

Abstract
Elevated levels of the carcinoembryonic antigen (CEA; CEACAM5) in the serum of colorectal cancer (CRC) patients represent a clinical biomarker that correlates with disease recurrence. However, a mechanistic role for soluble CEA (sCEA) in tumor progression and metastasis remains to be established. In this study, we report that sCEA acts as a paracrine factor, activating human fibroblasts by signalling through both the STAT3 and AKT1-mTORC1 pathways, promoting their transition to a cancer-associated fibroblast (CaF) phenotype. sCEA-activated fibroblasts express and secrete high levels of fibronectin, including cellular EDA+-fibronectin (Fn-EDA) that selectively promote the implantation and adherence of CEA-expressing cancer cells. Immunohistochemical analyses of liver tissues derived from CRC patients with elevated levels of sCEA reveal that the expression of cellular Fn-EDA co-registers with CEA-expressing liver metastases. Taken together, these findings indicate a direct role for sCEA as a human fibroblast activation factor, in priming target tissues for the engraftment of CEA-expressing cancer cells, through the differentiation of tissue-resident fibroblasts, resulting in a local change in composition of the extracellular matrix.
Keywords: Colorectal cancer, Carcinoembryonic antigen, Fibroblasts, Fibronectin, Metastasis
Introduction:
Metastatic cancers remain the primary cause of morbidity and mortality for cancer patients despite significant advances in the treatment of localized tumours [1]. An understanding of molecular determinants driving the progression of cancer to a metastatic stage remains limited, thus outlining the complexity of events leading to tumour implantation and the formation of micrometastases. Components responsible for intercellular and cell-matrix adhesion events involving tumour cells have been shown to serve an important role in the formation as well as expansion of metastatic foci [1–7]. Currently, few tumor-derived soluble factors have been identified as causal agents leading to the initiation or the progression of the metastatic cascade.
The carcinoembryonic antigen (CEA, CEACAM5 and CD66e) is a GPI-linked glycoprotein that is intimately linked to cancer metastasis [8–12]. CEA is a 180-kDa N-linked glycoprotein that is aberrantly over-expressed by epithelial cancers including cancers of the gastrointestinal tract, breast, lung, ovary and pancreas [8–12]. Clinically, high levels of CEA in the serum of patients correlate with a worsening prognosis and is associated with a high probability of subclinical metastases and a significant reduction in disease free and overall survival rates [8–11]. CEA is a cell adhesion molecule displayed on the surface of cancer cells that is involved in both homotypic and heterotypic interactions resulting in cellular adherence and aggregation [2, 3, 12–14]. Thus far, two classes of molecular interactions displaying nanomolar dissociation constants (Kds) have been characterized for CEA [3]. Specifically, the CEA IgV-like N domain homotypically associates with itself and its IgC-like A3 domain. The N domain also binds tightly to fibronectin (heterotypic interaction). These interactions were shown to be essential for the engraftment of disseminated CEA-expressing tumour cells as well as the formation and expansion of tumour foci in viva [3]. Additionally, CEA is shed by cancer cells into circulation [8–12]. Although, high circulating levels of soluble CEA is a clinically proven, independent prognostic marker of disease progression in CRC patients, there is no mechanism linking sCEA and metastasis. In view of the intimate association between CEA and fibronectin [3, 15–17], we hypothesize that elevated levels of circulating sCEA may promote local increases in fibronectin levels, thereby enhancing the adherence of CEA-expressing tumor cells. Since fibroblasts are the main cell type responsible for the maintenance of the extracellular matrix [18], we tested the effects of patient-derived sCEA on normal human fibroblasts. In the present study, we report that sCEA activates normal human fibroblasts and cause their differentiation and proliferation into a cancer-associated fibroblast (CaF)-like state, displaying the enhanced expression of total as well as cellular Fn isoforms. This enhanced pattern of cellular Fn expression co-registered with both α-SMA-expressing CaFs as well as CEA-positive liver metastases in liver biopsies derived from CRC patients.
Materials and Methods:
Cells and growth conditions.
Primary normal human neonatal dermal fibroblasts (ATCC PCS-201–010; thereafter referred to as HNNFbs), as well as the human carcinoma cell lines HT-29 (ATCC HTB-38), LS174T (ATCC CL-188) were used in this study. MC38 cells were kindly provided by Dr. J. Schlom (National Cancer Institute; Bethesda, Maryland) while HeLa (ATCC CCL-2) and HeLa cells stably transfected to express CEA (HeLa.CEA) were gifts from Dr. S. Gray-Owen (University of Toronto). All cells were cultured at 37°C in a humidified 5.0% CO2 atmosphere in Dulbecco’s modified Eagle’s medium supplemented with RPMI amino acid mixture (Thermofisher; Oakville, Ontario, Canada), L-glutamine, Sodium Pyruvate (110 mg/mL), D- Glucose (4.5 g/L), 10% fetal bovine serum, penicillin (100 U/mL), and dihydrostreptomycin (100 mg/mL).
Purification of CEA.
CEA was purified from colorectal carcinoma liver metastases using the PCA method, as previously described by Pritchard and Todd [19]. The purity of the eluted CEA (>95%) was ascertained by N-terminal sequence analysis, as well as by SDS-PAGE and Western Blot analysis using the CEA-specific mAb T84.66A3 (Supplementary Fig 1).
HNNFb activation by sCEA.
HNNFbs were stimulated by rinsing subconfluent monolayers with pre-warmed medium, followed by the addition of medium containing sCEA. Activation of HNNFbs was first assessed by examining changes in cellular morphology occurring following the addition of sCEA using light microscopy. Concurrently, changes in Paxillin phosphorylation levels by western blotting using a combination of mouse anti-human total Paxillin (mAb PAX227; BioLegend, San Diego, CA) and rabbit anti-phospho-Paxillin (Tyr 118; EMD Millipore, Etobicoke, Canada).
To generate conditioned media for use in investigating changes in tumor cell dynamics, subconfluent HNNFb monolayers were rinsed with pre-warmed medium, followed by the addition of either growth medium or medium containing sCEA (200 ng/mL). Following a 24-hour incubation, the conditioned media were collected from the HNNFb monolayers and clarified by high-speed ultracentrifugation to remove particulate cellular debris. The resulting clarified supernatants were carefully collected and frozen at −80°C until needed. The conditioned media from CEA-activated HNNFbs did contain traces of sCEA; a feature that recapitulates the microenvironment present in the interstitial spaces of metastatic cancer patients with high circulating sCEA levels.
Determination of the polarization state of sCEA-activated HNNFbs.
HNNFb cultures were stimulated with either TGF-β (5ng/mL), sCEA (200ng/mL) or BSA (200 ng/mL) for 24 hours followed by harvesting of cell cultures for the purpose of preparing lysates. Lysates prepared from stimulated and non-stimulated HNNFb cultures were resolved by SDS-PAGE and transferred onto nitrocellulose membranes. Changes to HNNFb polarity induced by the addition of sCEA was determined by monitoring differences in α-SMA (Sino Biological Inc; Beijing, China) and VEGF (Abcam; Toronto, ON) expression levels by immuno-blotting.
Intracellular signalling cascade induced following stimulation of HNNFbs with sCEA.
Semi-confluent HNNFb cultures were treated with sCEA (200 ng/mL) for 0, 15 minutes, 30 minutes, 1, 2, 4, 8 or 12 hours. Cell cultures were immediately lysed (following their respective stimulation time) in the presence of protease and phosphatase inhibitors then rapidly frozen at −80°C.
Intracellular signalling cascades affected by the addition of sCEA were monitored by measuring changes of the AKT-mTORC1/STAT3 total/phospho-protein ratios, from western blot analyses. Specifically, phosphorylated proteins for lysates of sCEA-stimulated HNNFb cells were resolved by SDS PAGE, electro-transferred to nitrocellulose membranes and detected using antibodies specific to AKT (Millipore), phospho-AKT (Thr 308; Millipore), mTOR (Biolegend), phospho-mTOR (Ser 2481; Millipore), Total S6 Kinase (Biolegend), phospho-S6 Kinase (Thr 412/Thr 389), as well as STAT-3 (Biolegend) and phospho-STAT3 (Ser 727; Cell Signaling Technology; Danvers, MA). Differences in phospho-protein signal intensities were determined by performing densitometric measurements of western blot bands, using ImageJ 1.44 (http://imagej.nih.gov/ij) followed by statistical analyses, as described below.
