

Abstract
Background and objectives
Fullerene-based compounds are a novel class of molecules being developed for a variety of biomedical applications, with nearly 1000 publications in this area in the last 4 years alone. One such compound, the e,e,e malonic acid C60 (C3), is a potent catalytic superoxide dismutase mimetic with neuroprotective efficacy in a number of animal models of neurologic disease, including Parkinsonian Macaca fascicularis monkeys.
Methods
To assess pharmacokinetics in mice, we synthesized and administered 14C-C3 to mice using various routes of delivery, including orally. To assess potential toxicity in primates, serial blood studies and electrocardiograms (ECGs) were obtained from monkeys treated with C3 (3 or 7 mg/kg/day) for two months.
Results and conclusions
The plasma half-life was 8.2 ± 0.2 hr, and there was wide tissue distribution, including uptake into brain. The compound was cleared by both hepatic and renal excretion. C3 was quite stable, with minimal metabolism of the compound even after 7 days of treatment. The LD50 in mice was 80 mg/kg for a single intraperitoneal injection, and was >30 mg/kg/day for sustained administration; therapeutic doses are 1–5 mg/kg/day. For primates, no evidence of renal, hepatic, electrolyte or hematologic abnormalities were noted, and serial ECGs demonstrated no alteration in cardiac electrical activity. Thus, doses of C3 that have therapeutic efficacy appear to be well tolerated after 2 years (mice) or 2 months (non-human primates) of treatment.
Keywords: Carboxyfullerene; C60; e,e,e malonic acid C60; C3; Neuroprotection; Anti-inflammatory; Pharmacokinetics; Toxicology
1. Introduction
C60 fullerenes are spherical all-carbon molecules whose discovery two decades ago earned Kroto, Curl, and Smalley the Nobel Prize in Chemistry. The unique chemical and electronic properties of the fullerene carbon lattice confer potent antioxidant and anti-inflammatory properties on C60 that have been exploited to develop potential therapeutics for a variety of diseases from cancer to Parkinson disease to aging. The parent C60 fullerene molecule, however, is not soluble in water and forms aggregates in aqueous solutions, driving the need to synthesize water-soluble derivatives. One such compound, the e,e,e tris malonic acid derivative of C60 (designated C3), is among the most extensively studied fullerene-based compounds (reviewed in [1]). The chemical structure of C3 as well as the contrast in solubility with native C60 are shown in Figure 1. C3 is an antioxidant with catalytic superoxide dismutase mimetic activity [2] which has neuroprotective efficacy in several mouse models of disease [1, 3–7]. In addition, chronic oral administration of C3 produced a modest increase in lifespan [3, 8] and prevented cognitive deficits [3] in aging mice, arguing against toxicity at therapeutic doses, even with long-term administration. In addition, chronic systemic treatment with C3 in a non-human primate model of Parkinson’s disease for two months demonstrated significant behavioral and neuroanatomical improvement [9]. In that study, C3 treatment was started after degeneration of the nigral dopaminergic neurons was already in process. To date, however, no studies have characterized the tissue distribution, pharmacokinetics, or toxicology of C3.
Figure 1.

Structure (A) of C3 (e,e,e, tris malonic acid C60) indicating the position of the 14C-labeled cyclopropane carbon, chosen because of its stable attachment to C60. Each molecule of C3 was labeled with exactly one 14C-cyclopropane carbon per molecule. (B) Comparison of unreacted C60 precursor (left) and the final C3 product in water (right); C60 is unsoluble in water and C3 is soluble up to 400 mg/ml.
Studies on pharmacokinetics and toxicity of other derivatives of fullerene C60 have yielded conflicting data on biodistribution and in vivo toxicity [10–12]. This almost certainly reflects the nature and location of functional groups attached to the C60 sphere that can dramatically affect physical properties like water solubility, protein binding and biological activities of the molecule. In addition, in most of these studies there was no characterization of the preparation being administered, including impurities, and for a number of these studies, subsequent work demonstrated that there was toxicity from unreacted material, or contaminants, not the C60-based compound itself. Because, C3 has shown efficacy in several standard animal models of CNS disease, we wanted to further characterize its toxicity and pharmacokinetics in two species, mice, and Macaque fascicularis monkeys.
2. Materials and methods
2.1 Materials
C60 carboxyfullerene was purchased from Materials, Technologies, Research, Ltd (Cleveland, OH, USA). 14C-malonic acid was purchased from Sigma Chemical Co. (St Louis, MO, USA). Silica gel (Merck grade 9385, 260–400, 60 A) was obtained from Aldrich Chemicals (St. Louis, MO, USA). Other reagents were purchased from Sigma Chemical Co. (St. Louis, MO, USA) and other standard sources. The detailed synthesis of e,e,e tris(dimethylmalonyl) -fullerene (C3) and 14C-C3 carboxyfullerene are described in a Supplemental File.
