

Abstract
Perfluoroalkyl acids (PFAAs) are ubiquitous and persistent environmental contaminants. Compounds such as perfluoroocanoic acid (PFOA), perfluorooctane sulfonate (PFOS), perfluorononanoic acid (PFNA), and perfluorohexane sulfonate (PFHxS) are readily found in the tissues of humans and wildlife. While PFOA and PFOS have been the subject of numerous studies since they were first described over a decade ago, less is known about the biological activity of PFHxS and PFNA. Most PFAAs are activators of peroxisome proliferator-activated receptor α (PPARα), although the biological effects of these compounds are likely mediated by other factors in addition to PPARα. To evaluate the effects of PFHxS and PFNA, male wild-type and PPARα-null mice were dosed by oral gavage with PFHxS (3 or 10 mg/kg/day), PFNA (1 or 3 mg/kg/day), or vehicle for 7 days, and liver gene expression was evaluated by full-genome microarrays. Gene expression patterns were then compared to historical in-house data for PFOA and PFOS in addition to the experimental hypolipidemic agent, WY-14,643. While WY-14,643 altered most genes in a PPARα-dependent manner, approximately 11–24% of regulated genes in PFAA-treated mice were independent of PPARα. The possibility that PFAAs regulate gene expression through other molecular pathways was evaluated. Using data available through a microarray database, PFAA gene expression profiles were found to exhibit significant similarity to profiles from mouse tissues exposed to agonists of the constitutive activated receptor (CAR), estrogen receptor α (ERα), and PPARγ. Human PPARγ and ERα were activated by all four PFAAs in trans-activation assays from the ToxCast screening program. Predictive gene expression biomarkers showed that PFAAs activate CAR in both genotypes and cause feminization of the liver transcriptome through suppression of signal transducer and activator of transcription 5B (STAT5B). These results indicate that, in addition to activating PPARα as a primary target, PFAAs also have the potential to activate CAR, PPARγ, and ERα as well as suppress STAT5B.
Keywords: perfluoroalkyl acid, transcript profiling, liver, ToxCast, peroxisome proliferator-activated receptor α, estrogen receptor alpha, constitutive activated receptor, peroxisome proliferator, activated receptor γ, STAT5B
1. Introduction
Perfluoroalkyl acids (PFAAs) are stable man-made chemicals that have been used for over 50 years in the manufacture of a wide range of industrial and consumer products. Health concerns related to this class of chemicals have been raised because widespread environmental distribution (Yamashita et al., 2005; Ellis et al., 2004; Wei et al., 2007; Boulanger et al., 2005) and bioaccumulation (reviewed by Houde et al., 2006) have been demonstrated. Contributing to these concerns are the long elimination half-lives in humans for certain PFAAs which can be in the order of years (Olsen et al., 2007). While perfluorooctanoic acid (PFOA) and perfluorooctane sulfonate (PFOS), the two most prevalent PFAAs in the environment, have attracted the greatest attention, additional PFAAs are known to contribute to the chemical body burden in humans. Data for 12 perfluorinated compounds, for example, were included in the most recent exposure report from the National Health and Nutrition Examination Survey (National Center for Health Statistics, 2014). This group included perfluorononanoic acid (PFNA) and perfluorohexane sulfonate (PFHxS), both of which have been measured in human sera at concentrations that are approximately 33–50% of that reported for PFOA (Kato et al., 2011). While serum levels of PFOS, and possibly PFOA, have begun to decline in the general population due to cessation of production by US manufacturers, levels of PFNA and PFHxS appear to be increasing (Kato et al., 2011; Olsen et al., 2011).
A number of PFAAs activate peroxisome proliferator-activated receptor α (PPARα) (Maloney and Waxman, 1999; Wolf et al., 2008a; Wolf et al., 2012). Like other PPARα activators, PFAAs induce liver tumors in rodents (Gonzalez and Shah, 2008). A comprehensive review concluded that the liver tumors that occur by this MOA are either not relevant or not likely to be relevant to humans (Corton et al., 2014). In addition to liver tumor formation, immune system modulation associated with exposure to PFOA and PFOS (reviewed by Dewitt et al., 2009) or PFNA (Rockwell et al., 2013) has been reported in animal studies. Further observations in laboratory animals include changes in mammary development and body composition related to altered endocrine function (reviewed by White et al., 2011), altered brain development (Johansson et al., 2008; Johansson et al., 2009; Onishchenko et al., 2011), and testicular toxicity (Shi et al., 2010) including in vitro effects of PFNA on either rat primary Sertoli cells or mixed testicular cells (Feng et al, 2010; Lindeman et al., 2011). Developmental toxicity has also been reported in rodents for a number of the long chain PFAAs (reviewed by Lau et al., 2007). PFNA was found to delay development and reduce neonatal survival in 129S1/SvlmJ mice based on a PPARα-dependent mode of action (Wolf et al., 2010), and this has also been shown in the CD-1 mouse (Das et al., 2015). Long term effects on blood pressure have been demonstrated in rats exposed to PFNA or PFOS during pregnancy as well (Rogers et al., 2014). PFHxS, on the other hand, has not been shown to be a reproductive or developmental toxicant in the rat (Butenhoff et al., 2009), although the number of laboratory studies conducted using PFHxS has been limited presumably due to compound availability.
The biological activity of PFAAs is not limited to activation of PPARα. There is evidence of PPARα-independent transcript regulation in mammals including activation of the constitutive androstane receptor (CAR) (Rosen et al., 2008a,b; Cheng and Klaassen 2008; Ren et al., 2009). Activation of estrogen receptor α (ERα) by various PFAAs has also been demonstrated in rainbow trout (Tilton et al., 2008; Benninghoff et al., 2011; Benninghoff et al., 2012) and was recently suggested in PFOA-exposed human primary hepatocytes (Buhrke et al, 2015). Bjork et al. (2011) utilized cultured primary rat and human hepatocytes and found that exposure to moderately high concentrations of either PFOA or PFOS could increase the expression of specific marker genes for not only PPARα, but CAR, pregnane X receptor (PXR), and liver X receptor (LXR). These investigators suggested that triggering these additional signaling pathways may be secondary to the metabolic remodeling which was initiated by activation of PPARα. Using a proteomic approach, Scharmach et al. (2012) was able to show that PFOA is an inhibitor of the HNF4α pathway in HepG2 cells. This same group came to a similar conclusion based on transcript analysis of human primary hepatocytes (Buhrke et al., 2015). A more recent study showed that human hepatocytes treated with PFOA or PFOS at a concentration relevant to occupational exposure caused a decrease in HNF4α protein without affecting HNF4α mRNA or causing cell death (Beggs et al., 2016). One of the more intriguing observations in treated Pparα-null mice is dysregulation of genes associated with fatty acid metabolism (Rosen et al., 2008a,b; Rosen et al., 2010). While such a result is generally associated with transactivation of PPARα, it is not clear why this outcome could occur in the absence of a functional PPARα. In the past, our group has speculated that activation of PPARγ or PPARβ could be involved (Rosen et al., 2008). Indeed, based on gene array data, Buhrke et al. (2015) suggested that PPARγ was activated in human primary hepatocytes exposed to PFOA. Teasing apart the effects of the various PPAR subtypes based on gene expression data, however, is difficult given the potential overlap in target genes among these nuclear receptors. Cell-based reporter assays have not provided convincing support for the role of PPARγ or PPARβ. Vanden Heuvel et al. (2006) and Taxvig et al. (2012) found only modest activation of PPARγ by either PFOA or PFOS, while other publications have generally been negative (Maloney and Waxman, 1999). Takacs and Abbott (2007) did observe significant activation of murine PPARβ by either PFOA or PFOS but neither compound was found to activate mouse or human PPARγ.
Here, we examine the transcript profiles of PFNA and PFHxS in the livers of both wild-type and Pparα-null mice. The goal of this study was to evaluate these PFAAs within the context of historical data generated by our group for PFOA and PFOS. The Nextbio database (Illumina, San Diego, CA) was utilized to allow for comparisons to microarray datasets associated with activation or inhibition of various nuclear receptors. ToxCast data (http://actor.epa.gov/dashboard/) was utilized as well. Emphasis was placed on identifying those effects that were independent of PPARα.
