

SUMMARY
Cell-penetrating peptides (CPPs) are short protein segments that can transport cargos into cells. Although CPPs are widely studied as potential drug delivery tools, their role in normal cell physiology is poorly understood. Early during infection, the L2 capsid protein of human papillomaviruses binds retromer, a cytoplasmic trafficking factor required for delivery of the incoming non-enveloped virus into the retrograde transport pathway. Here we show that the C-terminus of HPV L2 proteins contains a conserved cationic CPP that drives passage of a segment of the L2 protein through the endosomal membrane into the cytoplasm where it binds retromer, thereby sorting the virus into the retrograde pathway for transport to the trans-Golgi network. These experiments define the cell-autonomous biological role of a CPP in its natural context and reveal how a luminal viral protein engages an essential cytoplasmic entry factor.
Graphical Abstract
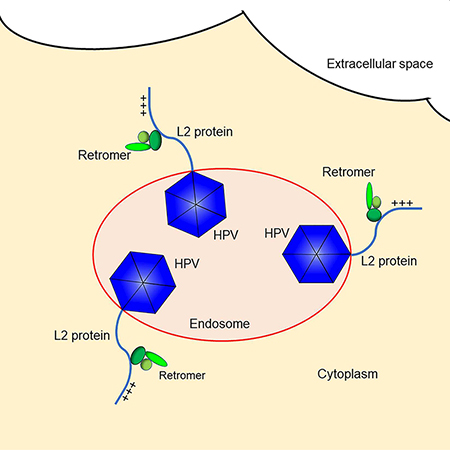
In Brief:
A conserved cell-penetrating peptide (CPP) encoded by the HPV genome drives non-enveloped viral entry across the endosomal membrane into the cytoplasm during infection
INTRODUCTION
Cell membranes pose formidable barriers to the passage of proteins and non-enveloped viruses. Unlike enveloped viruses, which deliver virion contents into the cytoplasm by membrane fusion, non-enveloped viruses need to cross or disrupt membranes during virus entry (Kumar et al., 2017). For most non-enveloped viruses, a lytic peptide released by proteolytic cleavage of a capsid protein physically disrupts the membrane or forms a large pore, allowing passage of the capsid into the cytoplasm. Human papillomaviruses (HPVs) are responsible for 5% of human cancer, most notably cervical cancer (Schiffman et al., 2016). These non-enveloped viruses have evolved a unique mode of intracellular trafficking during entry, in which viral material is sequestered in membrane-bound retrograde transport vesicles until late during entry (Calton et al., 2017; Day et al., 2013; DiGiuseppe et al., 2014; DiGiuseppe et al., 2016; Lipovsky et al., 2013). This mechanism is thought to protect the virion from cytoplasmic innate immune sensors until the viral DNA has reached safe haven in the nucleus, where viral genome expression and replication occur.
The HPV virion consists of the eight kilobase DNA genome in a capsid composed of 360 molecules of the L1 major capsid protein and up to 72 copies of the L2 minor capsid protein, which is largely buried in the L1 protein shell but plays an essential role in the trafficking of viral DNA to the nucleus (Buck et al., 2008; Campos, 2017; Kamper et al., 2006). HPV infection is initiated by the binding of L1 to heparan sulfate proteoglycans on the cell surface (Joyce et al., 1999), which triggers conformational changes in the capsid and proteolytic cleavage of L1 and the N-terminus of L2 (Cerqueira et al., 2015; Richards et al., 2006). The capsid is then thought to be transferred to a specific cell surface internalization receptor, whose identity remains controversial (Raff et al., 2013). After endocytosis, acidification of the endosomal lumen exposes the capsid to low pH, which is required for further progression of infection (Schelhaas et al., 2012; Smith et al., 2008). The protease γ-secretase is also required for proper HPV trafficking by facilitating association of L2 with membranes (Inoue et al., 2018; Zhang et al., 2014).
A key step in HPV infection is the entry of the capsid into the retrograde transport pathway. This pathway normally recycles cellular proteins in the endosome to the trans-Golgi network (TGN) for reuse. A genome-wide siRNA screen revealed that numerous retrograde transport factors are required for HPV entry and identified retromer as being essential for transport of the virion from the endosome to the TGN (Lipovsky et al., 2013). Retromer, a trimeric protein complex of Vps26, Vps29, and Vps35, resides in the cytoplasm where it binds to the cytoplasmic domain of cellular transmembrane (TM) proteins in the endosome membrane and sorts them into transport vesicles that bud from the endosome (Burd and Cullen, 2014). These vesicles are then transported to the TGN where membrane fusion deposits the protein cargo in the latter organelle. Retromer binds directly to conserved sites in the C-terminal segment of the HPV16 L2 protein (Figure 1A), and retromer knockdown or mutation of these binding sites causes the capsid to accumulate in the endosome and prevents it from reaching the TGN (Lipovsky et al., 2015; Popa et al., 2015). Thus, HPV is a novel class of retromer cargo.
Figure 1. The basic region can be replaced by a cationic cell-penetrating motif.
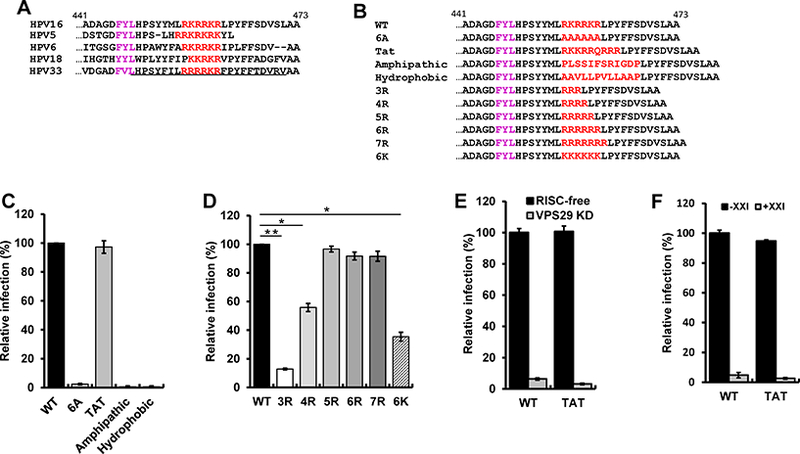
(A) Sequence of the C-terminus of the L2 protein of various HPV types. Basic amino acids (red) downstream of the major FYL retromer binding site (purple) are shown. Numbers indicate position in the HPV16 L2 protein. The membrane-destabilizing sequence in HPV33 L2 is underlined. (B) Sequence of the C-terminus of wild-type and mutant HPV16 L2 proteins. (C and D) HeLa S3 cells were infected with wild-type (WT) or mutant HPV16 PsV stocks containing equal numbers of the HcRed reporter plasmid (corresponding to MOI of one for wild-type). Forty-eight hpi, flow cytometry was used to determine the fraction of fluorescent cells. The results were normalized to the fraction of cells infected by wild-type. The mean results and standard deviation of three or more independent experiments for each sample are shown. *p<0.05; **p<0.01. (E) HeLa S3 cells were transfected with RISC-free control siRNA (black bars) or siRNA targeting Vps29 (grey bars), followed by infection at MOI of one with wild-type or L2-Tat HPV16 PsV. Infectivity was measured (n=3) and displayed as in panel (C). (F) HeLa S3 cells were untreated (black bars) or treated with 250 nM γ-secretase inhibitor XXI for one h at 37°C (grey bars), and then infected as in panel E. Infectivity was measured (n=3) and displayed as in panel C. See also Figure S1 and Table S1.
This model poses a topological challenge because the incoming virus is in the lumen of the endosome but retromer is in the cytoplasm. To resolve this, we and others proposed that the C-terminus of the L2 protein protrudes through the endosomal membrane into the cytoplasm so its binding sites are accessible to retromer (Campos, 2017; Popa et al., 2015). Indeed, the central portion of the L2 protein also binds to additional cytoplasmic proteins such as SNX17 and SNX27 required for HPV entry (Bergant Marusic et al., 2012; Pim et al., 2015), suggesting that much of the L2 protein is in the cytoplasm. In addition, the L2 protein contains an N-terminal hydrophobic segment that can act as a TM domain, implying that the L2 protein adopts a TM existence during infection (Bronnimann et al., 2013). This model is supported by antibody staining and protease sensitivity experiments showing that the bulk of the L2 protein is cytoplasmic by 18 hours after infection, other than a short N-terminal segment upstream of the putative TM domain (Calton et al., 2017; DiGiuseppe et al., 2015).
It is not known how the L2 protein accesses the cytoplasm during infection. Several years ago, Kamper et al. reported that peptides derived from the C-terminus of HPV16 or HPV33 L2 destabilized membranes at low pH, resulting in cell death (Kamper et al., 2006). Furthermore, this L2 segment was required for infection of fibroblasts by HPV16 and bovine papillomavirus 1, and pseudoviruses (PsVs) carrying mutations in this segment accumulated in an endo-lysosomal compartment. At the time of that report, papillomaviruses were thought to exit directly from the endosome into the cytoplasm, and the involvement of retromer and retrograde trafficking in infection was not known. Therefore, the contribution of this membrane-destabilizing sequence to cytoplasmic exposure of L2 was not explored.
The C-terminal segment of all sequenced papillomavirus L2 proteins contains a stretch of basic amino acids near the retromer binding sites (Figure 1A and Table S1). This basic segment is present in the membrane-destabilizing peptide identified by Kamper (Kamper et al., 2006) and is thought to act as a nuclear import signal during capsid assembly (Darshan et al., 2004). This segment resembles a class of peptides known as cationic cell-penetrating peptides (CPPs), which can transport proteins across the plasma membrane into cells (Guidotti et al., 2017). The prototype cationic CPP from the human immunodeficiency virus (HIV) Tat protein is RKKRRQRRR (where R, K, and Q represent arginine, lysine, and glutamine, respectively) and the basic sequence in HPV16 L2 is RKRRKR. Certain hydrophobic and amphipathic sequences also display cell-penetrating activity. The mechanisms of membrane penetration are not fully understood, but most cationic CPPs appear to undergo endocytosis (Kauffman et al., 2015). Although CPPs have been widely studied as agents to deliver molecules into cells for therapeutic purposes, there are few cases where the physiological role of cell-penetrating activity is known. In the nervous system, homeobox proteins such as Engrailed are secreted and then enter neighboring cells in a paracrine fashion to specify various aspects of neuronal function (Spatazza et al., 2013). The Tat protein can be secreted from HIV-infected cells, but it has not been conclusively shown that secreted Tat plays a role in virus replication or pathogenesis (Faust et al., 2017). A short basic sequence at the C-terminus of the HPV11 L1 major capsid protein displays CPP activity, but the role of this activity in HPV infection is not known (Lee and Lim, 2012).
Here, we show that the basic segment in HPV16 L2 has CPP activity that mediates passage of the C-terminus of the L2 protein through the endosomal membrane into the cytoplasm after virus endocytosis. This activity allows retromer to bind L2 and initiate retrograde transport of the capsid to the TGN. Thus, the physiologic role of this CPP is to mediate transfer of a protein segment from one cellular compartment to another, thereby enabling a protein-protein interaction essential for virus infection.
RESULTS
The C-terminal basic sequence of L2 can be replaced by a cationic cell-penetrating motif
Direct binding of retromer to a carboxy-terminal segment of the 473-residue HPV16 L2 protein is required for transport of the incoming virus from endosomes to the Golgi (Lipovsky et al., 2013; Popa et al., 2015). To test whether the basic amino acids in the C-terminus of the L2 protein function as a CPP to transfer a segment of the L2 protein into the cytoplasm, we first tested whether mutations in the basic segment inhibited infectivity. For these experiments, we used PsVs comprised of an HcRed reporter plasmid, wild-type HPV16 L1, and wild-type or mutant HPV16 L2 (Buck et al., 2005). PsV assembly was confirmed by electron microscopy, which showed no obvious morphologic differences between wild-type and mutant PsVs (Figure S1A). Each PsV stock was normalized to the number of encapsidated reporter plasmids and analyzed by SDS-polyacrylamide gel electrophoresis (SDS-PAGE). Wild-type and mutant PsVs displayed comparable purity and contained similar levels of L1 and L2 (Figure S1B).
To determine if the L2 basic segment is important for HPV16 infection, we replaced wild-type RKRRKR with six alanines to construct the 6A mutant (Figure 1B). HeLa and HaCaT cells were infected with wild-type HPV16 PsV at multiplicity of infection (MOI) of one or with mutant PsV containing an equivalent number of encapsidated reporter plasmids, and infection efficiency was measured two days later by flow cytometry for HcRed fluorescence. As shown in Figure 1C and Figure S1C, the 6A mutation abolished infectivity, showing that the basic sequence is essential for HPV16 infection. Next, we replaced the basic segment of L2 with the CPP domain of the HIV Tat protein to generate L2-Tat (Guidotti et al., 2017; Vives et al., 1997) (Figure 1B). Strikingly, HPV16 PsV with the Tat sequence in L2 infected cells as well as PsV containing wild-type L2 (Figure 1C). In contrast, an amphipathic or a hydrophobic CPP did not support infection. These results suggest that the basic segment of L2 acts as a cationic CPP to deliver the C-terminus of L2 containing retromer binding sites through a membrane into the cytoplasm and show that only the cationic class of CPP can support HPV infection.