In order to validate the involvement of the AKT-mTORC1/STAT3 signalling axis in HNNFb activation, sCEA-activated HNNFb cells were treated with the AKT-mTOR inhibitors pentoxifylline or Palomid-529 (Sigma-Aldrich) followed by analysis of the alterations of α-SMA and total Fn expression levels by immuno-blotting.
Influence of HNNFb-conditioned medium on CEA-expressing cancer cells
Growth medium derived from sCEA-stimulated and non-stimulated HNNFb cultures were collected and used to monitor the effect of secreted factors on the adhesion and proliferation of cancer cells in real time using an xCELLigence RTCA DP label-free, impedance-based cell-sensing instrument (ACEA Biosciences Inc.; San Diego, CA). Briefly, HeLa and CEA-expressing HeLa.CEA cells were suspended in culture medium and dispensed (2.0 × 104 cells per well) into 16-well sensor plates (E-plates; ACEA Biosciences Inc.) pre-equilibrated with growth medium (serum-free or supplemented with FBS (10%; v/v)) or conditioned medium from stimulated and non-stimulated HNNFb cultures to assess the effects of HNNFb stimulation by sCEA on the adhesion of CEA-expressing cancer cells. Changes in adherence and proliferation of CEA null and CEA-expressing colon cancer cells were also determined by dispensing LS174T, HT-29, MC38 cells (2.0 × 104 cells per well) into sensor plates (E-plates; ACEA Biosciences Inc.) pre-equilibrated with conditioned medium from stimulated and non-stimulated HNNFb cultures. Changes in relative impedance were measured at 1-min intervals, over the course of 48 hours [2–5, 20].
Analysis of HNNFb-conditioned medium by mass spectrometry.
Subconfluent HNNFb cultures, grown in 150 mm dishes, were rinsed with warm, serum-free, DMEM followed by incubation for 36 hours with either serum-free DMEM or medium containing sCEA (200 ng/mL). Media were collected and concentrated by ultrafiltration using a 3,000 molecular weight cut-off filter followed by exchange against ammonium bicarbonate.
Samples were then lyophilized and sent for mass spectrometry at the SPARC BioCentre (The Hospital for Sick Children; Toronto, Ontario, Canada). Identified proteins were then classified, and the list of the top 50 extracellular protein species analyzed for putative interactions using the STRING V 10.0 functional protein association network [21].
Impact of HNNFb-conditioned medium on FAK signalling.
Suspensions containing 5.0 X 106 HeLa or HeLa.CEA cells were dispensed into 6 well culture plates pre-equilibrated with either growth medium or HNNFb-conditioned medium, and incubated at 37°C for 30 minutes. Medium containing non-adherent cells was aspirated and plates were gently rinsed with PBS, followed by lysis of the remaining cells in the presence of protease and phosphatase inhibitors. Lysates of adhering HeLa and HeLa.CEA were first used to assess changes of Paxillin phosphorylation levels by western blotting using a combination of mouse anti-human total Paxillin (Biolegend) and rabbit anti-phospho-Paxillin (Tyr118; EMD 34 Millipore). Lysates of the adhering HeLa and HeLa.CEA were subsequently used in immuno-precipitation (IP). Specifically, lysates were pre-cleared with magnetic protein G beads (New England BioLabs; Ipswich, MA) followed by IP using anti-human integrin α5 (ITGA5; Biolegend) and blotting using mAbs specific to either focal adhesion kinase (FAK) or SRC (Millipore). In parallel experiments, FAK and SRC were immuno-precipitated from lysates of adhering HeLa and HeLa.CEA, followed by blotting using the phospho-tyrosine specific mAb 4G10.
Analysis of changes in Fibronectin expression.
Changes in the expression of total Fn or cellular Fn (Fn-EDA) by activated and non-stimulated HNNFbs was determined by western blotting using either rabbit anti-human fibronectin pAb (Sigma-Aldrich) or mAb IST-9 (Abcam; Cambridge, MA), respectively. Alterations of Fn expression, in response to sCEA activation of HNNFbs, was determined by ELISA. Briefly, lysates and culture media of non-stimulated and sCEA-stimulated HNNFbs were coated on 96-well microtiter plates (Costar; Oakville, ON) in 100 µl of sodium carbonate (pH 9.5) for 24 h at 37°C. Wells were washed 3 times with PBS-0.05% Tween 20 (PBST) to remove unbound proteins then blocked with 200 µl of 1% BSA in PBS for 60 min at room temperature. Plates were then washed three times with PBST and incubated for 1 h at room temperature with rabbit anti-human Fn (1:1000; Sigma-Aldrich) followed by three washes with 200 µl of PBST. The presence of bound anti-Fn antibodies was detected using horseradish peroxidase (HRP) coupled anti-Rabbit IgG secondary antibodies (diluted to 1:5000 in PBST; Bethyl; Montgomery, TX) in combination with 3,3’,5,5’-tetramethylbenzidine (TMB; Sigma) as the HRP substrate.
Inhibition of CEA-mediated tumor adherence in vitro.
Suspensions of HeLa.CEA (2.0 × 104 cells) were dispensed into sensor plates (E-plates; ACEA Biosciences Inc.) pre-equilibrated with either HNNFb conditioned medium or conditioned medium supplemented with cRGD (200 µM; Sigma-Aldrich), miltefosine (100 µM; Sigma-Aldrich), conA (80 µg/mL; Sigma-Aldrich) or anti-CEA N domain pAb (1:100 final dilution; 3). Loss of cell adherence was measured as a decrease in relative impedance recorded at 1-min intervals over the course of 24 hours [2–5, 20].
Immunohistochemistry and image analysis.
Liver tissue specimens were collected from 23 patients with late stage colorectal cancer undergoing liver resection at the Sunnybrook Health Sciences Center (supplementary Table 1). Specimens were collected with the consent of patients in accordance with the rules and regulations of the Sunnybrook Research Institute’s Research Ethics Board. The liver specimens were fixed in 10% neutral buffered formalin and processed overnight (Leica Tissue Processor; Leica Biosystems; Ontario, Canada). Specimens were then embedded in paraffin and sliced at 4 µm intervals in order to assess signal co-registration (which refers to the alignment of series of images to allow comparisons of marker signal distribution within macroscopic tissue samples). Liver tissues were subsequently stained for the presence of membrane-bound CEA (mCEA; mAb COL1; 1:200 dilution), alpha-smooth muscle actin (α-SMA; mAb 1A4; 1:200 dilution) as well as EDA+ Fibronectin (Fn-EDA; mAb IST-9; 1:200 dilution). The presence of bound antibodies was visualized using an UltraView Universal DAB (3,3’-diaminobenzidine) Detection Kit (Ventana Medical Systems, Inc.). Stained slides were scanned using a Leica SCN400 (Leica Biosystems) in brightfield scanning mode at an optical resolution of 0.5 micron/pixel. The tumors from all patient specimens stained positive for CEA.
For the quantification of DAB staining patterns observed by IHC, digitized images from scanned slides were analyzed using the Sedeen Viewer v5.0.3 (beta) (Pathcore Inc, Toronto, ON) [22]. Images corresponding to specimens stained using the anti-CEA, Fn and α-SMA antibodies were displayed side by side. Using the manual alignment tools in the viewer, all three sets of images were aligned and regions of interest were defined manually on each image [22]. Image processing and stain analysis were carried out using a software plug-in written using the Sedeen SDK provided by Pathcore. First, color deconvolution was performed to isolate the DAB stain (brown color) corresponding to the IHC marker from the digitized images as previously described [22]. A threshold was then determined by visual assessment; it was found that a threshold of 1.0 (where each stain image had been rescaled to the range 0.0–255.0) provided a good separation between stained nuclei and unstained background regions, with the stained nuclei corresponding to pixels with an intensity ≤ 1.0. The ratio of the stained area to the total area was then calculated for each region of interest and each slide and was expressed as a percentage [22]. The analysis was performed on the full resolution images without down sampling. Quantified DAB signals were expressed as % DAB staining corresponding to the percent of pixels within the image containing the DAB stain.
