4 with apparent Kd values similar to wild-type XLF (Fig. 5C and Supplementary Table 1). The fact that XLFL117A (hXLFL115A) interacts with XRCC4 and supports NHEJ, despite not bridging DNA in vitro, argues that DNA bridging by XLF-XRCC4 filaments is not essential for NHEJ.
Discussion
Despite years of study, the mechanism of DNA end synapsis during non-homologous end joining has remained elusive. To address this problem, we employed Xenopus egg extract, which contains the entire soluble egg proteome and supports highly efficient end joining that requires all four core NHEJ factors (Ku, DNA-PKcs, XLF, and XRCC4:LIG4). Using single-molecule imaging in egg extract, we show that the XLF-XRCC4 interaction is essential for close alignment of DNA ends within the SR-complex. We further demonstrate that a single XLF dimer, interacting with XRCC4 through both head domains, binds to DNA ends immediately prior to SR-complex formation. Our results shed light on the previously obscure role of XLF in NHEJ and indicate that end joining involves the juxtaposition of DNA ends in a stoichiometrically well-defined complex.
Our results suggest that within the SR-complex, a single XLF dimer bridges two XRCC4:LIG4 complexes (Fig. 5D). Formation of a (XLF)2:((XRCC4)2:LIG4)2 ternary complex is consistent with results from size-exclusion chromatography of XLF-XRCC4-LIG4 mixtures48. Such a complex was also proposed based on structural data suggesting that steric occlusion from full-length LIG4 may permit binding of only a single XLF dimer to XRCC4:LIG413,15. It remains unclear precisely how the XLF:XRCC4:LIG4 ternary complex facilitates SR-complex formation. One possibility is that DNA-binding motifs from XLF, XRCC4, and LIG4 directly contact DNA ends to bring them together (Fig. 5Di). Alternatively, assembly of the XLF:XRCC4:LIG4 subcomplex within the DNA-PK holoenzyme may induce a conformational transition of DNA-PK that aligns DNA ends (Fig. 5Dii). In support of the latter model, the SR-complex remains stable in the presence of XRCC4K104E, which dissociates much more rapidly from XLF than wild-type XRCC4, suggesting that XLF-XRCC4 interactions are required only for the initial formation of the SR-complex but not for its maintenance (Fig. 2E). Given that the lifetime of the SR-complex is non-exponentially distributed, it will be interesting to characterize the multiple kinetic steps involved in SR-complex dissociation and determine which factors influence these steps.
There are several conceptual problems associated with the XLF-XRCC4 filament model. First, end-bridging filaments of XLF-XRCC4 should block LIG4 and other end processing enzymes from gaining access to the DNA ends49. Second, it is unclear how the inner channel of these filaments could accommodate the large size of DNA-PKcs. Third, some mechanism would be needed to promote stable assembly of filaments near DNA ends while preventing their assembly on the much larger quantity of unbroken DNA in the cell. Fourth, phosphorylation of XLF and XRCC4 by DNA-PK inhibits filament formation, which would be expected to suppress, rather than promote filament assembly near DNA ends16,45. In contrast, none of these difficulties apply to a model in which XLF and XRCC4 form a short oligomer within a stoichiometrically defined synaptic complex.
Our experiments cannot rule out the possibility that XLF and XRCC4 form filaments in cells. For instance, other protein-protein interactions might concentrate XLF and XRCC4 within chromosomal DNA damage response foci in a manner that is not recapitulated on the relatively short DNA substrates in our assay. However, our observation of efficient NHEJ in the absence of XLF-XRCC4 filaments shows that these structures are not required and suggests a careful reappraisal of the filament model is timely. In vitro DNA bridging experiments with purified XLF and XRCC4 have generally employed buffers with low ionic strength (25–75 mM KCl). By contrast, we find that DNA pull-down by XLF-XRCC4 mixtures, as well as formation of large complexes measured by dynamic light scattering, is abolished at a more physiological salt concentration of 150 mM (Supplementary Fig. 1H-I). A more direct line of evidence supporting XLF-XRCC4 filament formation in cells comes from super-resolution imaging of fixed cells stained with anti-XLF and anti-XRCC4 antibodies27. XLF and XRCC4 appeared punctate in these experiments, and a subset of puncta exhibited an elongated morphology suggestive of filaments. However, further characterization of these structures is needed, as colocalization of points in super-resolution imaging does not prove the existence of physically continuous filaments. Also, it remains to be tested whether XLF-XRCC4 interaction is required for focus formation (i.e., by analysis of mutants).
Previously, it was shown that human XLFL115A rescues the DNA damage-sensitivity of a majority of XLF-deficient mammalian cell lines despite failing to bridge DNA in vitro, calling into question whether XLF-XRCC4 filaments are generally required for NHEJ17,47. Likewise, we observe that X. laevis XLFL117A (corresponding to hXLFL115A) and N-terminally Halo-tagged XLF support NHEJ in egg extract despite exhibiting no DNA-bridging activity in a pull-down assay (Fig. 3A-C; Fig. 5A-B; Supplementary Fig. 1H, 2A). Contrary to previous qualitative observations11, we find that hXLFL115A is not deficient in hXRCC4 binding (Fig. 5C; Supplementary Table 1). These results support the idea that, as in our cell-free extract, end joining in cells requires XLF-XRCC4 interaction but not DNA bridging by XLF-XRCC4 filaments.
In summary, a single XLF dimer, interacting through both head domains with XRCC4, promotes the close alignment of DNA ends. This suggests that rather than being tethered by protein filaments, DNA ends are precisely aligned within a stoichiometrically defined protein complex. Reconstitution and structural characterization of this complex will be an important direction for future work.