We then replaced RKRRKR with various numbers of arginines or six consecutive lysines (Figure 1B). Published studies show that a stretch of five or more arginines can efficiently act as CPPs, whereas fewer arginines and polylysine are less effective (Tunnemann et al., 2008). Infectivity of the mutant PsV with three arginines (3R) was minimal (~10%) and increased with the number of arginine residues, reaching a plateau with five arginines (Figure 1D and Figure S1). The mutant with six lysines was ~40% as infectious as PsV containing five or more arginines. These results demonstrate that the basic segment at the C-terminus of HPV16 L2 can be functionally replaced by arginine-rich sequences with known CPP activity, and that infectivity correlated with predicted CPP activity. These results provide strong genetic evidence that the basic segment of L2 acts as a CPP during HPV16 infection.
We next tested if HPV16 PsV containing L2-Tat utilized a similar entry pathway as wildtype HPV16 PsV. To determine if retromer was required for infection mediated by L2-Tat, we infected cells transfected with siRNA that knocked down the Vps29 retromer subunit (Lipovsky et al., 2013). Retromer knockdown dramatically inhibited infection by wild-type and L2-Tat PsV, indicating that the L2-Tat chimera requires retromer (Figure 1E). To determine if γ-secretase was required for PsV containing L2-Tat, we treated cells with a chemical inhibitor of γ-secretase, compound XXI, and measured infectivity. XXI caused near-complete inhibition of both wildtype and L2-Tat PsV infection, showing that the L2-Tat chimera also requires γ-secretase (Figure 1F). Thus, the L2-Tat chimeric HPV16 PsV displays two of the key entry requirements as wildtype HPV16 PsV, dependence on retromer and γ-secretase.
The basic sequence of L2 can translocate peptides and GFP fusion proteins into cells
We conducted several experiments to determine whether the basic segment of L2 has cell penetrating activity. First, we conjugated Alexa Fluor 488 onto the N-terminus of a 28-residue L2 peptide that terminates with the wild-type basic segment or the corresponding peptide with the 6A or the 3R mutation (Figure 2A, top). As assessed by confocal microscopy, the wild-type peptide entered cells, but the peptide containing 6A mutation did not (Figure 2A, bottom). Cells treated with the 3R peptide displayed less fluorescence signal than cells treated with wild-type peptide, suggesting that the 3R peptide is partially impaired for cell penetration.
Figure 2. The L2 basic sequence displays cell-penetrating activity.
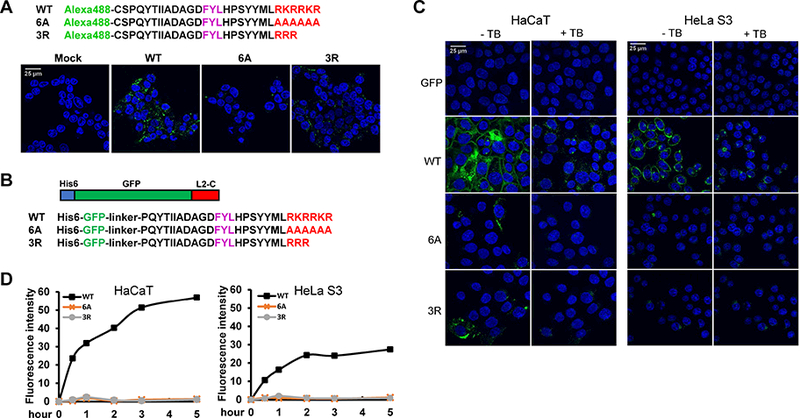
(A) Amino acid sequence of HPV16 L2 C-terminal peptides containing the wild-type (WT) basic sequence or the six alanine (6A) or three arginine (3R) mutations conjugated to Alexa Fluor 488. 293T cells were mock-treated or incubated with fluorescent peptides for three h at 37°C and examined by confocal microscopy. (B) Schematic diagram and C-terminal sequences of GFP-L2 fusion proteins. (C) HaCaT and HeLa S3 cells were incubated with GFP-L2 fusion proteins for three h. Cells were examined by confocal microscopy, then treated with trypan blue (+TB) to quench extracellular fluorescence, and the same fields were re-imaged. (D) HeLa S3 and HaCaT cells were incubated with GFP-L2 fusion proteins for various times. Cells were then treated with trypsin, and fluorescence was measured by flow cytometry. Mean fluorescent intensity (MFI) was plotted at the indicated time periods.
To confirm CPP activity of this L2 segment, we purified proteins from bacteria consisting of GFP fused in-frame to a 28-residue segment from the C-terminus of L2 terminating with either the wild-type or mutant basic segment (Figure 2B). Microscopic examination of HeLa and HaCaT cells incubated with GFP-L2 fusion proteins showed that the protein with the wild-type L2 segment generated a strong punctate fluorescence signal inside cells and at the cell periphery (Figure 2C). The 3R mutation greatly impaired cellular uptake and the 6A mutation abolished it. To determine if the fusion proteins were internalized, cells were treated with 0.04% trypan blue, a membrane-impermeable agent that quenches cell-surface GFP fluorescence but not intracellular fluorescence. As shown in Figure 2C, in cells incubated with the wild-type fusion protein, the signal at the cell periphery was eliminated by trypan blue, but the punctate signal persisted, demonstrating intracellular uptake of the protein. In contrast, the signal from the 3R mutant was largely eliminated. To quantify cellular fluorescence, flow cytometry was performed at 0.5 to 5 h post-incubation following trypsinization to remove any GFP fusion protein adsorbed to the cell surface. The wild-type fusion protein translocated into cells rapidly, whereas there was no internalization of 6A and 3R fusion proteins (Figure 2D). Thus, the wild-type and mutant basic segments of L2 displayed cell penetration activity that correlated with the infectivity of PsV containing these segments. These results demonstrate that the L2 CPP has cell-penetration activity and strongly suggest that this activity is required for HPV entry.
The L2 CPP is not essential for virus binding and internalization
To determine the role of the L2 CPP in HPV infection, we first conducted cell binding experiments. HeLa cells were incubated with either wild-type or mutant HPV16 PsV for two hours at 4°C, followed by washing to remove unbound viruses. Non-permeabilized cells were stained with an L1 antibody, and immunofluorescence was performed to detect viruses stably bound to cells. As shown in Figure 3A, similar amounts of wild-type and 3R mutant PsV bound cells. Thus, a mutation that inhibits the cell penetrating activity of L2 did not affect virus binding to cells, showing that L2 CPP activity was not required for binding. Unexpectedly, the 6A mutation resulted in a dramatic reduction of cell binding. Similar results were obtained when cell binding was assessed by flow cytometry or western blotting for L1 (Figures 3B and 3C). Because the cell binding defect of the 6A mutant conflicted with published studies showing efficient binding of capsids lacking L2 [e.g., (Kamper et al., 2006)], we also tested binding with PsV comprised of L1 only. As shown in Figure 3C, L1-only PsV bound cells to a similar level as complete PsV containing L1 plus wild-type L2, far better than PsV containing the 6A mutant. These results imply that although the L2 CPP is not required for cell surface binding, sequences in the C-terminus of L2 can modulate binding.
Figure 3. The basic region of HPV16 L2 is not essential for virus binding and internalization.
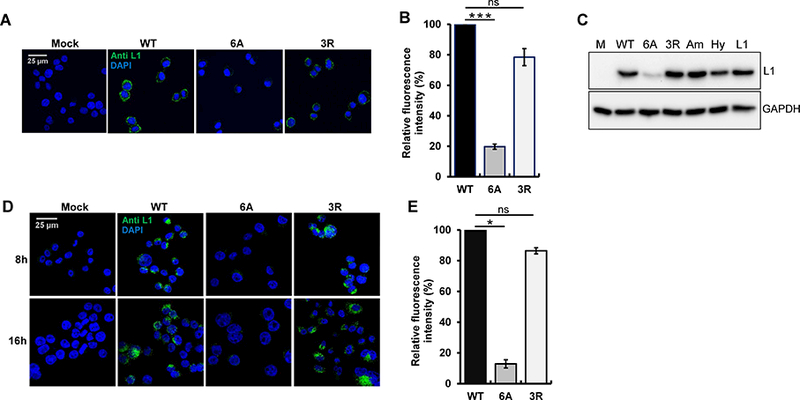
(A) HeLa-sen2 cells were mock-infected or incubated at 4°C for two h at MOI of 20 with wild-type, 6A, or 3R HPV16 PsV. Non-permeabilized cells were stained with anti-L1 antibody (green) and examined by confocal microscopy. (B) HeLa-sen2 cells were treated as described in panel A, detached with EDTA, and stained with anti-L1 antibody. MFI of the cells was measured by flow cytometry and normalized to cells incubated with wild-type HPV16 PsV. Mean results and standard deviation are shown (n=3). ***p<0.001; ns, not significant. (C) HeLa-sen2 cells were treated as described in panel A with the following PsVs: wild-type HPV16 (WT), 6A mutant, 3R mutant, mutant with amphipathic CPP (Am), mutant with hydrophobic CPP (Hy), and L1-only PsV (L1). After two h at 4ºC, cells were washed, lysed, and bound virus was assessed by SDS-PAGE and blotting for L1 (top panel). GAPDH is a loading control (bottom panel). (D) HeLa cells-sen2 were mock-infected or infected at MOI of 50 at 4°C for two h with wild-type, 6A or 3R HPV16 PsV and then washed and shifted to 37°C for eight or 16 h to allow internalization. Permeabilized cells were stained with anti-L1 antibody (green) and examined by confocal microscopy. (E) HeLa-sen2 cells were infected as described in panel D and harvested by trypsinization 6 h after shift to 37ºC. Permeabilized cells were stained with anti-L1 antibody and analyzed by flow cytometry. MFI of the cell populations was normalized to cells infected with wild-type HPV16 PsV. Mean results and standard deviation are shown (n=3). *p<0.05. See also Figure S4.
Because the 6A mutation affected binding, we also tested HPV16 PsV containing L2 with the amphipathic or hydrophobic cell-penetrating peptide in place of the wild-type sequence. As shown in Figure 3C, both of these mutants bound cells well, indicating that the severe infection defect of these mutants was not due to impaired cell surface binding.
We next examined the role of the L2 CPP in virus internalization. After incubation of cells with HPV16 PsVs at 4°C, we shifted cells to 37°C for eight or 16 hours to allow internalization, which was assessed by immunofluorescence and by flow cytometry. The 3R L2 mutant internalized into cells as well as wild-type, whereas the 6A mutant showed much less internalized L1, as expected because of its cell surface binding defect (Figures 3D and 3E). This result showed that the short basic segment of three arginines could support cell surface binding and virus internalization, even though it was not sufficient to restore infectivity or mediate membrane penetration. Thus, the L2 CPP is required for an intracellular event after cell binding and internalization.
The 3R mutant accumulates in endosomes and fails to reach the Golgi.
To examine the post-internalization defect of the 3R mutant, we used the proximity ligation assay (PLA) to determine the localization of incoming wild-type and 3R mutant HPV16 PsV. PLA is an immune assay used to test if proteins of interest are within 40 nm. PLA was performed with an anti-L1 antibody and an antibody that recognizes either EEA1, a marker of the early endosome, or the TGN marker, TGN46. The L2 double mutant (DM) mutant, which lacks retromer binding sites, was used as a control. As shown in Figure 4A, these antibodies did not generate a PLA signal in mock-infected cells. At eight hours post-infection (hpi), punctate intracellular EEA1/L1 PLA signal was observed in cells infected with wild-type PsV or either mutant and showed similar fluorescence intensity, confirming that the 3R mutant, like DM, efficiently entered cells and reached the endosome. At 16 hpi, the EEA1/L1 PLA signal of cells infected with wild-type PsV was significantly diminished, due to the departure of the virion from the endosome, whereas the signal for the DM in the endosome was markedly increased, reflecting endosomal accumulation due to the absence of retromer binding sites (Popa et al., 2015) (Figure 4A). Notably, the EEA1/L1 PLA signal of the 3R mutant at 16 hpi was increased ~ 3-fold compared to the eight-hour signal, similar to the increase seen with DM mutant. Thus, the 3R mutant accumulated in the endosome. As expected, little TGN46/L1 PLA signal was generated at eight hours after wild-type or mutant PsV infection (Figure 4B). At 16 hpi, cells infected with wild-type displayed abundant TGN46 PLA signal, reflecting delivery of L1 to this distal site. In contrast, 3R and DM mutants showed a greatly reduced TGN46/L1 PLA signal, indicating that these mutants did not arrive at the TGN. These results indicate that 3R mutation causes accumulation of the incoming virus particle in the endosome and prevents its delivery to the TGN, a phenotype similar to the mutant that cannot bind retromer. Together with the reduced infectivity of the 3R mutant and the impaired cell entry of 3R mutant fusion proteins, these data suggest that the trafficking defect displayed by 3R mutant is due to impaired endosomal membrane penetration and retromer association.