2. Materials and Methods
2.1. Animals and dosing
In-house studies were approved by the U.S. EPA ORD/NHEERL Institutional Animal Care and Use Committee. The facilities and procedures used followed the recommendations of the 1996 NRC “Guide for the Care and Use of Laboratory Animals”, the Animal Welfare Act, and the Public Health Service Policy on the Humane Care and Use of Laboratory Animals. Pparα-null mice (129S4/SvJae-Pparatm1Gonz/J, stock #003580) and wild-type mice (129S1/SvlmJ, stock #002448) were purchased from The Jackson Laboratory (Bar Harbor, ME) and maintained as an inbred colony on the 129/Sv background at the U.S. EPA, Research Triangle Park, NC. Animals were housed 4 per cage and allowed to acclimate for a period of one week prior to the conduct of the study. Commercial lab chow and municipal tap water were provided ad libitum. Animal facilities were controlled for temperature (20–24°C), relative humidity (40–60%), and kept under a 12 hr light-dark cycle. Pparα-null and wild-type male mice at 6–9 months of age were dosed by gavage for 7 consecutive days with either 0, 3, or 10 mg/kg PFHxS or 0, 1, or 3 mg/kg PFNA (#394459, Sigma-Aldrich, St, Louis, MO) in water. PFHxS (CAS # 355–46-4) was kindly provided by 3M Corp (St. Paul, MN). Dosing was based on previous work from our laboratory (PFNA, Das et al., 2015; PFHxS, based on a related chemical, PFOS, Rosen et al., 2010). All dose levels reflected exposures that produce hepatomegaly in wild-type adult mice without inducing overt toxicity. Four biological replicates consisting of individual animals were included in each dose group. Dose levels reflected exposures that produce hepatomegaly in adult mice without inducing overt toxicity. All dosing solutions were freshly prepared each day. At the end of the dosing period, animals were euthanized by CO2 asphyxiation and tissue was collected from the left lobe of the liver for preparation of total RNA as detailed below.
2.3. RNA preparation
Collected liver from PFNA and PFHxS treated mice (≤ 50 mg) was immediately placed in 1 ml RNAlater (Thermo Fisher/Life Technologies, Grand Island, NY) and stored at −20◦C. Total RNA preparations for microarray analysis were then prepared using the mirVANA miRNA isolation kit (Thermo Fisher/Life Technologies) according to the manufacturer’s protocol without further enrichment for small RNAs. All samples used in the study were quantified by determining the ratio of absorbance at 260 and 280 nm using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher, Waltham, MA). RNA quality was evaluated using a 2100 Bioanalyzer (Agilent, Palo Alto, CA) which calculates the RNA Integrity number. Only samples with an RNA Integrity number ≥ 7.5 were included in the study (2100 Expert software, version B.01.03).
2.4. Microarray data collection and analysis
In-house microarray hybridization and scanning was conducted at the U.S. EPA NHEERL Genomics Research Core Facility using Affymetrix 430_2 expression GeneChips according to the protocols recommended by the manufacturer (Manual #701021 rev. 5, Affymetrix, Santa Clara, CA). Sample labeling and microarray hybridization were conducted as a single block to minimize technical variation. In addition to PFHxS and PFNA (archived in Gene Expression Omnibus as GSE55756), three additional studies were analyzed to allow for examination of similarities and differences across five chemicals:
PFOA administered by gavage (3 mg/kg) for 7 days (GSE9786) (Rosen et al., 2008b).
PFOS administered by gavage (10 mg/kg) for 7 days (GSE22871) (Rosen et al., 2010).
WY-14,643 (0.1% w/w) mixed in the food and fed for 5 days (GSE8295) (Rakhshandehroo et al., 2007).
All studies were conducted using similar background genotypes of male mice. All but the WY study was conducted in the Lau lab. The microarray platform for all studies consisted of the Affymetrix mouse 430_2 chip. All Affymetrix .cel files were processed using the standard procedures described in Kuperschmidt et al. (2010). Robust multichip average was used to normalize the data. To address the potential unreliability of the fold-change metric at low intensity levels, genes with signals lower than a 20th percentile cutoff in both control and test groups were discarded. A Welch’s paired t-test was used to identify significantly altered genes with a p-value significance cutoff of 0.05 and a minimum absolute fold-change cutoff of 1.2. For comparisons of the gene lists to each other and for analysis in Ingenuity Pathways Analysis (IPA), the lists were further filtered for those genes with a false discovery rate of p < 0.05 and an absolute fold-change cutoff of ≥ 1.5.
2.5. Comparison of PFAA data to a gene expression database.
A rank-based nonparametric analysis strategy called the Running Fisher test implemented within the NextBio database (Illumina, San Diego, CA) was used to compare gene lists derived from PFAA treated mice to other gene lists in the database. This normalized ranking approach enables comparability across data from different studies, platforms, and analysis methods by removing dependence on absolute values of fold-change, and minimizing some of the effects of normalization methods used, while accounting for the level of genome coverage by the different platforms. The Running Fisher algorithm computes statistical significance of similarity between ranked fold-change values of two gene lists using a Fisher’s exact test (Kupershmidt et al., 2010). A p-value ≤ 1E-4 was selected as our cutoff for significance based on prior evaluation of the cutoff as predictive of activation of a number of transcription factors (Oshida et al., 2015a,b,c; Oshida et al., 2016a,b; Ryan et al., 2016). The PFAA gene lists were compared to gene lists from a number of studies that were analyzed in NextBio:
Livers of wild-type or CAR-null mice exposed to 100 mg/kg phenobarbital for three days (GSE40120).
Livers from ovariectomized wild-type and ERα-null mice exposed to the ERα non-steroidal agonist PPT (4,4’,4’’-(4-Propyl-[1H]-pyrazole-1,3,5-triyl)trisphenol) for 3 days (Pedram et al., 2013) ( GSE45346).
Livers from Lepr (db/db) mice fed for 18d 280umol/kg/d the PPARγ agonist rosiglitazone (Le Bouter et al., 2010) (GSE19896).
Mouse 3T3-L1 adipocytes exposed to four PPARγ full agonists (COOH, rosiglitazone, Full agonist2, Full agonist4) or an LXR agonist (“LXR agonist 1”) for 24 hours at 10uM (Tan et al., 2012) (GSE31222).
Mouse 3T3-L1 adipocytes were treated with a control siRNA or a siRNA targeting the Pparg gene (Schupp et al., 2009) (GSE14004)
2.6. Additional data analysis.
Liver weight was analyzed by genotype using one-way ANOVA followed by Tukey-Kramer honest significant difference (HSD) test to compare individual means (SAS JMP, ver.7.0). Significant probe sets were further evaluated for relevance to biological pathway and function using Ingenuity Pathway Analysis software (https://analysis.ingenuity.com). Hierarchical clustering and heat maps were generated using Eisen Lab Cluster and Treeview software (http://bonsai.hgc.jp/~mdehoon/software/cluster/software.htm). ToxCast data came from a Dashboard download (Dashboard_Export_08_31_15) from the ToxCast data website (http://actor.epa.gov/dashboard/) and filtered for data derived from exposures to the four PFAAs. The ability of the PFAAs to activate PPAR subtypes and ERα were evaluated using the trans-activation assays from Attagene, Inc. (Durham, NC) in which the ligand binding domains of each nuclear receptor were linked to the GAL4 DNA binding domain. The chemicals were also examined for ability to activate an estrogen responsive element (ERE) linked to a reporter gene. The ToxCast analysis pipeline fits dose response curves to data generated in over 700 in vitro high throughput screening assays (Judson et al 2010, Kavlock et al 2012). Constant, hill, and gain-loss models are fit to the data, and the best fitting model as determined by Akaike information criterion (AIC) is used to make hit calls and generate modeled AC50 values, hill slopes and maximal activities of predicted active chemicals. Baseline values for each assay are determined from DMSO (negative) controls, and a fold induction increase in signal cutoff (usually 3X the baseline median absolute deviation) is used to determine hit calls. Details of the analysis pipeline (“ToxCast Data Pipeline Overview”) are available on the ToxCast website at: http://www2.epa.gov/chemical-research/toxicity-forecaster-toxcasttm-data (accessed October 13, 2016).
3. Results
3.1. PFAA induction of hepatomegaly
Mice were treated for one week with daily gavage exposures of PFNA (1 and 3 mg/kg), PFHxS (3 and 10 mg/kg), or control vehicle. The effects of treatment on body and liver weights are shown in Tables 1 and 2. No change in body weight was observed with either compound. Increased relative liver weight was apparent in wild-type mice at both doses of PFNA or PFHxS, although the effect of PFHxS at the low dose was modest. In Pparα-null mice, liver weight increase was limited to the highest dose of either compound.
Table 1.