Methods
Plasmid construction
XLF and XRCC4:LIG4 expression constructs
Wild-type X. laevis XLF, XRCC4, and LIG4 plasmids are the same as those previously described38. A plasmid encoding human XRCC4 was obtained from Addgene (#13331) and was found to contain a single-nucleotide substitution resulting in the amino acid substitution I134T. This was restored to the reference sequence by site-directed mutagenesis, and the XRCC4 coding sequence was subcloned into a H10-SUMO expression vector. For human XLF, a bacterial codon-optimized gene block (Integrated DNA Technologies) was cloned into a H10-SUMO expression vector. Point mutants, epitope-tagged proteins, and C-terminally truncated XLF1−226 were generated by standard ‘round the horn site-directed mutagenesis50. A H10-SUMO-Halo-XLF expression plasmid was created by isothermal assembly of a Halo-encoding fragment and a PCR product of the original H10-SUMO-XLF plasmid. A dual expression vector for XRCC4 and H6-SUMO-LIG4K278R was constructed by amplifying XRCC4, H6-SUMO, and LIG4 from previously described plasmids38, and inserting the fragments in a pETDuet vector by isothermal assembly. All constructs were verified by Sanger sequencing.
Tandem XLF dimer
A custom Python script was used to generate a second codon-optimized XLF sequence (XLF’) as dissimilar as possible from our existing codon-optimized XLF sequence, to facilitate cloning and to prevent recombination between the two XLF sequences. Additional sequence encoding the linker GGGSGGGSGGGSGGG was appended at the 3’ end of the XLF’ sequence, and this combined fragment was ordered as a gBlock from Integrated DNA Technologies, inserted into a cloning vector, and sequence-verified. The XLF’ + linker sequence was then inserted by isothermal assembly immediately upstream of the initial XLF methionine in H10-SUMO-XLF38, yielding the tandem dimer H10-SUMO-XLF’-linker-XLF.
XLF heterodimers
A plasmid for expression of H10-tagged XLF (pTG355) was created by using ‘round the horn mutagenesis to delete the SUMO tag in H10-SUMO-XLF (pTG296). The alternative codon-optimized XLF sequence described above was inserted together with a T7 promoter and an N-terminal Flag-Avitag sequence into the H10-XLF plasmid, thereby generating a vector for dual expression of two versions of XLF with different affinity tags.
Protein purification
XLF
Plasmids encoding H10-SUMO-tagged wild-type XLF and XLF point mutants were transformed into E. coli BL21(DE3) pLysS cells. Cultures were grown in LB medium at 37°C to an OD600 of ~0.6, IPTG was added to a final concentration of 1 mM to induce expression, and cultures were grown an additional 3 h at 25–30°C. Cells were collected by centrifugation, and cell pellets were flash-frozen in liquid nitrogen and stored at −80°C. H10-SUMO-tagged proteins were purified essentially as described previously51. Briefly, cells were lysed by sonication in His-SUMO lysis buffer (20 mM Tris-HCl, pH 8, 1 M NaCl, 30 mM imidazole, 5 mM BME, 1 mM PMSF), and lysates were clarified by centrifugation for 1 h at 25,000 rpm in an SW-41 rotor at 4°C. Clarified lysates were incubated with NiNTA-agarose (Qiagen) for 1 h at 4°C to bind His-tagged proteins. NiNTA resin was washed extensively with lysis buffer, followed by “salt-reduction” buffer (20 mM Tris-HCl, pH 8, 350 mM NaCl, 30 mM imidazole, 5 mM BME). Protein was eluted with elution buffer (20 mM Tris-HCl, pH 8, 350 mM NaCl, 300 mM imidazole, 5 mM BME). Peak elution fractions were pooled, H6-tagged Ulp1 protease was added to cleave the H10-SUMO tag, and the protein was dialyzed overnight against His-SUMO dialysis buffer (20 mM Tris-HCl, pH 8, 350 mM NaCl, 10% glycerol, 10 mM imidazole, 5 mM BME). The dialysate was then incubated again with NiNTA resin for 1.5 h at 4°C to remove H10-SUMO, uncleaved H10-SUMO-XLF, and H6-Ulp1, and the flowthrough (containing untagged XLF) was collected. This was diluted with 1.36 volumes of SP buffer A (50 mM Na-MES, pH 6.5, 10% glycerol, 1 mM dithiothreitol [DTT]) and passed over SP Sepharose Fast Flow (Amersham) equilibrated with SP buffer A + 150 mM NaCl. The resin was washed with SP buffer A + 150 mM NaCl, and protein was eluted with SP buffer A + 300 mM NaCl. Protein aliquots in this buffer were flash-frozen in liquid nitrogen and stored at −80°C.
Halo-XLF
Halo-XLF was expressed as a H10-SUMO fusion and subjected to NiNTA purification as described for H10-SUMO-XLF above. The flowthrough after the second NiNTA step was subjected to size-exclusion purification on a HiPrep Sephacryl S-200 26/60 High Resolution column equilibrated with His-SUMO dialysis buffer (20 mM Tris-HCl, pH 8, 350 mM NaCl, 10% glycerol, 10 mM imidazole, 5 mM BME). Peak fractions were pooled and concentrated on a 15 ml 10 kDa molecular weight cutoff (MWCO) centrifugal concentrator.
XLF(1–226) truncation mutant
XLF truncation mutants were expressed as fusions to H10-SUMO in BL21(DE3)pLysS cells and subjected to the same initial His-SUMO purification protocol as full-length XLF. Flowthrough from the second NiNTA step was diluted with approximately 2.5 volumes of 20 mM Tris-HCl pH 8, 10% glycerol, 5 mM BME to reduce the NaCl concentration to ~100 mM. The diluted sample was passed over a Mono Q column equilibrated with 20 mM Tris-HCl pH 8, 100 mM NaCl, 10% glycerol, 5 mM BME. The flowthrough, containing the protein of interest, was concentrated using an Amicon 10 kDa MWCO centrifugal concentrator and dialyzed overnight against 20 mM Tris-HCl, pH 8, 350 mM NaCl, 10% glycerol, 5 mM BME. Human XLF1−224 was purified using the same protocol. However, it bound the MonoQ column and was eluted using a 100–1000 mM NaCl gradient.