Figure 4. The 3R mutant accumulates in endosomes.
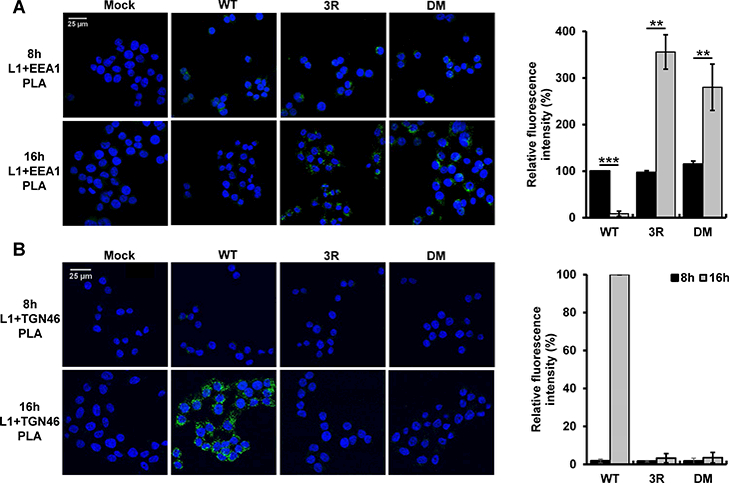
HeLa-sen2 cells were mock-infected or infected at 37ºC at MOI of 100 with wild-type, 3R or retromer binding site (DM) mutant HPV16 PsV. At eight and 16 hpi, PLA was performed with anti-L1 antibody and either an EEA1 (panel A) or a TGN46 (panel B) antibody. PLA signal is green. Multiple images obtained as in left panels were processed with BlobFinder software to determine the fluorescence intensity per cell. The graphs in the right panels show mean results and standard deviation (n=3), in which the results for the EEA1/L1 samples and the TGN46/L1 samples were normalized to those of cells infected with wild-type HPV16 PsV at eight and 16 hpi, respectively. Black bars, 8 hpi; grey bars, 16 hpi **p<0.01; ***p<0.001.
The 3R mutant is defective in accessing retromer during HPV infection but binds retromer in vitro
We next tested whether the 3R mutation impaired association between the capsid and retromer in infected cells. HeLa cells were infected with either wild-type or mutant HPV16 PsV, and PLA was performed with an anti-L1 antibody and an antibody recognizing Vps35, a subunit of retromer. As shown in Figure 5A, wild-type PsV generated abundant Vps35/L1 PLA signal at eight hpi, while the signal was diminished by 16 hours, after the virus exited from endosomes and dissociated from retromer. As expected, the PLA signal for DM mutant was very weak at both eight and 16 hours. Interestingly, we observed ~ 75% reduction in the PLA signal for the 3R mutant at eight hours, and this signal decreased further by 16 hours (Figures 5A and 5B), despite the accumulation of the mutant in the endosome at this later timepoint.
Figure 5. The 3R mutant is defective in accessing retromer during HPV infection.
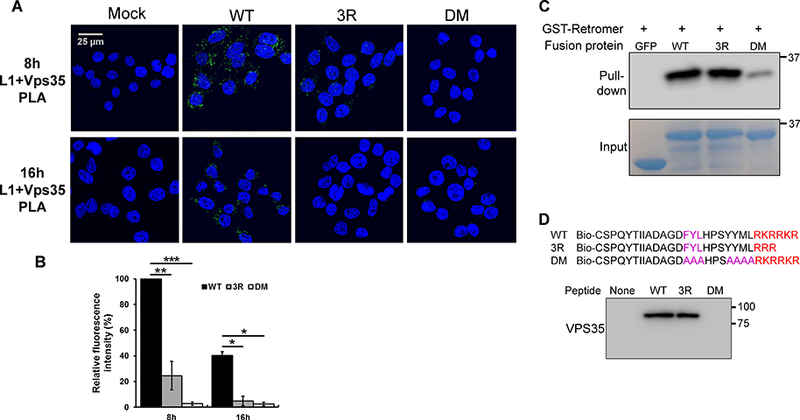
(A) HeLa-sen2 cells were infected as in Figure 4. At eight and 16 hpi, PLA was performed with anti-L1 antibody and an antibody recognizing Vps35 to assess L1 in proximity to retromer. (B) Multiple images obtained as in panel A were processed and displayed as in Figure 4 with the mutant samples normalized to that of cells infected with wild-type PsV at eight hpi. Black bars, wild-type; dark grey bars, 3R mutant; light grey bars, DM mutant. *p<0.05; **p<0.01; ***p<0.001. (C) GST-tagged retromer adsorbed to glutathione beads was incubated with GFP-L2 fusion proteins. After pull-down, proteins bound to retromer were separated by SDS-PAGE and detected by blotting with anti-GFP antibody (top panel). Bottom panel shows input GFP-L2 fusion proteins. (D) Top. Sequences of wild-type and mutant biotinylated peptides. Bottom. Biotinylated peptides were incubated with extracts of uninfected HeLa S3 cells, pulled-down with streptavidin, and separated by SDS-PAGE. Retromer associated with the peptide was detected by blotting for Vps35.
The 3R mutation could inhibit retromer association directly, by impinging on the retromer binding sites, or indirectly, by preventing the exposure of the binding sites in the cytoplasm. To determine if mutations in the L2 CPP directly inhibited binding to retromer, we performed in vitro pull-down experiments. GST-tagged retromer subunits were expressed in bacteria, purified, assembled into the trimeric retromer complex, and bound to glutathione beads. The purified GFP-L2 fusion proteins containing a wild-type L2 segment, the 3R mutation, or the DM mutations in the retromer binding sites were incubated with the retromer beads at 4ºC, pelleted, and subjected to western blotting with an anti-GFP antibody to detect the L2 fusion protein in the pellet. As expected, the wild-type protein bound retromer well and the DM mutant bound poorly (Figure 5C). Notably, the 3R mutant L2 fusion protein also bound retromer well, demonstrating that this mutation did not inhibit the intrinsic ability of L2 to bind retromer. We also incubated biotinylated wild-type and mutant C-terminal L2 peptides with detergent lysates of uninfected HeLa cells and performed streptavidin pull-down experiments and western blotting for Vps35. Similar to the results with purified fusion proteins, the 3R mutation did not affect retromer binding to the L2 peptide, whereas binding was abolished by the retromer binding site mutations (Figure 5D). Therefore, the 3R mutation does not directly interfere with retromer binding, and we conclude that the 3R mutant displays impaired retromer association in infected cells because it cannot protrude through the endosomal membrane and access retromer in the cytoplasm.
Split GFP assay detects CPP-mediated cytoplasmic exposure of the C-terminus of L2
To directly demonstrate membrane passage of the L2 C-terminus during virus entry, we adapted a split GFP assay (Cabantous et al., 2005; Milech et al., 2015). A protein consisting of GFP beta strands 1 to 10 (GFP1–10) does not fluoresce, nor does the 16-residue beta strand 11 of GFP (GFP11). However, when GFP11 is in the same cellular compartment as GFP1–10, GFP is reconstituted, generating a fluorescent signal. This approach was used to demonstrate cytoplasmic delivery of soluble fusion proteins linked to CPPs (Lonn et al., 2016; Milech et al., 2015).
We first constructed a clonal HaCaT cell line expressing GFP1–10 linked at its C-terminus to a nuclear export signal (NES) (Figure S2A). Immunofluorescence experiments with an anti-GFP antibody confirmed cytoplasmic expression of GFP1–10NES (Figure S2B). As expected, cells expressing this construct without GFP11 displayed minimal fluorescence (data not shown; see also Figures S2E, 6B and 6C). We then conducted control experiments to establish that GFP fluorescence in HaCaT/GFP1–10NES cells indicated the presence of GFP11 in the cytoplasm. We used CD8-cation-independent mannose phosphate receptor (CD8-CIMPR) fusion proteins consisting of the extracellular domain of CD8 fused to the TM and cytoplasmic domains of CIMPR (Popa et al., 2015; Seaman, 2007). We inserted seven tandem copies of GFP11 at either the N-terminus or the C-terminus of CD8-CIMPR to generate GFP11-CD8-CIMPR and CD8-CIMPR-GFP11, respectively (Figure S2C). When expressed, these proteins adopt a type 1 TM orientation with the GFP11 segment located in the extracellular/luminal space for GFP11-CD8-CIMPR and in the cytoplasm for CD8-CIMPR-GFP11 (Figure S2D). Neither construct fluoresced on its own when transfected into unmodified HaCaT cells (data not shown). We then transfected HaCaT/GFP1–10NES cells with a plasmid expressing GFP11-CD8-CIMPR or CD8-CIMPR-GFP11 and assessed fluorescence. As shown in Figure S2E, bright cytoplasmic fluorescence was observed after expression of CD8-CIMPR-GFP11 (containing cytoplasmic GFP11) but not after expression of GFP11-CD8-CIMPR. Additional control experiments showed that GFP11-CD8-CIMPR reconstituted fluorescence in cells expressing luminal GFP1–10 (Figures S2C, S2D, and S2F). Taken together, these results validate the split GFP reporter system by showing that fluorescence is reconstituted in HaCaT/GFP1–10NES cells only when the GFP11 segment is in the cytoplasm and establish that GFP1–10NES does not access the luminal space.
We next constructed fusion proteins consisting of seven tandem copies of GFP11 fused to a C-terminal segment of HPV16 L2 extending from 22 residues upstream of the CPP to the end of the basic sequence (Figure 6A). As a control protein, the L2 CPP was replaced with the HIV Tat CPP. Incubation of HaCaT/GFP1–10NES cells with purified GFP11 fusion proteins containing the wild-type L2 or Tat CPP generated a fairly uniform, cytoplasmic, reconstituted GFP signal (Figure 6B). Fluorescence was resistant to trypan blue treatment, showing that it was intracellular. Similarly, when the CPP from HPV16 L2 was replaced by the basic segment from HPV5, HPV18, or HPV31, reconstituted intracellular fluorescence was observed (Figure 6C), whereas GFP11 fusion proteins containing the 3R or 6A mutant HPV16 L2 CPP were markedly impaired for cytoplasmic delivery. These results demonstrate that the L2 CPP from several HPV types could deliver the C-terminus of a fusion protein into the cytoplasm of HaCaT cells.
Figure 6. Cell-penetrating peptides for several HPV types can deliver fusion proteins into the cytoplasm.
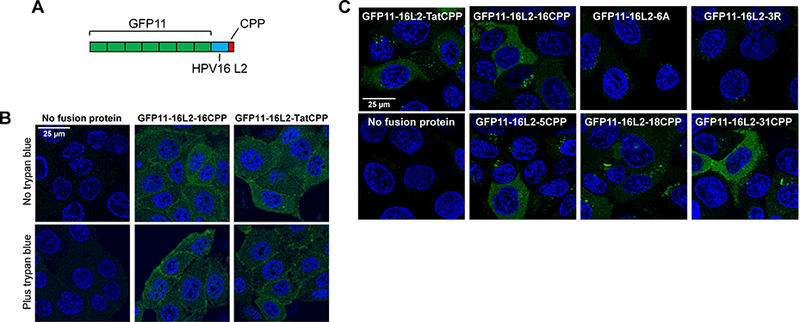
(A) Schematic diagram of GFP11 fusion proteins containing seven tandem copies of GPF11 (green) linked to amino acids 434 to 455 of HPV16 L2 (blue) and a CPP (red). (B) HaCaT/GFP1–10NES cells were mock-incubated or incubated with GF11–16L2–16CPP or GFP11–16L2-TatCPP for three h. In bottom panels, cells were then treated with trypan blue to quench extracellular fluorescence. Cells were examined by confocal microscopy. (C) Cells were treated as in panel B except there was no trypan blue treatment. GFP11 fusion proteins containing the following CCPs were used: HIV Tat, HPV16 (wild-type, 6A, and 3R mutants), HPV5, HPV18, and HPV31. See Figure S2.