Body and liver weight in WT and PPARα-null mice exposed to PFNA for 7 days.
| WT | PPARα-null | ||||||
|---|---|---|---|---|---|---|---|
| 0 mg/kg | 1 mg/kg | 3 mg/kg | 0 mg/kg | 1 mg/kg | 3 mg/kg | ||
| N | 4 | 4 | 4 | 4 | 4 | 4 | |
| Body Weight | 28.1±1.6 | 29.2±1.6 | 29.2±3.0 | 30.5±8.0 | 30.3±3.6 | 30.8±3.4 | |
| Liver Weight | 0.94±0.06 | 1.54±0.14* | 1.93±0.16* | 1.20±0.24 | 1.41±0.20 | 1.83±0.30* | |
| Relative Liver Weight | 0.034±0.002 | 0.053±0.002* | 0.066±0.004* | 0.040±0.006 | 0.047±0.006 | 0.059±0.004* | |
Significantly different than control (p≤0.05)
Table 2.
Body and liver weight in WT and PPARα-null mice exposed to PFHxS for 7 days.
| WT | PPARα-null | ||||||
|---|---|---|---|---|---|---|---|
| 0 mg/kg | 3 mg/kg | 10 mg/kg | 0 mg/kg | 3 mg/kg | 10 mg/kg | ||
| N | 4 | 4 | 4 | 4 | 4 | 4 | |
| Body Weight | 29.03±2.00 | 29.80±0.90 | 29.15±2.16 | 30.83±5.00 | 29.40±3.98 | 30.23±4.18 | |
| Liver Weight | 0.99±0.10 | 1.18±0.08 | 1.68±0.14* | 1.07±0.16 | 1.13±0.14 | 1.50±0.08* | |
| Relative Liver Weight | 0.034±0.004 | 0.040±0.004* | 0.058±0.001* | 0.035±0.002 | 0.039±0.006 | 0.050±0.008* | |
Significantly different than control (p≤0.05)
3.2. PFAA-regulated gene expression is dominated by PPARα-dependent effects
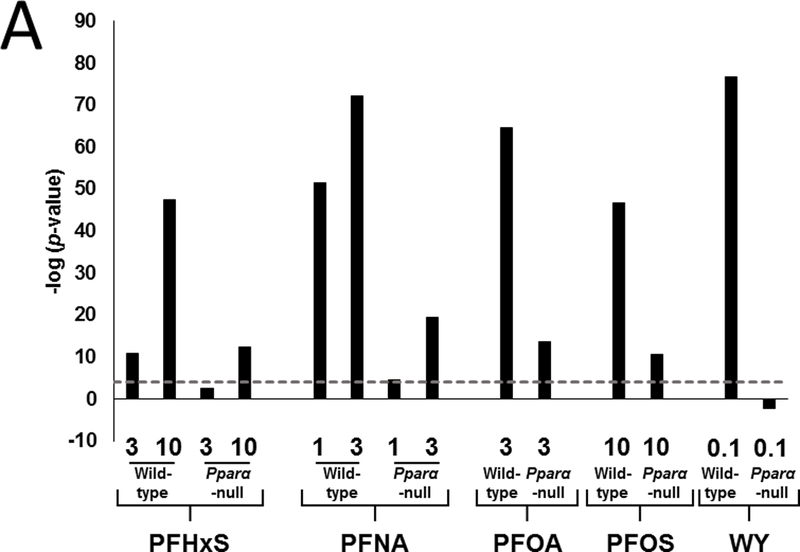
Changes in gene expression induced by PFHxS and PFNA were examined by microarray analysis. In order to facilitate the identification of factors which alter gene expression by other PFAAs, changes induced by PFOA and PFOS were examined using data that were previously collected by our lab under similar experimental conditions. In addition, a study in which wild-type and Pparα-null mice were treated with the specific PPARα agonist, WY-14,643 (WY) was included to allow PFAA-specific effects to be distinguished from those mediated by a more specific PPARα activator. Fig. 1A shows the number of genes that exhibited significant increased or decreased expression in each of the treatment groups (False Discovery Rate ≤ 0.05; |fold change| ≥ 1.5). Dose-dependent changes in differentially regulated genes were observed for PFHxS and PFNA in both wild-type and Pparα-null mice but fewer differentially regulated genes were observed in Pparα-null mice. PFOA also altered fewer genes in Pparα-null mice compared to wild-type mice. A greater percentage of total genes were altered by PFOS in Pparα-null mice compared to the other PFAAs. WY treatment altered the greatest number of genes in wild-type mice but also affected the fewest genes in Pparα-null mice. The complete list of genes altered by the treatments and pathway analysis is provided in Supplemental File 1.
Fig. 1. PPARα regulates the majority of PFAA-responsive genes.
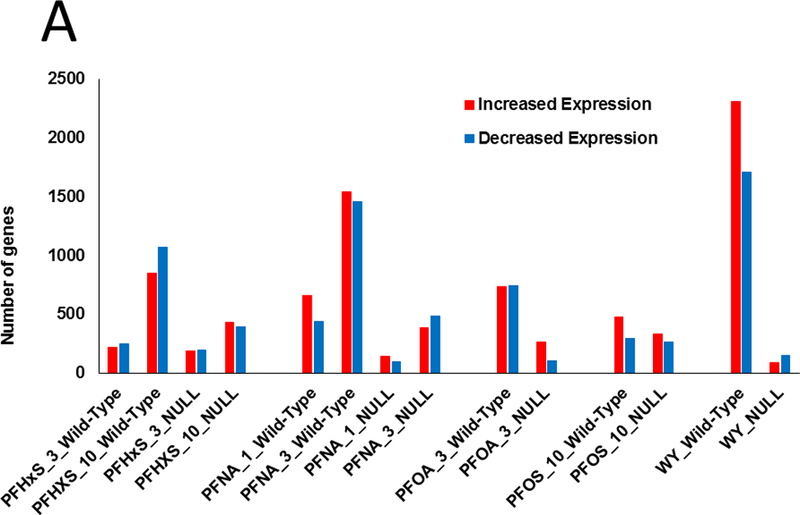
Significantly altered genes (FDR ≤ 0.05; |fold change| ≥ 1.5) were identified using procedures described in the Methods.
A. Number of genes with increased or decreased expression in each treatment group. Numbers refer to treatment in mg/kg/day.
B. Heat maps of the distribution of genes in wild-type and Pparα-null mice. Genes regulated by the PFAAs were divided into three classes: I, PPARα-dependent; II, PPARα-independent and similarly expressed in both genotypes; III, PPARα-independent and expressed only in Pparα-null mice. The heat maps are not proportional to the total number of genes that are altered by each treatment.
C. Percentage of genes that were determined to be PPARα-independent. Values were derived by calculating the number of genes in class II/number of genes in class I and II.
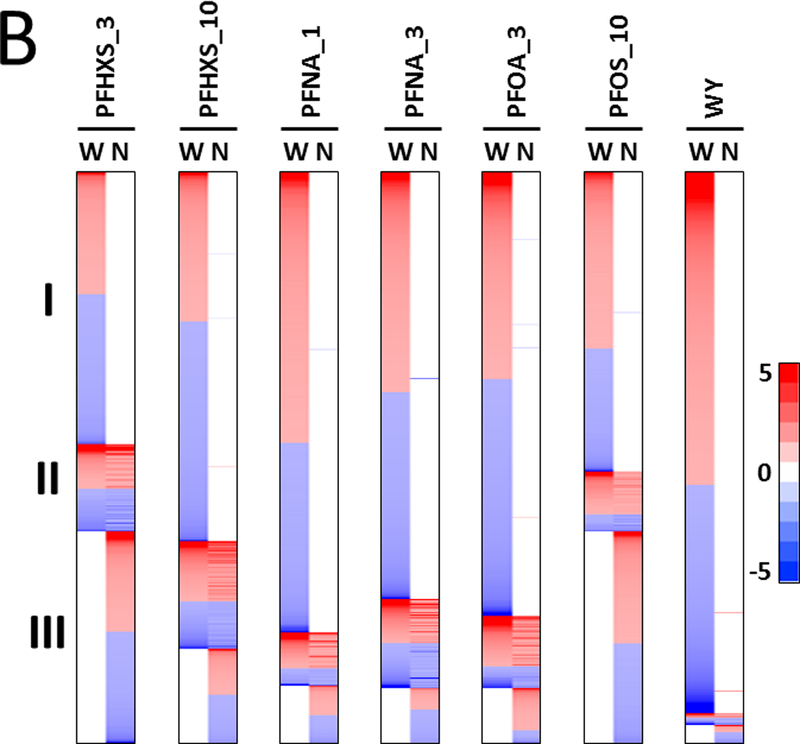
Earlier studies showed that PFOA and PFOS regulate the majority of genes in the mouse liver in a Pparα-dependent manner (Rosen et al., 2008a; Rosen et al., 2008b; Rosen et al., 2010). To determine if that was also the case for PFHxS and PFNA, gene expression was compared between the genotypes. Genes were divided into groups based on their expression behavior in the two genotypes. Class I genes were defined as completely dependent on PPARα for regulation in wild-type mice (i.e., no regulation in the same direction in Pparα-null mice compared to wild-type mice). Class II genes exhibited regulation in the same direction in both genotypes. Class III genes were regulated only in the Pparα-null mice. Genes were rank-ordered by fold-change in each group and summarized as heat maps for each chemical-dose treatment (Fig. 1B). Each chemical-dose pair altered the expression of different proportions of class I-III genes. The lowest dose of PFHxS regulated the smallest proportion of Pparα-dependent genes while WY regulated almost all genes in a Pparα-dependent manner. In the absence of PPARα, PFAA exposure led to a unique set of gene targets (class III genes). PFHxS and PFOS, the two sulfonated PFAAs, had the greatest proportion of class III genes relative to class II genes when compared to the non-sulfonated PFAAs.
