

Abstract
Reactive oxygen species (ROS) are byproducts of oxygen metabolism best known for their damaging potential, but recent evidence has exposed their role as secondary messengers, which regulate cell function through redox-activatable signaling systems. In immune cells, specifically in T cells, redox-sensitive signaling pathways have been implicated in controlling several functional domains; including cell cycle progression, T effector cell differentiation, tissue invasion and inflammatory behavior. T cells from patients with the autoimmune disease rheumatoid arthritis (RA) have emerged as a valuable model system to examine the functional impact of ROS on T cell function. Notably, RA T cells are distinguished from healthy T cells based on reduced ROS production and undergo “reductive stress”. Upstream defects leading to the ROSlow status of RA T cells are connected to metabolic reorganization. RA T cells shunt glucose away from pyruvate and ATP production towards the pentose phosphate pathway, where they generate NADPH and consume cellular ROS. Downstream consequences of the ROSlow conditions in RA T cells include insufficient activation of the DNA repair kinase ATM, bypassing of the G2/M cell cycle checkpoint and biased differentiation of T cells into IFN-γ and IL-17–producing inflammatory cells. Also, ROSlow T cells rapidly invade into peripheral tissue due to dysregulated lipogenesis, excessive membrane ruffling, and overexpression of a motility module dominated by the scaffolding protein Tks5. These data place ROS into a pinnacle position in connecting cellular metabolism and protective versus auto-aggressive T cell immunity. Therapeutic interventions for targeted ROS enhancement instead of ROS depletion should be developed as a novel strategy to treat autoimmune tissue inflammation.
Keywords: Reactive oxygen species, rheumatoid arthritis, reductive stress, glycolysis, NADPH, ATM, podosomes, tissue invasion, TKS5
Graphical abstract
T cells from patients with Rheumatoid Arthritis
-
-
Metabolically reprogrammed
-
-
Biased towards biosynthesis and proliferation
-
-
ROSlow; reductive stress

Introduction
Metabolism of oxygen in mitochondria inevitably leads to the production of “free radicals”, molecules with unpaired electrons that are highly reactive and promptly introduce chemical modifications of proteins, lipids, and nucleic acids. The term “oxidative stress’ implies that such chemical reactions induce loss-of-function; amplifying the decline of molecular fidelity associated with disease and aging [1, 2]. Out of this dogma grew a high interest in antioxidant therapy. In line with the concept, cells are equipped with antioxidant defense systems, which rapidly detoxify ROS and repair any associated damage. Cell-endogenous antioxidant systems protect the redox balance through chemical buffering (best known is the reduced/oxidized glutathione system) or detoxify through enzyme-catalyzed reactions (e.g., catalase, superoxide dismutase). However, free radicals are much more than unwanted side products that cause harm [3].
Considering that mitochondria are the major source of intracellular ROS [4], monitoring of free radicals emerges as an elegant strategy to collect information on the metabolic activity of individual cells and coordinate cellular function, metabolic needs and energy outputs. As the final electron acceptor in the mitochondrial electron transport chain, oxygen is reduced to water, unless oxygen is prematurely reduced to the superoxide radical. Thus, local production of this radical reflects intactness of the electron transport chain, metabolic pressure the cell is exposed to and overall mitochondrial activity [5]. The superoxide radical acts locally, triggering mitochondrial redox-sensitive transcription factors that respond to the metabolic needs and coordinate mitochondrial biogenesis, glycolytic flux, and other components of the metabolic machinery. The superoxide radical can also diffuse into the cytoplasm to activate cytoplasmic redox-sensitive kinases, including AMP-activated protein kinase (AMPK), ataxia-telangiectasia mutated (ATM) and pyruvate kinase M2 (PKM2). Through reversible oxidation and reduction of specific amino acids, often reactive cysteine residues, the mitochondrial “ROS signal” can thus target a wide variety of cellular processes. Redox-dependent signaling does not fall under the dogma of “oxidative stress” and may overall be the more important function of ROS in cells, tissues and organs [6].
This review will focus on the signaling effects of ROS in T cells and will utilize T cells from patients with rheumatoid arthritis (RA) as a model system to explore the functional implications of ROS in directing protective and pathogenic T cell functions. T cells were chosen as they represent key drivers of the chronic inflammatory process that characterizes the autoimmune syndrome RA and by “memorizing” prior encounters in their life cycle are responsible for the chronicity of the disease. RA T cells have a distinct metabolic signature, which has been directly linked to their pathogenic behavior [7, 8]. Specifically, RA T cells have downregulated glycolytic breakdown and shunt glucose into the pentose phosphate pathway (PPP), reducing ROS production and enhancing NADPH generation. The surplus of reductive elements in the cellular milieu triggers a number of downstream events that culminate in a tissue-invasive, pro-inflammatory effector T cell. The impact of low concentrations of cellular ROS on redox-sensitive signaling loops that drive pathogenic immunity may be applicable to other chronic inflammatory conditions.
Loss of Reactive Oxygen Species in Effector T Cells
Like many other autoimmune conditions, RA has a long period of preclinical abnormalities during which self-tolerance is already broken and T and B cells against self-antigens are already detectable, but joint inflammation has not yet ensued [9, 10]. To address the question whether T cells from RA patients have intrinsic defects that render them susceptible to “low threshold” activation, studies have focused on responsiveness of naïve CD4 T cells, T cells that have not been involved in memory and inflammatory responses. Such naïve T cells may best capture the “disease prone” period in the life cycle of T cells. These studies have revealed that naïve CD4 T cells from RA patients have a distinct metabolic organization that segregates them from naïve CD4 T cells in age-matched healthy individuals. The pinnacle defect appears to be the slowdown of glycolytic activity [11] and the preference for the generation of NADPH and biosynthetic precursor molecules [12], equipping the cell for synthetic and proliferative functions. This metabolic reorganization is tightly linked to a shift in the redox status away from oxidative to reductive conditions.
Naïve CD4 T cells need to undergo massive clonal expansion to generate sufficient numbers of immunocompetent cells. Clonal proliferation is coupled to the transition of naïve cells into effector and memory cells and cellular differentiation, eventually assigning the T cell to a functional lineage, such as the Th1, Th2, Th17, etc lineage [13, 14]. Lineage commitment is closely linked to the production of cytokines, typically lineage-defining cytokines, such as IFN-γ, IL-4, IL-17, IL-21, etc. It is through these cytokines that T cells mediate their protective and pathogenic functions. The process of massive clonal expansion requires rapid access to fuel, which is typically supplied by glucose. Accordingly, normal CD4 T cells will upregulate the glycolytic machinery, import glucose, break it down to pyruvate, transport pyruvate into the mitochondria and generate ATP, ROS and Krebs cycle intermediates [15, 16]. Instead, RA T cells respond to activation with transcriptionally repressing the key glycolytic enzyme 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3) [11] and reduce their autophagic activity [17] (Fig. 1). PFKFB3 catalyzes the generation of fructose-2,6-bisphosphate, the most potent allosteric activator of phosphofructokinase-1 (PFK-1), the rate-limiting enzyme of glycolysis. PFK-1 is responsible for the first committed step of glycolysis in an irreversible reaction. Thus, by controlling the levels of fructose 2,6-bisphosphate, PFKFB3 dynamically regulates glycolytic flux. Because of PFKFB3 repression in RA T cells, intracellular pyruvate concentrations are diminished and ATP production shrinks; indicative of reduced mitochondrial activity. In essence, the cell is in a state of energy deprivation [18]. In parallel, RA T cells that undergo transition into effector cells upregulate glucose-6-phosphate dehydrogenase (G6PD), the gate keeper enzyme to the PPP and produce high levels of nicotinamide adenine dinucleotide phosphate (NADPH) [12]. Slowing of mitochondrial activity and preference for the PPP creates a redox milieu that consumes ROS and protects reduced glutathione. When measured with a redox-sensitive indicator, RA T cells hold about 50% of the intracellular ROS compared to a healthy control cell [12]. In essence, in T cells, glucose shunting into the PPP favors reductive conditions over oxidative pressure (Fig. 1) and metabolic reorganization in RA T cells creates an experimental model to explore the functional outcomes in ROSlow T cells.