XLF tandem dimer
XLF tandem dimer constructs were expressed as fusions to H10-SUMO in BL21(DE3)pLysS cells and subjected to the His-SUMO purification described above for wild-type XLF. Flowthrough from the second NiNTA step was diluted with 1.36 volumes of buffer A (50 mM Na-MES, pH 6.5, 10% glycerol, 5 mM BME) and bound to a HiTrap SP HP column equilibrated with the same buffer containing 150 mM NaCl. The protein was eluted with a gradient of 150 mM to 1 M NaCl over 20 column volumes. Peak fractions were pooled, concentrated on a 10 kDa MWCO Amicon centrifugal concentrator, and subjected to size-exclusion purification on a Superdex 200 Increase 10/300 column equilibrated with 50 mM Na-MES, pH 6.5, 350 mM NaCl, 10% glycerol, 5 mM BME. Peak fractions were pooled and concentrated as above.
XLF heterodimers
BL21(DE3) cells were co-transformed with the BirA biotin ligase expression plasmid pBirAcm and a dual-expression plasmid encoding H10-XLF as well as Flag-Avitag-XLFWT (pTG448) or Flag-Avitag-XLFL117D (pTG449). After growing the cells to an OD600 of 0.4, expression was induced by addition of isopropyl β-D-1-thiogalactopyranoside (IPTG) to 1 mM, and the growth medium was supplemented with biotin to a final concentration of 25 μM. Cells were collected by centrifugation after 3 h of induction at 25–30°C, flash-frozen, and stored at −80°C. Frozen cell pellets were resuspended in 20 mM Tris-HCl, pH 8, 1 M NaCl, 30 mM imidazole, 5 mM β-mercaptoethanol (BME) and lysed by three 30-s cycles of sonication (1 s on, 1 s off). Lysates were clarified by centrifugation for 1 h at 25,000 rpm in an SW-41 rotor. Supernatants were incubated for 1 h at 4°C with 250 μl of NiNTA agarose (Qiagen) per liter of culture. Resin was washed with 40 column volumes (CV) of lysis buffer, and H10-tagged complexes were eluted with 20 CV of the same buffer containing 300 mM imidazole. Next, eluates were passed by gravity flow over SoftLink Avidin resin (Promega; 1 ml per liter of culture) to capture Avitagged H10-XLF:Flag-Avitag-XLF heterodimers. Resin was washed with 25 CV of 20 mM Tris-HCl pH 8, 1 M NaCl, 5 mM BME, followed by 5 CV of 20 mM Tris-HCl pH 8, 350 mM NaCl, 5 mM BME. We suspect that the use of high-salt buffers at this stage is important to remove nonspecifically bound H10-XLFWT:H10-XLFWT homodimer, as early WT:mutant heterodimer preparations purified under less stringent conditions exhibited variable and elevated activity (not shown). Bound protein was eluted in 1-CV increments with 20 mM Tris-HCl pH 8, 350 mM NaCl, 5 mM BME, 5 mM biotin, pausing for at least 5 min between elution steps to facilitate dissociation of bound protein. Protein was flash-frozen and stored at −80°C in elution buffer.
XRCC4
N-terminal H10-SUMO fusion constructs of X. laevis XRCC4 were transformed into BL21(DE3)pLysS E.coli cells. Cells were grown at 37°C to an OD600 of 0.6 and then induced with 1 mM IPTG for 3 hours at ~25–30°C. Cells were pelleted by centrifugation, flash frozen, and stored at −80°C. Cell pellets were then thawed and subjected to the His-SUMO purification used previously38.
Biotinylated XRCC4-Avitag
X. laevis XRCC4 with an N-terminal H10-SUMO tag and a C-terminal Avitag was expressed in BL21(DE3) E. coli cells co-transformed with the BirA biotin ligase-expressing plasmid pBirAcm. Cells were grown in LB medium supplemented with 100 μM biotin, and protein expression was induced with 1 mM IPTG for 3 h at ~25–30°C. Protein was subjected to the same initial H10-SUMO purification steps as XLF (see above). Biotinylated protein was then bound to SoftLink Avidin resin equilibrated with 20 mM Tris-HCl, pH 8, 150 mM NaCl, 10% glycerol, 1 mM DTT. The resin was washed with 20 CV of the same buffer, and protein was eluted with elution buffer (20 mM Tris-HCl, pH 8, 150 mM NaCl, 10% glycerol, 1 mM DTT, 5 mM biotin). Elution was performed in increments of 2 CV, pausing for 5 min between elutions. Peak fractions were pooled and dialyzed overnight against 20 mM Tris-HCl, pH 8, 350 mM NaCl, 10% glycerol, 1 mM DTT. Human XRCC4-Avitag was purified using the same protocol.
XRCC4:LIG4 complex
Wild-type and mutant XRCC4:LIG4 complexes were expressed in Sf9 cells and purified as described previously38. Catalytically inactive XRCC4:LIG4K278R complexes (for the experiments in Fig. 2D-E) were expressed from a pETDuet vector in Rosetta 2 (DE3) E. coli cells. Cultures were grown at 37°C to an OD600 of 0.6 and then placed on ice for 20 minutes. IPTG was added to a final concentration of 1 mM, and cultures were incubated with shaking for 16 hours at 17°C. Cells were pelleted by centrifugation, flash frozen, and stored at −80°C. Cell pellets were thawed and resuspended in 20 ml of lysis buffer (20 mM HEPES, pH 7.5, 400 mM NaCl, 20 mM imidazole, 10% glycerol, 1 mM DTT, 1 cOmplete, EDTA-free Protease Inhibitor tablet [Sigma Aldrich]) per liter of culture. The resuspension was sonicated for 2 minutes (5 s on, 10 s off). Lysate was clarified by 1 hour of centrifugation at 38,724 rcf at 4°C. Lysate was incubated at 4°C for 1 hour with 250 μl of NiNTA agarose (Qiagen) per liter of culture. The samples were then centrifuged at 500 rcf, the supernatant was discarded, and the Ni-NTA resin was resuspended in 10 ml of lysis buffer and poured into a gravity flow column. The resin was then washed with 20 ml of lysis buffer, and protein was eluted in 250 μl fractions with lysis buffer containing 250 mM imidazole. Fractions containing the protein of interest were pooled, supplemented with Ulp1 protease, and dialyzed into cleavage buffer (20 mM HEPES, pH 7.5, 150 mM NaCl, 1 mM DTT, 10% glycerol). The dialyzed sample was then incubated with 250 μl of NiNTA resin per liter of culture for 1.5 hours at 4°C. The flowthrough was collected, passed through a 0.22 μm filter, and diluted in cleavage buffer without NaCl to bring the final NaCl concentration to 50 mM. Protein was bound to a MonoQ 5/50-GL column (G.E. Lifesciences) equilibrated in Buffer A (cleavage buffer with 50 mM NaCl) and eluted with a 20 ml gradient of 0–60% Buffer B (cleavage buffer with 1 M NaCl). Peak fractions were pooled and further separated on a S200 16/600 200 pg sizing column (G.E. Lifesciences) equilibrated in cleavage buffer. Protein-containing fractions were pooled and concentrated using an Amicon 10 kDa MWCO centrifugal concentrator.