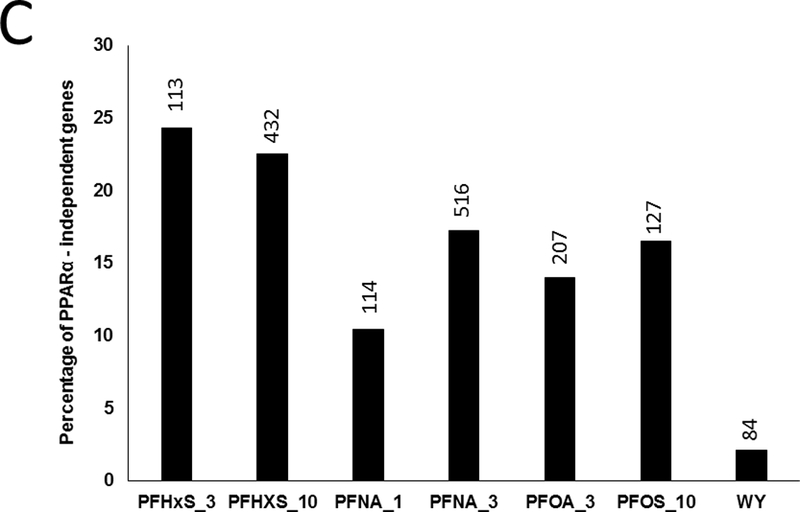
The percentage of PPARα-independent genes regulated specifically in wild-type mice (Class II genes) is summarized in Fig. 1C. This was expressed as the number of class II genes as a percent of the total number of differentially expressed genes in wild-type mice. Both doses of PFHxS altered the greatest percentage of PPARα-independent genes (24% and 22% for 3 and 10 mg/kg, respectively). The other PFAAs exhibited PPARα-independence between 10 and 17%. In contrast, only approximately 2% of WY-regulated genes were PPARα-independent. These results indicate that greater than ~75% of all genes regulated by PFAAs in wild-type mice are PPARα-dependent, and the degree of dependence varies by dose and chemical. Additionally, in the absence of PPARα, each PFAA regulated an additional set of genes unique from those regulated in wild-type mice.
3.3. Common activation of CAR by PFAAs
Earlier studies indicated that CAR is activated by exposure to PFOA and PFOS (Ren et al., 2009; Rosen et al., 2008b, 2010; Oshida et al., 2015b). If that is the case, similarities in the expression patterns between various PFAAs and the prototypical activator of CAR, phenobarbital, should be apparent. To test this hypothesis, gene lists derived from PFAA-treated mice were compared to lists derived from the livers of wild-type or CAR-null mice exposed to 100 mg/kg phenobarbital for three days (Chua and Moore, 2005). A Running Fisher Test was used to provide a similarity statistic for overlapping genes using pairwise comparisons. The same level of significance was used at that in previous studies (|–log(p-value)| ≥ 4; Oshida et al., 2015a,b,c). All PFAA treatment groups except the low dose of PFHxS and PFNA in Pparα-null mice exhibited significant similarity to wild-type mice treated with phenobarbital (Fig. 2A). In contrast, there was no positive similarity to phenobarbital treatment in Car-null mice across the PFAAs even though 633 genes were significantly altered by phenobarbital in Car-null mice. WY-treatment in wild-type or Pparα-null mice also did not exhibit similarity to phenobarbital treatment, as expected. Significant similarity between PFAA-treatment and a phenobarbital time course of up to 28 days in wild-type mice (Thomson et al., 2013) was also observed (data not shown).
Fig. 2. CAR activation by PFAAs is PPARα-independent.
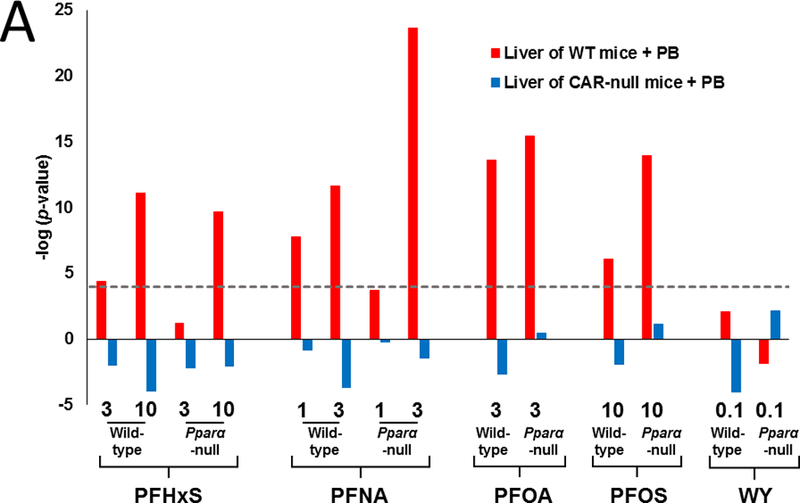
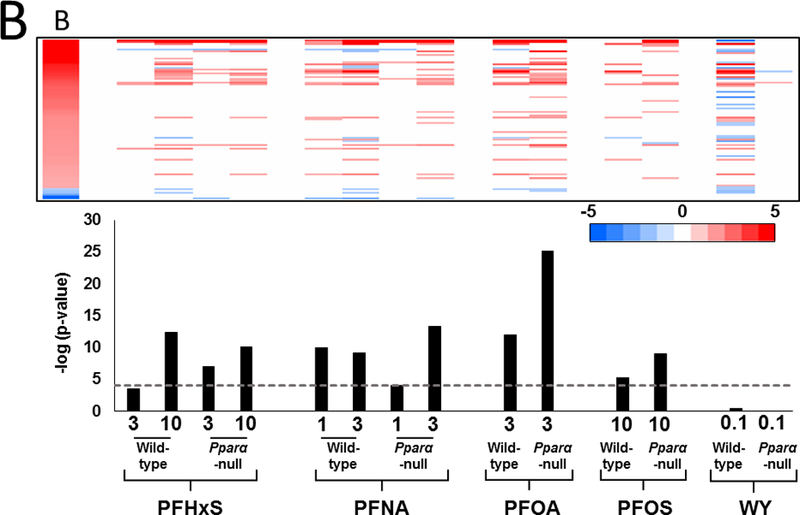
A. Lists of significant genes from PFAA-treated groups were compared to two gene lists derived from the livers of wild-type or Car-null mice exposed to 100 mg/kg phenobarbital for three days (from Chua and Moore, 2005). PFAA treatment groups are indicated in mg/kg/day. WY treatment groups are in %w/w.
B. Comparison of the PFAA gene lists to the CAR biomarker. (Top) Altered regulation of the genes in the CAR biomarker. The expression of the CAR biomarker genes are shown on the far left for reference (B). (Bottom) -Log(p-value)s of the similarity of the gene lists to the CAR biomarker.
PFAA gene lists were also compared to a previously described gene expression biomarker shown to accurately predict CAR activation (Oshida et al., 2015b). Fig. 2B (top) shows the expression of the 83 CAR biomarker genes after PFAA or WY treatment. Many genes in the biomarker set were regulated by PFAA treatment in a pattern that was similar to that of the CAR biomarker itself. The results of the statistical test are shown in Fig. 2B (bottom). All PFAA groups except low dose PFHxS in wild-type mice exhibited significant similarity to the CAR biomarker set (–log(p-value) ≥ 4). PFOA treatment in Pparα-null mice exhibited the greatest significance. These results demonstrate that all PFAAs examined activate CAR independently of PPARα and that in most cases except PFHxS, CAR is activated to greater extents in Pparα-null mice compared to wild-type mice at at least one dose level.