Figure 1. Metabolic reorganization favors reductive conditions in T cells.
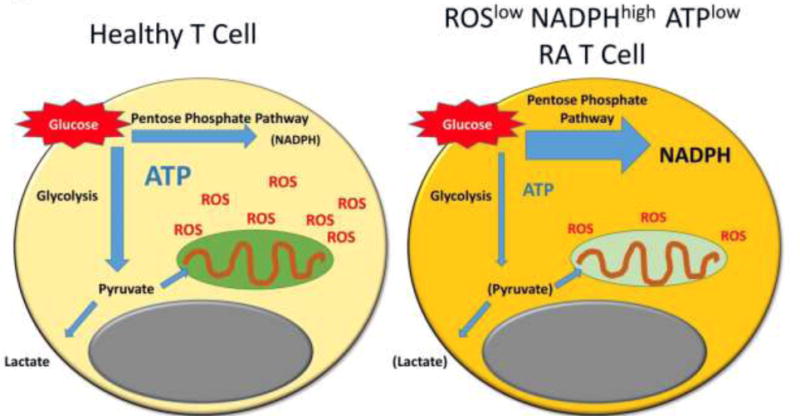
Healthy T cells undergoing activation have high energy and biosynthetic needs. They upregulate glucose import and glycolysis, driving mitochondrial activity and ROS production. T cells from patients with rheumatoid arthritis (RA) prefer shunting of glucose into the pentose phosphate pathway, thus reducing mitochondrial ROS production and enhancing NADPH generation. The outcome is a reduction of ATP and an intracellular milieu depleted of ROS. ROS, reactive oxygen species; ATP, adenosine triphosphate; NADPH, nicotinamide adenine dinucleotide phosphate
Ros-Dependent Functions in Effector T Cells
The cytoplasm contains multiple key signaling networks that are ultimately dependent on receiving ROS signals from the mitochondria. Prominent amongst the redox-activable signaling molecules are the energy sensor AMPK [19, 20], the cell cycle regulator and DNA repair kinase ATM [21, 22] and the glycolytic enzyme PKM2 [23, 24]. Based on the low ATP concentrations encountered in RA T cells (Fig. 1), one would expect that AMPK senses the energy-deprived state and initiates a program geared at ATP production, e.g. mitochondrial biogenesis and slowdown of cell building. This cellular adaptation appears to function insufficiently, but precise data on how AMPK responds to the metabolic reprogramming of RA T cells is not available.
ATM, a member of the phosphatidylinositol 3-kinase-related kinases (PIKKs) superfamily, is a serine/threonine protein kinase critical in orchestrating cellular responses to DNA double-strand breaks. Upon recognition of broken DNA, ATM coordinates a canonical DNA damage response, including slowing down of cell cycle passage, repair of the break and apoptotic death if damage cannot be repaired [25]. However, recent work emphasizes that ATM activation triggers a transcriptional program going beyond classical repair responses [26, 27]. Spontaneous T cell hyperactivation, unrestricted clonal expansion and excessive production of pro-inflammatory cytokines has been reported for mice deficient in the DNA repair cofactor ATM Interactor (ATMIN) [28]. ATMIN holds a key position in ATM-mediated signaling pathway choices [29]. DNA-damage induced signal transduction is critically important in T and B cell development, guiding the repair of programmed DNA lesions [30], but ATM remains a key player in regulating differentiation of peripheral T cells. ATM deficiency is known to reduce mitochondrial quality and interfere with nuclear-mitochondrial signaling [31]. Accordingly, ROS-dependent activation of ATM will impact a spectrum of cellular functions, not only DNA repair.
In healthy T cells, TCR-mediated stimulation induces rapid dimerization of the ATM molecule to prepare the molecule for phosphorylation [32]. ROSlow T cells in RA patients that are transitioning from naïve into effector state express lower amounts of ATM transcripts and the phosphorylated form of ATM is barely detectable [12]. The required dimerization of ATM is a typical consequence of oxidation [33, 34]. Thus, ROSlow RA T cells are essentially ATM deficient. Functional consequences include: (1) hyperproliferation, with the majority of cells passing through the cell cycle at a faster speed; (2) bypassing of the G2/M cell cycle checkpoint; and (3) accelerated conversion from the naïve into the memory state [7]. This phenotype of ROSlowATMlow RA T cells is in line with the conventional concept of how DNA damage responses affect the dynamics of cellular division (Fig. 2).
Figure 2. Functional consequences of ROSlow conditions in T cells.
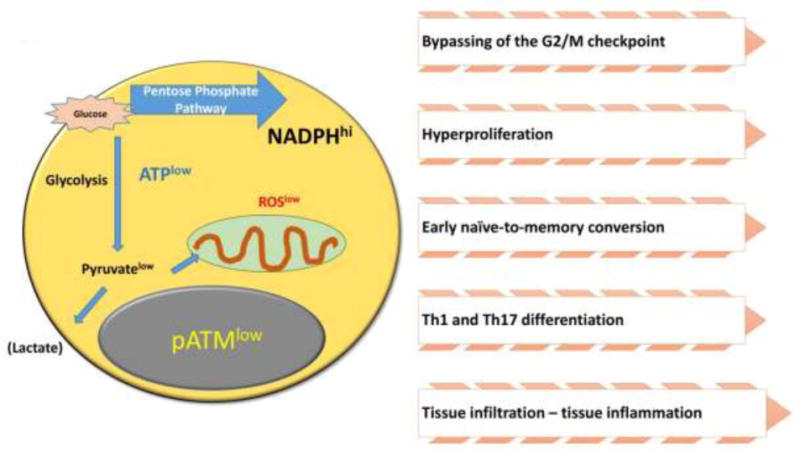
Beyond their role as damaging agents, ROS regulate cellular function through redox-activable signaling pathways. T cells from patients with rheumatoid arthritis (RA) consume intracellular ROS due to excess NADPH production, creating a reductive environment. A major target of ROS is the cell cycle kinase ATM, which regulates cell cycle behavior and differentiation. ATMlow RA T cells bypass the G2/M cell cycle checkpoint, hyperproliferate, differentiate into pro-inflammatory effector cells and infiltrate into peripheral tissues, where they cause sustained tissue inflammation.
In T cells, cell cycle progression is closely associated with polarization of the cells into functional subsets. Specifically, naïve T cells will differentiate into cytokine-secreting effector memory T cells. T cells producing IFN-γ have been named Th1 cells and together with IL-17–producing Th17 cells are considered pro-inflammatory effector cells. Culture of healthy naïve CD4 T cells in the presence of a ROS scavenger promotes lineage commitment to the Th1 and Th17 subset, whereas IL-4-producing Th2 cells and anti-inflammatory regulatory T cells seem unaffected [12]; phenocopying ROSlow RA T cells, that spontaneously commit to the Th1 and Th17 lineage in much higher frequencies (Fig. 2).
The critical function of ROS in determining pro- and anti-inflammatory effector functions has been tested in vivo. In a chimeric mouse model, in which human synovial tissue is engrafted and the mice are reconstituted with human T cells, T cells from RA patients can induce robust synovitis [12, 35]. Such chimeric mice have been treated with pro-oxidants: (A) buthionine sulphoximine (BSO), an inhibitor of gamma-glutamylcysteine synthase, which lowers glutathione concentrations in the tissue; and (B) menadione, a redox cycling agent, which forms a transient semiquinone to yield an extra electron to oxygen. Treatment with either BSO or menadione was highly efficient in suppressing tissue inflammation [12], directly implicating ROS in mediating anti-inflammatory actions.