2.2 Pharmacokinetic studies in mice
Mice were obtained from (Jackson Laboratory, Bar Harbor, ME, USA) at 8 weeks of age and housed in the vivarium barrier facility, under standard housing conditions, until 16 weeks of age (22–28 g), when used for experiments. All animal studies were approved by the Animal Care Programs at Washington University, and the University of California, San Diego, and are in accordance the PHS Guide for the Care and Use of Laboratory Animals, USDA Regulations, and the AVMA Panel on Euthanasia. 14C-C3 was administered to 10 week-old C57BL/6J male mice by i.v. (intravenous), i.p (intraperitoneal)., p.o. (per os), and s.c. (subcutaneous) routes, or by surgically implanted intraperitoneal mini-osmotic pumps (Alza Pharmaceuticals, Palo Alto, CA, USA). Mice were given a single bolus of 1.1 mg/kg 14C- C3, plus 9 X cold C3 as a carrier (to minimize loss of radiolabeled C3 on the syringe surface) in normal (0.9%) saline as the vehicle for the i.p., s.c. or i.v. (retro-orbital sinus) injections or in water for delivery by gastric gavage needle. Single day pumps (8 μl/hr, 24 hr duration) were loaded to deliver 17.5 mg/kg/day 14C-C3 (plus cold C3 as carrier). Seven day (1 μl/hr, 7-day duration) pumps were loaded to deliver 10 mg 14C- C3/kg/day (with no cold C3 carrier). Mice were anesthetized by inhaled metaphane prior to pump implantation. Blood (50–75 μl) was collected from the saphenous vein into heparinized capillary tubes at various times after drug administration. Each animal was bled 3 times and at least 3 blood samples were collected for each time point. Samples were either processed immediately or stored at −20° C overnight. Samples were analyzed by liquid scintillation counting (LSC). Animals were euthanized by pentobarbital injection followed by perfusion with ice-cold phosphate buffered saline. The following tissues and organs were collected at the time of euthanasia for determination of radioactivity: bone (femur), brain, subcutaneous fat, heart, kidney, liver, lung, muscle, small intestine, spleen, and testis. Samples were weighed immediately and stored at −80° C until further processing. Tissue samples were macerated and homogenized directly in scintillation cocktail and analyzed for radioactive content.
Although the cyclopropane carbon of C3 has previously been reported to be highly resistant to removal from the C60 molecule, we performed studies to confirm that the 14C label was retained on C3. First, we subjected C3 to electrospray mass spectroscopy (ESI-MS) and found that the carboxyl carbons, but not the cyclopropane carbons, could be stripped from the C3 molecule. In addition, when C3 was exposed to solutions at pH 1 or pH 10 for >24 h at 37°C, a small fraction of carboxylic acid head-groups were removed, but the cyclopropane carbons were intact.
To identify potential metabolites, HPLC analysis on plasma samples from mice injected with 14C-C3 was performed. Plasma was extracted into ethyl acetate using a two-phase extraction procedure [13]. The extraction was done at 4°C to minimize degradation during processing. The ethyl acetate fraction was evaporated and reconstituted in 0.1% trifluoroacetic acid (TFA) in water. The plasma extracts as well as aliquots of the original 14C-C3 sample were injected onto an Agilent 1100 HPLC system equipped with a C18 column. C3 and potential metabolites were separated using a step-wise gradient starting at 95:5; A (=0.1% TFA in water): B (=95% isopropyl alcohol, 0.005% TFA, 5% water) and going to 5:95; A:B (final) over 40 min. Fractions (0.5 min) were collected and radioactivity measured.
2.3 Mouse stroke model and surgical procedures
Mice obtained from Jackson Labs at 8 weeks of age were housed in the vivarium barrier facility as above, until 4 months of age, when used for experiments. Male C57BL/6J mice of 20–25 g weight were used to determine brain accumulation of C3 after disruption of the blood-brain barrier. Focal reversible cerebral ischemia was induced by direct occlusion of the middle cerebral artery (MCA) as previously described [14]. Briefly, mice were anesthetized with ketamine (75 mg/kg) and xylazine (5 mg/kg). The right MCA was exposed by a 0.4-cm vertical skin incision midway between the right eye and ear. After splitting the temporalis muscle, a 1.5–2mm burr hole was drilled at the junction of the zygomatic arch and the squamous bone. The right MCA was ligated distal to the lenticulostriate branches with an 11-0 suture under an operating microscope. Both common carotid arteries (CCAs) were then occluded using non-traumatic aneurysm clips (0.75X 4mm, from BRI, Rockville, MD, USA). Reduction in blood flow was confirmed by laser doppler. At the end of a 15 min ischemic period, the right MCA ligature and both CCA clips were released, and restoration of blood flow was confirmed visually and by laser doppler. Rectal temperature was recorded and maintained at 37.0 ± 0.5°C prior to and for >1 hour after MCA ligation. All mice were housed in an air-ventilated room at 24 ± 0.5°C. 14C-C3 (1.1 mg/kg) was injected i.p. at 16 or 24 hr post-surgery, the period during which blood brain barrier breakdown occurs in this model [14]. At 24 or 36 hr after ischemia, mice were anesthetized, decapitated, and the cortical hemispheres dissected. Tissue samples were frozen on dry ice, and the ipsilateral and contralateral cortices weighed.
2.4 C3 binding properties in mouse blood and plasma
Partitioning of C3 between plasma and cellular components of blood was determined as follows. Whole blood spiked with 14C- C3 or blood collected from animals dosed with radiolabeled C3 was centrifuged (1000 × g, 15 min), with the supernatant defined as the plasma fraction and the pellet as the cellular component. Cells were resuspended in phosphate buffered saline (PBS) and centrifuged again. The second PBS supernatant was defined as wash and the pellet as washed cells. The samples were analyzed by LSC. To examine the plasma protein binding of C3, plasma was prepared from freshly collected heparinized whole blood by centrifugation (1000 × g, 15 min). Radiolabeled C3 was added to saline (0.9%) or to plasma at 10 or 100 ug/ml and incubated for 1 hr. The samples were passed through a 3000 mwco Centricon (Millipore Corp., Bedford, MA, USA) filter (m.w. of C3 is 1027). Filtrate and retained solution were analyzed for radioactivity, with radioactivity in the filtrate defined as free C3 and that in the retained solution as protein-bound C3.