3.4. Similarities between the PFAA profiles and those altered by estrogen receptor agonists
There is evidence that PFAAs affect the activity of the estrogen receptor (Tilton et al., 2008; Benninghoff et al., 2011; Benninghoff et al., 2012). Assessment of estrogen as well as androgen receptor activity was evaluated using available ToxCast data. All ToxCast assays were evaluated for dose-response trans-activation assays carried out in the human hepatocyte HepG2 cell line showed that all PFAAs, except PFHxS, activate ERα (Table 3, top). ERα activation was also suggested for all compounds based on a cis-activation assay in which activation of an ERE-linked reporter was driven by an exogenously expressed ERα (Table 3, bottom). By contrast, none of the PFAAs were found to activate the androgen receptor (data not shown).
Table 3.
Activation of ERα in trans-activation assays by PFAAs
| Assay | Chemical | Hit Call 1 | Modeled Maximum 2 | Maximum SD 3 | Modeled AC50 4 | AC50 SD 5 |
|---|---|---|---|---|---|---|
| ATG_ERE_CIS_up | PFNA | 1 | 1.898 | 0.124 | 1.490 | 0.047 |
| PFOS | 1 | 0.707 | 0.070 | 1.257 | 0.069 | |
| PFOA | 1 | 2.034 | 0.029 | 1.529 | 0.007 | |
| PFOS-K | 1 | 1.848 | 0.762 | 1.514 | 0.222 | |
| PFOA, ammonium salt | 1 | 1.708 | 0.052 | 1.356 | 0.013 | |
| PFHS-K | 1 | 1.381 | 0.449 | 1.986 | 0.177 | |
| ATG_ERa_TRANS_up | PFNA | 1 | 2.566 | 0.228 | 1.330 | 0.089 |
| PFOS | 1 | 2.256 | 0.070 | 1.590 | 0.026 | |
| PFOA | 1 | 4.278 | 1.538 | 1.674 | 0.210 | |
| PFOS-K | 1 | 2.159 | 1.683 | 1.555 | 0.397 | |
| PFOA, ammonium salt | 1 | 2.874 | 0.382 | 1.650 | 0.121 | |
| PFHS-K | 0 | NA | NA | NA | NA |
= Hit call: 1 = active, 0 = inactive;
= top asymptote predicted by model (log10 fold induction);
= standard deviation of modeled maximum;
= modeled half maximal concentration (log10 μM);
= standard deviation of AC50.
We also compared our PFAA gene expression profiles to those induced by the ERα non-steroidal agonist PPT (4,4’,4’’-(4-Propyl-[1H]-pyrazole-1,3,5-triyl)trisphenol) following a 3 day exposure in ovariectomized wild-type and ERα-null mice (Pedram et al., 2013). Using the Running Fisher Test, most PFAA treatment groups from wild-type mice exhibited significant similarity to the profile induced by PPT in the livers of wild-type mice (Fig. 3A). Based on the data in Pparα-null mice, this response was partially or completely dependent upon PPARα for all of the PFAAs except PFHxS and WY (Fig. 3A, blue bars). Many of the comparisons to the PPT-treated ERα-null mice exhibited a negative relationship with the PFAA data.
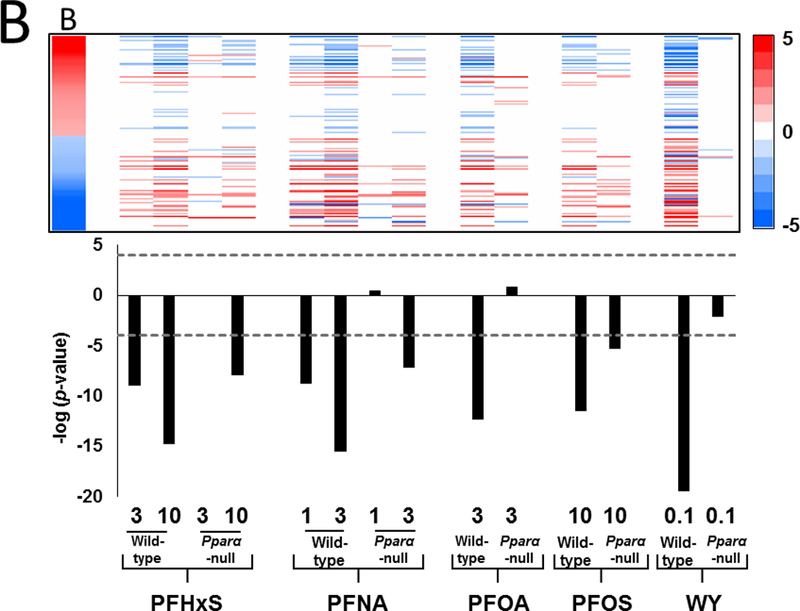
Fig. 3. Feminization of the liver transcriptome by PFAAs.
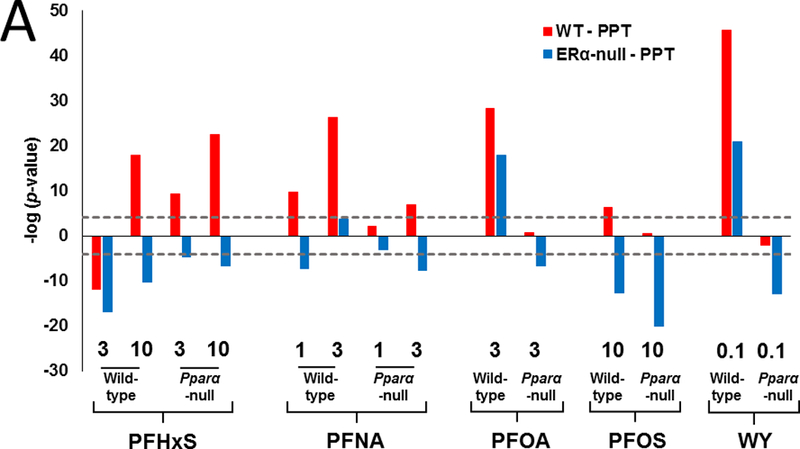
A. Similarity to the profile from PPT-treated wild-type but not PPT-treated ERα-null mice. Lists of significant genes from PFAA-treated groups were compared to the lists derived from the livers of wild-type or ERα-null mice exposed to 100 ug PPT intraperitoneally daily for 3 days (from Pedram et al., 2013).
B. Comparison of the PFAA-regulated genes to the STAT5B biomarker. The STAT5B biomarker previously described in Oshida et al. (2016a,b) consists of 144 STAT5B-dependent genes. The gene lists from each of the treatment groups were compared to the biomarker as described in the Methods. (Top) Heat map showing the expression of STAT5B biomarker genes after PFAA treatment. The expression of the STAT5B biomarker genes are shown on the far left for reference (B). In the biomarker the male-dominated upregulated genes are red and female-dominated down-regulated genes are blue. (Bottom) The –log(p-value)s are derived from the Running Fisher test of significance of the correlation in the expression pattern between the biomarker and the gene list.
It is possible that the observed transcript similarity between PPT and the PFAAs may be due to factors that are downstream of ERα activation. Therefore, we also evaluated whether PFAA treatment had an effect on the growth hormone (GH)-regulated transcription factor STAT5B. This transcription factor determines sexually-dimorphic differences in hepatic gene expression in mice (Udy et al., 1997). In previous studies, we characterized a gene expression biomarker that accurately predicts activation or suppression of STAT5B. A group of male-dominant up-regulated genes were identified that positively correlated with activation of STAT5B, whereas, a second group of female-dominant down-regulated genes were associated with disruption of STAT5B (Oshida et al., 2016a,b). Evaluation of this biomarker in PFAA-treated mice indicated decreased expression of the male-specific genes along with increased expression of the female-specific genes (Fig. 3B, upper panel). PFHxS at 3 mg/kg, PFNA at 1 mg/kg, PFOA, and WY were found to regulate this group of genes in a PPARα-dependent manner, while the other PFAAs lost some but not all significance in Pparα-null mice (Fig. 3B, lower panel). Taken together, the results indicate that there were changes in the gene expression profiles of PFAA-treated mice indicative of feminization of the liver.