These data add a different perspective to the conventional paradigm that ROS are tissue destructive and maintain inflammation through damaging proteins, membranes and DNA. Given the hypoxic conditions in chronically inflamed joints, ROS have been suspected to act as amplifiers of tissue injury [36, 37]. Nevertheless, antioxidants have not found their way into treatment of RA, but most success has been achieved with blocking pro-inflammatory cytokines. Anti-inflammatory functions of ROS have also been described in animal models of arthritis, where contrary to the prevailing paradigm disease-promoting genetic polymorphisms function by lowering ROS production [38, 39]. Holmdahl and colleagues have delineated the anti-inflammatory role of ROS in numerous disease models and have identified a common underlying mechanism, ROS-mediated suppression of macrophages that are mediators of chronic inflammation [40–42].
Thus, metabolic reconditioning can directly influence T cell behavior and sway adaptive immune responses towards sustained inflammation instead of resolution. ROS represent the mediator between metabolic status and functional differentiation. Here, reduction of ROS availability drives differentiating T cells towards disease-inducing functions (Fig. 2).
Dna Damage in ROSlow T cells
A major target of ROS is the nuclear DNA, where reactive molecules can damage the DNA nucleobases and the sugar phosphate backbone, causing a variety of DNA lesions [43–45]. The relevance of oxidative DNA damage is exemplified in hereditary human diseases, such as Xeroderma Pigmentosum, Cockayne Syndrome and Fanconi Anemia [46], all of which occur due to defects in repairing oxidative modifications of chromosomal DNA. To damage DNA, ROS must reach the nucleus and must be present in high concentrations. In most healthy cells, antioxidant defense systems are efficient enough to protect the nucleus from such deleterious effects. Subcellular distribution of intracellular ROS may also be a major determinant of outcome. ROSlow T cells from patients with RA offer a model system to examine the impact of reduced ROS concentrations on genome intactness.
In ROSlow RA T cells, the overall load of ROS is reduced, but at the same time ROS-dependent repair mechanisms are dismantled. Comparative examination of naïve and memory T cell populations in RA patients and age-matched healthy individuals have yielded evidence for a high load of DNA double-strand breaks in the patients [47, 48] (Fig. 3). These data suggest that genome integrity is ultimately determined by the repair system and that the role of ROS in activating repair enzymes outweighs their direct actions in DNA damage. Forced overexpression of ATM in ROSlow ATMlow RA T cells was sufficient to prevent double-strand break accumulation [47]. Interestingly, the lack of ATM in such T cells was associated with high expression of the DNA repair enzyme DNA-PKcs [48]; possibly the attempt of substituting DNA-PKcs activity for failing ATM action. ATM and DNA-PKcs belong to the phosphatidylinositol 3-kinase-related kinases (PIKK) family, a group of atypical kinases critical for DNA damage responses, nutrient-dependent signaling, and nonsense-mediated mRNA decay [49–51]. DNA-PKcs has been implicated in mediating apoptotic death of RA T cells [48].
Figure 3. Impact of impaired redox signaling on DNA repair, telomere intactness and cellular aging.
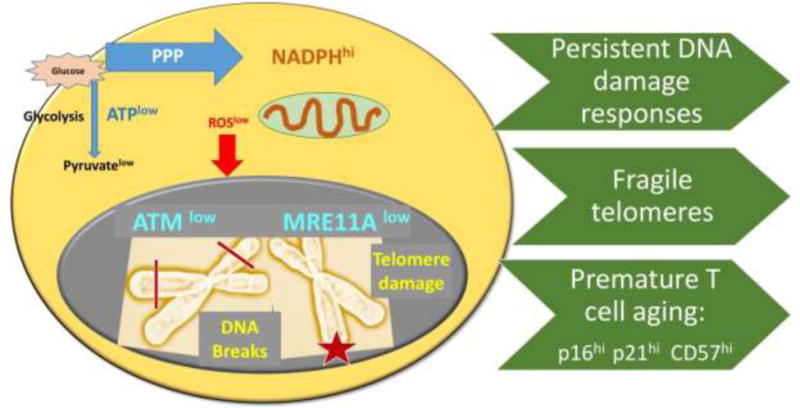
ATM-dependent DNA repair pathways function optimally in a ROS rich environment. ROSlow RA T cells lack proper function of the kinase ATM and the nuclease MRE11A. Consequences include inappropriate repair of DNA double strand breaks and insufficient repair of telomeric ends. With persistent DNA damage responses and fragile telomeres, RA T cells enter the aging program prematurely. RA T cells express a typical aging signature, including p16hi, p21hi, CD57hi.
ATM does not function alone but is part of a large protein complex that assembles around a DNA break. Prior to repair, the broken DNA ends need to be processed to enable religation; such as trimming of damaged overhangs, removal of fragments, etc. Here, the MRN complex [52–54] composed of three proteins, Mre11, Rad50 and Nbs1 has an important role in the initial processing of double-strand DNA breaks prior to repair by homologous recombination or non-homologous end joining. Quantitative studies in T cells from healthy individuals and RA patients have revealed that the inability to properly activate ATM is combined with dysfunction of the nuclease MRE11A (Fig. 3). Protein expression of MRE11A in human T cells is an age-dependent process, showing age-related decline in all individuals, but much earlier and much more profound in patients with RA.
MRE11A is a nuclease with both endo- and exonucleolytic activity [55, 56]. In human T cells, MRE11A accumulates at the telomeric ends [57] of chromosomes. RA T cells recruit distinctly low amounts of MRE11A to their telomeres and, associated with the deficiency of MRE11A, display a variety of telomeric damage lesions, including telomere fragility, apposition, fusion, and complete loss of end signals [35] (Fig. 3). In studies published more than a decade ago, RA T cells had been reported to have age-inappropriate shortening of telomeric sequences [58, 59]. Recent data suggest that this phenotype, which is indicative of premature aging, may not simply reflect proliferative pressure but may result from metabolic reorganization in the T cells, which lose redox-activatable signaling systems controlling the DNA repair machinery.
In contrast to the prevailing paradigm, which classifies ROS as drivers of cellular and organismal aging, emerging concepts emphasize that ROS-dependent signaling keeps cells young. ROSlow RA T cells fulfill many criteria identifying them as prematurely aged [18, 60, 61]. While they are not in irreversible cell cycle block, a characteristic of senescent cells, they have defects in nutrient sensing, are pro-inflammatory, escape from peripheral tolerance mechanisms and fail to properly repair their DNA. They share this phenotype with individuals born with mutated ATM that suffer from the progeroid syndrome Ataxia-telangiectasia (AT) [62, 63]. AT patients are known to have T cells that enter the aging process early in life [64]. Prematurity of immune aging may contribute to the cancer predisposition of AT patients. While sharing the deficiency of ATM signaling and DNA repair capacity, AT and RA patients arrive at this defect through distinct pathways. In RA patients, shunting of glucose into the PPP lies upstream of insufficient ATM activation and can be attributed to insufficient redox signaling (Figs. 1, 2). In contrast, the DNA repair defect due to mutated ATM eventually undermines antioxidant defense systems and is combined with oxidative pressure [65, 66].