2.5 Excretion of C3 in mice
Urine samples were collected from 30 min up to 48 hr after i.p. dosing with radiolabeled C3, and up to 7 days after implanting a 7-day Alzet pump. Radioactivity was determined per μl urine. Data were transformed to the amount of 14C- C3 excreted per ml urine at the time of collection. Fecal material was collected from 0–48 hr after dosing and extracted for radioactivity by dissolving fecal material in acidified aqueous methanol followed by 3 extractions with ethyl acetate (3:1 vol:vol).
2.6 Evaluation of the effects of C3 on physiology, hematology and serum chemistry in Macaca fascicularis monkeys
A group of 15 male M. fascicularis monkeys (age 5–10 years) which were enrolled in a vehicle-controlled study testing C3 in a model of Parkinson disease, using intracarotid 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP 0.25 mg/kg) injection [9], also served here as subjects to evaluate the effect of C3 on hematology, blood chemistry, and heart function. Care was taken throughout the study to keep all investigators blinded to the treatment group. As we previously reported [9], monkeys were fasted overnight, anesthetized with ketamine, intubated and given inhaled isoflurane to maintain anesthesia. An 80 cm catheter was guided from the femoral artery into the internal carotid artery utilizing fluoroscopic guidance. MPTP (0.25 mg/kg) was then infused into the animal. At 10 days following MPTP, monkeys were implanted with Alzet pumps subcutaneously to deliver a chronic infusion of either C3 (3–5 mg/kg/day) or food coloring for a total treatment duration of 2 months (1 pump exchange was performed after one month). Following the 2 month treatment period with either C3 or food coloring, monkeys given MPTP had blood drawn for hematology and serum chemistry. Two additional control monkeys, which were not lesioned with MPTP, were given C3 (7mg/kg/day) for 1 month. Electrocardiograms were recorded at baseline (prior to MPTP), after MPTP, after 1 month of placebo or C3, and after 2 months of placebo or C3, from all animals.
2.7 Pharmacokinetics Studies in monkeys
We had the fortuitous opportunity to determine pharmacokinetics of C3 in two monkeys which were naïve to any previous experimental procedures, including MPTP lesioning. Each was fasted overnight; anesthesia was induced with ketamine (10 mg/kg i.m.) and maintained with inhalation of isoflurane (1–3%). Alzet osmotic pumps loaded to deliver 7 mg/kg/day for one month were implanted subcutaneously between the scapulae. Blood was drawn at 1.5, 3, 24 h, 1 week, and 1 month after implantation. Plasma was separated, and the samples were run in triplicate on an ELISA plate prepared with anti-C60 antibody which also recognizes C3 [15, 16]. Non-specific background was determined on plasma lacking C3.
2.8 Pharmacokinetic calculations
The area under the curve (AUC) was calculated from the blood level radioactivity from animals using the area function of Sigma Plot (SPSS, Inc, Chicago. IL, USA). The first order rate constant (k) was determined with the regression analysis component of Sigma Plot software. Other parameters were as follows: plasma half-life, t1/2 = ln2/k; clearance, Cl = dose/AUC; Volume of Distribution, V = dose/(k x AUC).
2.9 Animal Care and Use
The study was approved by the Institutional Animal Care and Use Committee (IACUC) of Washington University in St. Louis and the nonhuman primate studies were conducted in the Nonhuman Primate Facility at the Washington University School of Medicine. All procedures were designed to conform to suggestions of the 2006 Weatherall Report with steps taken to ameliorate subject suffering in accordance with the requirements of the National Institutes of Health (NIH, USA). In the event of mild to moderate pain or discomfort, a primate would be treated with Buprenex, 0.01–0.03 mg/kg for up to three days, per the discretion of the staff veterinarian. No animals were knowingly exposed to potential infection and humane endpoints were pre-defined and applied, if necessary, to reduce subject discomfort. The macaques were housed in appropriate animal facilities with 12-hour day and 12-hour night cycles. They were provided with food and water ad libitum and toys and movies as enrichment activities. All animals were housed in the same USDA-approved facility room in cages that met or exceeded the Weatherall Report’s requirements. The monkeys were monitored daily by the researchers, animal care staff, and a veterinarian, to check their general health and welfare. All routine animal care procedures have been described in detail [17].
3. Results
3.1 Kinetics of 14C-C3 in mouse blood
Peak blood levels occurred approximately 1 hr after administration of drug by i.p., s.c. or p.o. (Table 1, Figure 2a). However, following i.v. administration, there was an initial 1 hr phase of rapid elimination followed by a second slower phase that was similar to that observed with i.p. s.c., or p.o. administration (Figure 2a). Blood levels of C3 from 3–24 hr, regardless of route of administration, followed first order decay kinetics with a plasma half-life of 8.2 ± 0.2 hr (Table 1, Figure 2a). As discussed below, we found that >99 % of blood C3 was in the plasma, therefore the reported blood levels reflect plasma levels for C3.
Table 1.
Pharmacokinetic parameters of 14C-C3 in C57B6 mice
| Parametera | dose | t1/2 | AUC | k | Cl | V | Tmax | Cmax |
|---|---|---|---|---|---|---|---|---|
| Units | mg/kg | hr | μg-hr/mL | 1/hr | mL/kg-min | l/kg | min | μg/mL |
| i.p. | 1.1 | 8.0 ± 0.6 | 13.9 ± 1.4 | 0.087 ± 0.007 | 1.32 ± 0.13 | 0.91 ± 0.12 | 60 | 1.3 ± 0.1 |
| i.v. | 1.1 | 8.2 ± 0.2 | 15.2 ± 1.4 | 0.085 ± 0.002 | 1.20 ± 0.11 | 0.85 ± 0.08 | 5 | 4.0 ± 0.2 |
| i.v.0-1b | 1.1 | 0.8 ± 0.1 | 0.90 ± 0.12 | |||||
| p.o. | 1.1 | 0.36 | 60 | 0.03 |
Parameters and formulas are described in the methods section.
values in the i.v.0-1 row are from data collected within the first hour after C3 injection. Values are mean ± SEM (n=6).
i.p. = intraperitoneal, i.v. = intravenous, and p.o. = per os,
AUC = area under the curve, Cl = systemic clearance, V= volume of distribution
Figure 2.