3.5. PFAAs induce fatty acid β-oxidation genes in Pparα-null mice similar to that of PPARγ agonists
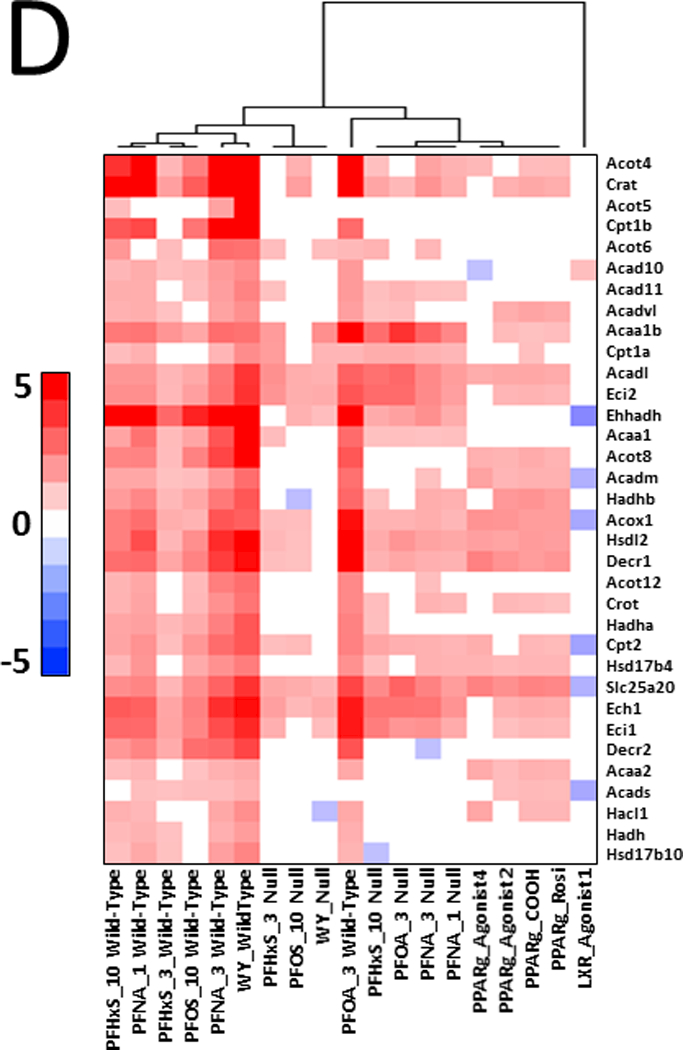
Our analysis above indicated that there are genes similarly regulated by PFAAs in both genotypes of mice (class II genes). Fig. 4 shows one-way hierarchical clustering of 67 common class II genes that exhibited increased expression in at least 11 of the 12 PFAA treatment groups. A number of genes in this group encode for proteins involved in fatty acid β-oxidation including Acox1, a gene often used as a marker of PPARα activation. These results indicate that not all genes known to be regulated by PPARα are necessarily suitable indicators of PPARα activation. Also included in this group were genes regulated by CAR (Cyp2c55, Cyp2b10, Cyp2c65, Gstm3), which segregated to a different hierarchical clade due to down-regulation of these transcripts in wild-type mice by WY.
Fig. 4. Genes regulated similarly by PFAAs in wild-type and Pparα-null mice.
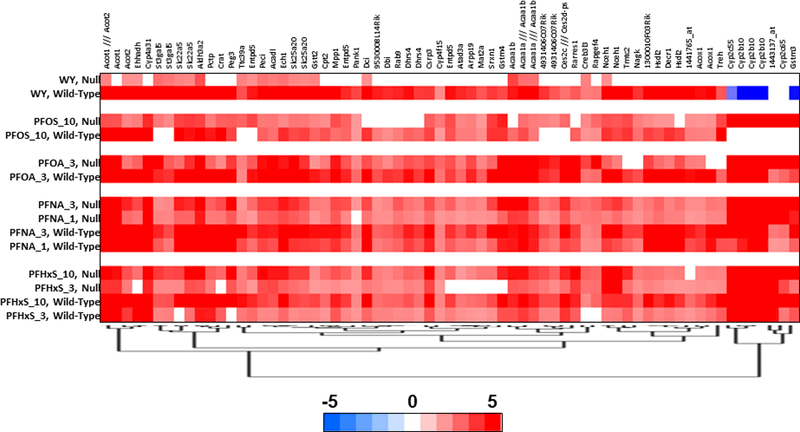
Genes regulated by PFAAs were filtered for those that exhibited regulation in 11 or 12 out of the 12 PFAA treatment groups and clustered by one-dimension hierarchical clustering.
A number of genes commonly regulated across PPAR subtypes are associated with fatty acid β-oxidation (DeLuca et al., 2000; Rosen et al. 2008a,b; Rosen et al., 2010); hence, teasing apart the activity of the various PPAR subtypes based on transcript data can be a challenge. To address this, we initially evaluated ToxCast high throughput screening data for evidence of either PPARγ or PPARβ activation. All four PFAAs represented by 6 discrete chemicals were examined in trans-activation assays carried out in HepG2 cells. AC50 values derived from these experiments were positive for activation of PPARα and PPARγ in human cells (Table 4). In contrast, activation of PPARβ was not observed (data not shown).
Table 4.
PPAR subtype activation from ToxCast Attagene assays.
| Assay | Chemical | Hit Call 1 | Modeled Maximum 2 | Maximum SD 3 | Modeled AC50 4 | AC50 SD 5 |
|---|---|---|---|---|---|---|
| ATG_PPARa_TRANS_up | PFNA | 1 | 3.315 | 0.119 | 1.157 | 0.038 |
| PFOS | 1 | 1.765 | 0.074 | 1.770 | 0.013 | |
| PFOA | 1 | 3.564 | 0.229 | 1.339 | 0.052 | |
| PFOS-K | 1 | 1.263 | 0.193 | 1.234 | 0.133 | |
| PFOA, ammonium salt | 1 | 2.876 | 0.170 | 0.828 | 0.105 | |
| PFHS-K | 1 | 1.580 | 0.118 | 1.051 | 0.063 | |
| ATG_PPARg_TRANS_up | PFNA | 1 | 3.282 | 0.063 | 1.638 | 0.017 |
| PFOS | 1 | 2.745 | 0.075 | 1.426 | 0.147 | |
| PFOA | 1 | 3.406 | 0.992 | 1.695 | 0.174 | |
| PFOS-K | 1 | 1.829 | 0.194 | 1.347 | 0.116 | |
| PFOA, ammonium salt | 1 | 2.647 | 0.149 | 1.339 | 0.052 | |
| PFHS-K | 1 | 1.987 | 0.220 | 1.484 | 0.161 |
= Hit call, 1 = active, 0 = inactive;
= top asymptote predicted by model (log10 fold induction);
= standard deviation of modeled maximum;
= modeled half maximal concentration (log10 μM);
= standard deviation of AC50.