ABNORMAL MIGRATORY BEHAVIOR OF ROSLOW T CELLS
A key functional task of RA T cells is their ability to infiltrate into the tissue and establish organized lymphoid structures [67, 68]. This task marks the transition from the preclinical to the clinical phase of RA. Once RA T cells leave the blood stream, transmigrate through the endothelial cell layer, and enter the subendothelial matrix, they must be able to maneuver through the extravascular space to reach their destination. Human T cells need to form invasive podosomes to palpate the surface and form transcellular pores through the endothelium to migrate out of the vasculature into the tissue site [69, 70]. It is believed that human T cells use the amoeboid migration mode to move in porous matrices, but do not employ matrix proteolysis [71]. To understand whether the ROSlow state of RA T cell is in any way related to the key pathogenic task of enabling tissue invasion of pro-inflammatory T cells, a T cell motility module was defined [72, 73]. Metabolically intact healthy T cells and metabolically reprogrammed RA T cells could be distinguished based on a package of 10 genes, all related to the formation of membrane protrusions involved in tissue invasion. Expression of Tks5, also known as SH3PXD2A, was directly related to the metabolic activity of RA T cells. Tks5 is a known Src kinase substrate and functions as a tether for transport vesicles to build membrane extensions at the leading edge of a migrating cell [74, 75]. Two metabolic interventions could change the expression of Tks5 and revert the spontaneous hypermigratory phenotype of RA T cells; supplementing pyruvate to enhance mitochondrial function and mitochondrial ROS production and inhibiting fatty acid synthesis [72]. Obviously, fatty acids serve as precursor molecules of phospholipids, which built the lipid bilayers of cell membranes. Dynamic restructuring of cell membranes is a prerequisite of generating invasive membrane protrusions. Fatty acid synthesis [76, 77] relies on acetyl-CoA and NADPH, two metabolic intermediates favored under conditions of restricted mitochondrial activity and high flux into the PPP (Figs. 1, 4).
Figure 4. ROS deficiency promotes cellular hypermotility and tissue invasiveness.
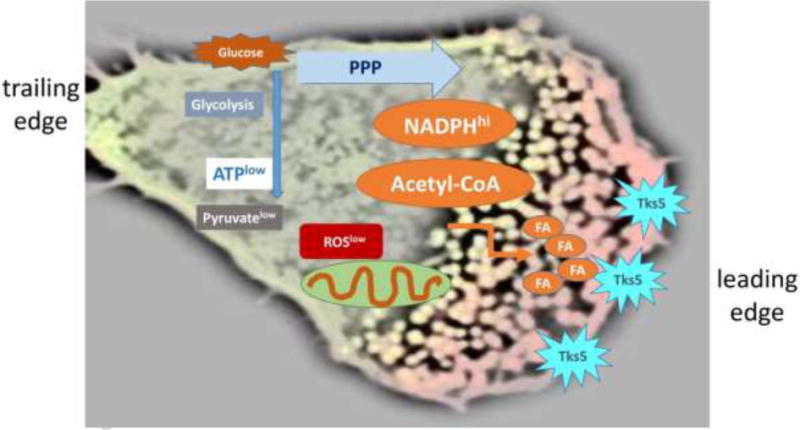
Metabolic reorganization in RA T cells results in a NADPHhi, acetyl-CoAhi, ATPlow, pyruvatelow, ROSlow environment. Excess NADPH and acetyl-CoA favor lipogenesis, lipid droplet deposition, membrane ruffling and hypermotility. The adaptor molecule Tks5, which focalizes actin polymerization, is a marker of T cell podosomes. Podosomes are formed at the cell’s leading edge, where they facilitate tissue invasiveness. FA, fatty acid.
Detailed metabolic analysis of ROSlow RA T cells has yielded important insights into the nutrient streams and the composition of metabolic intermediates: due to the shunting of glucose into the PPP, NADPH and reduced glutathione outweigh intracellular ROS [7, 78]. Depressed glycolytic breakdown reduces pyruvate production and essentially starves the mitochondria. Inactive mitochondria not only lessen the release of mitochondrial ROS, they enable accumulation of acetyl-CoA and fail to contribute to the breakdown of fatty acid storage through β-oxidation (Fig. 4). With excess acetyl-CoA and NADPH, ROSlow RA T cells have ideal conditions to enhance fatty acid synthesis and dynamically restructure their plasma membranes. Hypermotility in RA T cells is associated with the formation of lipid droplets which are deposited in the cytoplasm and can be visualized in circulating as well as tissue-resident T cells populating chronic inflammatory lesions [72] (Fig. 4).
The tight connection between T cell motility and pathogenic behavior was exemplified in experiments that measured the tissue-inflammatory potential of T cells in a human synovium-SCID chimeric model. In this model system, human synovial membrane is engrafted into immunocompromised mice. These mice are reconstituted with human peripheral blood mononuclear cells. T cells from RA patients are capable of inducing robust synovitis, whereas healthy normal T cells cannot break self-tolerance and stay out of the synovial tissue. Knockdown of Tks5 was sufficient to rescue the tissue-invasive phenotype of patient-derived T cells [72]. Vice versa, forced overexpression of Tks5 was sufficient to render healthy T cells tissue invasive. Metabolic interference in such chimeric mice proved the mechanistic relationship of glycolytic flux, fatty acid synthesis, ROS production, podosome formation and tissue-invasive behavior. Metabolic manipulations that increased intracellular ROS levels were adequate to counteract the phenotype of hypermotile T cells and suppressed tissue inflammation.
Thus, the emerging paradigm perceives ROS as powerful metabolic signaling molecules, reporting on the activity of mitochondria and the intensity of glycolytic flux. Metabolic constellations have immediate impact not only on energy generation and the building of biosynthetic precursor molecules, but also control core cellular functions, including the ability of T cells to transmigrate from the blood into tissue. The advantages of utilizing ROS are obvious-they directly correlate with mitochondrial function, they can be tightly controlled in their subcellular distribution, they can target essential signaling pathways and they are short-lived.
ROS as Effector Molecules in Anti-inflammatory T cells
Redox sensing in T cells is not limited to the regulation of complex effector functions, but redox-activatable signaling cascades are also triggered as part of T cell activation [79, 80] through the antigen receptor. Similar to their role in connecting metabolic needs with cellular behavior, ROS participate in early signaling events after antigen recognition [81]. The subcellular source of such signaling ROS are believed to originate from NADPH oxidases [82]. NADPH-oxidase derived superoxide is considered limiting in ensuring the development of antigen-specific CD4 T cell responses [83]. ROS generated by NADPH oxidases in CD8 T cells have been implicated in tuning JNK and NFκB signaling and ultimately regulate IFN-γ production and expression of the ATPase CD39 [84]. Excessive production of NADPH-derived ROS in T cells has been reported for patients with the autoimmune disease systemic sclerosis, with NADPH oxidase inhibition providing relief from unopposed T cell activation [85].
In keeping with the theme that ROS are linked to anti-inflammatory T cell responses, a recent study described that NADPH oxidase 2 (NOX2) is a critical effector molecule in immune-inhibitory CD8 T regulatory cells (Treg) [86, 87] (Fig. 5). CD8 Treg express CCR7 and are stationed in lymphoid tissues. They represent a small population in the circulation. Upon activation, they assemble NOX2 complexes in their membranes and form NOX2-containing exosomes. CD8 Treg interacting with close-by CD4 T cells transfer NOX2 onto such CD4+ neighbors and effectively inhibit their activation and proliferation (Fig. 5). In an in vivo model system, lack of NOX2+ CD8 Tregs caused overall expansion of CD4 T cells, implicating ROS in controlling the compartment size of CD4 T cells [86]. Lack of this critical function has been reported for two scenarios: (1) in healthy elderly, the inducibility of CD8 Tregs is impaired, in line with the concept that immune aging is associated with increased susceptibility to inflammatory disease; and (2) in patients with the autoimmune syndrome giant cell arteritis NOX2 CD8 Treg are essentially lacking [88]; predisposing such patients to difficult-to-control CD4 immunity. Thus, ROS production from NADPH may provide a protective mechanism to avoid tissue-destructive inflammation and participate in the broader regulation of T cell immunity.
Figure 5. ROS as anti-inflammatory effector molecules.