Blood levels of 14C-C3 in C57BL/6J mice: (a) following a single bolus of 14C-C3 (1.1 mg/kg) given by i.v., i.p., s.c. or p.o. routes of administration, (b) during and after infusion of 14C-C3 at 17.5 mg/kg/day for one day via intraperitoneal Alzet osmotic pump, (c) during infusion at 10 mg/kg/day for 7 days via intraperitoneal pump. Values are mean ± SEM (n=6).
i.v. = intravenous, i.p, = intraperitoneal, p.o. = per os, s.c. = subcutaneous
The effect of chronic administration by mini-osmotic pump on blood levels of C3 was then investigated. Mice were implanted with 1-day or 7-day pumps and blood samples collected and analyzed for radioactivity (Figure 2b and 2c). Levels of C3 increased for 24 hr following 1-day pump implantation, after which the blood level declined with first order decay (Figure 2b). At 48 hr after the 1-day pump was depleted, blood levels were less than 6% of the peak blood concentration. Following implantation of the 7-day pump, C3 blood levels, as expected, increased at a fairly constant linear rate of 0.96 μg/day (Figure 2c).
3.2 Behavior of C3 in mouse blood and plasma
To determine to what extent C3 partitions into blood cells versus plasma, the distribution and plasma protein binding of C3 was evaluated in blood collected from C3-treated animals and in naïve blood spiked with 14C- C3. More than 99% of 14C- C3 partitioned into the plasma fraction and was separable from the cell pellet by centrifugation. To measure C3 plasma protein binding, plasma and saline were spiked with 10 or 100 μg/ml 14C- C3 and centrifuged through 3000 mwco centricon filter units (C3 m.w. = 1027). Both concentrations of C3 behaved identically. After centrifugation, 73% of the 14C-C3 was in the filtrate of the saline control compared to 13% of the plasma filtrate indicating that 87 ± 0.1 % (mean ± SEM) of C3 was protein bound.
Plasma samples obtained at 2 h, 8 h (one plasma half-life) and 18 h (about two plasma half-lives) after i.p. administration of 14C-C3 were analyzed by HPLC to determine stability of the 14C-label on C3 in vivo. Radioactivity was extracted from plasma, with an efficiency of 96 ± 2%. The HPLC chromatograms for eluted radioactivity are shown in Figure 3.
Figure 3.
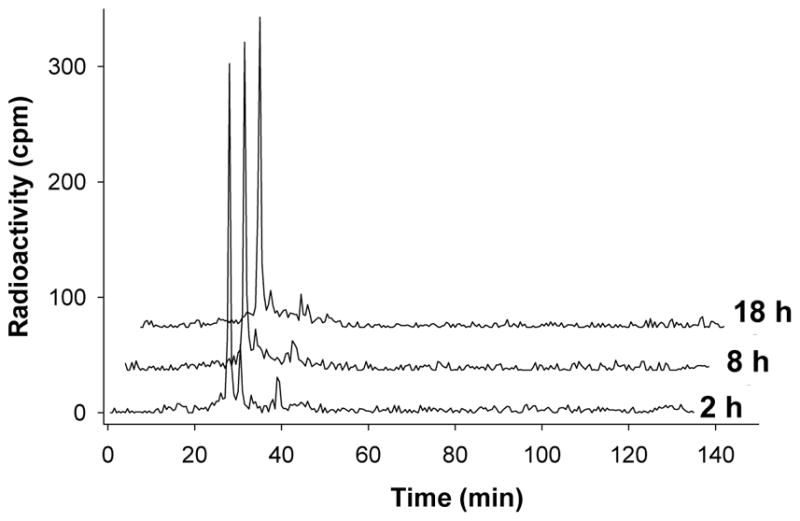
HPLC radiochromatograms of a 14C-C3 standard, and extracted plasma samples taken at 2 h and 18 h after a single 1.1 mg/kg i.p. administration of 14C-C3 in mice. Similar amounts of total radioactivity were chromatographed for each sample. C3 elutes at 27 min, with the two minor mono-decarboxylation products, present in the original preparation, appearing at 31 and 40 min, respectively. Both the 2 h and 18 h plasma samples were essentially identical to the original C3 stock. i.p, = intraperitoneal
3.3 Tissue distribution of C3 in mice
After a single intraperitoneal (i.p.) dose of 14C- C3, radioactivity distributed to all tissues examined (Figure 4a), with highest uptake into kidney and liver. There was no difference in distribution between i.p. only i.v. routes of administration (data not shown). On the other hand, animals with 1-day implanted pumps showed tissue radioactivity levels that were higher than in animals which received a single i.p. or i.v. bolus, suggesting that chronic delivery can increase loading into tissues (note scale). When adjusted for differences in dose, tissue levels of radioactivity in animals with surgically implanted 7-day pumps were 5 times higher than that in animals given a single bolus dose (Figure 4b; only data from 7-day pumps are not shown). Surprisingly, muscle and heart had the lowest levels of radioactivity of all the tissues examined, while liver and kidney exhibited the highest levels. The half-life for renal elimination was calculated to be approximately 150 hr compared to the shorter plasma clearance t1/2 of 8.2 ± 0.2 hr. The amount of radioactivity remaining in the whole liver 144 hr after a single i.p. administration of 1.1 mg/kg was about 10.6% of the administered dose and that in each kidney was about 2.2% of the dose.