Statistical analyses.
Collected data sets were tested for normality, to confirm Gaussian distribution, using the Shapiro-Wilk normality test. Data sets were compared using either the Mann-Whitney-U-test or the Student t-test. Time-dependent, cellular adhesion data recorded using the xCelligence RTCA DP instrument were analyzed using the RTCA Data Analysis Software 1.0 (ACEA Biosciences). Analysis of statistical significance and plotting of graphs were all done using PRISM software (Graph Pad Software for Science). Statistical significance was acceptable when P ≤ 0.05.
Results:
Patient-derived serum CEA activates human naïve fibroblasts
We began by investigating whether soluble CEA isolated from the serum of cancer patients (sCEA), could activate naïve human neonatal fibroblasts (HNNFbs) in vitra. First, changes in cellular morphology in serum-deprived HNNFbs occurred following their incubation for 24 hours with either sCEA or TGF-β, but not with BSA (Fig 1A and B). This morphological change was accompanied by a significant increase in Paxillin phosphorylation, a marker of cytoskeletal rearrangement (Fig 1C) within 8 hours following the addition of sCEA. Importantly, the addition of either TGF-β or sCEA to HNNFbs resulted in the increased expression of α-SMA while the stimulation of fibroblasts with sCEA led to a significant increase in VEGF levels (Fig. 1D). Together, these findings suggest that sCEA may act as a tumor-derived fibroblast activating factor causing the differentiation of naïve fibroblasts into a CaF (cancer-associated fibroblast)-like phenotype.
Figure 1. Affinity-purified serum CEA (sCEA) derived from cancer patients activates naïve human neonatal fibroblasts (HNNFbs).
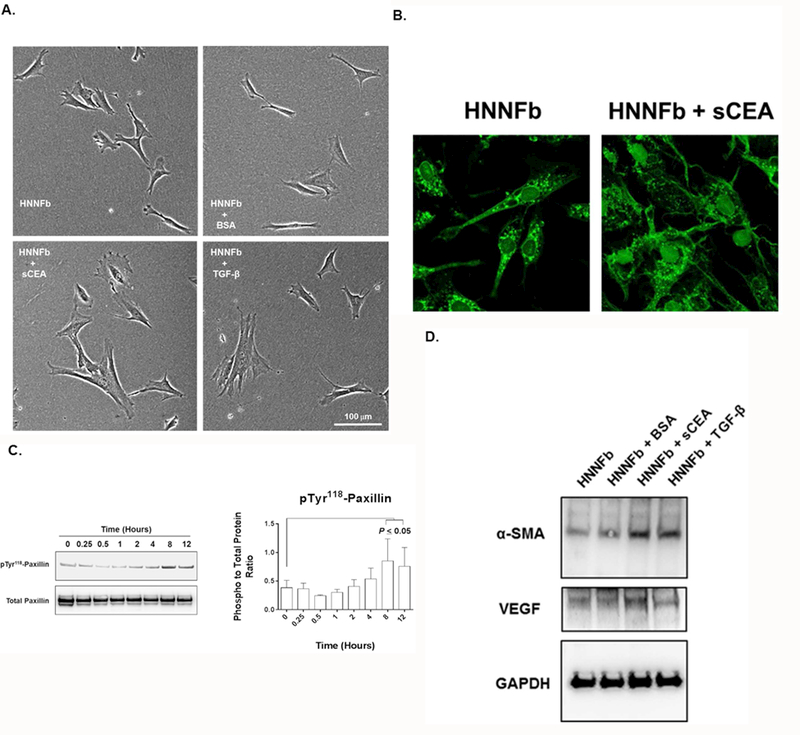
A. sCEA induces changes of fibroblast cellular morphology. Photomicrograph of semi-confluent HNNFb cultures stimulated by TGF-β (5pg/mL), sCEA or BSA (200 ng/mL) for 24 hours. Note the change in cellular morphology of TGF-β and sCEA treated HNNFb cultures. B. Fluorescence images of untreated and sCEA-treated HNNFbs stained with FITC-phalloidin. C. Stimulation of HNNFbs with sCEA triggers an increase in paxillin phosphorylation. D. Activation of HNNFbs with sCEA results in the upregulation of α-SMA and VEGF expression.
Next, we investigated whether the AKT-mTORC1 signalling pathway, a pathway known to regulate fibroblast cellular differentiation [23], was involved in the activation of HNNFbs by sCEA. A statistically significant upregulation of AKT phosphorylation in HNNFbs was observed within 15 minutes of exposure to sCEA (Fig. 2A), which was accompanied by an increase in detectable mTOR (within 30–60 minutes), S6 kinase, and STAT 3 phosphorylation (Fig. 2A). The involvement of the AKT-mTORC1 signalling pathway was further confirmed through the pharmacological inhibition of the pathway by the co-addition of either palomid-529 or pentoxifylline with sCEA. Both agents were found to downregulate α-SMA expression levels (Fig 2B). Taken together, these findings suggest that sCEA activates normal fibroblasts and favors their transition to a CaF-like phenotype by signalling through the AKT-mTORC1/STAT3 signalling pathway.
Figure 2. sCEA activates fibroblasts through the AKT-mTOR C1/STAT3 signalling pathways.
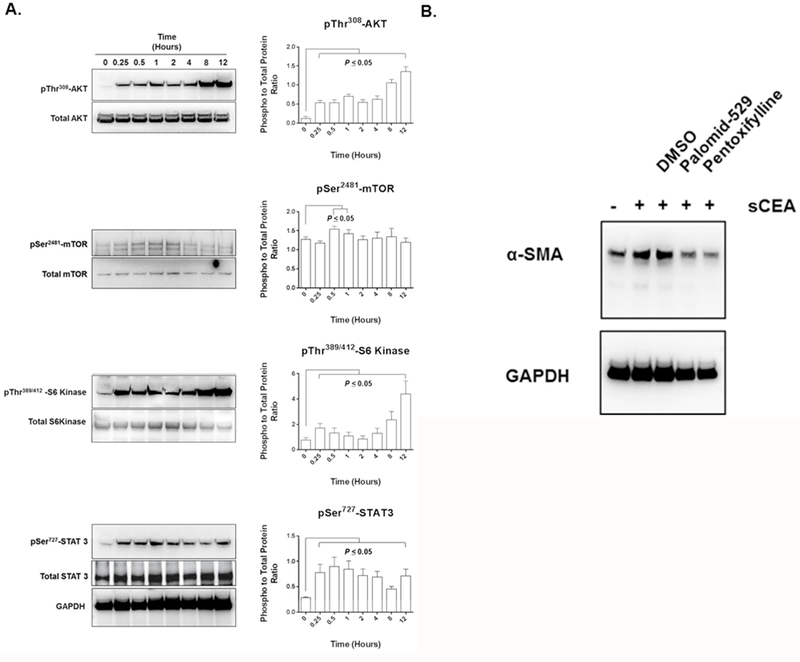
A. Addition of sCEA to HNNFb cultures activates a signalling through the AKT-mTOR C1 as well as the STAT3 signalling axes. B. Pharmacological reversal of α-SMA expression in sCEA-activated HNNFbs upon the addition of AKT-mTOR C1 inhibitors palomid-529 or pentoxifylline.