Regulatory T cells, formerly known as suppressor T cells, inhibit the function of other immune cells and are critically important in protecting from unopposed immunity and inflammation. A specialized subset of regulatory CD8 T cells produces membrane exosomes that are loaded with the NADPH oxidase 2. NOX2+ CD8 Treg release such exosomes onto CD4 effector T cells. The uptake of NOX2-containing microparticles creates a ROS-rich environment and blocks TCR-mediated signaling. As an end result, ROS paralyze T cell function.
Summary and Conclusions
This review has attempted to summarize available data on the functional implications of intracellaur ROS concentrations in human T cells. ROS, formerly considered harmful reagents, have recently been implicated in gain-of-function activities by targeting vital signaling pathways. In the cytoplasm, several critical signaling cascades are redox-activatable, directly linking cellular function with mitochondrial output. ROS produced as byproducts of the mitochondrial electron transport chain serve as sensitive measures of metabolic activity, which in turn is adapted to activating signals obtained from the cell’s environment. In T cells, activating signals lead to massive cellular expansion, imposing high needs for biosynthetic precursors to build new cells. In essence, ROS function as communicators between the metabolic machinery, cell cycle behavior and effector functions.
Defining ROS as critical contributors to the seamless integration of cellular networks collides with the long-held paradigm that ROS are fundamentally destructive, the source of tissue damage and critical in the organismal aging program. The most likely explanation for such opposing functional activities lies in the range of concentrations that ROS can span. Redox-sensitive transcription factors and cellular proteins respond with reversible biochemical modifications to low concentrations of ROS that are locally released. If ROS are generated in high amounts and antioxidant defense systems fail to remove them promptly, they have an opportunity to damage lipids, proteins and DNA. Enzymatically generated ROS, e.g. during respiratory burst, are intentionally concentrated to destroy their targets.
Recognition that ROS serve fundamental functions in cellular health have led to the unanticipated scenario that dysfunction is associated with lack of ROS. “Reductive stress” instead of oxidative stress has added new dimensions to the study of disease. A model system that has been particularly fruitful are T cells isolated from patients with RA. Recognized for their key function as drivers of pathogenic immune responses that manifest with tissue-injurious inflammation, RA T cells have recently attracted attention because they are metabolically reprogrammed. Specifically, they shunt glucose into the PPPs and away from glycolytic breakdown. This prepares RA T cells for high synthetic function, allowing them to massively proliferate to sustain inflammation. Shunting glucose into the PPP also generates a reductive environment with excess of NADPH and acetyl-CoA, which promotes lipogenesis and lipid droplet deposition. Reduced generation of pyruvate starves the mitochondria and further reduces ROS production. ROS deficiency results in a number of functional outcomes, which all promote and perpetuate pathogenic T cell function and inflammation. Underlying defects have been defined and include insufficient activation of redox-sensitive enzymes; including the cell cycle regulator and DNA repair kinase ATM. Despite their ROSlow status, RA T cells suffer from accumulation of damaged DNA and uncapped telomeres; emphasizing the critical role of ROS-driven repair pathways, whose failure is more important than the loss of ROS-mediated DNA damage. The reductive cellular milieu fosters lipogenesis despite the relative energy deficiency and mobilizes a program of membrane rebuilding needed for tissue invasion.
Exploiting ROS-activatable signaling systems for therapeutic purposes can possibly be achieved, if ROS concentrations can be fine-tuned, even at the low end of the concentration scale. Modifying the subcellular distribution of ROS could also be a fruitful approach. Given the impact of cellular metabolism on ROS production and spreading, redox-sensitive signaling events could be targeted by metabolic interference. For example, ROS generation could be enhanced in RA T cells by intensifying mitochondrial activity.
Obviously, further work is needed to understand the origin of the metabolic re-organization that leads to ROS deficiency and disruption of redox-dependent signaling pathways. Most of the studies in RA T cells have focused on the naïve population, which lives in secondary lymphoid organs, far away from the chronic inflammatory lesions that are sustained by unleashed T cells. How the metabolic circuitry in tissue-residing T cells is related to redox biology and functional outcome is much less understood. Since metabolites lie at the root of mitochondrial activity and ROS production, the tissue environment and the supply of glucose, amino acids, glutamine and lipids are almost certainly major determinants in promoting or dampening mitochondrial activity. Finally, consideration needs to be given to cell-type specific effects. Whereas long-lived T cells from RA patients have a signature of slowed glycolysis, short-lived macrophages from these patients have a strongly upregulated glycolytic machinery, generate excess ROS, oxidize cytoplasmic enzymes and function as hyperinflammatory cells; implicating ROS “signals” in multiple aspects of the disease process.
Highlights.
T cells undergoing activation use mitochondrial ROS to guide cellular behavior.
Pro-inflammatory T cells in rheumatoid arthritis are metabolically reprogrammed.
RA T cells fail to produce ROS and are under “reductive stress”.
ROSlow T cells insufficiently activate the kinase Ataxia telangiectasia mutated (ATM).
ATMlow T cells bypass the G2/M cell cycle checkpoint and maldifferentiate.
ROSlow T cells are hypermobile and tissue-invasive due to membrane ruffling.
Targeted ROS enhancement instead of ROS depletion may be immunomodulatory.
Acknowledgments
This work was supported by the National Institutes of Health (R01 AR042527, R01 HL117913, R01 AI108906 and P01 HL129941 to CMW and R01 AI108891, R01 AG045779, U19 AI057266, R01 AI129191 I01 BX001669 to JJG), the Praespero Foundation and the Cahill Discovery Fund.
List of Abbreviations
- AMPK
AMP-activated protein kinase
- AT
Ataxia-telangiectasia
- ATM
ataxia-telangiectasia mutated
- ATMIN
ATM Interactor
- ATP
adenosine triphosphate
- BSO
buthionine sulphoximine
- DNA-PKcs; NADPH
nicotinamide adenine dinucleotide phosphate
- PIKKs
phosphatidylinositol 3-kinase-related kinases
- PKM2
pyruvate kinase M2
- PFKFB3
6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3
- PPP
pentose phosphate pathway
- RA
rheumatoid arthritis
- ROS
reactive oxygen species
- Treg
T regulatory cells
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare no competing financial interests.