Figure 4.

Tissue distribution of 14C-C3. Following single-dose i.p. (a), or chronic 7-day i.p. pump administration (b) of radiolabeled C3, tissues were collected, homogenized and analyzed for radioactivity. Data were transformed to μg 14C-C3 per gm tissue and normalized to dose for comparison.
3.4 Delivery of C3 to mouse brain
We examined distribution of C3 into brain under normal conditions and after disruption of the blood-brain barrier, since systemic administration of C3 has shown neuroprotective effects [5, 18, 19] Following i.p. administration of compound in mice, levels of C3 in brain were highest at 3 hr, the earliest time point examined, and declined by half at 6 hr (Figure 5a). Similar to other tissues examined, radioactivity in the brains of mice with compound delivered by the 7-day osmotic pump was higher than i.p. administration (Figure 5b). To determine whether increased permeability of the blood brain barrier would enhance delivery of C3 to brain, we measured C3 uptake in mice subjected to focal cerebral ischemia using a model that disrupts blood brain barrier on the ipsilateral (ischemic) side [14]. After ischemia, C3 levels in the ipsilateral hemisphere were twice as high as in the undamaged contralateral cortex (Figure 5c). Accumulation of 14C-C3 in the ischemic hemisphere was even greater than in uninjured brain, indicating that disruption of the blood-brain barrier can significantly increase brain penetration, a feature which might be used to advantage to deliver higher concentrations of compound to injured tissue.
Figure 5.

Distribution of 14C-C3 to the brain: following i.p. (a) or chronic 7-day pump administration (b) of radiolabeled C3. Data was transformed to percent-administered dose 14C-C3 per gm tissue for comparison. After i.p. administration, 14C-C3 concentration in the brain peaked at 3 hr (a). 14C-C3 uptake into the brain was increased by chronic administration (b). Focal brain ischemia was surgically induced by 10–15 min arterial restriction followed by i.p. administration of 14C-C3 (c). Samples consisting of the focal lesion area (ipsilateral) and contralateral normal tissue were collected, homogenized and subjected to liquid scintillation counting. 14C-C3 levels in the damaged tissue were higher than in undamaged tissue. Data represents sample mean± SEM (n=6). i.p, = intraperitoneal
3.5 Mouse Excretion of C3
The urinary excretion of 14C-C3 was assayed for 24 hr after i.p. administration in mice. The concentration of radioactivity in urine showed 2 phases of decay (Figure 6). The initial phase lasted for 3 hr after compound administration, corresponding to the period of accumulation seen in blood (Figure 2). The second phase of decreasing concentration was tracked for 24 hr after dosing. The 2 phases of decay suggested elimination from 2 compartments, most likely plasma initially, then kidney.
Figure 6.
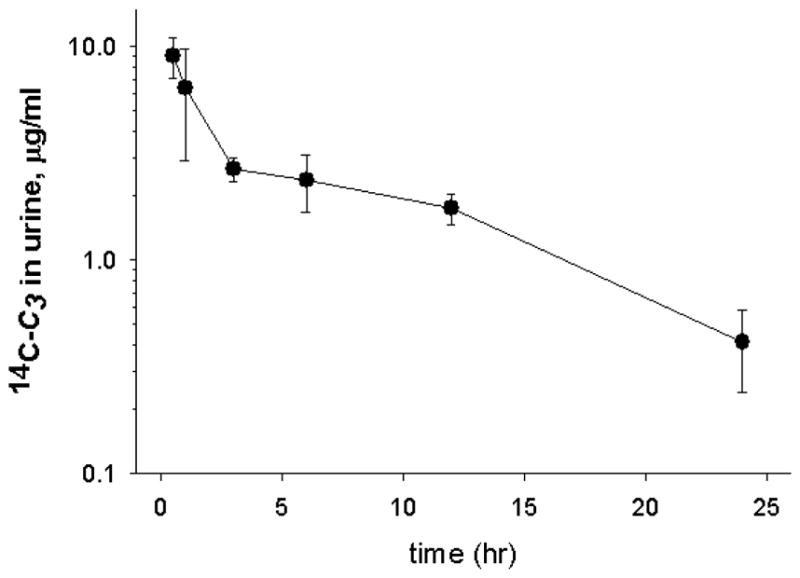
Urinary excretion of 14C-C3 in the C57BL/6J mouse. Urine samples were collected at various times during the 24 hr following a single i.p. administration of 14C-C3 (1.1 mg/kg). Samples were analyzed directly for radioactivity by scintillation counting. Data points represent mean ± SEM (n=6).
N.B.: 0 hour represents 1hour after final administration of C3 or 1 hour after removal of pump.
Levels in liver were high after i.p. administration. Fecal extractions showed that 12 ± 2.7 % of the administered dose was excreted by liver during the first 24 hr, and 6.6 ± 2.2% was excreted from 24–48 hr suggesting that a substantial amount of drug was eliminated hepatically. Urinary excretion was approximately 5.5% of the administered dose in the first 24 hr following i.p. administration. The urine concentration of radioactivity remained relatively constant at 23 ± 1.8 ug/ml during chronic administration via 7-day pump.
3.6 Evaluation of toxicity of C3 in mice
We determined that the LD50 for C3 in mice was 80 mg/kg after, a single i.p. injection. Additionally, we evaluated chronic delivery of 30 mg/kg/day for 2 months, which produced no deaths, thus the LD50 was > 30 mg/kg/day for 2 months.