Medium recovered from sCEA-activated fibroblasts selectively promotes the expansion of CEA-expressing colorectal cancer cells
We have recently shown that CEA-expressing tumor cells bind tightly to immobilized Fn through their CEA IgV-like N domain [3]. This high affinity interaction (Kd ~16 nM for their monomeric interaction) was postulated to be a factor favoring the implantation of CEA-expressing tumor foci [3]. Since fibroblasts secrete fibronectin and its variants, we explored the relevance of activating fibroblasts with sCEA and observed if the resulting conditioned medium increased the adherence of CEA-expressing human colon cancer cells LS174T and HT29 to plates in relation to medium from un-stimulated fibroblasts. As predicted, CEA-expressing tumor cells adhered more rapidly (0–8 hours; Fig 3 A) and proliferated more quickly (8–48 hours; Fig 3B) in plates pre-treated with medium recovered from sCEA-activated fibroblasts, whereas CEA null murine MC38 colonic carcinoma cells did not (Fig 3).
Figure 3. Medium from sCEA-stimulated HNNFb cultures favors the adherence and expansion of CEA-expressing colonic carcinoma cell lines.
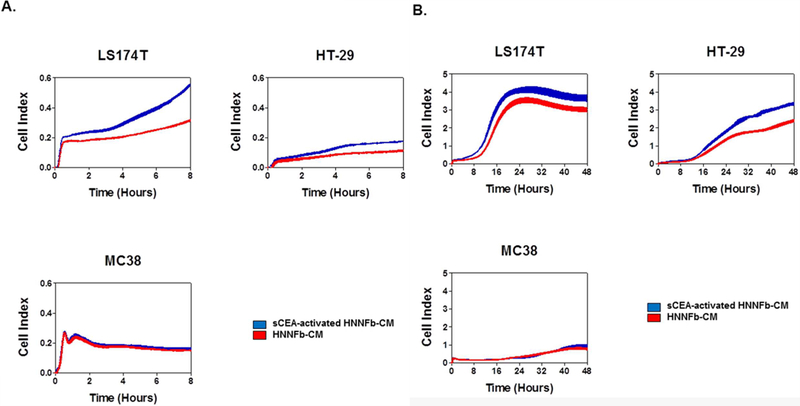
Medium from sCEA-activated fibroblasts promotes a more rapid adherence of CEA-expressing colorectal cancer cells relative to medium recovered from naïve fibroblasts (0–8 hours; panel A) as well as their increased proliferation and expansion (8–48 hours; panel B). CEA-expressing human colon cancer LS174T and HT-29 were dispensed into E-plate wells pre-treated with medium from either naïve or sCEA-activated fibroblasts. The impact of incubating cancer cells in medium from either naïve or sCEA-activated fibroblasts was monitored as changes in relative electrical impedance (CI) at 1-min intervals, for 48 hours.
Since the majority of human colonic carcinoma cell lines express higher levels of CEA, we investigated the focused role of membrane-bound CEA on cancer cells in this adhesion event by expressing CEA in HeLa cells; a cell line that does not endogenously express CEA or its related CEACAMs [5, 24]. Expression of the full-length CEA on the surface of these cells resulted in augmenting their fibronectin-binding ability (Supplementary Figure 2). Therefore, we used CEA-expressing HeLa transfectants and compared their adhesion to CEA null HeLa cells to assess if activated HNNFbs specifically facilitated the cellular adhesion of CEA-expressing cells (Fig. 4). As observed for CEA-expressing colorectal cancer cells (Fig 3), the incubation of CEA-expressing HeLa cells into wells pre-treated with medium derived from either naïve or sCEA-activated HNNFbs resulted in a rapid increase in cellular adhesion, with cells adhering better when incubated in medium derived from activated fibroblasts when compared to untreated or serum-alone treated HeLa cells (Fig 4A).In contrast, CEA− parental HeLa cells displayed a lower, monotonic increase in adherence following their exposure to conditioned medium from fibroblasts (Fig 4A). The observed slow increase in cellular adherence for parental HeLa cells suggested a role for other adhesion factors such as integrins. HeLa and HeLa.CEA cells were thus incubated in either normal or fibroblast-conditioned medium then lysed and cellular complexes incorporating α5 integrin immune-precipitated using an anti-human α5 integrin mAb. Recovered complexes from adhering CEA-expressing HeLa cells displayed higher levels of SRC and FAK recruitment and phosphorylation than HeLa cells alone (Fig 4B,C) as well as elevated levels of phospho-paxillin (Fig 4D). These observations confirm that sCEA-activated fibroblasts/CaFs secrete factors that promote the adherence and proliferation of CEA-expressing colorectal cancer cells. A previously postulated mechanistic model suggested that CEA facilitated cancer cell adhesion by improving membrane fluidity and favoring a cluster formation with integrins [15, 16]. We first compared the relative importance of disrupting CEA or integrins interactions on cellular adherence. Specifically, HeLa.CEA incubated in fibroblast-conditioned medium containing either anti-CEA N domain pAb [3] or cRGD [25] respectively, resulted in a selective loss of CEA or integrin adhesion functions reducing cellular adherence by half (Fig.4E). CEA-expressing HeLa cells were also incubated in fibroblast-conditioned medium supplemented with either ConA [26] or miltefosine [27, 28] in order to limit molecular clustering events on the cell surface. As shown in Figure 4E, interfering with overall clustering events involving cell surface components dramatically reduced cellular adherence by > 90% (Fig 4E). The importance of this mechanism was further confirmed using an in viva model of tumor implantation, wherein the blocking of either the CEA N domain, β1integrin or of the clustering of cell surface components with miltefosine, resulted in disrupting in vivo cellular engraftment events (Supplementary Figure 3). Taken together, these observations highlight the importance of the CEA/integrin-Fn axis and the role it plays in ensuring efficient cellular engraftment.
Figure 4. Medium from sCEA-activated fibroblasts preferentially augments the adherence of CEA-expressing cells.
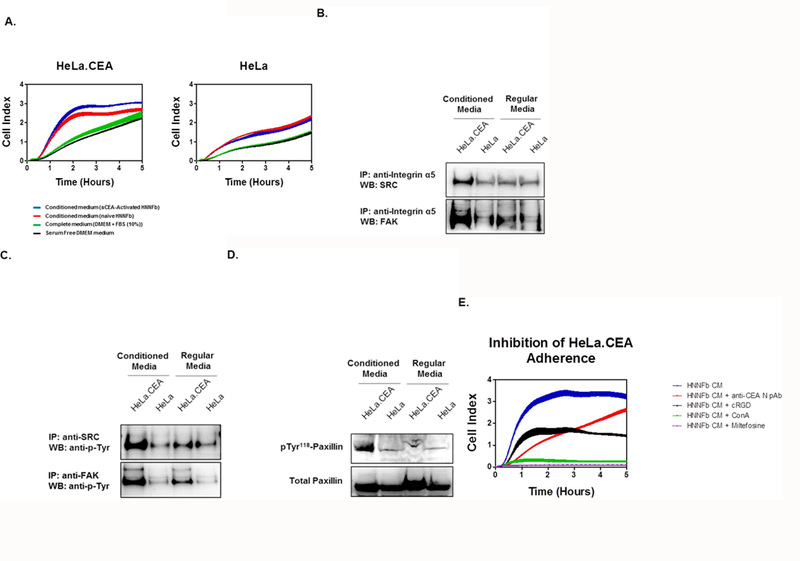
A. sCEA-activated fibroblasts promote the adherence of CEA-expressing HeLa (HeLa.CEA) cells, but not of parental HeLa cells. Cells (2.0 × 104) were dispensed to E-plate wells pre-treated with serum-free, complete medium (negative control) or medium recovered from either naïve or sCEA-activated fibroblasts. Cellular adhesion is reported as averaged Cell Index (CI) values (± SEM). B. Incubation of CEA-expressing cells in fibroblast-conditioned media promotes integrin signalling and recruitment of FAK and SRC. Suspensions of HeLa or HeLa.CEA (5 × 106) cells were incubated in either regular medium or fibroblast-conditioned medium for 30 minutes. Adhering cells were lysed in RIPA buffer followed by immuno-precipitation using an anti-human integrin α5 mAb and immuno-blotting for either SRC or FAK. C. Incubation of CEA-expressing cells in fibroblast-conditioned medium promotes FAK and SRC phosphorylation. Lysates of adhering cells were immuno-precipitated using anti-human FAK or SRC mAb and immuno-blotted for phospho-tyrosine using mAb 4G10. D. Incubation of CEA-expressing cells in fibroblast-conditioned medium favors a substantial increase in paxillin phosphorylation. Lysates of adhering cells were immuno- blotted for both total and phospho-paxillin. E. Disruption of the Fn-CEA/integrin interactions inhibits cellular adherence. Addition of cRGD, anti-CEA N pAb, miltefosine or ConA to adhering HeLa.CEA cells reduces cellular adhesion. HeLa cells (2.0 × 104) were dispensed to E-plate wells containing medium from either naïve or sCEA-activated fibroblasts. The impact of adding these inhibitors was monitored in real time as changes in relative electrical impedance (CI) at 1-min intervals, for 5 hours.