References
- 1.Sies H, Berndt C, Jones DP. Oxidative stress. Annu Rev Biochem. 2017;86:715–748. doi: 10.1146/annurev-biochem-061516-045037. [DOI] [PubMed] [Google Scholar]
- 2.Scialo F, Fernandez-Ayala DJ, Sanz A. Role of mitochondrial reverse electron transport in ROS signaling: Potential roles in health and disease. Front Physiol. 2017;8:428. doi: 10.3389/fphys.2017.00428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sack MN, Fyhrquist FY, Saijonmaa OJ, Fuster V, Kovacic JC. Basic biology of oxidative stress and the cardiovascular system: Part 1 of a 3-Part Series. J Am Coll Cardiol. 2017;70(2):196–211. doi: 10.1016/j.jacc.2017.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wong HS, Dighe PA, Mezera V, Monternier PA, Brand MD. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetics conditions. J Biol Chem. 2017;292(41):16804–16809. doi: 10.1074/jbc.R117.789271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drose S, Brandt U. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv Exp Med Biol. 2012;748:145–169. doi: 10.1007/978-1-4614-3573-0_6. [DOI] [PubMed] [Google Scholar]
- 6.Butt JN. Explorations of time and electrochemical potential: opportunities for fresh perspectives on signalling proteins. Biochem Soc Trans. 2014;42(1):47–51. doi: 10.1042/BST20130256. [DOI] [PubMed] [Google Scholar]
- 7.Weyand CM, Goronzy JJ. Immunometabolism in early and late stages of rheumatoid arthritis. Nat Rev Rheumatol. 2017;13(5):291–301. doi: 10.1038/nrrheum.2017.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang Z, Matteson EL, Goronzy JJ, Weyand CM. T-cell metabolism in autoimmune disease. Arthritis Res Ther. 2015;17:29. doi: 10.1186/s13075-015-0542-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mankia K, Emery P. Preclinical rheumatoid arthritis: Progress toward prevention. Arthritis Rheumatol. 2016;68(4):779–788. doi: 10.1002/art.39603. [DOI] [PubMed] [Google Scholar]
- 10.Ramwadhdoebe TH, Hahnlein J, Maijer KI, van Boven LJ, Gerlag DM, Tak PP, van Baarsen LG. Lymph node biopsy analysis reveals an altered immunoregulatory balance already during the at-risk phase of autoantibody positive rheumatoid arthritis. Eur J Immunol. 2016;46(12):2812–2821. doi: 10.1002/eji.201646393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang Z, Fujii H, Mohan SV, Goronzy JJ, Weyand CM. Phosphofructokinase deficiency impairs ATP generation, autophagy, and redox balance in rheumatoid arthritis T cells. J Exp Med. 2013;210(10):2119–2134. doi: 10.1084/jem.20130252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang Z, Shen Y, Oishi H, Matteson EL, Tian L, Goronzy JJ, Weyand CM. Restoring oxidant signaling suppresses proarthritogenic T cell effector functions in rheumatoid arthritis. Sci Transl Med. 2016;8(331):331ra338. doi: 10.1126/scitranslmed.aad7151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shih HY, Sciume G, Poholek AC, Vahedi G, Hirahara K, Villarino AV, Bonelli M, Bosselut R, Kanno Y, Muljo SA, O’Shea JJ. Transcriptional and epigenetic networks of helper T and innate lymphoid cells. Immunol Rev. 2014;261(1):23–49. doi: 10.1111/imr.12208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu J, Paul WE. CD4 T cells: fates, functions, and faults. Blood. 2008;112(5):1557–1569. doi: 10.1182/blood-2008-05-078154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Desdin-Mico G, Soto-Heredero G, Mittelbrunn M. Mitochondrial activity in T cells. Mitochondrion. doi: 10.1016/j.mito.2017.10.006. pii: (2017) S1567-7249(1517)30192-30197. [DOI] [PubMed] [Google Scholar]
- 16.Yong CS, Abba Moussa D, Cretenet G, Kinet S, Dardalhon V, Taylor N. Metabolic orchestration of T lineage differentiation and function. FEBS Lett. 2017;591(19):3104–3118. doi: 10.1002/1873-3468.12849. [DOI] [PubMed] [Google Scholar]
- 17.Yang Z, Goronzy JJ, Weyand CM. The glycolytic enzyme PFKFB3/phosphofructokinase regulates autophagy. Autophagy. 2014;10(2):382–383. doi: 10.4161/auto.27345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weyand CM, Yang Z, Goronzy JJ. T-cell aging in rheumatoid arthritis. Curr Opin Rheumatol. 2014;26(1):93–100. doi: 10.1097/BOR.0000000000000011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hart PC, Mao M, de Abreu AL, Ansenberger-Fricano K, Ekoue DN, Ganini D, Kajdacsy-Balla A, Diamond AM, Minshall RD, Consolaro ME, Santos JH, Bonini MG. MnSOD upregulation sustains the Warburg effect via mitochondrial ROS and AMPK-dependent signalling in cancer. Nat Commun. 2015;6:6053. doi: 10.1038/ncomms7053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaminskyy VO, Zhivotovsky B. Free radicals in cross talk between autophagy and apoptosis. Antioxid Redox Signal. 2014;21(1):86–102. doi: 10.1089/ars.2013.5746. [DOI] [PubMed] [Google Scholar]
- 21.Paull TT. Mechanisms of ATM Activation. Annu Rev Biochem. 2015;84:711–738. doi: 10.1146/annurev-biochem-060614-034335. [DOI] [PubMed] [Google Scholar]
- 22.Stankovic-Valentin N, Drzewicka K, Konig C, Schiebel E, Melchior F. Redox regulation of SUMO enzymes is required for ATM activity and survival in oxidative stress. EMBO J. 2016;35(12):1312–1329. doi: 10.15252/embj.201593404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shirai T, Nazarewicz RR, Wallis BB, Yanes RE, Watanabe R, Hilhorst M, Tian L, Harrison DG, Giacomini JC, Assimes TL, Goronzy JJ, Weyand CM. The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J Exp Med. 2016;213(3):337–354. doi: 10.1084/jem.20150900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luo W, Semenza GL. Emerging roles of PKM2 in cell metabolism and cancer progression. Trends Endocrinol Metab. 2012;23(11):560–566. doi: 10.1016/j.tem.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blackford AN, Jackson SP. ATM, ATR, and DNA-PK: The trinity at the heart of the DNA damage response. Mol Cell. 2017;66(6):801–817. doi: 10.1016/j.molcel.2017.05.015. [DOI] [PubMed] [Google Scholar]
- 26.Bednarski JJ, Sleckman BP. Lymphocyte development: integration of DNA damage response signaling. Adv Immunol. 2012;116:175–204. doi: 10.1016/B978-0-12-394300-2.00006-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guleria A, Chandna S. ATM kinase: Much more than a DNA damage responsive protein. DNA Repair (Amst) 2016;39:1–20. doi: 10.1016/j.dnarep.2015.12.009. [DOI] [PubMed] [Google Scholar]