HPV CPP drives the C-terminus of L2 into the cytoplasm during infection
The experiments described above showed that fusion proteins containing the HPV L2 CPP could enter the cytoplasm. We next determined whether the C-terminus of L2 carried in the intact virion protruded into the cytoplasm during infection. We inserted seven tandem copies of GFP11 at two different positions in the C-terminus of the HPV16 L2 protein, between the FYL retromer binding site and the CPP (L2-GFP11-CPP) or at the extreme C-terminus of L2 (L2-CPP-GFP11) (Figures 7A and S3A), and tested the activity of PsV stocks containing L2 with the inserted GFP11 segments. Inserted GFP11 did not impair HPV16 PsV infectivity in HeLa cells (Figure S3B). In HaCaT/GFP1–10NES cells, GFP11 had a modest (<50%) inhibitory effect on infectivity, and infection remained sensitive to γ-secretase inhibition (Figure S3C). We then infected HaCaT/GFP1–10NES cells at high MOI with HPV16 PsV with or without GFP11-tagged L2 protein and examined the cells by confocal microscopy. As shown in Figure 7B, fluorescence was not detectable in HaCaT/GFP1–10NES cells infected with wild-type HPV16 PsV lacking the GFP11 insert, confirming that the GFP1–10NES protein did not fluoresce on its own. Similarly, as expected, infection of unmodified HaCaT cells with HPV16 PsV containing GFP11-tagged L2 did not generate a fluorescent signal (data not shown). In contrast, infection of HaCaT/GFP1–10NES cells with PsV containing GFP11 inserted at either C-terminal position in L2 resulted in reconstituted cytoplasmic fluorescence in ~60–90% of cells, demonstrating that the C-terminus of L2 protrudes into the cytoplasm during infection (Figure 7B). Reconstituted fluorescence showed either a punctate distribution or a more uniform distribution throughout the cytoplasm (or both) and was evident at a low level by 1.5 hpi and increased by 3 hpi (Figures 7B and S4A). Notably, reconstituted fluorescence did not display peripheral, cell-surface localization at any timepoint examined.
Figure 7. Reconstitution of split GFP by infection with PsV containing L2-GFP11.
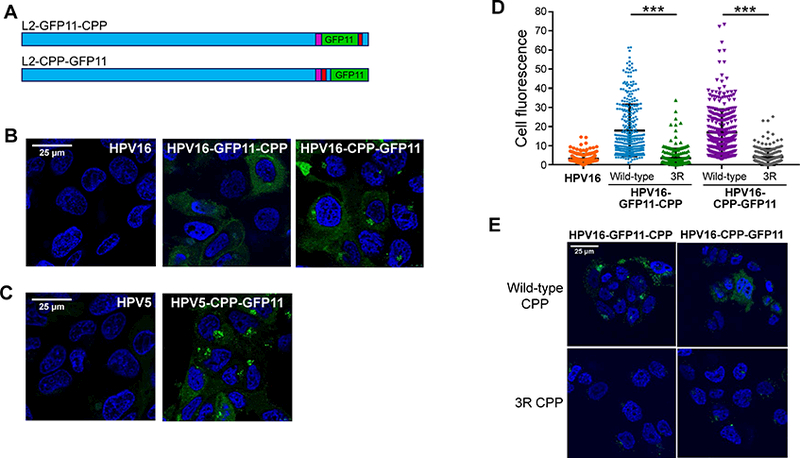
(A) Schematic diagrams of L2 protein with tandem GFP11 (green) inserted upstream of RKRRKR (L2-GFP11-CPP) or at the C-terminus of L2 (L2-CPP-GFP11). The bulk of the L2 protein is shown in blue; CPP in red; and retromer binding sites in purple. See Figure S3A for sequences. (B) Clonal HaCaT/GFP1–10NES cells were infected at MOI of 2000 with untagged HPV16 PsV or with a PsV containing GFP11-tagged L2. Three hpi, cells were examined by confocal microscopy for GFP fluorescence. (C) As in panel (B), except cells were infected with HPV5 PsV containing untagged or GFP11-tagged L2. (D) Fluorescence of cells in images as in panel B was quantified for ~250 cells for each condition. Results show the fluorescence intensity of individual cells plotted in arbitrary units from three independent experiments. ***p<0.0001. (E) Cells were infected as described in panel B with GFP11-tagged PsV containing the wild-type or the 3R mutant CPP. Three hpi, cells were examined by confocal microscopy for GFP fluorescence. See Figures S2 to S5.
We also produced PsV with GFP11 fused to the C-terminus of the L2 protein from HPV5, a β-type HPV that infects skin and is associated with skin cancer. As shown in Figure 7C, reconstituted cytoplasmic GFP was also generated by HPV5-GFP11-CPP PsV infection. Thus, early during infection by two distinct HPV types, the C-terminus of at least a fraction of L2 molecules protruded into the cytoplasm.
To test if the L2 CPP was required for membrane protrusion, PsV were produced in which the wild-type CPP in L2-GFP11-CPP and L2-CPP-GFP11 was replaced with three arginines (Figure S3A). As expected, the 3R mutation did not inhibit PsV internalization but blocked infection (Figures S3B and S4B). In HaCaT/GFP1–10NES cells infected with these mutant PsVs, there was significantly less reconstituted fluorescence compared to cells infected with PsV with wild-type CPP (Figures 7D and 7E). This result provides direct evidence that the L2 CPP is required for cytoplasmic exposure of the L2 C-terminus during infection.
To determine if L2 protrusion required cell cycle progression, we treated cells with aphidocolin, which causes S-phase arrest and blocks HPV infection by inhibiting translocation of the incoming virus into the nucleus late during entry (Calton et al., 2017). As shown in Figure S5, aphidocolin inhibited infection by PsV containing GFP11 but did not decrease the reconstituted GFP signal, showing that cell cycle progression and nuclear membrane breakdown are not required for exposure of the C-terminus of L2 in the cytoplasm during infection.
DISCUSSION
In this paper, we show that a short sequence of basic amino acids near the C-terminus of the HPV L2 protein acts as a CPP to transfer a segment of the L2 protein into the cytoplasm where it binds retromer to initiate retrograde transport of the incoming virion to the TGN. First, we showed that the basic segment of HPV16 L2 is required for efficient infection of epithelial cells and can be replaced with the cationic CPP from HIV Tat. Like wild-type HPV16 PsV, PsV containing the Tat CPP required retromer for infection. Five or more consecutive arginine residues restored full infectivity, whereas fewer arginines or six lysines were less active, consistent with the known cell-penetrating activities of these sequences. We then used peptide and protein transduction assays to demonstrate that the basic segment of HPV16 L2 displayed CPP activity. Importantly, a truncated sequence of three arginines was defective for CPP activity and failed to support infection. The preferential uptake of the 3R peptide compared to the 3R fusion protein may reflect the higher concentration of molecules used in the peptide experiments. Taken together, these experiments demonstrated that L2 CPP activity was required for HPV infection.
HPV16 PsV containing a mutant CPP consisting of three arginines was internalized, showing the L2 CPP was not required for cell binding or endocytosis. However, in contrast to L1-only PsV, which bound cells well, the 6A mutant bound poorly. Thus, cell binding can be modulated by the L2 protein. The simplest explanation for these findings is that L1 is sufficient for binding and that the 6A mutation causes a subtle change in the structure of the capsid that inhibits L1 from binding to its receptor.
The 3R CPP mutant was defective for retromer engagement, exit of PsV from the endosome, and trafficking to the TGN. The same phenotype is caused by mutations in the retromer binding sites or by retromer knockdown. However, the CPP mutation did not directly impair the ability of L2 to bind retromer, implying that during infection the retromer binding sites in the mutant were not in the same cellular compartment as retromer. To directly assay cytoplasmic exposure of L2, we developed a split GFP assay, in which fluorescence is reconstituted when a short segment of GFP at the C-terminus of L2 on incoming PsV encounters GFP1–10 in the cytoplasm. Cytoplasmic exposure of HPV16 and HPV5 L2 was detectable early during infection and was impaired by replacing the CPP with three arginines. These findings show that the L2 CPP mediates passage of the C-terminus of the L2 protein into the cytoplasm where it can engage retromer and enter the retrograde trafficking pathway. The presence of multiple L2 molecules in each virion ensures a high local concentration of CPPs upon infection even at low MOI, which may be important for membrane penetration (Kauffman et al., 2015). The split GFP assay should be useful in investigating chemicals and mutations that inhibit HPV trafficking and in elucidating the molecular mechanism of this unusual process.
In addition to reconstituted fluorescence concentrated in discrete intracellular locations, many cells displayed a uniform signal throughout the cytoplasm (e.g., Figure 7B), suggesting that the C-terminal segment of some L2 molecules are cleaved or released from the virion after protrusion through the membrane or that transport vesicles containing PsV rapidly distribute throughout the cytoplasm. Western blotting of infected HaCaT/GFP1–10NES cells failed to reveal shorter species of wild-type L2 or L2 linked to GFP11 at the time we measure L2 protrusion (Figure S4C). In addition, we recently reported that a small fraction of C-terminally FLAG-tagged L2 is cleaved by γ-secretase by eight hpi, but cleavage is not required for infectivity (Inoue et al., 2018). Taken together, these results indicate that few L2 molecules are cleaved early during infection. The origin and significance of the diffuse, reconstituted GFP signal remains to be determined.
The rapid generation of reconstituted GFP fluorescent signal is consistent with the report that the L2 protein binds to the SNX17 as early as two hpi (Bergant et al., 2017). In addition, by one and a half to three hpi, intracellular L1 is detectable by immunofluorescence (Fig. S4D) and by western blotting (Campos et al., 2012), providing additional evidence that HPV internalization can occur rapidly. The Campos laboratory used a related approach to determine when the L2-vDNA complex translocates across the limiting membrane to access the nucleus (Calton et al., 2017). They fused the ~30 kDa BirA biotin ligase to the C-terminus of L2 and used biotinylation of a cytoplasmic target protein as a biochemical indicator of L2 translocation. Biotinylation required cell cycle progression and was detected only after the virus reached the nucleus and the cells underwent mitosis, beginning approximately 8–10 hpi. In contrast, cytoplasmic exposure of L2 assessed by reconstituted GFP occurred earlier and did not require cell cycle progression. Thus, biotinylation indicates that L2 molecules have been released from retrograde transport vesicles in the terminal stages of trafficking, whereas GFP reconstitution indicates the protrusion of L2 molecules into the cytoplasm to engage the retrograde trafficking machinery.
The physiological role of most CPPs is not known. Naturally-occurring CPPs are usually studied as small peptide fragments removed from their protein of origin, and in many cases cell-penetrating activity may be the fortuitous consequence of a basic amino acid sequence that does not normally penetrate membranes when present in its host protein. Our results show that CPP-driven membrane penetration by L2 plays an important role in HPV infection. We propose that after the L2 CPP of endocytosed virus protrudes through the endosomal membrane into the cytoplasm, most of L2 passes through the membrane. If passage is arrested by its N-terminal TM domain, L2 will adopt a type 1 TM orientation with its N-terminus in the endosomal lumen and most of the protein exposed in the cytoplasm. The exposed C-terminus then binds to essential entry factors including retromer, which sorts the virus into retrograde transport vesicles. It should be pointed out, however, that it is possible that not all L2 molecules insert into membranes or bind cytoplasmic factors.
The effect of the L2 CPP on the membrane is relatively subtle and localized, in contrast to more drastic membrane disruption caused by other non-enveloped viruses, which are deposited into the cytoplasm. This allows the HPV virion to be retained in transport vesicles throughout trafficking, thereby sequestering it from cytoplasmic immune sensors during cell entry. The L2 protein may protrude into the cytoplasm in a sequential fashion, with the C-terminus being exposed prior to the middle of the protein. Thus, cellular proteins may bind L2 sequentially, first retromer to the C-terminus of L2 and later proteins such as SNX17 to the middle portion of L2. Such ordered binding may be important for the assembly of the protein complexes necessary for proper trafficking.
The presence of a C-terminal basic region in all papillomavirus L2 proteins implies that the essential role of the L2 CPP has been maintained since the papillomaviruses emerged more than 250 million years ago. The sequence of the basic segment is variable, consistent with the relatively relaxed sequence requirements for CPPs. The 353 sequenced L2 proteins in the papillomavirus PaVe sequence database (https://pave.niaid.nih.gov/#home) contain 164 different C-terminal basic sequences, including a 10-residue poly-arginine stretch in three canine viruses, and many more CPPs are likely to exist in as-yet-uncharacterized papillomaviruses because most of these different basic sequences have been identified in only a single virus type (Table S1).
Moreover, sequences flanking the core basic amino acids may influence membrane penetration activity. The multitude of extant papillomavirus CPPs thus represents the results of a mutational analysis carried out over evolutionary time, revealing hundreds of presumably non-toxic sequences that should be evaluated for cargo-carrying activity for practical uses. In addition, analysis of these sequences may reveal new aspects of CPP action. For example, the ten most common L2 basic segments terminate in a C-terminal RKR sequence, implying that there is a biological advantage in maintaining RKR at the C-terminus of these L2 CPPs (Table S1 and Figure S4E).