Mass spectrometry analysis identified a number of proteins secreted into the medium by naïve and sCEA-activated HNNFbs. The identified proteins species were then organized, with the top 50 extracellular protein species analyzed using the STRING V 10.0 functional protein association network [21]. This analysis led to the identification of fibronectin (Fn) as a dominant extracellular matrix factor produced by HNNFbs (Suppl Table 2). In particular, mass spectrometry detected 2 adjacent tryptic peptides (GLAFTDVDVDSIK and IAWESPQGQVSR) located in the extracellular domain A (EDA) of a fibronectin isoform termed Fn-EDA. EDA represents an alternative splice domain of Fn inserted between the 11th and 12th FN3 modules of Fn. The Fn-EDA isoform is produced by HNNFbs in response to sCEA stimulation and is an isoform associated with cancer progression [29–31].
Western blots and ELISA assays confirmed that the exposure of HNNFbs to sCEA results in a quantitative increase in Fn secretion (Fig. 5). These findings correlate with the increase in CEA-expressing tumor cell adherence observed in the presence of medium derived from sCEA-treated HNNFbs in contrast to medium from naïve HNNFbs or BSA-treated fibroblasts (Fig. 5A). However, Fn expression remains lower than the level observed in TGF-β treated cells (positive control; Fig 5A). Importantly, the observed increases in Fn production and secretion by sCEA-activated fibroblasts were dose dependent (Fig 5, panels B-D). Addition of either palomid-529 or pentoxifylline reversed the sCEA-induced increase in Fn production by HNNFbs thereby confirming the relevance of the AKT-mTORC1 signalling pathway for the sCEA-induced increase in Fn production by naïve HNNFbs (Fig 5D). Collectively, these results support the notion that sCEA enhances the expression and secretion of Fn by activated fibroblasts.
Figure 5. Dose-dependent production and secretion of Fn by HNNFbs following stimulation with sCEA.
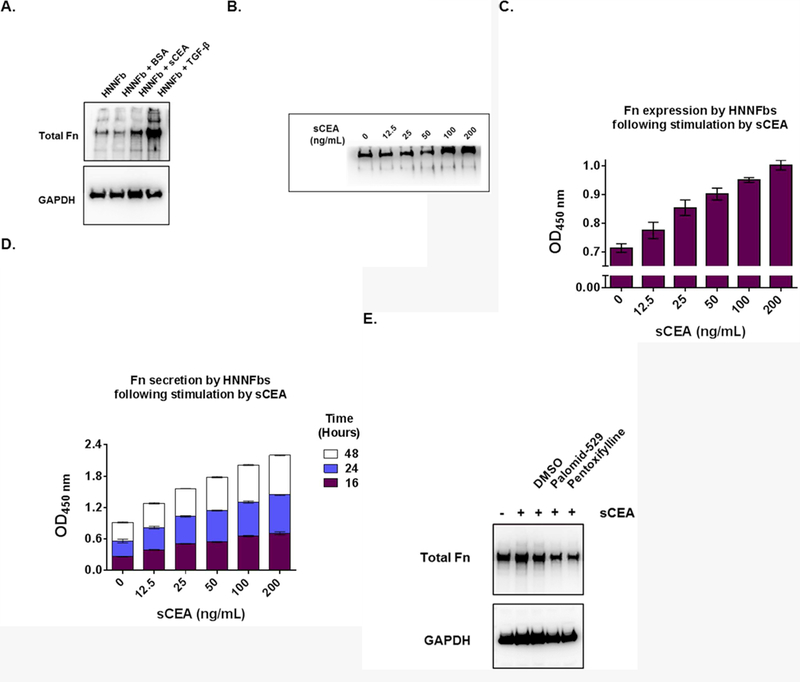
A. Activation of HNNFbs by either sCEA (200 ng/mL) or TGF-β (5pg/mL) specifically upregulate Fn expression in a dose-dependent manner. B. Addition of sCEA induces a dose-dependent upregulation of Fn expression in fibroblasts. Immuno-blot analysis of total Fn expression by HNNFbs stimulated with increasing concentrations of sCEA for 24 hours. C. Increased production in total Fn as measured by ELISA, of HNNFbs cultures stimulated by escalating concentrations of sCEA for 24 hours. D. Rise in total Fn secreted by sCEA-activated HNNFb cultures as a function of time. E. AKT-mTOR C1 inhibitors palomid-529 or pentoxifylline reduce the production of Fn by sCEA-activated HNNFbs.
Characterization of the cellular Fn species upregulated by sCEA stimulation of fibroblasts
Thus far, we have presented evidence to support the notion that the addition of sCEA to naïve fibroblasts favored their transition to a CaF-like phenotype, and that in turn allowed for an upregulation in Fn expression which favored the selective adhesion of CEA-expressing cancer cells. However, fibroblasts express several different Fn isoforms, ranging from circulating soluble Fn to cellular Fn (cFn), the main form observed in tumor cells. As previously mentioned, mass spectrometry identified Fn-EDA as a cFn isoform produced by HNNFbs following sCEA stimulation. This cFn isoform is known to be deposited into the ECM and to play a role in cancer progression [29–31]. We therefore stimulated HNNFbs with sCEA and observed a specific upregulation of Fn-EDA production by HNNFbs (Fig 6A). Similarly to total Fn (Fig. 5A), the expression of Fn-EDA increased with rising amounts of sCEA (Fig 6B). Importantly, histochemical staining of liver biopsies from colorectal cancer patients (with metastatic disease) displayed high cellular Fn-EDA levels relative to normal human liver tissues (Fig 6C). A quantitative analysis of Fn-EDA levels observed in liver biopsies of 23 patients indicated a strong correlation between Fn-EDA expression and the occurrence of liver metastases. This correlation was independent of the age or sex of CRC patients (Fig 6D). Moreover, the cellular Fn-EDA signals co-registered with both membrane-bound CEA (tumor) and α-SMA (CaF) staining (Fig 6 panels E and F). Image analysis quantifying the DAB signal deposition in the liver specimens, taken from all the studied colorectal carcinoma patients, revealed an increase in Fn-EDA expression in both tumor and non-tumor parts of the liver (Fig 6 G).
Figure 6. Activation of fibroblasts by sCEA upregulates the production and deposition of cellular Fn-EDA.