Biotin-Avitag-Halo
N-terminally SUMO-tagged Avitag-Halo protein was expressed in BL21(DE3) cells containing the pBirAcm plasmid, which encodes the BirA biotin ligase. It was subjected to the same His-SUMO purification procedure as for XLF, without the final ion exchange step.
Flag-XLF and H10-XLF (for monomer exchange experiment)
A Flag-XLF expression plasmid was transformed into BL21(DE3) pLysS cells, which were grown in 100 ml of LB to an OD600 of 0.4, induced with 1 mM IPTG for 3 h at 25–30°C, and collected by centrifugation. Cells were lysed in 20 mM Tris-HCl pH 7.5, 150 mM NaCl, without reducing agent. The lysate was clarified by centrifugation at 16,000 rcf in a 4°C microcentrifuge and incubated with 50 μl of αFlag M2 affinity gel (Sigma-Aldrich) for 1 h at 4°C. The gel was washed 3 times with 1.4 ml of lysis buffer, and protein was eluted by overnight incubation at 4°C with 100 μg/μl of 3x Flag peptide (Sigma-Aldrich). Protein was dialyzed against storage buffer (20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 10% glycerol, 5 mM BME) using a 7 kDa molecular weight cut-off dialysis cassette, aliquotted, flash-frozen, and stored at −80°C. H10-XLF was expressed in BL21(DE3) pLysS cells under the same growth and induction conditions used for H10-SUMO-XLF. Initial NiNTA purification followed the same steps as for H10-SUMO-XLF. The protein was dialyzed overnight against 20 mM Na-MES, pH 6.5, 150 mM NaCl, 10% glycerol, 5 mM BME, bound to a Mono S column, and eluted with a gradient of the same buffer with 150 mM to 1 M NaCl. Peak fractions were pooled and dialyzed overnight against 20 mM Tris-HCl pH 7.5, 350 mM NaCl, 10% glycerol, 1 mM DTT. Protein was concentrated with a centrifugal concentrator, aliquotted, flash-frozen, and stored at −80°C.
Test of monomer exchange between XLF dimers
To assess whether monomers are exchanged between XLF dimers in solution (Supplementary Fig. 4D), N-terminally Flag- and H10-tagged XLF proteins were mixed at final concentrations of 8 μM and 20 μM, respectively, in a 5 μl volume of 20 mM Tris-HCl pH 7.5, 250 mM NaCl, 10% glycerol, 2.5 mM BME, 0.5 mM DTT. Control reactions contained only one protein or the other. Reactions were incubated for 3 hours at room temperature in a humidified chamber. 100 μl of His-SUMO lysis buffer was added (20 mM Tris-HCl, pH 8, 1 M NaCl, 30 mM imidazole, 5 mM BME), and tubes were centrifuged for 10 min at 16,000 rcf in a 4°C microcentrifuge. No pellet was visible. After withdrawing 15 μl of supernatant for analysis, 80 μl of supernatant was incubated for 1 h at 4°C with 10 μl of NiNTA-agarose resin pre-washed with His-SUMO lysis buffer. Supernatant was collected, and resin was washed 2 times with 500 μl of His-SUMO lysis buffer, aspirating thoroughly after each wash. 80 μl of His-SUMO elution buffer was added (20 mM Tris-HCl, pH 8, 350 mM NaCl, 300 mM imidazole, 5 mM BME), and protein was eluted for 30 min at 4°C. Input, supernatant, and eluate samples were separated on a 4–15% pre-cast SDS-PAGE gel (BioRad), transferred to a polyvinylidine fluoride (PVDF) membrane for 1 h at 100 V at 4°C, and blocked with 5% powdered nonfat milk in PBST (1x phosphate buffered saline with 0.05% Tween 20). Membranes were probed overnight with 1:5000 anti-H6 antibody (Sigma-Aldrich) or 1:5000 anti-FLAG M2 antibody (Sigma-Aldrich), washed three times with PBST, and probed for 1 h with 1:20,000 horseradish peroxidase (HRP)-conjugated rabbit anti-mouse IgG (H+L) secondary antibody (Jackson ImmunoResearch Cat. #315–035-003). After washing again three times with PBST, the anti-H6 blot was incubated with chemiluminescent HRP substrate (HyGLO Quick Spray, Denville Scientific), and the anti-Flag blot was incubated with enhanced chemiluminescent substrate (Pierce). Both blots were imaged on an Amersham Imager 600 (GE Healthcare Life Sciences).
Preparation of Sulfo-Cy5-Halo substrate
8 μl of 100 mM Cy5-NHS ester in DMSO was combined with 3.2 μl of 250 mM HaloTag amine (O2) ligand (Promega) and 38.8 μl of 100 mM sodium bicarbonate and allowed to react overnight. The products were separated by reversed-phase HPLC on a semi-preparative C18 column using a gradient of 20–100% acetonitrile in water. Product identity was verified by mass spectrometry. Peaks corresponding to the correct product were pooled, and solvent was evaporated overnight using a speed-vac. The compound was redissolved in DMSO, and the concentration was determined by measuring absorbance at 650 nm of a 1:100 dilution in water, assuming an extinction coefficient of 250,000 M−1cm−1 for Cy5.