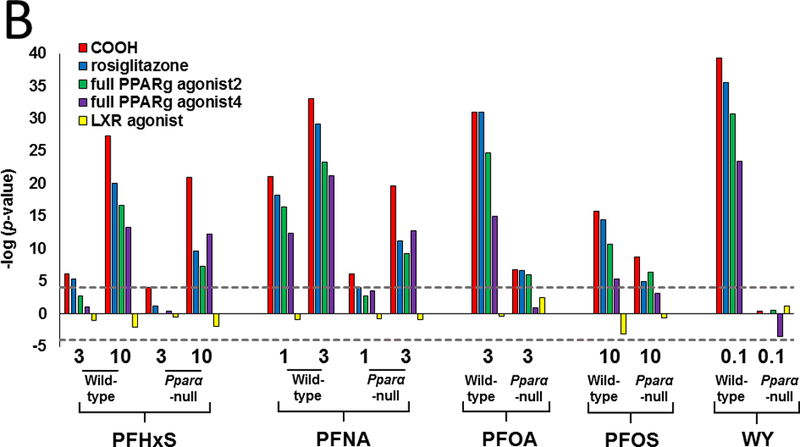
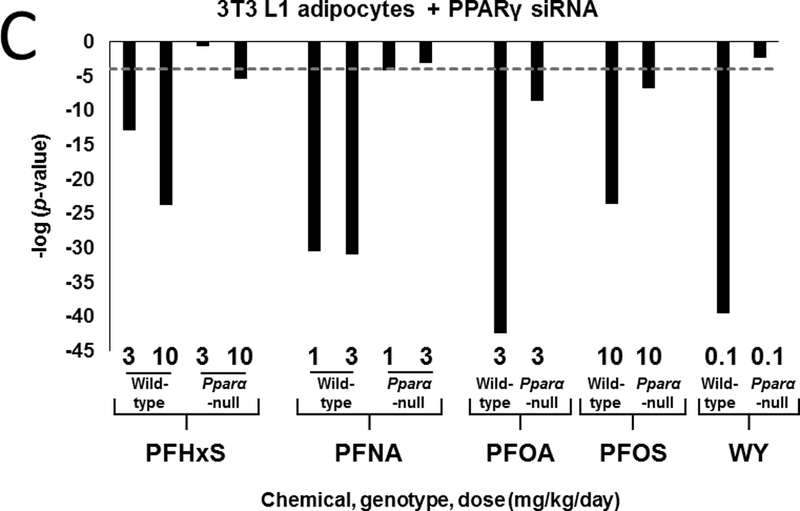
We further hypothesized that the pattern of gene expression in treated mice would be similar to activators of PPARγ. To test this hypothesis, gene lists from PFAA-treated mice were compared to gene expression profiles derived from three experiments. In the first comparison, the PPARγ agonist rosiglitazone was administered to Lepr (db/db) mice and evaluated for liver gene expression (Le Bouter et al., 2010). The profile from the rosiglitazone-treated mice exhibited significant similarity (-log(p-value) ≥ 4) to profiles from PFAA-treated wild-type mice (Fig. 5A). Although diminished, significance was also observed in most treated Pparα-null mice. Only WY or PFHxS (3 mg/kg) treatment in Pparα-null mice lacked significance. In a second experiment, comparisons were made to genes altered in mouse 3T3-L1 adipocytes by four PPARγ full agonists following a 24 hour exposure (Tan et al., 2012). Treatment with each of the PFAAs or WY exhibited similarity to all four PPARγ agonists in wild-type mice but not to a LXR agonist included as a negative control (Fig. 5B). All PFAAs retained similarity to most, if not all of the PPARγ agonists in Pparα-null mice, although the significance was not as great as in the wild-type groups. WY treatment in Pparα-null mice lacked similarity. In a third experiment, profiles were compared to those observed in 3T3-L1 cells after interference with PPARγ signaling by RNAi against the Pparg gene (Schupp et al., 2009). A negative relationship was found in all treated wild-type mice along with similar but muted effects in Pparα-null mice exposed to PFHxS, PFOA, and PFOS (Fig 5C). PFNA (1 mg/kg) was marginally significant in Pparα-null mice (-log(p-value) ~ −4), whereas, WY treatment did not reach significance in the nulls. Finally, we compared the gene expression data from PFAA-treated mice to direct targets of PPARs. We focused on 34 mitochondrial and peroxisomal fatty acid β-oxidation genes, most if not all of which are regulated by PPARs through interactions with peroxisome proliferator response elements (PPREs) (Kersten, 2014). Using two-dimensional unsupervised hierarchical clustering, Pparα-null mice treated with three of the PFAAs (high dose PFHxS, PFOA, both doses of PFNA) segregated with all four of the PPARγ agonists (Fig 5D). Low dose PFHxS, PFOS and WY treatment in Pparα-null mice segregated with the wild-type groups rather than the PPARγ agonists. Taken together, the results from these analyses support the plausibility of genes being regulated by PPARγ in PFAA-treated Pparα-null mice.
Fig. 5. The pattern of regulation of fatty acid metabolism genes in Pparα-null mice by PFAA exhibits similarity to that induced by PPARγ agonists.
A. Similarity between PFAA-regulated genes and those regulated by the PPARγ agonist rosiglitazone. The gene lists derived from PFAA-treated mice were compared to the profiles from livers of db/db mice treated with rosiglitazone (Le Bouter et al., 2010).
B. Similarity between PFAA-regulated genes and those regulated by four PPARγ agonists. Lists of significantly altered genes derived from exposure to one of the four indicated PPARγ agonists or a LXR agonist were compared to the PFAA groups. Mouse 3T3-L1 adipocytes were exposed to 10 uM of the indicated PPARγ agonists or LXR agonist for 24 hrs (Tan et al., 2012).
C. The PFAA profiles exhibit negative correlation to a profile of knockdown of the Pparg gene. The PFAA profiles were compared to the profile of 3T3-L1 cells treated with a siRNA against Pparg (Schupp et al., 2009).
D. Similarity in the expression pattern of fatty β-oxidation genes between PFAAs in Pparα-null mice and PPARγ agonists. The expression of fatty acid β-oxidation genes was compared between the groups by two-dimensional hierarchical clustering and shows greater similarity in the expression pattern between the PPARγ agonists and PFAA from Pparα-null mice than from wild-type mice. Only the hierarchical tree of one dimension is shown.
4. Discussion
PFHxS, PFNA, PFOA and PFOS represent environmental contaminants readily identified in human biomonitoring data (National Report on Human Exposure to Environmental Chemicals, 2014). PFOA and PFOS are no longer being manufactured in the US while PFHxS and PFNA continue to be used as replacements. To gain insight into common and chemical-specific effects, we examined the gene expression profiles associated with these PFAAs in the murine liver and compared them to prototypical activators of PPARα, CAR, ERα, and PPARγ using a microarray database. In addition, ToxCast data was mined to further evaluate these compounds for PPARα-independent effects. While transcriptional effects of PFOA and PFOS have been reported earlier, the present analysis is the first comprehensive examination of gene expression changes caused by PFHxS and PFNA and is the first comprehensive comparison of the transcriptional effects of all four chemicals.
4.1. PPARα mediates most of the transcriptional effects of PFAAs in the liver
PPARα-dependent and -independent transcriptional effects were considered across four PFAAs. For each treatment group, regulated genes were divided into three classes based on regulation in wild-type or Pparα-null genotypes. PPARα-independence in wild-type mice was estimated by considering the number of similarly-regulated genes in both genotypes (defined as class II genes) divided by the total number of genes regulated in wild-type mice. PFHxS exhibited the greatest percentage of PPARα-independent genes (24% and 22% for 3 and 10 mg/kg, respectively) (Fig. 1). PPARα-independence for the other PFAAs was estimated to be between 10 and 17%. In contrast, only 2% of the genes regulated by WY were PPARα-independent. These results indicate that the majority of genes (> ~75%) regulated by PFAAs in wild-type mice are PPARα-dependent, and that the degree of dependence is contingent upon chemical and dose. Other chemicals have been profiled in wild-type and Pparα-null mice, but only one, di(2-ethyl)hexyl phthalate (DEHP), has been similarly evaluated for PPARα-independence. Ren et al. (2010) found that only ~6% of genes were regulated by DEHP in a PPARα-independent manner. Based on these results, it is likely that other environmental contaminants identified as activators of PPARα (unlike marketed or experimental drugs designed specifically to target PPARα such as WY) regulate PPARα-independent molecular pathways, albeit to different extents.
The fact that PPARα is the major regulator of PFAA effects in the mouse liver leads to the question of human relevance of the PPARα mode of action as detailed in a number of reviews (Klaunig et al., 2003; Corton et al., 2014). Prior studies have shown that there is lower expression of the receptor compared to mouse and rat livers (summarized in Corton et al., 2014). Recently, Thomas et al. extensively characterized the mRNA and protein levels of a truncated splice variant of PPARα (Pparα-tr) in 150 human liver samples and found that on average there was ~3-fold lower expression of PPARα-tr protein compared to PPARα-wt and that the truncated form can suppress the expression of proliferative and pro-inflammatory genes (Thomas et al., 2015). Expression of the PPARα-tr protein which is not observed in responsive species could help explain the inability of human primary hepatocytes to mount a proliferative response to PPARα activators (Corton et al., 2014). A large body of evidence including these differences in the PPARα expression and activity does not support the human relevance of rodent liver tumors that are mediated by PPARα, and for compounds such as the PFAAs shifts the focus of research to other pathways altered by PFAAs in the liver that may be more relevant for human toxicity.
4.2. Role for CAR in mediating PFAA effects
Many genes regulated by PFAAs encode for enzymes catalyzing xenobiotic metabolism, indicating involvement of one or more transcription factors which could include PXR, Ah receptor (AhR), NFE2L2 (Nrf2), and CAR. As previously reported, there is no evidence that PFAAs activate AhR (Oshida et al., 2015a). Nrf2 is a principal regulator of the oxidative stress response and activation of this factor has been previously suggested in mice exposed to perfluorodecanoic acid (PFDA) (Maher et al., 2008). Oxidative stress has also been reported at higher micromolar concentrations of PFOA in HepG2 cells (Yao and Zhong, 2005), although Eriksen et al. (2010) observed only modest effects for various PFAAs in HepG2 cells. Oxidative stress has been reported in the murine liver following relatively high PFOA exposure levels as well (Yang et al., 2014). Activation of CAR by PFOA and PFOS has previously been demonstrated in either rodents or by in vitro studies (Cheng and Klaassen, 2008; Ren et al., 2009; Elcombe et al., 2010; Bjork et al., 2011; Elcombe et al., 2012). In the current study, when comparisons were made with the gene expression profiles of wild-type or Car-null mice exposed to phenobarbital, significant similarity was observed only to those profiles from PB-treated wild-type mice (Fig. 2A). A CAR gene expression biomarker that accurately predicts CAR modulation (Oshida et al., 2015b) was used to show CAR activation by all four PFAAs but not by WY (Fig. 2B). Furthermore, significance of the overlap with the CAR biomarker was found to increase in Pparα-null mice for PFNA, PFOA and PFOS compared to treated wild-type mice. The phenomenon of increased activation of CAR in the absence of PPARα has been discussed in Corton et al. (2014) and may be due to a number of overlapping mechanisms including competition for shared co-activators.