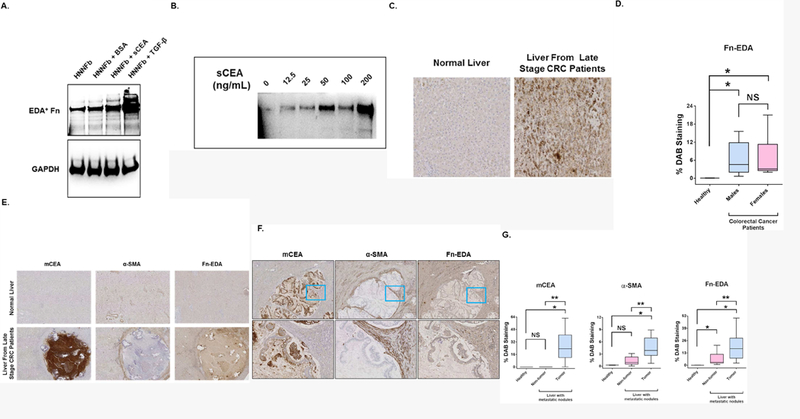
A. Activation of HNNFbs by either sCEA (200 ng/mL) or TGF-β (5pg/mL) specifically upregulate Fn-EDA expression. B. Addition of sCEA induces a dose-dependent upregulation of Fn-EDA expression in fibroblasts. Semi-confluent HNNFbs cultures were stimulated with sCEA for 24 hours followed by western blot analysis of Fn-EDA expression using mAb IST-9. C. Immunohistochemical (IHC) detection of increases in cellular Fn-EDA in liver biopsies from late stage CRC patients, but not in normal liver. D. Quantification of DAB (3,3’-diaminobenzidine) signals of cellular Fn-EDA in stained specimens taken from 23 late stage CRC patients with metastatic disease. DAB signals (defined as % DAB staining) were quantified using the Sedeen software image analysis package. E. Fn-EDA IHC DAB signals co-register with the membrane-bound CEA [mCEA] and α-SMA signals [fibroblasts] in liver biopsies from late stage CRC patients with metastatic disease. F. Enlargement of a representative hepatic tumor nodule highlighting the co-registration of DAB signals for mCEA together with α-SMA and Fn-EDA signals. G. Quantification of DAB signals of mCEA, α-SMA and cellular (EDA+) Fn in stained specimens taken from 23 colorectal cancer patients with metastatic disease. DAB signals were quantified using the Sedeen software image analysis package (Pathcore). Signals registered from the tumor-free as well as the tumor afflicted areas of the stained liver specimens were compared with signals quantified from liver specimens taken from healthy patients. NS; not statistically significant. * P < 0.05 when compared to signals registered from stained healthy liver specimens. ** P < 0.05 when compared to non-tumor areas within liver specimens. Statistical significance was determined using the Mann-Whitney test.
Discussion:
The monitoring of sCEA levels in colorectal cancer (CRC) patients represents a proven independent prognostic marker of disease progression [8–12]. It is the most common test performed in pre- and post-operative follow-ups of CRC patients, as elevated sCEA levels directly correlate with disease recurrence [8–11,]. Surprisingly, no functional role linking serum levels of CEA to disease progression has ever been established.
Earlier work by Hostetter et al. [32] demonstrated that repeated systemic injection of soluble CEA into athymic nude mice, increased the metastatic potential of a weakly metastatic colorectal cancer cell line by augmenting the number of tumor nodules establishing in the liver. The underlying mechanism was not elucidated at the time, but the increase in the number of hepatic tumor nodules was attributed to CEA acting as an attachment factor [32]. Subsequent work identified the association of CEA (through residues 108–115) with heterogeneous nuclear RNA-binding protein M (hnRNP M; also referred to as CEA receptor) expressed by macrophages/Kupffer cells and endothelial cells, and postulated that this interaction may account for the pro-metastatic attributes of CEA [33–35]. We thus performed transcriptome analyses of HNNFbs in the presence and absence of sCEA as part of the present study, and observed that these cells did not express messenger RNAs for hnRNP M or any CEACAM known to interact with CEA [Abdul-Wahid et al., unpublished observations]. These results suggest that hnRNP M or CEACAMs are not part of the mechanism of activation of HNNFbs by sCEA, but that sCEA activated HNNFbs through a yet unknown CEA/CEA receptor complex.
We have previously shown that the IgV-like N domain of CEA binds avidly to fibronectin, promoting the implantation of disseminated CEA-expressing tumor cells [3]. Based on this finding, we postulated an alternate hypothesis whereby sCEA would play a direct role in activating tissue-resident fibroblasts to alter their local environment by increasing Fn deposition; a key event that would lead to the seeding of circulating CEA+ tumor cells and the subsequent formation of metastatic tumor foci. Fibroblasts are a heterogeneous cell population, ubiquitously distributed through organs and connective tissues in the body [8]. Under normal physiological conditions, fibroblasts maintain the integrity of the ECM and play a regulatory role in epithelial differentiation, wound healing and tissue inflammation [18, 31, 36, 37]. However, in the context of the tumor microenvironment, cancer-associated fibroblasts (CAFs) represent a major cellular constituent of the stroma [18, 38] that plays a critical role in the expansion of nascent tumor nodules [18, 39]. Studies suggest that cancer-activated fibroblasts display pro-metastatic characteristics such as the enhancement of tumor migration through the release of soluble factors [40, 41] or the creation of a niche conducive to cancer growth and initiation of angiogenesis [42]. The data presented in this study suggest that sCEA activates resting fibroblasts in a paracrine manner, at concentrations typically observed in patients with metastatic disease [8–11]. Specifically, the addition of sCEA induced a change of cellular morphology of naïve human fibroblasts (Figure 1 A and B), as well as an upregulation in expression levels of α-SMA, a widely used marker for identifying CAFs (Figure 1D). Furthermore, we also noted an upregulation in VEGF (Figure 1D) and Fn expression levels (Figure 6) following fibroblast stimulation with sCEA. Together, these findings suggest that sCEA induces the activation of fibroblasts and promotes their transition to a type 2 Cancer-associated Fibroblast (CaF) phenotype, a differentiated cancer-associated fibroblast that promotes the onset of tumor metastasis [38].
To assess the role of sCEA as a paracrine factor in signalling fibroblasts to differentiate into CAFs, we investigated the intracellular signaling pathway engaged by the sCEA-stimulated HNNFbs. It is currently understood that fibroblasts can be activated and differentiated via one of two signalling pathways. In one instance, fibroblasts can be specifically activated and differentiated as a result of TGF-β stimulation through the SMAD signalling pathway [18, 38, 36]. Alternatively, fibroblasts can be activated by several known factors through the STAT3 and AKT1-mTORC1 signaling pathways [23, 43–45]. In this study, we found that sCEA stimulated fibroblast differentiation through the AKT1-mTORC1/STAT3 signalling axis (Fig.2A), a signalling pathway typically engaged in pro-fibrotic fibroblasts [23]. Pro-fibrotic fibroblasts represent a pro-pathological subtype within the spectrum of fibroblast activation states, in that they play a role in the dysregulated deposition of extracellular matrix proteins in interstitial spaces [23]. This finding was substantiated by the proteomic analysis of sCEA-activated HNNFbs conditioned medium, which indicated an upregulation in the secretion of soluble protein factors, including Fn and its isoform Fn-EDA (Suppl Table 2).
Given the importance of fibronectin to the selective adherence of CEA-expressing tumor cells [3, 15–17], we explored the role of sCEA on the expression and secretion of Fn by the stimulated fibroblasts and found that exposure of naïve fibroblasts to sCEA concentrations ≥ 12.5 ng/mL resulted in a dose dependent increase in the production of Fn (Fig.5), including the EDA-expressing isoform of cFn (Fig.6). The increase in Fn production by sCEA-stimulated HNNFbs resulted in an improvement in the adherence and growth of the CEA-expressing human colon cancer cell lines HT-29 and LS174T (Fig. 3). Likewise, CEA-expressing HeLa cells incubated in sCEA-activated fibroblast conditioned medium resulted in an enhancement in their cellular adherence (Fig 4A). In contrast, the conditioned medium did not yield an improvement in the adherence of parental (CEA-) HeLa cells (Fig 4A). Furthermore, adherent CEA-expressing cells displayed higher levels of SRC and FAK recruitment and phosphorylation (Fig 4 panels B and C) accompanied by elevated levels of phospho-paxillin (Fig 4D). This observed enhancement of cellular adherence to Fn-enriched surfaces by CEA-expressing cells, may also reflect an increase in membrane fluidity associated with the over-expression of CEA on their surface [15, 16] in combination with the high affinity of the CEA IgV-like N domain to bind to fibronectin (Kd 16 ± 3 nM) [3]. Disrupting the association of CEA-expressing cells to Fn (with CEA or integrin blocking agents) or by limiting the clustering of components on the cell surface (through the addition of ConA or miltefosine) led to a reduction in cellular adherence in vitra (Fig 4E), as well as the disruption of tumor engraftment in viva (Supplementary Figure 3). These findings were supported by the observed presence of α-SMA+ CaFs at the perimeter of CEA+ tumor foci in liver sections of CRC patients with metastatic disease, as highlighted by immunohistochemistry (IHC; Fig. 6E). Importantly, IHC-derived, DAB (3,3’-diaminobenzidine) staining patterns for the Fn isoform, Fn-EDA and CEA-expressing metastatic foci co-registered in these same sections (Fig.6E), adding further support to our postulated hypothesis. Quantitative image analyses of DAB signal levels (% DAB staining) for membrane-bound CEA (mCEA), αSMA and Fn-EDA signals from these IHC images indicated a strong co-registration of all 3 markers in sections of liver metastases recovered from CRC patients (Fig.6 panels F and G). It has been reported that the expression of Fn-EDA normally increases after wounding, and is upregulated in malignant cells [46–48]. The EDA domain has also been shown to mediate cell differentiation [49], to promote cell adhesion and spreading as compared to Fn [50] and to enhance Fn-EDA binding to α5β1integrin [50].