Fluorescent labeling of Halo-XLF
A 2-fold molar excess of Cy5- or Alexa Fluor 488-Halo ligand in dimethylsulfoxide (DMSO) was added to Halo-XLF protein in its storage buffer. The mixture was allowed to react for 10 min while being centrifuged at 16,000 rcf at room temperature. Labeled protein was separated from free dye on a Superdex 200 Increase 10/300 column equilibrated with 20 mM Tris-HCl, pH 8, 350 mM NaCl, 10% glycerol, 5% β-mercaptoethanol. Peak fractions were pooled and concentrated using an Amicon 10 kDa MWCO centrifugal concentrator. Protein concentrations were measured by absorbance at 280 nm, assuming an extinction coefficient of 82390 M−1cm−1. Dye concentrations were measured by absorbance at 650 nm for Cy5 (assuming an extinction coefficient of 250,000 M−1cm−1) or 495 nm for AF488 (assuming an extinction coefficient of 73,000 M−1cm−1). For AF488-labeled protein, the absorbance at 280 nm due to protein was determined by subtracting AF488 absorbance at 280 nm from total absorbance, using the correction factor (AF488 absorbance at 280 nm) = 0.11 * (AF488 absorbance at 495 nm). Labeled fraction of monomer was obtained by dividing the molar concentration of dye by the molar concentration of protein. Measurements of labeled fraction obtained using a NanoDrop spectrophotometer agreed with those calculated from dual-wavelength absorbance measurements of labeled protein peaks during size-exclusion chromatography.
Fluorescent labeling of XLF with Cy5-maleimide
130 μl of 53 μM wild type X. laevis XLF was incubated for 1 hour at room temperature with 50 μM sulfo-Cyanine5 maleimide (Lumiprobe) in 20 mM Tris-HCl, pH 7.5, 350 mM NaCl, 10% glycerol. The labeling reaction was quenched by the addition of dithiothreitol (DTT) at a final concentration of 5 mM. Labeled protein was separated from free dye on a S200 Increase 10/300 GL sizing column (GE Healthcare Life Sciences). Protein-containing fractions were collected, and the Cy5:XLF ratio was determined by measuring the absorbance of each on a Nanodrop spectrophotometer. Using an extinction coefficient of 250,000 M−1cm−1 for Cy5 and an extinction coefficient of 24,450 M−1cm−1 for XLF, the dye-to-protein ratio was measured to be 0.77. This same procedure was performed in parallel for a AF488-labeled XLF sample and a mock-labeled XLF sample.
Preparation of Xenopus egg extracts
Xenopus egg extract was prepared as previously described52. The female frogs used to produce oocytes are cared for by the Center for Animal Resources and Comparative Medicine at Harvard Medical School (AAALAC accredited). Work performed for this study was in accordance with the rules and regulations set by AAALAC. The Institutional Animal Care and Use Committee (IACUC) approved the work.
Antibodies and immunodepletion
XLF and XRCC4 antibodies were the same as those described previously38. Immunodepletion experiments were performed as described previously37,38. For rescue experiments in Fig. 1C-D and Fig 5B, immunodepleted extract was supplemented with 500 nM XLF or 50 nM XRCC4:LIG4 complex.
Bulk end joining assays
Bulk end joining assays were performed as described previously38. The final concentration of radiolabeled linear substrate DNA in bulk end joining assays was approximately 1 ng/μl (~0.5 nM). Integrated intensity of gel bands (Fig. 4C and Supplementary Fig. 4F) was quantified in Fiji.
tdXLF and XLF heterodimer titration experiments
To test the dependence of end joining rate on the protein concentration of XLF tandem dimer (tdXLF; Fig. 4B-D) and heterodimer (Supplementary Fig. 4E-F) constructs), extract was depleted of endogenous XLF, and the depleted extract was supplemented with ATP regeneration system and closed-circular carrier DNA as described previously 37. Radiolabeled linear substrate DNA was added to extract on ice to a final concentration of ~1 ng/μl, and a portion of this extract mixture was supplemented with XLF at the indicated maximum concentration. The same fractional volume of XLF storage buffer was added to the remaining extract mixture. Extract supplemented with XLF was then serially diluted with extract mixture, while keeping tubes in a PCR block at 0°C. Following serial dilutions, the temperature of the block was raised to 22°C for 5 min, and then lowered to 0°C to stop the reactions. One volume of reaction stop solution/loading dye with proteinase K37 was immediately added to all tubes. Products were analyzed by gel electrophoresis as described previously37.
Microscope and flowcell construction
Samples were imaged using a through-objective total internal reflection fluorescence (TIRF) microscope built around an Olympus IX-71 inverted microscope body. 488 nm, 532 nm, and 641 nm laser beams (Coherent Sapphire 488, Coherent Sapphire 532, and Cube 641) were expanded, combined using dichroic mirrors, expanded again, and focused on the rear focal plane of an oil immersion objective (Olympus UPlanSApo, 100x, NA 1.40). The focusing lens was placed on a vertical translation stage to permit manual adjustment of the TIRF angle. Emission light was separated from excitation light using a multi-pass dichroic mirror, and laser lines were further attenuated using a StopLine 488/532/635 notch filter (Semrock). A home-built beamsplitter (see Figure 6 of ref. 37) was used to separate Alexa Fluor 488 and Cy3 emission from Cy5 emission; these two channels were imaged on separate halves of an electron-multiplying charge-coupled device (EMCCD) camera (Hamamatsu, ImageEM 9100–13), which was operated at maximum EM gain. An automated microstage (Mad City Labs) was used to position the sample and move between fields of view.