The experiments reported here, together with earlier published reports by us and others [reviewed in (Campos, 2017)], strongly suggest that the L2 protein is an inducible TM protein. In the intact capsid, which lacks membranes, L2 cannot have any TM character. However, once the L2 CPP inserts into the membrane and protrudes into the cytoplasm, the L2 protein appears to adopt a TM existence. Thus, CPPs can not only transport proteins into cells or between compartments, but can also transform a soluble protein into a TM one. Cellular and other viral proteins may also be transferred between intracellular compartments or transition from a soluble to a TM state, with important consequences for their biological activities. Our results suggest that a primary role of CPPs in biology may not be to transport proteins into cells in a paracrine fashion but rather to act intracellularly to mediate the transfer of proteins or protein segments between cellular compartments and to convert soluble proteins into TM proteins.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Daniel DiMaio (daniel.dimaio@yale.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
HeLa S3 cells
HeLa S3 cells, purchased from American Type Culture Collection (ATCC), are a clonal derivative of the parent human female HeLa cell line. Cells were cultured at 37ºC in DMEM (Sigma Aldrich) with HEPES and L-glutamine, supplemented with 10% fetal bovine serum (FBS) and 100 units/mL penicillin streptomycin, in 5% CO2. For infectivity experiments, 1×105 HeLa S3 cells were seeded in 12-well plates and cultured in the same medium for 16 h before infection. For cellular uptake experiments, 5×104 HeLa S3 cells were seeded in eight-chambered glass coverslips and cultured in the same medium overnight. Cell line was authenticated by confirming expression of HPV18 E6/E7 oncogene expression. All cell lines were periodically tested for mycoplasma contamination.
HeLa-sen2 cells
HeLa-sen2 cells, a cloned derivative of the parent human female HeLa cell line, were generated by our lab (Goodwin et al., 2000). Cells were cultured at 37ºC in DMEM (Sigma Aldrich) with HEPES and L-glutamine, supplemented with 10% FBS and 100 units/mL penicillin streptomycin, in 5% CO2. For immunofluorescence and PLA experiments, 5 ×104 cells were seeded on coverslips and cultured in the same medium for 16 h before infection. Cell line was authenticated by confirming expression of HPV18 E6/E7 oncogene expression.
HaCaT cells
HaCaT cells purchased from AddexBio Technologies are spontaneously transformed keratinocytes from human male histologically normal skin. HaCaT cells were cultured at 37ºC in DMEM with HEPES and L-glutamine, supplemented with 10% FBS and 100 units/mL penicillin streptomycin, in 5% CO2. To generate HaCaT/GFP1–10NES stable cell line, 3 ×104 HaCaT cells were seeded in 24-well plates and cultured in the same medium for 16 h before transduction. HaCaT/GFP1–10NES cells stably expressing GFP1–10NES were cultured at 37ºC in DMEM with HEPES and L-glutamine, supplemented with 10% FBS, 100 units/mL penicillin streptomycin, and 1 μg/mL puromycin (Sigma Aldrich), in 5% CO2.
HEK293T/17 cells
HEK293T/17 (293T) cells used for making lentiviruses were purchased from ATCC. 293T is a female human embryonic kidney (HEK) cell line, carries a stably expressed Simian virus 40 (SV40) genome, and expresses large T antigen. Clone 17 was selected specifically for its high transfectability. 293T cells were cultured at 37ºC in DMEM with HEPES and L-glutamine, supplemented with 10% FBS and 100 units/mL penicillin streptomycin, in 5% CO2. For producing lentiviruses, 2×106 cells were seeded in 100 mm dishes and cultured in the same medium overnight. Cell line was authenticated by confirming expression of nuclear SV40 large T antigen.
293TT cells
293TT cells, obtained from Christopher Buck (NIH), were generated by introducing SV40 Large T antigen cDNA into female HEK293T cells to increase Large T antigen expression. 293TT were cultured at 37ºC in DMEM with HEPES and L-glutamine, supplemented with 10% FBS, 100 units/mL penicillin streptomycin, and 250 μg/mL hygromycin, in 5% CO2. To produce PsVs, 8×106 cells were seeded in 150 mm dishes and cultured in the same medium for 16 h before transfection. Cell line was authenticated by confirming expression of nuclear SV40 large T antigen.
METHOD DETAILS
Producing and characterizing pseudovirus
Plasmids used for producing pseudoviruses
293TT cells were used for HPV PsV production with packaging plasmids designated p16sheLL and p5sheLL, expressing both L1 and L2 for complete HPV16 or HPV5 PsV production, or p16L1-GFP for L1-only PsV production (Buck et al., 2005). The HPV16 L2 C-terminal mutants were constructed in p16sheLL by using the Q5 site-directed mutagenesis protocol to design primers, and Phusion High-Fidelity DNA polymerase (New England Biolabs) to amplify plasmids with mutations or known CPPs in the basic segment of the L2 protein: cationic (RKKRRRQRRR), amphipathic (PLSSIFSRIDG), and hydrophobic (AAVLLPVLLAAP), from HIV Tat, hepatitis B virus large surface antigen, and the K-fibroblast growth factor signal peptide, respectively (Lin et al., 1995; Oess and Hildt, 2000; Vives et al., 1997). The L1 and L2 genes in each mutant were sequenced to verify mutations.
Pseudovirus production
HPV PsVs were produced by co-transfecting 293TT cells with pCAG-HcRed (Addgene) and wild-type or mutant p16sheLL, p5sheLL, or p16L1-GFP. The packaged PsVs were purified by density gradient centrifugation in OptiPrep (Axis-shield) (Buck et al., 2005). Encapsidated HcRed genomes were quantified by qPCR as described (Popa et al., 2015). Briefly, purified PsVs were treated with DNase I from RNase-free DNase set (Qiagen) to remove unencapsidated DNA associated with capsids. Reporter plasmid was isolated using a DNeasy Blood and Tissue Kit (Qiagen), and the copy number of encapsidated reporter plasmids was determined by qPCR using primers for the HcRed gene in comparison to a standard curve.
Pseudovirus characterization
To characterize purified pseudovirus, 5 μL of pseudovirus solution was placed on freshly glow-discharged copper grids (formvar/carbon-coated, 200 mesh, Electron Microscopy Services, Hatfield, PA). After 2 min, the grids were rinsed twice with droplets of deionized water, stained by 2% aqueous uranyl acetate for 2 min, and then the excess staining solution was blotted off. The grids were allowed to air dry for 15 min. The samples were examined in a FEI Tecnai F20 transmission electron microscope at 200kV. Images were acquired using a FEI Eagle CCD camera (4Kx4K). Virus stocks containing ~1×107 PsV genomes were also analyzed by SDS-PAGE, followed by staining with Coomassie brilliant blue.
Infectivity
1×105 HeLa S3 or HaCaT cells in 12-well plates were incubated with PsVs at infectious MOI of approximately one as assessed by flow cytometry for reporter gene expression after wild-type infection. The number of packaged reporter plasmids required to achieve this MOI for wild-type PsV in each cell line was quantified by qPCR, and an equivalent number of genomes in mutant PsV were used to infect cells. Approximately five-fold more virus was used to attain the same MOI in HaCaT cells. Cells were analyzed by flow cytometry on a Stratedigm-13 flow cytometer to determine the fraction of cells displaying HcRed fluorescence 48 hpi. To measure dependence on retromer, 1×105 HeLa S3 cells in a 12-well plate were transfected 48 hpi prior to infection with 10 pmol siRNAs targeting Vps29 (Dharmacon) or Vps35 (Dharmacon) by using RNAiMAX (Thermo Fisher) according to manufacturer’s protocol. Cells transfected with RISC-free siRNA were used as a control. Forty-eight hpi, infectivity was measured by flow cytometry for reporter gene expression. To measure dependence on γ-secretase, HeLa S3 cells were pretreated for 1 h with 250 nM γ-secretase inhibitor, compound XXI (Millipore), which was retained in the medium for the duration of the experiment. Cells were infected at MOI of one. Forty-eight hpi, infectivity was measured by flow cytometry for HcRed expression. To measure the effect of aphidicolin, HeLa S3 cells were pretreated for 24 h with 6 μM aphidicolin (Santa Cruz), which was retained in the medium for the duration of the experiment. Cells were infected and analyzed as above.
Cell binding and internalization
Cell binding assays
To measure cell binding by microscopy, 5×104 HeLa-sen2 cells grown on glass coverslips were infected with wild-type PsV at MOI of 20 or mutant PsV containing the same number of encapsidated reporter plasmids. Cells were incubated with PsV at 4°C for 2 h and then washed three times with phosphate-buffered saline (PBS). Cells were fixed for 15 min at room temperature with 4% paraformaldehyde (Electron Microscopy Sciences), washed with PBS and reacted with anti-L1 rabbit polyclonal serum (obtained from J. Schiller, NIH), followed by fluorescently-tagged donkey anti-rabbit secondary antibody (Thermo Fisher). Images were acquired with a Leica SP5 inverted fluorescence microscope.
To measure cell surface binding by flow cytometry, 7.5×105 HeLa-sen2 cells in six-well plates were incubated with wild-type PsV at MOI of 20 or mutant PsV containing the same number of reporter plasmids at 4°C for 2 h and then washed three times with PBS. Cells were harvested by treatment of 0.5 mM EDTA on ice for 15 min. Samples were fixed in ice-cold methanol and stained with rabbit polyclonal anti-L1 antibody (obtained from J. Schiller, NIH), and incubated with corresponding Alexa Fluor secondary antibodies (Thermo Fisher). Fluorescence intensity was assayed on a Stratedigm-13 flow cytometer.
Cell binding was also determined by immunoblotting. 1×105 HeLa-sen2 cells grown on 12-well plates were incubated with wild-type PsV at MOI of 20 or mutant PsV containing the same number of encapsidated reporter plasmids at 4°C for two h and then washed three times with cold PBS. Cells were harvested, and viruses bound to cells were detected by Western blotting with the BD Biosciences anti-L1 antibody.
Internalization assays
To measure internalization, HeLa-sen2 cells were incubated with PsV at MOI of 50 at 4°C for 2 h, followed by extensive washing in PBS to remove loosely bound PsV. Cells were then shifted to 37°C to initiate internalization. After 8 h, samples were fixed by 4% Paraformaldehyde, permeabilized with 1% Saponin (Sigma Aldrich) and reacted with anti-L1 antibody (BD Biosciences), followed by donkey antimouse secondary antibody (Thermo Fisher). Cells were analyzed by a Leica SP5 inverted fluorescence microscope. In the experiment shown in Figure S4D, cells were infected with wild-type HPV16 PsV and stained with the antibody 33L1–7 (obtained from Martin Sapp, LSU), which recognizes L1 after virus is internalized and capsid disassembly has begun (DiGiuseppe et al., 2015).
To measure internalization by flow cytometry, 7.5×105 HeLa-sen2 cells in six-well plates were incubated at 4°C for 2 h with wild-type PsV at MOI of 50 or mutant PsV containing the same number of reporter plasmids and then washed three times with PBS, followed by additional incubation at 37°C for 6 h to initiate internalization. Cells were then harvested by trypsinization to remove PsV on the cell surface. Samples were fixed in ice-cold methanol and stained with anti-L1 antibody (BD Biosciences) and incubated with Alexa Fluor-labeled secondary antibody (Thermo Fisher). Fluorescence intensity was assayed on a Stratedigm-13 flow cytometer.
In all fluorescence imaging experiments, cells were also stained with DAPI or Hoescht 33342 to visualize nuclei (blue). Images were acquired with a Leica SP5 inverted fluorescence microscope, and a single confocal Z-plane is shown in each panel.
Proximity ligation assay
For proximity ligation assay (PLA), HeLa-sen2 cells were infected with wild-type PsV at MOI of 100-to-200 or with mutant PsV containing the same number of encapsidated reporter plasmids. Infected cells were fixed, permeabilized at various times post-infection, and incubated with pairs of antibodies, one recognizing L1 (BD Biosciences) and the other recognizing a cellular marker or a retromer subunit. PLA was performed with Duolink reagents (Sigma Aldrich) according to the manufacturer’s directions as described (Lipovsky et al., 2015). Briefly, cells were incubated in a humidified chamber with a pair of suitable PLA antibody probes diluted 1:5 and processed for ligation and amplification with fluorescent substrate at 37°C. Approximately 200 cells in each sample were imaged with a Leica SP5 inverted fluorescence microscope. The images were processed by Fiji and quantitatively analyzed by BlobFinder software to measure total fluorescence intensity in each sample. The average fluorescence intensity per cell in each sample was normalized to the control sample as indicated in each experiment. Similar results were obtained in three independent experiments for each antibody pair.