In summary, sCEA is a circulating fibroblast activation factor secreted by CEA-expressing tumor cells that acts in a paracrine fashion to stimulate the local differentiation of naïve fibroblasts into CAFs, activating them to secrete factors such as Fn-EDA that promote the implantation of CEA+ tumor cells. Importantly, the activation and differentiation of human fibroblasts by patient-derived sCEA occur at concentrations that clinically correlate with disease recurrence in CRC patients.
Supplementary Material
Novelty and impact statement:
Despite its use in managing colorectal cancer patients, the biological significance of elevated circulating CEA remains to be elucidated. Herein, we show that soluble CEA activates normal fibroblasts, and promotes their transition to a CaF-like state. This event engenders a change in the matricellular landscape of target tissues thereby selectively favoring the implantation of metastatic CEA-expressing cells. These findings offer an insight into a yet-unknown mechanism in which CEA directly participates in the metastatic cascade.
Acknowledgements:
The authors wish to thank Drs. Scott Gray-Owen and J. Schlom for providing the HeLa/HeLa.CEA and the MC38 cell lines, respectively. Funding: This work was financially supported by the Ontario Institute for Cancer Research (OICR), the Canadian Breast Cancer Foundation (CBCF) and The Canadian Institutes of Health Research (CIHR). The authors of this study declare no conflict of interest.
Footnotes
Competing Interests:
Authors declare no conflict of interests.
References:
- 1.Psaila B, Lyden D. The metastatic niche: adapting the foreign soil. Nat Rev Cancer 2009; 9: 285–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abdul-Wahid A, Huang EH, Lu H, Flanagan J, Mallick AI, Gariépy J. A focused immune response targeting the homotypic binding domain of the carcinoembryonic antigen blocks the establishment of tumor foci in vivo. Int J Cancer 2012; 131: 2839–2851 [DOI] [PubMed] [Google Scholar]
- 3.Abdul-Wahid A, Huang EH, Cydzik M, Bolewska-Pedyczak E, Gariépy J. The Carcinoembryonic Antigen IgV-like N domain plays a critical role in the implantation of metastatic tumour cells. Mol Oncol 2014; 8: 337–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abdul-Wahid A, Cydzik M, Prodeus A, Alwash M, Stanojcic M, Thompson M, Huang EH, Shively JE, Gray-Owen SD, Gariépy J. Induction of antigen-specific TH 9 immunity accompanied by mast cell activation blocks tumor cell engraftment. Int J Cancer 2016; 139: 841–853 [DOI] [PubMed] [Google Scholar]
- 5.Orava EW, Abdul-Wahid A, Huang EH, Mallick AI, Gariépy J. Blocking the attachment of cancer cells in vivo with DNA aptamers displaying anti-adhesive properties against the carcinoembryonic antigen. Mol Oncol 2013; 7: 799–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klein CA. Parallel progression of primary tumours and metastases. Nat Rev Cancer 2009; 9: 302–312 [DOI] [PubMed] [Google Scholar]
- 7.Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer 2003; 3: 453–458 [DOI] [PubMed] [Google Scholar]
- 8.Duffy MJ, van Dalen A, Haglund C, Hansson L, Holinski-Feder E, Klapdor R, Lamerz R, Peltomaki P, Sturgeon C, Topolcan O. Tumor markers in colorectal cancer: European Group on Tumor Markers (EGTM) guidelines for clinical use. Eur J Cancer 2007; 43: 1348–1360 [DOI] [PubMed] [Google Scholar]
- 9.Moertel CG, O’Fallon JR, Go VL, O’Connell MJ, Thynne GS. The preoperative carcinoembryonic antigen test in the diagnosis, staging, and prognosis of colorectal cancer. Cancer 1986; 58: 603–610 [DOI] [PubMed] [Google Scholar]
- 10.Thirunavukarasu P, Talati C, Munjal S, Attwood K, Edge SB, Francescutti V. Effect of Incorporation of Pretreatment Serum Carcinoembryonic Antigen Levels Into AJCC Staging for Colon Cancer on 5-Year Survival. JAMA Surg 2015; 150: 747–755 [DOI] [PubMed] [Google Scholar]
- 11.Locker GY, Hamilton S, Harris J, Jessup JM, Kemeny N, Macdonald JS, Somerfield MR, Hayes DF, Bast RC Jr., ASCO. ASCO 2006 update of recommendations for the use of tumor markers in gastrointestinal cancer. J Clin Oncol 2006; 24: 5313–5327 [DOI] [PubMed] [Google Scholar]
- 12.Beauchemin N, Arabzadeh A. Carcinoembryonic antigen-related cell adhesion molecules (CEACAMs) in cancer progression and metastasis. Cancer Metastasis Rev 2013; 32: 643–671 [DOI] [PubMed] [Google Scholar]
- 13.Benchimol S, Fuks A, Jothy S, Beauchemin N, Shirota K, Stanners CP. Carcinoembryonic antigen, a human tumor marker, functions as an intercellular adhesion molecule. Cell 1989; 57: 327–334 [DOI] [PubMed] [Google Scholar]
- 14.Zhou H, Fuks A, Alcaraz G, Bolling TJ, Stanners CP. Homophilic adhesion between Ig superfamily carcinoembryonic antigen molecules involves double reciprocal bonds. J Cell Biol 1993; 122: 951–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ordonez C, Zhai AB, Camacho-Leal P, Demarte L, Fan MM, Stanners CP. GPI-anchored CEA family glycoproteins CEA and CEACAM6 mediate their biological effects through enhanced integrin alpha5beta1-fibronectin interaction. J Cell Physiol 2007; 210: 757–765 [DOI] [PubMed] [Google Scholar]
- 16.Camacho-Leal P, Zhai AB, Stanners CP. A co-clustering model involving alpha5beta1 integrin for the biological effects of GPI-anchored human carcinoembryonic antigen (CEA). J Cell Physiol 2007; 211: 791–802 [DOI] [PubMed] [Google Scholar]
- 17.Blumenthal RD, Hansen HJ, Goldenberg DM. Inhibition of adhesion, invasion, and metastasis by antibodies targeting CEACAM6 (NCA-90) and CEACAM5 (Carcinoembryonic Antigen). Cancer Res 2005; 65: 8809–8817 [DOI] [PubMed] [Google Scholar]
- 18.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer 2006; 6: 392–401 [DOI] [PubMed] [Google Scholar]
- 19.Pritchard DG, Todd CW. Purification of Carcinoembryonic Antigen by Removal of Contaminating Mucopolysaccharides. Cancer Res 1976; 36: 4699–4701 [PubMed] [Google Scholar]
- 20.Cydzik M, Abdul-Wahid A, Park S, Bourdeau A, Bowden K, Prodeus A, Kollara A, Brown TJ, Ringuette MJ, Gariépy J. Slow binding kinetics of secreted protein, acidic, rich in cysteine-VEGF interaction limit VEGF activation of VEGF receptor 2 and attenuate angiogenesis. FASEB J 2015; 29: 3493–3505 [DOI] [PubMed] [Google Scholar]