Coverslips were functionalized with a mixture of methoxypolyethylene glycol (mPEG) and biotin-mPEG, and flowcells were constructed following a previously described protocol38,53.
Single-molecule imaging and analysis
See Supplementary Note 1.
Bio-layer interferometry (BLI) binding assays
BLI experiments were performed on an Octet RED384 (ForteBio, Menlo Park, California) using streptavidin-coated Dip and Read Biosensors (ForteBio) and 384 well plates. 100 nM of biotinylated C-terminally Avi-tagged XRCC4 was bound for 5 min in a binding buffer consisting of 20 mM Tris-HCl, pH 7.5, 125 mM KCl, 10 mg/mL bovine serum albumin (Calbiochem), and 5 mM BME. To test for nonspecific binding of XLF1−226, reference tips were incubated in buffer alone. The tips were washed with buffer for 60 s to obtain a baseline reading and then transferred to wells containing XLF1−226 or point mutants thereof (2 μM of each protein unless otherwise noted) for 120 s. After measuring association, tips were moved to wells containing buffer, and XLF dissociation was measured for 180 s. The response for each experimental condition shown in Fig. 1A is the average of three replicates, and each replicate is the average of three sequential rounds of association and dissociation. The standard deviation is represented as shading around each trace. The data shown in Supplementary Fig. 1A are representative results from a single experiment. For the titration experiments shown in Supplementary Fig. 1B,E-F, the background subtracted association-dissociation curve for each concentration was fit to a 1:1 model using Graphpad (La Jolla, California) Prism software and the built-in fitting tools. Data generated from fits for which R2 < 0.98 were not included in subsequent analysis. The resulting kon, koff, and apparent Kd values were averaged. This was performed for two replicates for each condition. The reported values are averages from the two replicates, and the reported error represents the minimum and maximum values. The same fitting and analysis procedure was used to generate the curves shown in Fig. 5C and the values in Supplementary Table 1. Those data represent two experimental replicates, with the error representing the minimum and maximum values.
Dynamic light scattering
DLS experiments were performed on a Wyatt/Protein Solutions (Santa Barbara, California) DynaPro-99-E-50 Dynamic Light Scattering Module with a TC100–830 Temperature Controlled Microsampler. The data shown in Supplementary Fig. 1C and 1I were collected at 20°C with an acquisition time of 10 s and a read interval of 1 s. Sample measurement began ~1 min after assembling and mixing the specified proteins in a filtered buffer containing 20 mM HEPES, pH 8, 75 mM KCl, 0.5 mM EDTA, 1 mM DTT, and 5% glycerol. The Dynamics V6 software (Wyatt/Protein Solutions) was used to calculate hydrodynamic radii using the built-in parameters for a 5% glycerol solution. For each condition, three replicates were averaged to generate the data shown in Supplementary Fig. 1C and 1I. The shaded region around each trace represents the standard error of the mean.
Bulk protein-DNA pull-down assays
DNA pull-down assays (Supplementary Fig. 1 panels D and H and Supplementary Fig. 3E) were performed as previously described25. A biotinylated 1000 bp DNA fragment was generated by PCR (template pTG064; primers oTG414 and oTG042F) using Pfu polymerase. The 500 bp DNA was generated by PvuII digestion of pTG024. These DNA substrates were then gel-purified by electroelution as described previously37. M-280 streptavidin-coated magnetic Dynabeads (Thermo Fisher Scientific) were washed 3 times with one volume of binding buffer (20 mM HEPES, pH 8, 75 mM KCl, 0.5 mM EDTA, 1 mM DTT, 5% glycerol, and 400 μg/ml BSA (New England Biolabs)), using a magnet to retain beads, and then resuspended in one volume of the same buffer. Beads were incubated with 1000 bp DNA at a final concentration of 20 ng/μl for five minutes at 20–25°C. One volume of a high-salt buffer (binding buffer with 850 mM KCl) was used to wash the beads twice before resuspending in the wash buffer. The 500 bp DNA was then added at a final concentration of 20 ng/μl. 10 μl of the bead/DNA suspension was added to each 40 μl reaction assembled in binding buffer. Indicated proteins were added at a final concentration of 2 μM, and the reactions were incubated at room temperature for 30 minutes. After using a magnet to capture the beads, the supernatant was collected, and the beads were then washed twice in binding buffer and resuspended in the same buffer without BSA. 10 μl of each sample was set aside and separated on a 4–15% SDS-PAGE gel (BioRad). An Amersham Imager 600 was used to visualize Cy5-labeled XLF. The samples were then treated with 40 ng proteinase K for 1 hour at 50°C, and DNA was separated on a 1.5% agarose gel.
Differential Scanning Fluorimetry (DSF)
DSF protein thermal shift assays were performed using a QuantStudio 6 Flex Real-Time PCR System (Applied Biosystems, Foster City, California). 20 μl reactions containing 10 μg of the specified protein were assembled in MicroAmp FAST optical 96-well reaction plates (Life Technologies) and covered with MicroAmp Optical Adhesive Film (Life Technologies). SYPRO Orange dye was included in the reactions using the Protein Thermal Shift Dye Kit (Applied Biosystems), following the manufacturer’s instructions. Protein samples were diluted in storage buffer (20 mM Tris-HCl, pH 8, 10 mM imidazole, 350 mM NaCl, 10% glycerol). The temperature was raised from 25 to 99°C at 3°C per minute, and the fluorescent dye was excited and measured at 470 nm and 587 nm respectively. To determine the melting temperatures, emission signals were plotted as a function of temperature and these thermal denaturation curves were fit with the Boltzmann equation using the Protein Thermal Shift software (Life Technologies). The average of three experimental replicates is shown for each condition. The error bars represent the standard error of the mean. A 2-tailed, unpaired t-test with unequal variance was performed to determine whether the melting temperatures of the mutants were significantly different from the wild type proteins.