Peptide and fusion protein cellular uptake
Preparation of peptides
Peptides were purchased from ABclonal Technology at >95% purity. L2-WT peptide (sequences shown in Figure 2A) was resuspended in 30% acetic acid and DMSO, and then dissolved in sterile deionized water with 0.01% sodium azide. The 3R and 6A peptides were initially solubilized in a small amount of DMSO and then dissolved in sterile deionized water with 0.01% sodium azide. Peptide stocks (~5 mg/mL) were aliquoted and stored at −20°C. Peptides were labell ed by treatment with Alexa Fluor 488 C5 Maleimide (Thermo Fisher) at 1:2 ratio in the presence of TCEP (tris-(2-carboxyethyl)phosphine) (Thermo Fisher) overnight at 4°C, followed by dialysis. The Bradfor d assay was used to determine the concentration of conjugated peptide.
Preparation of fusion proteins
To generate the construct expressing GFP-L2 fusion proteins, DNA encoding the L2 C-terminal segment from wild-type or mutant HPV16 L2 (amino acids 434–461) were inserted into BsrGI and NotI restriction sites in plasmid pET Biotin His6 GFP LIC cloning vector (H6-msfGFP) (Addgene). His6-GFP-L2 fusion proteins were expressed in bacteria and purified using the His GraviTrap column (GE Healthcare). Purified proteins were exchanged into PBS buffer by dialysis and quantified by bicinchoninic acid protein assay.
Cell uptake assays
5×104 293T cells grown on eight-chambered glass coverslips were incubated with 30 μΜ fluorescently-labeled peptides (ABclonal Technology) for 3 h at 37°C or mock-treated. 5×104 HaCaT and HeLa S3 cells grown on eight-chambered glass coverslips were incubated with 2.5 μΜ wild-type or mutant His-tagged GFP-L2 fusion proteins for 3 h at 37°C. After treatment, cells were washed with PBS three times, stained with Hoechst 33342, and examined by confocal microscopy. To observe internalized fusion proteins, cells were treated for 10–15 min with 0.04% trypan blue, washed with PBS, and examined by confocal microscopy. To quantify the cellular fluorescence by flow cytometry, 1×105 HaCaT and HeLa S3 cells grown on 12-well plates were incubated with 2.5 μΜ wild-type or mutant GFP-L2 fusion proteins for 0.5–5 h at 37°C. Cells were washed three times, har vested by trypsinization, and analyzed with a Stratedigm-13 flow cytometer for GFP fluorescence.
Retromer pull-down experiments
Binding to fusion proteins
For fusion protein pull-down experiments, human Vps26, Vps29, and GST-tagged Vps35 subunits were expressed individually in E. coli, and the trimeric retromer complex assembled in vitro was immobilized on GSH resin (GE Health) as described (Harrison et al., 2014). Ten μg wild-type or mutant GFP-L2 fusion protein was incubated with assembled 15 μg retromer trimer immobilized on GSH resin for 2 h at 4°C in 20 mM HEPES pH 8.0, 50 mM NaCl, 5 mM MgCl2, 1 mM DTT, and 0.1% Triton X-100. Beads were centrifuged and washed twice in HEPES buffer, suspended in SDS loading buffer, boiled, and subjected to SDS-PAGE and anti-GFP immunoblotting. Input proteins were subjected to SDS-PAGE and stained with Coomassie brilliant blue.
Binding to peptides
For peptide pull-down experiments, N-terminal biotinylated L2 peptides (sequences shown in Figure 5D) were purchased from ABclonal Technology at >95% purity. Peptides were dissolved and stored as described above. HeLa S3 cells plated in six-well plates were lysed at ~80% confluency with 500 μL RIPA-MOPS buffer (20 mM morpholinepropanesulfonic acid [pH 7.0], 150 mM NaCl, 1% Nonidet P-40, 1 mM EDTA, 1% deoxycholic acid, 0.1% sodium dodecyl sulfate [SDS]) supplemented with 1× HALT protease and phosphatase inhibitor cocktail (Thermo Fisher). The lysate was centrifuged at 14,000 rpm for 20 min, and the supernatant was incubated with 10 μg of a biotinylated peptide for 2 h at 4°C. Fifty μL of streptavidin agarose beads slurry (Thermo Fisher) was added, and the mixture was gently rocked for 45 min at 4°C. Beads were recovered by centrifugati on and washed four times with RIPA-MOPS buffer supplemented with NaCl to a final concentration of 0.4 M. Samples were analyzed by SDS-PAGE and immunoblotting with antibody recognizing Vps35 (Abcam).
Split GFP assay
Plasmid expressing GFP1–10NES
The full-length GFP gene in pLenti CMV GFP Puro (658–5) contained on a BamHI and SalI fragment was replaced with the DNA segment encoding GFP1–10 amplified from pCMV-mGFP1–10 (Van Engelenburg and Palmer, 2010), a gift from Amy Palmer (University of Colorado) to generate pLenti CMV.GFP1–10. To construct pLentiCMV GFP1–10NES, oligonucleotides encoding the nuclear export signal (NES) sequence LPPLERLTLD (Fischer et al., 1995) were inserted in-frame at the C-terminus of GFP1–10 in pLentiCMV GFP1–10.
Plasmids used to validate split GFP assay
Plasmid pCD8-CIMPR expresses a CD8-CIMPR fusion protein containing the extracellular domain of CD8 fused to the TM and mutant cytoplasmic domains of the cation-independent mannose phosphate receptor (CIMPR), which contains an endocytosis motif and three alanines replacing the WLM retromer binding site (Popa et al., 2015; Seaman, 2007). The In-Fusion HD cloning Kit (Clontech) was used to insert seven tandem copies of DNA encoding GFP11 (RDHMVLHEYVNAAGIT) in-frame into pCD8-CIMPR either immediately downstream of the CD8 signal peptide sequence or at the C-terminus of the fusion protein to generate pGFP11-CD8-CIMPR and pCD8-CIMPR-GFP11, respectively. Each GFP11 repeat was separated by GGSGG linker sequences. The sequences of the relevant portions of the encoded CD8-CIMPR fusion proteins are listed in Table S2, with GFP11 sequences highlighted in green. Plasmids CNX-S1–10(N) and CNX-S1–10(C) were obtained from Bernard Moss (NIH) and designated here GFP1–10-CNX and CNX-GFP1–10, respectively. GFP11 fusion proteins were expressed in E.coil.
Plasmids expressing GFP11 fusion proteins containing HPV16 L2 C-terminal segment with various CPPs were generated by insertion of the DNA segment encoding seven tandem copies of GFP11, a segment of HPV16 L2, and the basic region from HIV Tat or various HPV types into BsrGI and NotI restriction sites in plasmid pET Biotin His6 GFP LIC cloning vector (H6-msfGFP) (Addgene). GFP11 fusion proteins were purified using Ni-NTA agarose (~0.2 ml per L of bacteria) (Qiagen). The purification process was monitored by SDS-PAGE gel electrophoresis.
Plasmids to produce GFP11-tagged PsV
To construct p16sheLL-GFP11-CPP for producing PsVs containing the GFP11 inserted into the HPV16 L2 protein immediately upstream of the basic segment, DNA encoding the tandem GFP11 repeats was synthesized and cloned in-frame into L2 in p16sheLL by using the In-Fusion HD cloning Kit (Clontech). p16sheLL-CPP-GFP11 and p5sheLL-CPP-GFP11 with the GFP11 repeats at the extreme C-terminus of L2 was constructed by first inserting Nsil and AvrlI restriction sites immediately upstream of the L2 stop codon in p16sheLL and p5sheLL, respectively. The DNA segment encoding the GFP11 repeats was then inserted into this plasmid between these sites. Site-directed mutagenesis of p16sheLL-GFP11-CPP and p16sheLL-CPP-GFP11 was used to construct p16sheLL-GFP11–3R and p16sheLL-3R-GFP11, respectively, both of which contain three arginines in place of the wild-type CPP. The sequences of the relevant portions of the encoded L2-GFP11 fusion proteins with wild-type CPP are in Table S2.
Generating cells expressing GFP1–10NES
To generate stable cell lines expressing cytoplasmic GFP1–10NES, lentiviruses were produced by cotransfecting 293T cells in 100 mm dishes with 6 μg of pLentiCMV GFP1–10NES, 4.5 μg of lentiviral packaging plasmid psPAX2 and 1.5 μg envelope plasmid pMD2.G. Forty-eight h later, the lentiviral supernatant was harvested, filtered and stored at −80°C for later use. Cells stably expressing GFP1– 10NES were constructed by infecting HaCaT cells with lentivirus and selecting for 48 h in medium containing 1 μg/mL puromycin. Single cells were then plated in a 96-well plate, and monoclonal cell lines were isolated. Cells were used for approximately one month and discarded. To test the expression of GFP1–10NES, 5×104 clonal HaCaT/GFP1–10NES stable cells grown on glass coverslips were fixed for 15 min at room temperature with 4% Paraformaldehyde (Electron Microscopy Sciences), permeabilized with 1% Saponin (Sigma Aldrich) for 1 h, and incubated with anti-GFP mouse antibody (Santa Cruz) and Alexa Fluor 488 donkey anti-mouse IgG (H&L) secondary antibody (Thermo Fisher). Images were acquired with a Leica SP5 inverted fluorescence microscope.
Validation of split GFP assay
The split GFP assay was validated by fluorescence microscopy. 5×104 clonal HaCaT/GFP1–10NES cells in eight-chambered glass slides were transfected with 0.25 μg of pGFP11-CD8-CIMPR or pCD8-CIMPR-GFP11. Twenty-four h later, live cells were examined by fluorescence microscopy. To demonstrate the functionality of GFP11 in GFP11-CD8-CIMPR, 5×104 293T cells in eight-chambered glass slides were cotransfected with pGFP11-CD8-CIMPR and a plasmid expressing a calnexin TM fusion protein containing luminal GPF1–10 or cytoplasmic GFP1–10 (GFP1–10-CNX and CNX-GFP1–10, respectively) (Figures S2C and S2D) (Hyun et al., 2015). Twenty-four h later, live cells were examined by fluorescence microscopy.
To test if the L2 CPP from different HPV types could deliver the C-terminus of a fusion protein into the cytoplasm by using the split GFP assay, 5×104 clonal HaCaT/GFP1–10NES cells in eight-chambered glass slides were incubated with 15 μg of GFP11 fusion proteins for 3 h at 37°C. After t reatment, cells were washed with PBS three times, stained with Hoechst 33342, and examined by a Leica SP5 inverted fluorescence microscope. Some samples were treated with trypan blue.
Determining the protrusion of L2 C terminus
To demonstrate cytoplasmic protrusion of L2 of PsV by using the split GFP assay, 3×104 clonal HaCaT/GFP1–10NES cells seeded in eight-chambered glass slides were incubated for 1.5 or 3 h at MOI of ~2000 with HPV16 or HPV5 PsV containing wild-type L2 or L2 containing inserted GFP11 and a wildtype or 3R mutant L2 CPP. Live cells were analyzed by a Leica SP5 inverted fluorescence microscope and processed with Fiji. In the experiment shown in Figure S5, cells were pretreated with 6 μΜ aphidocolin (Santa Cruz), which was left in the medium for the duration of the experiment. To determine whether L2 with or without GFP11 is cleaved during infection, 3×105 clonal HaCaT/GFP1–10NES cells seeded in 12-well plates were incubated at MOI of ~1,000 with HPV16 PsV containing wild-type L2 or L2 containing inserted GFP11. After 1.5, 3, or 6 h., cells were washed once with PBS at pH 7.2, once with PBS at pH 10.75 to remove surface-bound virions, and twice with PBS at pH 7.2, harvested by trypsinization, and lysed in 80 μL sample buffer (2% SDS, 100 mM DTT, 150 mM Tris-HCL, 10% Glycerol, 0.02% Bromophenol Blue). Cell lysates (20 μL) were subjected to SDS-PAGE and anti-L2 immunoblotting with anti-L2 antibody (Santa Cruz), which recognizes residues 40–150 of L2. Membrane was stripped and reprobed for GAPDH as a loading control.
Statistical analysis
Data in figure panels reflect three or more independent experiments performed on different days. In all figures, data are presented as mean ± SD. Statistical tests and specific p-values used are indicated in the figure legends.
QUANTITATION AND STATISTICAL ANALYSIS
Quantitation of PLA assay
To quantify PLA signals, BlobFinder was used to perform a single cell analysis, which assigns each signal to the closest cell and quantifies the signal intensity per cell for each cell in the image (Allalou and Wahlby, 2009). The PLA fluorescence signal intensity of ~200 cells for each experiment was averaged. The average intensity for EEA1/L1 and VPS35/L1 samples was normalized to cells infected with wild-type PsV at 8 hpi to yield the relative fluorescence intensity; the average intensity for TGN46/L1 samples was normalized to cells infected with wild-type PsV at 16 hpi. The relative fluorescence intensity of three individual experiments were averaged and plotted.