3.7 Plasma levels of C3, blood chemistry, and heart function in monkeys
Plasma levels of C3 in two monkeys that received C3, 7 mg/kg/day for 28 days, were analyzed by an ELISA developed by Dr. Bernard Erlanger, Columbia University, based on an anti-fullerene antibody that recognized C3. The results from these two monkeys and the expected plasma levels for this dose based on the mouse data are shown in Table 2. Using allometric scaling calculations between mice and monkeys receiving the same dose of drug, we observed the expected higher plasma concentration in monkeys (Table 2). We then measured effects of C3 on blood chemistry (Figure 7) and hematology (Figure 8) parameters in monkeys chronically treated with C3 (3 mg/kg/day, 60 days) and compared to placebo. These monkeys were part of a study using unilateral intracarotid MPTP injection as a model of Parkinson Disease, and both sets of monkeys underwent identical procedures throughout the study. There was no difference in any measure between experimental groups, and all values were within the normal range for this species [20]. ECGs were also performed, and no differences in electrical activity or ECG intervals were observed between the two treatment groups (Table 3).
Table 2.
Plasma concentrations of C3 in M. fascicularis monkeys
| Time point | Blood concentration (μg/mL) (n=6) | Mouse levels (at same dose) |
|---|---|---|
| 1.5 h | 2.53 ± 0.21 | 1 |
| 3 h | 3.36 ± 0.45 | 1.5 |
| 24 h | 3.42 ± 0.25 | 2 |
| 1 week | 9.96 ± 0.40 | 5 |
Plasma levels of C3 measured in two M. fascicularis given C3 (7 mg/kg/day) by chronic subcutaneous infusion via osmotic pumps. Using allometric scaling to compare expected blood levels between monkeys and mice receiving the same administered dose, we used the following calculations. The Km for mice was 4.5, based on the 35 g mice used here, and for monkeys, was 9.5 [23]. Thus, the same dose should produce 9.5/4.5=2.1 times higher levels in monkeys compared to mice. The expected concentrations of C3 in monkey plasma based on data from mice given a similar dose (in the last column) and are predicted well by these scaling calculations [23]. Values are mean ± SEM (n=6).
Figure 7.

Blood chemistry studies in Macaque fascicularis monkeys. Blood was drawn just before induction of anesthesia (P) prior to injection of MPTP; 7 days after MPTP just before initiating treatment with C3 or placebo (M); after 1 month of treatment with C3 or placebo (1mo), and after 2 months of treatment with C3 or placebo (2mo).
MPTP = 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine
Figure 8.

Hematology studies in Macaque fascicularis monkeys. Blood was obtained as in Figure 7. N.B.: 0 hour represents 1hour after final administration of C3 or 1 hour after removal of pump.
Table 3.
EKG intervals in Macaque fascicularis monkey treated with C3 or placebo for two months. Mean ± SD, n=5 each.
| Placebo | C3-treatment | |
|---|---|---|
| Heart Rate (range, bpm) | 69–135 | 83–103 |
| Intervals (sec) | ||
| PR | 0.08±.00 | 0.08±.00 |
| QRS | 0.05±.02 | 0.05±.01 |
| Q-T | 0.19±.03 | 0.20±.04 |
4. Discussion
Fullerenes and carbon nanotubes are being developed for a broad range of biological applications; including Gd-endohedral (Gd@C60) MRI contrast agents, anti-cancer therapeutics, and neuroprotective agents. Many fullerene derivatives have antioxidant properties, and a small number of compounds, including C3, are highly effective catalytic superoxide dismutase mimetic antioxidants [16]. The C3 compound is also amphipathic, demonstrating excellent solubility in water (> 400 mM as the sodium salt), but with good entry into biological lipid bilayers [5]. Thus, we expected that C3 would show good tissue uptake. Consistent with this, we found that absorption of C3 into blood after both s.c. or i.p. injections was rapid, with peak blood levels observed 1 hr after injection. In addition, peak levels for both routes of administration were similar. After the first hour, all four routes of drug administration used for this study (i.v., i.p., s.c. and p.o.) produced plasma levels that followed similar first order kinetics, with a plasma t½ of 8.2 ± 0.2 hr. This is relatively similar to the 6.8 hr t½ reported for another water-soluble C60 derivative, MSCAD-C60 [11].
Elimination of C3 from blood also conformed to first-order kinetics, suggesting that there was little redistribution of compound from tissue compartments back into plasma. The observation that chronic delivery of drug did not modify the t½ indicated that clearance was not due to an inducible transport system.
Oral bioavailability of C3 as an aqueous solution was ~10% of the i.v. administration. However, many if not most, drugs require either formulation or administration without (or sometimes with) food for optimal GI bioavailability. We did not attempt to modify the oral delivery system to improve gut uptake of C3, but speculate that administration before feeding would increase enteric absorption of C3, a standard industry method for increasing enteric absorption of drugs with some degree of protein binding, as C3 exhibits.
After single-dose and chronic administration, C3 exhibited broad tissue distribution with detectable levels in all tissues analyzed. However, the highest tissue uptake was by liver (25% at 3 h) and kidney (20% at 3 h). These data are consistent with two previous pharmacokinetic studies of other fullerene derivatives that found accumulation predominantly in liver and kidney [11, 12]. In the report by Cagle (1999) their polyhydroxylated holmium metallofullerene preparation also accumulated significantly in bone [12], but we found only trace amounts of C3 in bone, even with chronic administration. Excretion of C3 through these two routes accounted for 17.5% of the administered dose during the 24 hr following i.p. administration, indicating that a substantial percentage of C3 is cleared by both liver and kidney. Fecal excretion was an important route of clearance, suggesting that the liver accumulation of C3 we observed reflected compound that was in the process of being excreted by the biliary system. During 7-day pump administration, animals also excreted an estimated 10–20% of the administered dose per day in the urine. C3 continued to be excreted by renal and hepatic routes after blood levels had become undetectable, with nearly 100% of the administered dose cleared 48 h after the last dose.