- 28.Prochazkova J, Sakaguchi S, Owusu M, Mazouzi A, Wiedner M, Velimezi G, Moder M, Turchinovich G, Hladik A, Gurnhofer E, Hayday A, Behrens A, Knapp S, Kenner L, Ellmeier W, Loizou JI. DNA Repair Cofactors ATMIN and NBS1 Are Required to Suppress T Cell Activation. PLoS Genet. 2015;11(11):e1005645. doi: 10.1371/journal.pgen.1005645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang T, Penicud K, Bruhn C, Loizou JI, Kanu N, Wang ZQ, Behrens A. Competition between NBS1 and ATMIN controls ATM signaling pathway choice. Cell Rep. 2012;2(6):1498–1504. doi: 10.1016/j.celrep.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 30.Prochazkova J, Loizou JI. Programmed DNA breaks in lymphoid cells: repair mechanisms and consequences in human disease. Immunology. 2016;147(1):11–20. doi: 10.1111/imm.12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fang EF, Kassahun H, Croteau DL, Scheibye-Knudsen M, Marosi K, Lu H, Shamanna RA, Kalyanasundaram S, Bollineni RC, Wilson MA, Iser WB, Wollman BN, Morevati M, Li J, Kerr JS, Lu Q, Waltz TB, Tian J, Sinclair DA, Mattson MP, Nilsen H, Bohr VA. NAD+ replenishment improves lifespan and healthspan in ataxia telangiectasia models via mitophagy and DNA repair. Cell Metab. 2016;24(4):566–581. doi: 10.1016/j.cmet.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ditch S, Paull TT. The ATM protein kinase and cellular redox signaling: beyond the DNA damage response. Trends Biochem Sci. 2012;37(1):15–22. doi: 10.1016/j.tibs.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kozlov SV, Waardenberg AJ, Engholm-Keller K, Arthur JW, Graham ME, Lavin M. Reactive oxygen species (ROS)-activated ATM-dependent phosphorylation of cytoplasmic substrates identified by large-scale phosphoproteomics screen. Mol Cell Proteomics. 2016;15(3):1032–1047. doi: 10.1074/mcp.M115.055723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perry JJ, Tainer JA. All stressed out without ATM kinase. Sci Signal. 2011;4(167):pe18. doi: 10.1126/scisignal.2001961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Y, Shen Y, Hohensinner P, Ju J, Wen Z, Goodman SB, Zhang H, Goronzy JJ, Weyand CM. Deficient activity of the nuclease MRE11A induces T cell aging and promotes arthritogenic effector functions in patients with rheumatoid arthritis. Immunity. 2016;45(4):903–916. doi: 10.1016/j.immuni.2016.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khojah HM, Ahmed S, Abdel-Rahman MS, Hamza AB. Reactive oxygen and nitrogen species in patients with rheumatoid arthritis as potential biomarkers for disease activity and the role of antioxidants. Free Radic Biol Med. 2016;97:285–291. doi: 10.1016/j.freeradbiomed.2016.06.020. [DOI] [PubMed] [Google Scholar]
- 37.Kundu S, Ghosh P, Datta S, Ghosh A, Chattopadhyay S, Chatterjee M. Oxidative stress as a potential biomarker for determining disease activity in patients with rheumatoid arthritis. Free Radic Res. 2012;46(12):1482–1489. doi: 10.3109/10715762.2012.727991. [DOI] [PubMed] [Google Scholar]
- 38.Hultqvist M, Olsson LM, Gelderman KA, Holmdahl R. The protective role of ROS in autoimmune disease. Trends Immunol. 2009;30(5):201–208. doi: 10.1016/j.it.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 39.Gelderman KA, Hultqvist M, Olsson LM, Bauer K, Pizzolla A, Olofsson P, Holmdahl R. Rheumatoid arthritis: the role of reactive oxygen species in disease development and therapeutic strategies. Antioxid Redox Signal. 2007;9(10):1541–1567. doi: 10.1089/ars.2007.1569. [DOI] [PubMed] [Google Scholar]
- 40.Holmdahl R, Sareila O, Olsson LM, Backdahl L, Wing K. Ncf1 polymorphism reveals oxidative regulation of autoimmune chronic inflammation. Immunol Rev. 2016;269(1):228–247. doi: 10.1111/imr.12378. [DOI] [PubMed] [Google Scholar]
- 41.Hultqvist M, Olofsson P, Wallner FK, Holmdahl R. Pharmacological potential of NOX2 agonists in inflammatory conditions. Antioxid Redox Signal. 2015;23(5):446–459. doi: 10.1089/ars.2013.5788. [DOI] [PubMed] [Google Scholar]
- 42.Sareila O, Kelkka T, Pizzolla A, Hultqvist M, Holmdahl R. NOX2 complex-derived ROS as immune regulators. Antioxid Redox Signal. 2011;15(8):2197–2208. doi: 10.1089/ars.2010.3635. [DOI] [PubMed] [Google Scholar]
- 43.Markkanen E. Not breathing is not an option: How to deal with oxidative DNA damage. DNA Repair (Amst) 2017;59:82–105. doi: 10.1016/j.dnarep.2017.09.007. [DOI] [PubMed] [Google Scholar]
- 44.Cadet J, Davies KJA, Medeiros MH, Di Mascio P, Wagner JR. Formation and repair of oxidatively generated damage in cellular DNA. Free Radic Biol Med. 2017;107:13–34. doi: 10.1016/j.freeradbiomed.2016.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shafirovich V, Geacintov NE. Removal of oxidatively generated DNA damage by overlapping repair pathways. Free Radic Biol Med. 2017;107:53–61. doi: 10.1016/j.freeradbiomed.2016.10.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Taniguchi T, D’Andrea AD. Molecular pathogenesis of Fanconi anemia: recent progress. Blood. 2006;107(11):4223–4233. doi: 10.1182/blood-2005-10-4240. [DOI] [PubMed] [Google Scholar]
- 47.Shao L, Fujii H, Colmegna I, Oishi H, Goronzy JJ, Weyand CM. Deficiency of the DNA repair enzyme ATM in rheumatoid arthritis. J Exp Med. 2009;206(6):1435–1449. doi: 10.1084/jem.20082251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shao L, Goronzy JJ, Weyand CM. DNA-dependent protein kinase catalytic subunit mediates T-cell loss in rheumatoid arthritis. EMBO Mol Med. 2010;2(10):415–427. doi: 10.1002/emmm.201000096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lempiainen H, Halazonetis TD. Emerging common themes in regulation of PIKKs and PI3Ks. EMBO J. 2009;28(20):3067–3073. doi: 10.1038/emboj.2009.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434(7033):605–611. doi: 10.1038/nature03442. [DOI] [PubMed] [Google Scholar]
- 51.Abraham RT. PI 3-kinase related kinases: 'big' players in stress-induced signaling pathways. DNA Repair (Amst) 2004;3(8–9):883–887. doi: 10.1016/j.dnarep.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 52.van den Bosch M, Bree RT, Lowndes NF. The MRN complex: coordinating and mediating the response to broken chromosomes. EMBO Rep. 2003;4(9):844–849. doi: 10.1038/sj.embor.embor925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Williams RS, Williams JS, Tainer JA. Mre11-Rad50-Nbs1 is a keystone complex connecting DNA repair machinery, double-strand break signaling, and the chromatin template. Biochem Cell Biol. 2007;85(4):509–520. doi: 10.1139/O07-069. [DOI] [PubMed] [Google Scholar]
- 54.Hohl M, Kwon Y, Galvan SM, Xue X, Tous C, Aguilera A, Sung P, Petrini JH. The Rad50 coiled-coil domain is indispensable for Mre11 complex functions. Nat Struct Mol Biol. 2011;18(10):1124–1131. doi: 10.1038/nsmb.2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Deshpande RA, Lee JH, Arora S, Paull TT. Nbs1 converts the human Mre11/Rad50 nuclease complex into an endo/exonuclease machine specific for protein-DNA adducts. Mol Cell. 2016;64(3):593–606. doi: 10.1016/j.molcel.2016.10.010. [DOI] [PubMed] [Google Scholar]
- 56.Shibata A, Moiani D, Arvai AS, Perry J, Harding SM, Genois MM, Maity R, van Rossum-Fikkert S, Kertokalio A, Romoli F, Ismail A, Ismalaj E, Petricci E, Neale MJ, Bristow RG, Masson JY, Wyman C, Jeggo PA, Tainer JA. DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol Cell. 2014;53(1):7–18. doi: 10.1016/j.molcel.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Porro A, Feuerhahn S, Lingner J. TERRA-reinforced association of LSD1 with MRE11 promotes processing of uncapped telomeres. Cell Rep. 2014;6(4):765–776. doi: 10.1016/j.celrep.2014.01.022. [DOI] [PubMed] [Google Scholar]
- 58.Schonland SO, Lopez C, Widmann T, Zimmer J, Bryl E, Goronzy JJ, Weyand CM. Premature telomeric loss in rheumatoid arthritis is genetically determined and involves both myeloid and lymphoid cell lineages. Proc Natl Acad Sci U S A. 2003;100(23):13471–13476. doi: 10.1073/pnas.2233561100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fujii H, Shao L, Colmegna I, Goronzy JJ, Weyand CM. Telomerase insufficiency in rheumatoid arthritis. Proc Natl Acad Sci U S A. 2009;106(11):4360–4365. doi: 10.1073/pnas.0811332106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goronzy JJ, Weyand CM. Successful and maladaptive T cell aging. Immunity. 2017;46(3):364–378. doi: 10.1016/j.immuni.2017.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goronzy JJ, Weyand CM. Immune aging and autoimmunity. Cell Mol Life Sci. 2012;69(10):1615–1623. doi: 10.1007/s00018-012-0970-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Carney EF, Srinivasan V, Moss PA, Taylor AM. Classical ataxia telangiectasia patients have a congenitally aged immune system with high expression of CD95. J Immunol. 2012;189(1):261–268. doi: 10.4049/jimmunol.1101909. [DOI] [PubMed] [Google Scholar]