- 21.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, von Mering C. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 2015; 43: D447–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martel AL, Hosseinzadeh D, Senaras C, Zhou Y, Yazdanpanah A, Shojaii R, Patterson ES, Madabhushi A, Gurcan MN. An Image Analysis Resource for Cancer Research: PIIP—Pathology Image Informatics Platform for Visualization, Analysis, and Management. Cancer Res 2017; 77: e83–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang S, Wilkes MC, Leof EB, Hirschberg R. Noncanonical TGF-beta pathways, mTORC1 and Abl, in renal interstitial fibrogenesis. Am J Physiol Renal Physiol 2009; 298: F142–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patel PC, Lee HS, Ming AY, Rath A, Deber CM, Yip CM, Rocheleau JV, Gray-Owen SD. Inside-out signaling promotes dynamic changes in the carcinoembryonic antigen-related cellular adhesion molecule 1 (CEACAM1) oligomeric state to control its cell adhesion properties. J Biol Chem 2013; 288: 29654–29669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ruoslahti E, Pierschbacher MD. New perspectives in cell adhesion: RGD and integrins. Science 1987; 238: 491–497 [DOI] [PubMed] [Google Scholar]
- 26.Gray JA, Sheffler DJ, Bhatnagar A, Woods JA, Hufeisen SJ, Benovic JL, Roth BL. Cell-type specific effects of endocytosis inhibitors on 5-hydroxytryptamine(2A) receptor desensitization and resensitization reveal an arrestin-, GRK2-, and GRK5-independent mode of regulation in human embryonic kidney 293 cells. Mol Pharmacol 2001; 60: 1020–1030 [DOI] [PubMed] [Google Scholar]
- 27.Moreira RA, Mendanha SA, Hansen D, Alonso A. Interaction of miltefosine with the lipid and protein components of the erythrocyte membrane. J Pharm Sci 2013; 102: 1661–1669 [DOI] [PubMed] [Google Scholar]
- 28.Bäumer W, Wlaź P, Jennings G, Rundfeldt C. The putative lipid raft modulator miltefosine displays immunomodulatory action in T-cell dependent dermal inflammation models. Eur J Pharmacol 2010; 628: 226–232 [DOI] [PubMed] [Google Scholar]
- 29.Rybak JN, Roesli C, Kaspar M, Villa A, Neri D. The extra-domain A of fibronectin is a vascular marker of solid tumors and metastases. Cancer Res 2007; 67: 10948–10957 [DOI] [PubMed] [Google Scholar]
- 30.Borgia B, Roesli C, Fugmann T, Schliemann C, Cesca M, Neri D, Giavazzi R. A proteomic approach for the identification of vascular markers of liver metastasis. Cancer Res 2010; 70: 309–318 [DOI] [PubMed] [Google Scholar]
- 31.White ES, Baralle FE, Muro AF. New insights into form and function of fibronectin splice variants. J Pathol 2008; 216: 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hostetter RB, Augustus LB, Mankarious R, Chi KF, Fan D, Toth C, Thomas P, Jessup JM. Carcinoembryonic antigen as a selective enhancer of colorectal cancer metastasis. J Natl Cancer Inst 1990; 82: 380–385 [DOI] [PubMed] [Google Scholar]
- 33.Zimmer R, Thomas P. Mutations in the carcinoembryonic antigen gene in colorectal cancer patients: implications on liver metastasis. Cancer Res 2001; 61: 2822–2826 [PubMed] [Google Scholar]
- 34.Thomas P, Forse RA, Bajenova O. Carcinoembryonic antigen (CEA) and its receptor hnRNP M are mediators of metastasis and the inflammatory response in the liver. Clin Exp Metastasis 2011; 28: 923–932 [DOI] [PubMed] [Google Scholar]
- 35.Bramswig KH, Poettle M, Unseld M, Wrba F, Uhrin P, Zimmermann W, Zielinski CC, Prager GW. Soluble carcinoembryonic antigen activates endothelial cells and tumor angiogenesis. Cancer Res 2013; 73: 6584–6596 [DOI] [PubMed] [Google Scholar]
- 36.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol 2002; 3: 349–363 [DOI] [PubMed] [Google Scholar]
- 37.Parsonage G, Filer AD, Haworth O, Nash GB, Rainger GE, Salmon M, Buckley CD. A stromal address code defined by fibroblasts. Trends Immunol 2005; 26: 150–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Augsten M Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front Oncol 2014; 4: 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hasegawa T, Yashiro M, Nishii T, Matsuoka J, Fuyuhiro Y, Morisaki T, Fukuoka T, Shimizu K, Shimizu T, Miwa A, Hirakawa K. Cancer-associated fibroblasts might sustain the stemness of scirrhous gastric cancer cells via transforming growth factor-β signaling. Int J Cancer 2014; 134: 1785–1795 [DOI] [PubMed] [Google Scholar]
- 40.Olaso E, Santisteban A, Bidaurrazaga J, Gressner AM, Rosenbaum J, Vidal-Vanaclocha F. Tumor-dependent activation of rodent hepatic stellate cells during experimental melanoma metastasis. Hepatology 1997; 26: 634–642 [DOI] [PubMed] [Google Scholar]
- 41.Cornil I, Theodorescu D, Man S, Herlyn M, Jambrosic J, Kerbel RS. Fibroblast cell interactions with human melanoma cells affect tumor cell growth as a function of tumor progression. Proc Natl Acad Sci U S A 1991; 88: 6028–6032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olaso E, Salado C, Egilegor E, Gutierrez V, Santisteban A, Sancho-Bru P, Friedman SL, Vidal-Vanaclocha F. Proangiogenic role of tumor-activated hepatic stellate cells in experimental melanoma metastasis. Hepatology 2003; 37: 674–685 [DOI] [PubMed] [Google Scholar]
- 43.Turkson J, Bowman T, Garcia R, Caldenhoven E, De Groot RP, Jove R. Stat3 activation by Src induces specific gene regulation and is required for cell transformation. Mol Cell Biol 1998; 18: 2545–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bromberg JF, Horvath CM, Besser D, Lathem WW, Darnell JE. Stat3 activation is required for cellular transformation by v-src. Mol Cell Biol 1998; 18: 2553–2558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol 2007; 7: 41–51 [DOI] [PubMed] [Google Scholar]
- 46.Ffrench-Constant C Alternative splicing of fibronectin – many different proteins but few different functions. Exp Cell Res 1995; 221: 261–271 [DOI] [PubMed] [Google Scholar]
- 47.Ou JJ, Wu F, Liang HJ. Colorectal tumor derived fibronectin alternatively spliced EDA domain exerts lymphangiogenic effect on human lymphatic endothelial cells. Cancer Biol Ther 2010; 9: 186–191 [DOI] [PubMed] [Google Scholar]
- 48.Ou J, Li J, Pan F, Xie G, Zhou Q, Huang H, Liang H. Endostatin suppresses colorectal tumor-induced lymphangiogenesis by inhibiting expression of fibronectin extra domain A and integrin alpha9. J Cell Biochem 2011; 112: 2106–2114 [DOI] [PubMed] [Google Scholar]
- 49.Jarnagin WR, Rockey DC, Koteliansky VE, Wang SS, Bissell DM. Expression of variant fibronectins in wound healing: cellular source and biological activity of the EIIIA segment in rat hepatic fibrogenesis. J Cell Biol 1994; 127: 2037–2048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Manabe R, Ohe N, Maeda T, Fukuda T, Sekiguchi K. Modulation of cell-adhesive activity of fibronectin by the alternatively spliced EDA segment. J Cell Biol 1997; 139: 295–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.