We were specifically interested in determining brain uptake of C3 because several groups have reported neuroprotection by C3 (reviewed in [1]), including protection after MPTP-induced brain injury in nonhuman primates [9]. C3 was detected in brain after single dose i.p. or chronic pump administration (Figure 5), and levels were higher than levels achieved by several drugs known to cross the blood brain barrier, and to have central nervous system (CNS) activity. For example, the brain concentration of the serotonin-selective reuptake inhibitor antidepressant sertraline (Zoloft) in rats after 5 mg/kg p.o. was ~0.5–1% of the administered dose [21, 22]. In comparison, after a single bolus of C3, the concentration in brain was 2% of delivered dose, which increased to approximately 4% during chronic systemic delivery. Disruption of the blood brain barrier by mild focal cerebral ischemia further increased uptake to 10% of the administered dose.
To estimate a therapeutic window, we determined an LD50 for C3 in mice. This was 80 mg/kg for a single i.p. injection and > 30 mg/kg/day for chronic delivery lasting 2 months (no deaths occurred at this dose, higher doses were not tested). Biologically effective doses of C3 have been on the order of 5–10 mg/kg for a single dose, or 0.5–10 mg/kg/day (which is 0.02–0.4 mg/kg/hr) for chronic delivery (for up to 24 months). Thus, C3 appears to have a broad therapeutic window. Here we did not explore embryotoxicity of C3, information that would be important for studies in reproductive-age organisms, but a previous study in which C3 was administered to pregnant mouse dams for 30 days failed to find any effects on fertility, pregnancy, viability or anatomy of the offspring [2].
Examination of the impact of chronic C3 treatment (3–7 mg/kg) on M. fascicularis monkeys showed that blood chemistry, hematology and ECGs were unchanged and remained within the normal range during a 1–2 month treatment period with C3. These doses are within the biologically effective range in mice. Additionally, we found the plasma half-life of C3 was 8 ± 1 h, which was similar to that in mice. Data from these primate studies may be a valuable tool for determining treatment strategies for evaluating the effectiveness of C3 against an array of injury and disease models in non-human primates and possibly humans.
In summary, C3 has favorable pharmacokinetics and a wide therapeutic index. C3 distributes into all tissues studied, including brain, albeit at much lower levels than kidney and liver, which are the main sites of excretion. Chronic administration of drug provided higher brain levels of C3 than bolus injection as did disruption of the blood brain barrier. Information from this study may assist in rational drug design to increase delivery to the target tissues (e.g. brain) and should also help guide further in vivo studies investigating therapeutic efficacy and structure-activity relationships of C3 and related malonic C60 compounds.
Supplementary Material
Key points.
C3 has favorable pharmacokinetics and a wide therapeutic index. C3 distributes into all tissues studied, including brain. Kidney and liver are the main sites of excretion.
No evidence of renal, hepatic, electrolyte or hematologic abnormalities were noted, and no alteration in cardiac electrical activity was observed.
The LD50 in mice was 80 mg/kg for a single intraperitoneal injection, and was >30 mg/kg/day for sustained administration; therapeutic doses are 1–5 mg/kg/day.
The doses of C3 that have therapeutic efficacy appear to be well tolerated after 2 years (mice) or 2 months (non-human primates) of treatment.
Acknowledgments
Funding: This study was supported by NIH/NINDS grants NS37688, NS050425, NS39913, NS41509 and NS058714; the American Parkinson Disease Association (APDA) Advanced Research Center at Washington University, the Greater St. Louis Chapter of the APDA and the Barnes-Jewish Hospital Foundation (Elliot Stein Family Fund for PD Research and the Jack Buck Fund for PD Research), the Larry L. Hillblom Foundation, the Hartke Fund and a Paul Beeson Physician Scholars Award (LLD). These sources of funding had no influence on any aspects of the research presented.
Footnotes
Conflict of interest: All of authors (Joshua I. Hardt; Joel S. Perlmutter; Christopher J. Smith; Kevin L. Quick; Subhasish K. Chakraborty and Laura L. Dugan) have no conflicts of interest.