Size-exclusion chromatography with multi-angle light scattering (SEC-MALS)
SEC-MALS was performed on an Agilent 1260 Infinity Isocratic Liquid Chromatography System equipped with a Wyatt Dawn Heleos II Multi-Angle Light Scattering (MALS) detector and a Wyatt Optilab T-rex Refractive Index Detector. An Agilent AdvanceBio 300 column was pre-equilibrated overnight with running buffer (50 mM Na-MES, pH 6.5, 350 mM NaCl, 10% glycerol, 5 mM BME) at a 0.2 ml/min flow rate and then allowed to equilibrate to a flow rate of 0.4 ml/min for 1 h. 30 μl of a 50 μM solution of tdXLFWT-WT was separated on the column. As a control, 100 μl of 2 mg/ml BSA fraction V (OmniPur) in the same buffer without BME was separated on the column, yielding monomer, dimer, and trimer peaks of the expected sizes.
Statistics and Reproducibility
A 2-tailed unpaired t-test with unequal variances was used to determine significance (p < 0.05) in Supplementary Figures 1E, F, and G. The Bonferroni correction was applied in the case of Supplementary Figure 1G. A log-rank test was used to determine significance (p < 0.05) in Figure 2E. The Lilliefors test for exponential distribution (MATLAB function ‘lillietest’) was applied to the data shown in Figure 2E. For figures that include representative images, the number of experimental replicates of the corresponding experiments are: two for Figures 1C and 1D, three for Figure 3B-C, three for Figure 5A, two for Fig 5B, two for Supplementary Figure 1A, two for Supplementary Figure 2B, three for Supplementary Figure 1D, two for Supplementary Figure 1H, five for Supplementary Figure 2A, two for Supplementary Figure 3D, two for Supplementary Figure 3E, two for Supplementary Figure 4D, and six for Supplementary Figure 4E-F. Exact experimental conditions were varied slightly between replicates of some experiments, while employing appropriate internal controls in all cases. Similar results were obtained among replicates for all experiments listed above.
Supplementary Material
Acknowledgements
We would like to thank Ethan van Arnam for assistance with purification of Cy5 Halo ligand, Benjamin Stinson for providing improved FRET analysis code and generating the pETDuet vector containing XRCC4 and LIG4, Karl Schmitz for advice on generating an “orthogonal” codon-optimized XLF sequence, Kelly Arnett (HMS Center for Macromolecular Interactions) for assistance with biophysical assays, and Steve Harrison and Simon Jenni for assistance with dynamic light scattering. We would also like to thank members of the Loparo and Walter labs for helpful discussions. This work was funded by a National Institutes of Health grant R01GM115487 (to J.J.L) and the Howard Hughes Medical Institute (J.C.W).
Footnotes
Author Contributions
All authors designed experiments and wrote the manuscript. T.G.W.G and S.M.C. performed experiments and data analysis.
Competing Interests
The authors declare no competing interests.
References
- 1.Walker JR, Corpina RA & Goldberg J Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature 412, 607–14 (2001). [DOI] [PubMed] [Google Scholar]
- 2.Dobbs TA, Tainer JA & Lees-Miller SP A structural model for regulation of NHEJ by DNA-PKcs autophosphorylation. DNA Repair (Amst). 9, 1307–14 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jette N & Lees-Miller SP The DNA-dependent protein kinase: A multifunctional protein kinase with roles in DNA double strand break repair and mitosis. Prog Biophys Mol Biol 117, 194–205 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jiang W et al. Differential phosphorylation of DNA-PKcs regulates the interplay between end-processing and end-ligation during nonhomologous end-joining. Mol. Cell 58, 172–85 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Critchlow SE, Bowater RP & Jackson SP Mammalian DNA double-strand break repair protein XRCC4 interacts with DNA ligase IV. Curr. Biol 7, 588–598 (1997). [DOI] [PubMed] [Google Scholar]
- 6.Grawunder U et al. Activity of DNA ligase IV stimulated by complex formation with XRCC4 protein in mammalian cells. Nature 388, 492–495 (1997). [DOI] [PubMed] [Google Scholar]
- 7.Buck D et al. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell 124, 287–99 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Ahnesorg P, Smith P & Jackson SP XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell 124, 301–13 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Junop MS et al. Crystal structure of the Xrcc4 DNA repair protein and implications for end joining. EMBO J. 19, 5962–70 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y et al. Crystal structure of human XLF/Cernunnos reveals unexpected differences from XRCC4 with implications for NHEJ. EMBO J. 27, 290–300 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Andres SN, Modesti M, Tsai CJ, Chu G & Junop MS Crystal structure of human XLF: a twist in nonhomologous DNA end-joining. Mol. Cell 28, 1093–101 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Hammel M, Yu Y, Fang S, Lees-Miller SP & Tainer JA XLF regulates filament architecture of the XRCC4·ligase IV complex. Structure 18, 1431–42 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ropars V et al. Structural characterization of filaments formed by human Xrcc4-Cernunnos/XLF complex involved in nonhomologous DNA end-joining. Proc. Natl. Acad. Sci. U. S. A 108, 12663–8 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hammel M et al. XRCC4 protein interactions with XRCC4-like factor (XLF) create an extended grooved scaffold for DNA ligation and double strand break repair. J. Biol. Chem 286, 32638–50 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malivert L et al. Delineation of the Xrcc4-interacting region in the globular head domain of cernunnos/XLF. J. Biol. Chem 285, 26475–83 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roy S et al. XRCC4’s interaction with XLF is required for coding (but not signal) end joining. Nucleic Acids Res. 40, 1684–94 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roy S et al. XRCC4/XLF interaction is variably required for DNA repair, and is not required for Ligase IV stimulation. Mol. Cell. Biol (2015). doi: 10.1128/MCB.01503-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu H, Pannicke U, Schwarz K & Lieber MR Length-dependent binding of human XLF to DNA and stimulation of XRCC4.DNA ligase IV activity. J. Biol. Chem 282, 11155–62 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Gu J, Lu H, Tsai AG, Schwarz K & Lieber MR Single-stranded