- 63.Exley AR, Buckenham S, Hodges E, Hallam R, Byrd P, Last J, Trinder C, Harris S, Screaton N, Williams AP, Taylor AM, Shneerson JM. Premature ageing of the immune system underlies immunodeficiency in ataxia telangiectasia. Clin Immunol. 2011;140(1):26–36. doi: 10.1016/j.clim.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 64.Kraus M, Lev A, Simon AJ, Levran I, Nissenkorn A, Levi YB, Berkun Y, Efrati O, Amariglio N, Rechavi G, Somech R. Disturbed B and T cell homeostasis and neogenesis in patients with ataxia telangiectasia. J Clin Immunol. 2014;34(5):561–572. doi: 10.1007/s10875-014-0044-1. [DOI] [PubMed] [Google Scholar]
- 65.Erttmann SF, Hartlova A, Sloniecka M, Raffi FA, Hosseinzadeh A, Edgren T, Rofougaran R, Resch U, Fallman M, Ek T, Gekara NO. Loss of the DNA damage repair kinase ATM impairs inflammasome-dependent anti-bacterial innate immunity. Immunity. 2016;45(1):106–118. doi: 10.1016/j.immuni.2016.06.018. [DOI] [PubMed] [Google Scholar]
- 66.Mongiardi MP, Stagni V, Natoli M, Giaccari D, D’Agnano I, Falchetti ML, Barila D, Levi A. Oxygen sensing is impaired in ATM-defective cells. Cell Cycle. 2011;10(24):4311–4320. doi: 10.4161/cc.10.24.18663. [DOI] [PubMed] [Google Scholar]
- 67.Takemura S, Braun A, Crowson C, Kurtin PJ, Cofield RH, O’Fallon WM, Goronzy JJ, Weyand CM. Lymphoid neogenesis in rheumatoid synovitis. J Immunol. 2001;167(2):1072–1080. doi: 10.4049/jimmunol.167.2.1072. [DOI] [PubMed] [Google Scholar]
- 68.Seyler TM, Park YW, Takemura S, Bram RJ, Kurtin PJ, Goronzy JJ, Weyand CM. BLyS and APRIL in rheumatoid arthritis. J Clin Invest. 2005;115(11):3083–3092. doi: 10.1172/JCI25265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sage PT, Varghese LM, Martinelli R, Sciuto TE, Kamei M, Dvorak AM, Springer TA, Sharpe AH, Carman CV. Antigen recognition is facilitated by invadosome-like protrusions formed by memory/effector T cells. J Immunol. 2012;188(8):3686–3699. doi: 10.4049/jimmunol.1102594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Carman CV, Sage PT, Sciuto TE, de la Fuente MA, Geha RS, Ochs HD, Dvorak HF, Dvorak AM, Springer TA. Transcellular diapedesis is initiated by invasive podosomes. Immunity. 2007;26(6):784–797. doi: 10.1016/j.immuni.2007.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cougoule C, Van Goethem E, Le Cabec V, Lafouresse F, Dupre L, Mehraj V, Mege JL, Lastrucci C, Maridonneau-Parini I. Blood leukocytes and macrophages of various phenotypes have distinct abilities to form podosomes and to migrate in 3D environments. Eur J Cell Biol. 2012;91(11–12):938–949. doi: 10.1016/j.ejcb.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 72.Shen Y, Wen Z, Li Y, Matteson EL, Hong J, Goronzy JJ, Weyand CM. Metabolic control of the scaffold protein TKS5 in tissue-invasive, proinflammatory T cells. Nat Immunol. 2017;18(9):1025–1034. doi: 10.1038/ni.3808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tsokos GC. Fat T cells go to the joint. Nat Immunol. 2017;18(9):955–956. doi: 10.1038/ni.3810. [DOI] [PubMed] [Google Scholar]
- 74.Jacob A, Linklater E, Bayless BA, Lyons T, Prekeris R. The role and regulation of Rab40b-Tks5 complex during invadopodia formation and cancer cell invasion. J Cell Sci. 2016;129(23):4341–4353. doi: 10.1242/jcs.193904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Oikawa T, Oyama M, Kozuka-Hata H, Uehara S, Udagawa N, Saya H, Matsuo K. Tks5-dependent formation of circumferential podosomes/invadopodia mediates cell-cell fusion. J Cell Biol. 2012;197(4):553–568. doi: 10.1083/jcb.201111116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bazin R, Ferre P. Assays of lipogenic enzymes. Methods Mol Biol. 2001;155:121–127. doi: 10.1385/1-59259-231-7:121. [DOI] [PubMed] [Google Scholar]
- 77.Edmunds LR, Sharma L, Kang A, Lu J, Vockley J, Basu S, Uppala R, Goetzman ES, Beck ME, Scott D, Prochownik EV. c-Myc programs fatty acid metabolism and dictates acetyl-CoA abundance and fate. J Biol Chem. 2014;289(36):25382–25392. doi: 10.1074/jbc.M114.580662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Weyand CM, Zeisbrich M, Goronzy JJ. Metabolic signatures of T-cells and macrophages in rheumatoid arthritis. Curr Opin Immunol. 2017;46:112–120. doi: 10.1016/j.coi.2017.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jackson SH, Devadas S, Kwon J, Pinto LA, Williams MS. T cells express a phagocyte-type NADPH oxidase that is activated after T cell receptor stimulation. Nat Immunol. 2004;5(8):818–827. doi: 10.1038/ni1096. [DOI] [PubMed] [Google Scholar]
- 80.Devadas S, Zaritskaya L, Rhee SG, Oberley L, Williams MS. Discrete generation of superoxide and hydrogen peroxide by T cell receptor stimulation: selective regulation of mitogen-activated protein kinase activation and fas ligand expression. J Exp Med. 2002;195(1):59–70. doi: 10.1084/jem.20010659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Williams MS, Kwon J. T cell receptor stimulation, reactive oxygen species, and cell signaling. Free Radic Biol Med. 2004;37(8):1144–1151. doi: 10.1016/j.freeradbiomed.2004.05.029. [DOI] [PubMed] [Google Scholar]
- 82.Kwon J, Shatynski KE, Chen H, Morand S, de Deken X, Miot F, Leto TL, Williams MS. The nonphagocytic NADPH oxidase Duox1 mediates a positive feedback loop during T cell receptor signaling. Sci Signal. 2010;3(133):ra59. doi: 10.1126/scisignal.2000976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Padgett LE, Tse HM. NADPH oxidase-derived superoxide provides a third signal for CD4 T cell effector responses. J Immunol. 2016;197(5):1733–1742. doi: 10.4049/jimmunol.1502581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bai A, Moss A, Rothweiler S, Longhi MS, Wu Y, Junger WG, Robson SC. NADH oxidase-dependent CD39 expression by CD8(+) T cells modulates interferon gamma responses via generation of adenosine. Nat Commun. 2015;6:8819. doi: 10.1038/ncomms9819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Amico D, Spadoni T, Rovinelli M, Serafini M, D’Amico G, Campelli N, Svegliati Baroni S, Gabrielli A. Intracellular free radical production by peripheral blood T lymphocytes from patients with systemic sclerosis: role of NADPH oxidase and ERK1/2. Arthritis Res Ther. 2015;17:68. doi: 10.1186/s13075-015-0591-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wen Z, Shimojima Y, Shirai T, Li Y, Ju J, Yang Z, Tian L, Goronzy JJ, Weyand CM. NADPH oxidase deficiency underlies dysfunction of aged CD8+ Tregs. J Clin Invest. 2016;126(5):1953–1967. doi: 10.1172/JCI84181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Berger CT, Hess C. Neglected for too long? - CD8+ Tregs release NOX2-loaded vesicles to inhibit CD4+ T cells. J Clin Invest. 2016;126(5):1646–1648. doi: 10.1172/JCI87429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Watanabe R, Hosgur E, Zhang H, Wen Z, Berry G, Goronzy JJ, Weyand CM. Pro-inflammatory and anti-inflammatory T cells in giant cell arteritis. Joint Bone Spine. 2017;84(4):421–426. doi: 10.1016/j.jbspin.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]