References
- 1.Dugan LL, Lovett E, Quick KL, Lotharius J, Lin TT, O’Malley KL. Fullerene-based antioxidants and neurodegenerative disorders. Parkinsonism Relat Disord. 2001;7:243–6. doi: 10.1016/s1353-8020(00)00064-x. [DOI] [PubMed] [Google Scholar]
- 2.Ali SS, Hardt JI, Quick KL, Sook Kim-Han J, Erlanger BF, Huang TT, et al. A biologically effective fullerene (C(60)) derivative with superoxide dismutase mimetic properties. Free Radic Biol Med. 2004;37:1191–202. doi: 10.1016/j.freeradbiomed.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 3.Quick KL, Ali SS, Arch R, Xiong C, Wozniak D, Dugan LL. A carboxyfullerene SOD mimetic improves cognition and extends the lifespan of mice. Neurobiol Aging. 2008;29:117–28. doi: 10.1016/j.neurobiolaging.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 4.Dugan LL, Gabrielesen JK, Yu SP, Lin T-S, Choi DW. Buckminsterfullerenol free radical scavengers reduce ex-citotoxic and apoptotic death of cultured cortical neurons. Neurobiol Dis. 1996;3:129–35. 305–10. doi: 10.1006/nbdi.1996.0013. [DOI] [PubMed] [Google Scholar]
- 5.Dugan LL, Turetsky DM, Du C, Lobner D, Wheeler M, Almli CR, et al. Carboxyfullerenes as neuroprotective agents. Proc Natl Acad Sci U S A. 1997;94:9434–9. doi: 10.1073/pnas.94.17.9434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang YL, Shen CK, Luh TY, Yang HC, Hwang KC, Chou CK. Blockage of apoptotic signaling of transforming growth factor-beta in human hepatoma cells by carboxyfullerene. Eur J Biochem. 1998;254:38–43. doi: 10.1046/j.1432-1327.1998.2540038.x. [DOI] [PubMed] [Google Scholar]
- 7.Lin AM, Fang SF, Lin SZ, Chou CK, Luh TY, Ho LT. Local carboxyfullerene protects cortical infarction in rat brain. Neurosci Res. 2002;43:317–21. doi: 10.1016/s0168-0102(02)00056-1. [DOI] [PubMed] [Google Scholar]
- 8.Ali SS, Xiong C, Lucero J, Behrens MM, Dugan LL, Quick KL. Gender differences in free radical homeostasis during aging: shorter-lived female C57BL6 mice have increased oxidative stress. Aging Cell. 2006;5:565–74. doi: 10.1111/j.1474-9726.2006.00252.x. [DOI] [PubMed] [Google Scholar]
- 9.Dugan LL, Tian L, Quick KL, Hardt JI, Karimi M, Brown C, et al. Carboxyfullerene neuroprotection postinjury in Parkinsonian nonhuman primates. Ann Neurol. 2014;76:393–402. doi: 10.1002/ana.24220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamago S, Tokuyama H, Nakamura E, Kikuchi K, Kananishi S, Sueki K, et al. In vivo biological behavior of a water-miscible fullerene: 14C labeling, absorption, distribution, excretion and acute toxicity. Chem Biol. 1995;2:385–9. doi: 10.1016/1074-5521(95)90219-8. [DOI] [PubMed] [Google Scholar]
- 11.Rajagopalan P, Wudl F, Schinazi RF, Boudinot FD. Pharmacokinetics of a water-soluble fullerene in rats. Antimicrob Agents Chemother. 1996;40:2262–5. doi: 10.1128/aac.40.10.2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cagle DW, Kennel SJ, Mirzadeh S, Alford JM, Wilson LJ. In vivo studies of fullerene-based materials using endohedral metallofullerene radiotracers. Proc Natl Acad Sci U S A. 1999;96:5182–7. doi: 10.1073/pnas.96.9.5182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quick KL, Dugan LL. Superoxide stress identifies neurons at-risk in a model of ataxia-telangiectasia. Annals Neurology. 2001;49:627–35. [PubMed] [Google Scholar]
- 14.Majid A, He YY, Gidday JM, Kaplan SS, Gonzales ER, Park TS, et al. Differences in vulnerability to permanent focal cerebral ischemia among 3 common mouse strains. Stroke. 2000;31:2707–14. doi: 10.1161/01.str.31.11.2707. [DOI] [PubMed] [Google Scholar]
- 15.Chen BX, Wilson SR, Das M, Coughlin DJ, Erlanger BF. Antigenicity of fullerenes: antibodies specific for fullerenes and their characteristics. Proc Natl Acad Sci USA. 1998;95:10809–13. doi: 10.1073/pnas.95.18.10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ali SS, Hardt JI, Quick KL, Kim-Han JS, Erlanger BF, Huang TT, et al. A biologically effective fullerene (C60) derivative with superoxide dismutase mimetic properties. Free Radic Biol Med. 2004;37:1191–202. doi: 10.1016/j.freeradbiomed.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 17.Tian L, Karimi M, Loftin SK, Brown CA, Xia H, Xu J, et al. No Differential Regulation of Dopamine Transporter (DAT) and Vesicular Monoamine Transporter 2 (VMAT2) Binding in a Primate Model of Parkinson Disease. PLoS ONE. 2012;7:e31439. doi: 10.1371/journal.pone.0031439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lotharius J, Dugan LL, O’Malley KL. Distinct mechanisms underlie neurotoxin-mediated cell death in cultured dopaminergic neurons. J Neurosci. 1999;19:1284–93. doi: 10.1523/JNEUROSCI.19-04-01284.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quick KL, Ali SS, Arch RH, Xiong C, Wozniak DF, Dugan LL. A carboxyfullerene SOD mimetic improves cognition and extends the lifespan of mice. Neurobiol Aging. doi: 10.1016/j.neurobiolaging.2006.09.014. In press. [DOI] [PubMed] [Google Scholar]
- 20.Matsuzawa T, Nagai Y. Comparative haematological and plasma chemistry values in purpose-bred squirrel, cynomolgus and rhesus monkeys. Comparative Haematology International. 1994;4:43–8. [Google Scholar]
- 21.Wiener HL, Kramer HK, Reith ME. Separation and determination of sertraline and its metabolite, desmethylsertraline, in mouse cerebral cortex by reversed-phase high-performance liquid chromatography. J Chromatogr. 1990;527:467–72. doi: 10.1016/s0378-4347(00)82133-7. [DOI] [PubMed] [Google Scholar]
- 22.Tremaine LM, Stroh JG, Ronfeld RA. Characterization of a carbamic acid ester glucuronide of the secondary amine sertraline. Drug Metab Dispos. 1989;17:58–63. [PubMed] [Google Scholar]
- 23.Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. Journal of Basic and Clinical Pharmacy. 2016;7:27–31. doi: 10.4103/0976-0105.177703. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.