DNA ligation and XLF-stimulated incompatible DNA end ligation by the XRCC4-DNA ligase IV complex: influence of terminal DNA sequence. Nucleic Acids Res. 35, 5755–62 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsai CJ, Kim SA & Chu G Cernunnos/XLF promotes the ligation of mismatched and noncohesive DNA ends. Proc. Natl. Acad. Sci. U. S. A 104, 7851–6 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tadi SK et al. PAXX Is an Accessory c-NHEJ Factor that Associates with Ku70 and Has Overlapping Functions with XLF. Cell Rep. 17, 541–555 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Riballo E et al. XLF-Cernunnos promotes DNA ligase IV-XRCC4 re-adenylation following ligation. Nucleic Acids Res. 37, 482–92 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mahaney BL, Hammel M, Meek K, Tainer JA & Lees-Miller SP XRCC4 and XLF form long helical protein filaments suitable for DNA end protection and alignment to facilitate DNA double strand break repair. Biochem. Cell Biol 91, 31–41 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu Q et al. Non-homologous end-joining partners in a helical dance: structural studies of XLF-XRCC4 interactions. Biochem. Soc. Trans 39, 1387–92, suppl 2 p following 1392 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Andres SN et al. A human XRCC4-XLF complex bridges DNA. Nucleic Acids Res. 40, 1868–78 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brouwer I et al. Sliding sleeves of XRCC4-XLF bridge DNA and connect fragments of broken DNA. Nature 535, 566–9 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Reid DA et al. Organization and dynamics of the nonhomologous end-joining machinery during DNA double-strand break repair. Proc. Natl. Acad. Sci. U. S. A 112, E2575–2584 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Di Virgilio M & Gautier J Repair of double-strand breaks by nonhomologous end joining in the absence of Mre11. J. Cell Biol 171, 765–71 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Postow L et al. Ku80 removal from DNA through double strand break-induced ubiquitylation. J. Cell Biol 182, 467–479 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Labhart P Nonhomologous DNA end joining in cell-free systems. Eur. J. Biochem 265, 849–61 (1999). [DOI] [PubMed] [Google Scholar]
- 31.Labhart P Ku-dependent nonhomologous DNA end joining in Xenopus egg extracts. Mol. Cell. Biol 19, 2585–93 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sandoval A & Labhart P Joining of DNA ends bearing non-matching 3’-overhangs. DNA Repair (Amst). 1, 397–410 (2002). [DOI] [PubMed] [Google Scholar]
- 33.Taylor EM et al. The Mre11/Rad50/Nbs1 complex functions in resection-based DNA end joining in Xenopus laevis. Nucleic Acids Res. 38, 441–54 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu S & Peng A Non-homologous end joining repair in Xenopus egg extract. Sci. Rep 6, 27797 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thode S, Schäfer A, Pfeiffer P & Vielmetter W A novel pathway of DNA end-to-end joining. Cell 60, 921–928 (1990). [DOI] [PubMed] [Google Scholar]
- 36.Pfeiffer P & Vielmetter W Joining of nonhomologous DNA double strand breaks in vitro. Nucleic Acids Res. 16, 907–924 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Graham TGW, Walter JC & Loparo JJ Ensemble and Single-Molecule Analysis of Non-Homologous End Joining in Frog Egg Extracts. Methods Enzymol. 591, 233–270 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Graham TGW, Walter JC & Loparo JJ Two-Stage Synapsis of DNA Ends during Non-homologous End Joining. Mol. Cell 61, 850–8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.DeFazio LG, Stansel RM, Griffith JD & Chu G Synapsis of DNA ends by DNA-dependent protein kinase. EMBO J. 21, 3192–200 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hammel M et al. Ku and DNA-dependent protein kinase dynamic conformations and assembly regulate DNA binding and the initial non-homologous end joining complex. J. Biol. Chem 285, 1414–1423 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cary RB et al. DNA looping by Ku and the DNA-dependent protein kinase. Proc. Natl. Acad. Sci. U. S. A 94, 4267–72 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spagnolo L, Rivera-Calzada A, Pearl LH & Llorca O Three-Dimensional Structure of the Human DNA-PKcs/Ku70/Ku80 Complex Assembled on DNA and Its Implications for DNA DSB Repair. Mol. Cell 22, 511–519 (2006). [DOI] [PubMed] [Google Scholar]
- 43.Cottarel J et al. A noncatalytic function of the ligation complex during nonhomologous end joining. J. Cell Biol 200, 173–86 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang JL et al. Dissection of DNA double-strand-break repair using novel single-molecule forceps. Nat. Struct. Mol. Biol 25, 482–487 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Normanno D et al. Mutational phospho-mimicry reveals a regulatory role for the XRCC4 and XLF C-terminal tails in modulating DNA bridging during classical non-homologous end joining. Elife 6, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lilliefors HW On the Kolmogorov-Smirnov Test for the Exponential Distribution with Mean Unknown. J. Am. Stat. Assoc 64, 387 (1969). [Google Scholar]
- 47.Fattah FJ et al. A role for XLF in DNA repair and recombination in human somatic cells. DNA Repair (Amst). 15, 39–53 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ochi T et al. Structural insights into the role of domain flexibility in human DNA ligase IV. Structure 20, 1212–22 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Menon V & Povirk LF XLF/Cernunnos: An important but puzzling participant in the nonhomologous end joining DNA repair pathway. DNA Repair (Amst). 58, 29–37 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods References
- 50.Hemsley A, Arnheim N, Toney MD, Cortopassi G & Galas DJ A simple method for site-directed mutagenesis using the polymerase chain reaction. Nucleic Acids Res. 17, 6545–51 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Graham TGW et al. ParB spreading requires DNA bridging. Genes Dev. 28, 1228–38 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lebofsky R, Takahashi T & Walter JC DNA replication in nucleus-free Xenopus egg extracts. Methods Mol. Biol 521, 229–52 (2009). [DOI] [PubMed] [Google Scholar]
- 53.Tanner NA et al. Single-molecule studies of fork dynamics in Escherichia coli DNA replication. Nat. Struct. Mol. Biol 15, 998 (2008). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.