Quantitation of split GFP assay
Quantitation of reconstituted GFP in HaCaT/GFP1–10NES cell line was performed using Fiji software. An outline was drawn around each cell, area, integrated density, and mean fluorescence measured, along with several adjacent background readings. The corrected total cellular fluorescence (CTCF) = Integrated density - (Area of selected cell × Mean fluorescence of background readings), was calculated (Burgess et al., 2010). CTCF of ~200–300 cells were quantified and plotted using GraphPad Prism 7. Statistical analysis (student t tests) were performed.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-L1 Protein of Human Papilloma Virus clone CAMVIR-1 (RUO) |
BD Biosciences | Cat#554171; RRID:AB_395284 |
| Mouse monoclonal anti-GFP (B-2) | Santa Cruz Biotechnology | Cat#sc-9996; RRID:AB_627695 |
| Rabbit polyclonal antibody anti-TGN46 | Abcam | Cat#ab50595; RRID:AB_2203289 |
| Rabbit monoclonal anti-VPS35 [EPR11501(B)] | Abcam | Cat#ab157220; RRID:AB_2636885 |
| Mouse monoclonal anti-VPS35 | Abcam | Cat#ab57632; RRID:AB_946126 |
| Rabbit polyclonal anti-L1 | J. Schiller, NIH | N/A |
| Mouse antibody 33L1–7 | Martin Sapp, LSU | N/A |
| Rabbit monoclonal antibody EEA1 (C45B10) | Cell Signaling Technology | Cat#3288; RRID:AB_2096811 |
| Mouse monoclonal anti-GAPDH (6C5) | Santa Cruz Biotechnology | Cat#sc-32233; RRID:AB_627679 |
| Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fisher | Cat#A-21202; RRID:AB_141607 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fisher | Cat#A-21206; RRID:AB_2535792 |
| Bacterial and Virus Strains | ||
| One Shot Stbl3 Chemically Competent E. coli | Thermo Fisher | Cat#C737303 |
| One Shot TOP10 Chemically Competent E. coli | Thermo Fisher | Cat#C404010 |
| Rosetta 2(DE3) Competent Cells-Novagen | Millipore | Cat#71397 |
| p16sheLL | (Buck et al., 2008) | Cat#37320; Addgene |
| p5sheLL | (Buck et al., 2008) | Cat#46953; Addgene |
| p16L1-GFP | (Buck et al., 2005) | Cat#89910; Addgene |
| Chemicals, Peptides, and Recombinant Proteins | ||
| γ-Secretase Inhibitor XXI, Compound E | Millipore | Cat#565790; CAS:209986–17-4 |
| Aphidicolin | Santa Cruz | Cat#sc-201535; CAS:38966–21-1 |
| Protein A/G PLUS-Agarose | Santa Cruz | Cat#sc-2003 |
| Paraformaldehyde | Electron Microscopy Sciences | Cat#15710; CAS:30525–89-4 |
| Saponin | Sigma Aldrich | Cat#47036; CAS:8047–15-2 |
| Hoechst33324, Trihydrochloride, Trihydrate | Thermo Fisher | Cat#H3570 |
| Trypan Blue | Sigma Aldrich | Cat#T6146; CAS:72–57-1 |
| Puromycin dihydrochloride | Sigma Aldrich | Cat#P7255; CAS:58–58-2 |
| OptiPrep | Axis-Shield | Cat#AXS-1114542 |
| Halt Protease and Phosphatase Inhibitor Single-Use Cocktail, EDTA-Free | Thermo Fisher | Cat#78443 |
| Alexa Fluor 488 C5 Maleimide | Thermo Fisher | Cat#A10254 |
| MOPS (morpholinepropanesulfonic acid) | AmericanBio | Cat#AB01270–00100; CAS:1132–61-2 |
| Nonidet P-40 | Sigma Aldrich | Cat#74385; CAS:9016–45-9 |
| SDS (20%) | AmericanBio | Cat#AB01922–00500 |
| EDTA, 0.5 M, pH 8.0 | AmericanBio | Cat#AB00502–01000 |
| Desoxycholic acid sodium salt | Sigma Aldrich | Cat#D6750; CAS:302–95-4 |
| Lipofectamine® RNAiMAX Transfection Reagent | Thermo Fisher | Cat#13778100 |
| Premium Grade TCEP-HCl | Thermo Fisher | Cat#PG82080 |
| RNase A, DNase and protease-free | Thermo Fisher | Cat#EN0531 |
| Streptavidin Agarose | Thermo Fisher | Cat#20349 |
| Glutathione Sepharose | GE Healthcare | Cat#17075601 |
| His GraviTrap Columns | GE Healthcare | Cat#45–000-006 |
| Ni-NTA Agarose | Qiagen | Cat#30210 |
| Phusion High-Fidelity DNA Polymerase | New England Biolabs | Cat#M0530L |
| Q5 Site-Directed Mutagenesis Kit | New England Biolabs | Cat#E0554S |
| T4 DNA Ligase | New England Biolabs | Cat#M0202L |
| PCR Nucleotide Mix | Sigma Aldrich | Cat# 11581295001 |
| Dulbecco’s Modified Eagle’s Medium - high glucose | Sigma Aldrich | Cat#D5671 |
| Fetal Bovine Serum | Atlanta Biologicals | Cat#S11150 |
| L-Glutamine | Thermo Fisher | Cat#25030081 |
| Pen Strep (Penicillin Streptomycin) | Thermo Fisher | Cat#15140122 |
| Trypsin-EDTA (0.25%) | Thermo Fisher | Cat#25200056 |
| Dulbecco’s Phosphate Buffered Saline (DPBS), with MgCl2 and CaCl2 |
Sigma Aldrich | Cat#D8662 |
| B-PER™ Bacterial Protein Extraction Reagent | Thermo Fisher | Cat#78243 |
| Lysozyme | Thermo Fisher | Cat#90082 |
| DNAse I solution | Thermo Fisher | Cat#90083 |
| CSPQYTIIADAGDFYLHPSYYMLRKRRKR | ABclonal Technology | N/A |
| CSPQYTIIADAGDFYLHPSYYMLAAAAAA | ABclonal Technology | N/A |
| CSPQYTIIADAGDFYLHPSYYMLRRR | ABclonal Technology | N/A |
| Biotin-SPQYTIIADAGDFYLHPSYYMLRKRRKR | ABclonal Technology | N/A |
| Biotin-SPQYTIIADAGDFYLHPSYYMLAAAAAA | ABclonal Technology | N/A |
| Biotin-SPQYTIIADAGDFYLHPSYYMLRRR | ABclonal Technology | N/A |
| Biotin-SPQYTIIADAGDAAAHPSAAAARKRRKR | ABclonal Technology | N/A |
| GFP-L2-WT | This paper | N/A |
| GFP-L2–6A | This paper | N/A |
| GFP-L2–3R | This paper | N/A |
| GFP11–16L2–16CPP | This paper | N/A |
| GFP11–16L2-TatCPP | This paper | N/A |
| GFP11–16L2–6A | This paper | N/A |
| GFP11–16L2–3R | This paper | N/A |
| GFP11–16L2–5CPP | This paper | N/A |
| GFP11–16L2–18CPP | This paper | N/A |
| GFP11–16L2–31CPP | This paper | N/A |
| Critical Commercial Assays | ||
| Duolink® In Situ PLA Probe Anti-Mouse PLUS | Sigma-Aldrich | Cat#DUO92001 |
| Duolink® In Situ PLA Probe Anti-Rabbit MINUS | Sigma-Aldrich | Cat#DUO92005 |
| Duolink® In Situ Detection Reagents Green | Sigma-Aldrich | Cat#DUO92014 |
| In-Fusion HD cloning Kits | Clontech | Cat#638909 |
| RNase-Free DNase Set | Qiagen | Cat#79254 |
| DNeasy Blood and Tissue Kits | Qiagen | Cat#69504 |
| iQ™SYBR Green Supermix | Bio-rad | Cat#1708880 |
| BCA Protein Assay Kit | Thermo Fisher | Cat#23225 |
| Experimental Models: Cell Lines | ||
| Human: HaCaT | AddexBio Technologies |
Cat#T0020001 |
| Human: HeLa-sen2 | (Goodwin et al., 2000) | N/A |
| Human: HeLa S3 | ATCC | Cat#CCL-2.2; RRID:CVCL 0058 |
| Human: HaCaT/GFP1–10NES | This paper | N/A |
| Human: 293TT | (Buck et al., 2005) | RRID:CVCL_1D85 |
| Human: HEK293T/17 | ATCC | Cat#CRL-11268; RRID:CVCL_1926 |
| Oligonucleotides | ||
| siGENOME Human VPS29 siRNA | Dharmacon | Cat# D-009764–03 |
| siGENOME Human VPS35 siRNA | Dharmacon | Cat# D-010894–01 |
| siGENOME RISC-Free Control | Dharmacon | Cat# D-001220–01 |
| HcRed Forward primer: GCACCCAGAGCATGAGAAT | This paper | N/A |
| HcRed Reverse primer: TCGTAGGTGGTGGTTCTCT | This paper | N/A |
| Recombinant DNA | ||
| p16sheLL-CPP-GFP11 | This paper | N/A |
| p16sheLL-GFP11-CPP | This paper | N/A |
| p16sheLL-3R-GFP11 | This paper | N/A |
| p16sheLL-GFP11–3R | This paper | N/A |
| p16sheLL.W | This paper | N/A |
| p16sheLL.6A | This paper | N/A |
| p16sheLL.3R | This paper | N/A |
| p16sheLL.4R | This paper | N/A |
| p16sheLL.5R | This paper | N/A |
| p16sheLL.6R | This paper | N/A |
| p16sheLL.7R | This paper | N/A |
| p16sheLL.Amphipathic | This paper | N/A |
| p16sheLL.Hydrophobic | This paper | N/A |
| p5sheLL-CPP-GFP11 | This paper | N/A |
| pCAG-HcRed | (Matsuda and Cepko, 2004) | Cat#11152; Addgene |
| pET Biotin His6 GFP LIC cloning vector (H6-msfGFP) | Scott Gradia, UC Berkeley |
Cat#29725; Addgene |
| pH6-msfGFP-WT | This paper | N/A |
| pH6-msfGFP-6A | This paper | N/A |
| pH6-msfGFP-3R | This paper | N/A |
| pQlinkG GST-TEV-Vps35 | (Harrison et al., 2014) | N/A |
| pET Vps29 | (Harrison et al., 2014) | N/A |
| pET Vps26 | (Harrison et al., 2014) | N/A |
| pLenti CMV GFP Puro (658–5) | (Campeau et al., 2009) |
Cat#17448; Addgene |
| pLentiCMV GFP1–10 | This paper | N/A |
| pLentiCMV GFP1–10NES | This paper | N/A |
| pCMV-mGFP1–10 | (Van Engelenburg and Palmer, 2010) | N/A |
| pCD8-CIMPR | (Popa et al., 2015; Seaman, 2007) | N/A |
| pGFP11-CD8-CIMPR | This paper | N/A |
| pCD8-CIMPR-GFP11 | This paper | N/A |
| pCNX-S1–10(N) | (Hyun et al., 2015) | N/A |
| pCNX-S1–10(C) | (Hyun et al., 2015) | N/A |
| psPAX2 | Didier Trono, EPFL | Cat#12260; Addgene |
| pMD2.G | Didier Trono, EPFL | Cat#12259; Addgene |
| pGFP11–16L2–16CPP | This paper | N/A |
| pGFP11–16L2-TatCPP | This paper | N/A |
| pGFP11–16L2–6A | This paper | N/A |
| pGFP11–16L2–3R | This paper | N/A |
| pGFP11–16L2–5CPP | This paper | N/A |
| pGFP11–16L2–18CPP | This paper | N/A |
| pGFP11–16L2–31CPP | This paper | N/A |
| Software and Algorithms | ||
| FlowJo | FLOWJO, LLC |
https://www.flowio.com/solutions/flowjo RRID:SCR_008520 |
| Fiji | National Institutes of Health, USA |
http://fiii.sc RRID:SCR_002285 |
| GraphPad Prism | GraphPad Software |
https://www.graphpad. com/ RRID:SCR_002798 |
| BlobFinder | The Centre for Image Analysis at Uppsala University |
http://www.cb.uu.se/~ amin/BlobFinder/ RRID:SCR_015788 |
| Other | ||
| Thinwall Polypropylene Tubes | Beckman Coulter | Cat#326819 |
| μ-Slide 8 Well | ibidi | Cat#80826 |
| Slide-A-Lyzer™ Dialysis Cassettes, gamma-irradiated, 10,000 MWCO | Thermo Fisher | Cat#66453 |
| Slide-A-Lyzer™ G2 Dialysis Cassettes, 2K MWCO, 0.5 mL | Thermo Fisher | Cat#87717 |
Supplementary Material
Highlights.
Cell-penetrating peptide transfers HPV L2 capsid protein across membranes
Reveals how a non-enveloped virus in the endosome can bind to a cytoplasmic factor
Intracellular biological role of a CPP in its natural context is defined
The HPV virome contains hundreds of natural sequences with presumed CPP activity
Acknowledgments
We thank Bernard Moss, John Schiller, Martin Sapp, and Amy Palmer for reagents, and Priti Kumar and Donald Engelman for helpful discussions. We thank Jan Zulkeski for assistance in preparing this manuscript. This work was supported by grants from the National Institutes of Health to D.D. (AI102876 and CA016038).
Footnotes
Declaration of Interests
P.Z., G.M.D.S., and D.D. are inventors on a provisional patent application related to this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allalou A, and Wahlby C (2009). BlobFinder, a tool for fluorescence microscopy image cytometry. Comput Meth Progr Biomed 94, 58–65. [DOI] [PubMed] [Google Scholar]
- Bergant M, Peternel S, Pim D, Broniarczyk J, and Banks L (2017). Characterizing the spatio-temporal role of sorting nexin 17 in human papillomavirus trafficking. J Gen Virol 98, 715–725. [DOI] [PubMed] [Google Scholar]
- Bergant Marusic M, Ozbun MA, Campos SK, Myers MP, and Banks L (2012). Human papillomavirus L2 facilitates viral escape from late endosomes via sorting nexin 17. Traffic 13, 455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronnimann MP, Chapman JA, Park CK, and Campos SK (2013). A transmembrane domain and GxxxG motifs within L2 are essential for papillomavirus infection. J Virol 87, 464–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck CB, Cheng N, Thompson CD, Lowy DR, Steven AC, Schiller JT, and Trus BL (2008). Arrangement of L2 within the papillomavirus capsid. J Virol 82, 5190–5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck CB, Pastrana DV, Lowy DR, and Schiller JT (2005). Generation of HPV pseudovirions using transfection and their use in neutralization assays. Methods Mol Med 119, 445–462. [DOI] [PubMed] [Google Scholar]
- Burd C, and Cullen PJ (2014). Retromer: a master conductor of endosome sorting. Cold Spring Harb Perspect Biol 6, pii: a016774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess A, Vigneron S, Brioudes E, Labbe JC, Lorca T, and Castro A (2010). Loss of human Greatwall results in G2 arrest and multiple mitotic defects due to deregulation of the cyclin B-Cdc2/PP2A balance. Proc Natl Acad Sci USA 107, 12564–12569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabantous S, Terwilliger TC, and Waldo GS (2005). Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat Biotechnol 23, 102–107. [DOI] [PubMed] [Google Scholar]
- Calton CM, Bronnimann MP, Manson AR, Li S, Chapman JA, Suarez-Berumen M, Williamson TR, Molugu SK, Bernal RA, and Campos SK (2017). Translocation of the papillomavirus L2/vDNA complex across the limiting membrane requires the onset of mitosis. PLoS Pathog 13, e1006200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO, Campisi J, Yaswen P, Cooper PK, and Kaufman PD (2009). A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS One 4, e6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos SK (2017). Subcellular trafficking of the papillomavirus genome during initial infection: the remarkable abilities of minor capsid protein L2. 10.20944/preprints201711.0007.v1. Viruses 9. [DOI] [PMC free article] [PubMed]
- Campos SK, Chapman JA, Deymier MJ, Bronnimann MP, and Ozbun MA (2012). Opposing effects of bacitracin on human papillomavirus type 16 infection: enhancement of binding and entry and inhibition of endosomal penetration. J Virol 86, 4169–4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerqueira C, Samperio Ventayol P, Vogeley C, and Schelhaas M (2015). Kallikrein-8 proteolytically processes human papillomaviruses in the extracellular space to facilitate entry into host cells. J Virol 89, 7038–7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darshan MS, Lucchi J, Harding E, and Moroianu J (2004). The L2 minor capsid protein of human papillomavirus type 16 interacts with a network of nuclear import receptors. J Virol 78, 12179–12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day PM, Thompson CD, Schowalter RM, Lowy DR, and Schiller JT (2013). Identification of a role for the trans-Golgi network in human papillomavirus 16 pseudovirus infection. J Virol 87, 3862–3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGiuseppe S, Bienkowska-Haba M, Hilbig L, and Sapp M (2014). The nuclear retention signal of HPV16 L2 protein is essential for incoming viral genome to transverse the trans-Golgi network. Virology 458–459, 93–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGiuseppe S, Keiffer TR, Bienkowska-Haba M, Luszczek W, Guion LG, Muller M, and Sapp M (2015). Topography of the human papillomavirus minor capsid protein L2 during vesicular trafficking of infectious entry. J Virol 89, 10442–10452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGiuseppe S, Luszczek W, Keiffer TR, Bienkowska-Haba M, Guion LG, and Sapp MJ (2016). Incoming human papillomavirus type 16 genome resides in a vesicular compartment throughout mitosis. Proc Natl Acad Sci USA 113, 6289–6294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust TB, Binning JM, Gross JD, and Frankel AD (2017). Making sense of multifunctional proteins: human immunodeficiency virus type 1 accessory and regulatory proteins and connections to transcription. Annu Rev Virol 4, 241–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer U, Huber J, Boelens WC, Mattaj IW, and Luhrmann R (1995). The HIV-1 Rev activation domain is a nuclear export signal that accesses an export pathway used by specific cellular RNAs. Cell 82, 475–483. [DOI] [PubMed] [Google Scholar]
- Goodwin EC, Yang E, Lee CJ, Lee HW, DiMaio D, and Hwang ES (2000). Rapid induction of senescence in human cervical carcinoma cells. Proc Natl Acad Sci USA 97, 10978–10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidotti G, Brambilla L, and Rossi D (2017). Cell-penetrating peptides: from basic research to clinics. Trends Pharmacol Sci 38, 406–424. [DOI] [PubMed] [Google Scholar]
- Harrison MS, Hung CS, Liu TT, Christiano R, Walther TC, and Burd CG (2014). A mechanism for retromer endosomal coat complex assembly with cargo. Proc Natl Acad Sci USA 111, 267–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun SI, Maruri-Avidal L, and Moss B (2015). Topology of endoplasmic reticulum-associated cellular and viral proteins determined with split-GFP. Traffic 16, 787–795. [DOI] [PubMed] [Google Scholar]
- Inoue T, Zhang P, Zhang W, Goodner-Bingham K, DiMaio D, and Tsai B (2018). gamma-secretase promotes membrane insertion of the human papillomavirus L2 capsid protein during virus infection. J Cell Biol doi.org/10.1083/jcb.201804171, July 13, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce JG, Tung JS, Przysiecki CT, Cook JC, Lehman ED, Sands JA, Jansen KU, and Keller PM (1999). The L1 major capsid protein of human papillomavirus type 11 recombinant virus-like particles interacts with heparin and cell-surface glycosaminoglycans on human keratinocytes. J Biol Chem 274, 5810–5822. [DOI] [PubMed] [Google Scholar]
- Kamper N, Day PM, Nowak T, Selinka HC, Florin L, Bolscher J, Hilbig L, Schiller JT, and Sapp M (2006). A membrane-destabilizing peptide in capsid protein L2 is required for egress of papillomavirus genomes from endosomes. J Virol 80, 759–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffman WB, Fuselier T, He J, and Wimley WC (2015). Mechanism matters: a taxonomy of cell penetrating peptides. Trends Biochem Sci 40, 749–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar CS, Dey D, Ghosh S, and Banerjee M (2017). Breach: host nembrane penetration and entry by nonenveloped viruses. Trends Microbiol pii: S0966–842X(17)30215–9. [DOI] [PubMed] [Google Scholar]
- Lee JE, and Lim HJ (2012). LDP12, a novel cell-permeable peptide derived from L1 capsid protein of the human papillomavirus. Mol Biol Rep 39, 1079–1086. [DOI] [PubMed] [Google Scholar]
- Lin YZ, Yao SY, Veach RA, Torgerson TR, and Hawiger J (1995). Inhibition of nuclear translocation of transcription factor NF-kappa B by a synthetic peptide containing a cell membrane-permeable motif and nuclear localization sequence. J Biol Chem 270, 14255–14258. [DOI] [PubMed] [Google Scholar]
- Lipovsky A, Popa A, Pimienta G, Wyler M, Bhan A, Kuruvilla L, Guie MA, Poffenberger AC, Nelson CD, Atwood WJ, et al. (2013). Genome-wide siRNA screen identifies the retromer as a cellular entry factor for human papillomavirus. Proc Natl Acad Sci USA 110, 7452–7457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipovsky A, Zhang W, Iwasaki A, and DiMaio D (2015). Application of the proximity-dependent assay and fluorescence imaging approaches to study viral entry pathways MiMB: Membrane Trafficking (BLTang, ed) 1270, 437–451. [DOI] [PubMed] [Google Scholar]
- Lonn P, Kacsinta AD, Cui XS, Hamil AS, Kaulich M, Gogoi K, and Dowdy SF (2016). Enhancing endosomal escape for intracellular delivery of macromolecular biologic therapeutics. Sci Rep 6, 32301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda T, and Cepko CL (2004). Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proc Natl Acad Sci USA 101, 16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milech N, Longville BA, Cunningham PT, Scobie MN, Bogdawa HM, Winslow S, Anastasas M, Connor T, Ong F, Stone SR, et al. (2015). GFP-complementation assay to detect functional CPP and protein delivery into living cells. Sci Rep 5, 18329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oess S, and Hildt E (2000). Novel cell permeable motif derived from the PreS2-domain of hepatitis-B virus surface antigens. Gene Ther 7, 750–758. [DOI] [PubMed] [Google Scholar]
- Pim D, Broniarczyk J, Bergant M, Playford MP, and Banks L (2015). A novel PDZ domain interaction mediates the binding between human papillomavirus 16 L2 and sorting nexin 27 and modulates virion trafficking. J Virol 89, 10145–10155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popa A, Zhang W, Harrison MS, Goodner K, Kazakov T, Goodwin EC, Lipovsky A, Burd CG, and DiMaio D (2015). Direct binding of retromer to human papillomavirus type 16 minor capsid protein L2 mediates endosome exit during infection. PLoS Pathog 11, e1004699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff AB, Woodham AW, Raff LM, Skeate JG, Yan L, Da Silva DM, Schelhaas M, and Kast WM (2013). The evolving field of human papillomavirus receptor research: a review of binding and entry. J Virol 87, 6062–6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards RM, Lowy DR, Schiller JT, and Day PM (2006). Cleavage of the papillomavirus minor capsid protein, L2, at a furin consensus site is necessary for infection. Proc Natl Acad Sci USA 103, 1522–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schelhaas M, Shah B, Holzer M, Blattmann P, Kuhling L, Day PM, Schiller JT, and Helenius A (2012). Entry of human papillomavirus type 16 by actin-dependent, clathrin- and lipid raft-independent endocytosis. PLoS Pathog 8, e1002657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffman M, Doorbar J, Wentzensen N, de Sanjose S, Fakhry C, Monk BJ, Stanley MA, and Franceschi S (2016). Carcinogenic human papillomavirus infection. Nat Rev Dis Primers 2, 16086. [DOI] [PubMed] [Google Scholar]
- Seaman MNJ (2007). Identification of a novel conserved sorting motif required for retromer-mediated endosome-to-TGN retrieval. J Cell Sci 120, 2378–2389. [DOI] [PubMed] [Google Scholar]
- Smith JL, Campos SK, Wandinger-Ness A, and Ozbun MA (2008). Caveolin-1-dependent infectious entry of human papillomavirus type 31 in human keratinocytes proceeds to the endosomal pathway for pH-dependent uncoating. J Virol 82, 9505–9512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spatazza J, Di Lullo E, Joliot A, Dupont E, Moya KL, and Prochiantz A (2013). Homeoprotein signaling in development, health, and disease: a shaking of dogmas offers challenges and promises from bench to bed. Pharmacol Rev 65, 90–104. [DOI] [PubMed] [Google Scholar]
- Tunnemann G, Ter-Avetisyan G, Martin RM, Stockl M, Herrmann A, and Cardoso MC (2008). Live-cell analysis of cell penetration ability and toxicity of oligo-arginines. J Pept Sci 14, 469–476. [DOI] [PubMed] [Google Scholar]
- Van Engelenburg SB, and Palmer AE (2010). Imaging type-III secretion reveals dynamics and spatial segregation of Salmonella effectors. Nat Meth 7, 325–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vives E, Granier C, Prevot P, and Lebleu B (1997). Structure-activity relationship study of the plasma membrane translocating potential of a short peptide from HIV-1 Tat protein. Lett Pept Sci 4, 429–436. [Google Scholar]
- Zhang W, Kazakov T, Popa A, and DiMaio D (2014). Vesicular trafficking of incoming human papillomavirus type 16 to the Golgi apparatus and endoplasmic reticulum requires gamma-secretase activity. mBio 5, e01777–01714. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.