4.3. Feminization of the liver transcriptome by PFAAs
There is increasing evidence that PFAAs activate ERα. Our evaluation of ToxCast trans- and cis-activation assays indicated that ERα is activated by all four PFAAs (Table 3). Using a human reporter gene assay, Benninghoff et al. (2011) found that PFOA, PFNA, PFDA, perfluoro-n-undecanoic acid (PFUnDA), and PFOS significantly increase human ERα-dependent transcriptional activation at concentrations ranging from 10–1000 nM. This group also showed that all PFAAs weakly bind to rainbow trout liver ERα with half maximal inhibitory concentration (IC50) values of 15.2–289 μM. PFHxS, PFOS and PFOA induced ERα trans-activation in the human breast cancer cell line, MCF-7, as well (Kjeldsen and Bonefeld-Jørgensen, 2013). Furthermore, Du et al. (2013) found that PFOS acts as an ERα agonist in either in an in vitro reporter cell line or a zebrafish-based in vivo assay. In contrast, PFOA treatment of a stable human cell line containing an ERα-dependent luciferase reporter construct (the human ovarian carcinoma cell line BG1-Luc4E2 recently renamed VM7Luc4E2 cells because they were found to be a variant of MCF-7 cells; https://ntp.niehs.nih.gov/iccvam/methods/endocrine/bg1luc/bg1luc-vm7luc-june2016–508.pdf) caused no change in ERα-dependent luciferase activity (Yao et al., 2014). It is possible that study differences were due to the underlying differences in the expression of the ER subtypes in these two MCF-7 variants.
Only one rodent study tested the hypothesis that ERα is perturbed upon PFAA exposure. Exposure to PFOA at up to 1 mg/kg in the mouse uterotrophic assay had no effect on uterine weight (Yao et al., 2014). We compared PFAA gene expression profiles to those from the livers of mice treated with the nonsteroidal ERα agonist, PPT (Pedram et al., 2013). Profiles from PFAA-treated wild-type mice exhibited significant similarity to those from PPT-treated wild-type mice but not PPT-treated ERα-null mice indicating that a set of genes regulated by PFAAs overlapped with those that were ERα-dependent (Fig. 3A). Interestingly, WY exhibited similarity in wild-type mice as well, an effect that became greatly diminished in Pparα-null mice. These data suggest that PFAAs alter gene expression in the mouse liver in a manner similar to ERα agonists. The observation that WY induced comparable changes to that of the ERα agonists suggests that this effect may be common to activators of PPARα and may not be specific to PFAAs. Assessment of ER activation in zebrafish using clofibrate, a well described hypolipidemic activator of PPARα, does not support this conclusion as clofibrate did not act like an ER activator (Tilton et al., 2008; Benninghoff et al, 2012). Given that PFOA did not act like a typical ER agonist in the uterotrophic assay (Yao et al., 2014), but exhibited some ER-like effects in the liver, it is possible that the effects are tissue-specific for an estrogen-like response. Despite the estrogen-like response in the liver at the transcriptional level, there is a lack of more objective cellular and tissue measures of estrogenicity in other estrogen-responsive tissues.
A murine gene expression biomarker which accurately predicts activation of STAT5B was recently characterized by our group. STAT5B functions downstream of GH to regulate sexually-dimorphic gene expression in the liver. Exposure to testosterone activates STAT5B while exposure to estrogens or genetic interference with GH function results in down-regulation of STAT5B and feminization of the liver in male mice (Oshida et al., 2016a,b). In the current study, we determined that all four PFAAs caused significant suppression of STAT5B in wild-type mice. Significance was found to be either partially (PFHxS, PFNA, PFOS) or completely (PFOA, WY) PPARα-dependent (Fig. 3B). Hence, PFAAs interfere with GH signaling as reflected by suppression of STAT5B. Chemical-induced feminization of the liver could occur by disruption of one or more sensitive nodes along the GH hypothalamic-pituitary-liver axis. In one model, antagonism could come about directly in the liver through physical interactions between STAT5B and PPARα. Circumstantial evidence for this model comes from observations of genes co-regulated by PPARα and STAT5B in which absence of one transcription factor leads to increased regulation by the other (Shipley and Waxman, 2004; Stauber et al., 2005). Alternatively, feminization could occur through changes in circulating hormones. Treatment of male mice or rats with estradiol antagonizes hepatic expression of male-specific cytochrome P450 genes (Ishii et al., 2006) and, when administered to neonatal castrated rats, estradiol induces female-specific liver enzymes (Dannan et al., 1986). High doses of DEHP, for example, increased estradiol levels (Eagon et al., 1994) and caused feminization (Oshida et al., 2016b). Feminization may also occur through decreases in testosterone levels as is observed after castration. PFOA has been shown to decrease testosterone and increase estradiol in the serum of male rats, effects at least partially associated with induction of hepatic aromatase (Cook et al., 1992). Such changes may be due in part to CAR, since CAR regulates a number of cytochrome P450 genes that metabolize testosterone to hydroxylated metabolites (Waxman, 1988). Male Car-null mice have serum levels of testosterone that are 2.5-fold higher than wild-type mice along with decreased constitutive hepatic expression of testosterone hydroxylases Cyp2b9 and Cyp2b10 (Cho et al., 2014). A role for cytochrome P450 activity in decreasing testosterone production is plausible based on suppression of STAT5B with parallel activation of CAR.
4.4. Role of PPARγ in mediating the transcriptional effects of PFAAs in the liver
In previous studies, we hypothesized that either PPARγ or PPARβ may regulate genes in Pparα-null mice exposed to PFOS or PFOA (Rosen et al., 2008a,b; Rosen et al., 2010). Our current study indicates that all four compounds similarly regulate genes involved in lipid homeostasis in both genotypes, including those involved in fatty acid β-oxidation, lipid catabolism, lipid synthesis, and lipid transport. Our studies provided plausibility to regulation of genes by PPARγ. We compared the expression profiles of PFAA-treated mice to those of PPARγ agonists administered to either Lepr (db/db) mice or to 3T3-L1 adipocytes. Significant similarity was observed between the PPARγ agonist profiles and PFAAs in both wild-type and Pparα-null mice (Fig. 5A,B). Profiles in PFAA-treated mice also exhibited negative correlation to 3T3-L1 adipocytes that were treated by RNAi to reduce PPARγ activity (Fig. 5C). As a final test, we focused on a set of fatty acid β-oxidation genes, most if not all reported to be regulated in a positive manner by PPAR subtypes through one or more PPREs (Kersten, 2014). Using two-dimensional hierarchical unsupervised clustering, groups from Pparα-null mice treated with PFOA, PFNA, and PFHxS segregated with PPARγ agonists (Fig. 5D).
Evidence against a role for PPARγ in mediating the effects in PFAA-treated mice have also come from studies using reporter cell lines. These studies have been largely negative, with the exception of modest activation of PPARγ by PFOA under high exposure conditions (Vanden Heuvel et al. 2006, Taxvig et al., 2012, Maloney and Waxman 1999). Likewise, Takacs and Abbott (2007) did not report activation of either human or mouse PPARγ by PFOA or PFOS, although significant effects on murine PPARβ were observed. In the current study, however, EPA ToxCast data showed activation of human PPARγ but not PPARβ by all four PFAAs (Table 4). Our group also found that expression of the Pparg gene was increased in both wild-type and Pparα-null mice after PFOA exposure while expression of Pparb was unchanged (Das et al., 2016). Additional studies indicate the involvement of PPARγ in mediating the inhibitory effects of PFOA during leukocyte activation in rats (Griesbacher et al., 2008) and in altering spontaneous differentiation of rat embryonic neural stem cells by PFOS (Wan Ibrahim et al., 2013). In summary, there is compelling evidence that PPARγ may be a target of PFAAs in liver and other tissues.
5.0. Conclusions
We have examined the hepatic gene expression profiles of PFNA and PFHxS in wild-type and Pparα-null mice. In order to carry out a comprehensive analysis of PFAA transcriptional effects, historical in-house data for PFOA and PFOS were also included as were data for WY-14,643, a recognized activator of PPARα. These microarray profiles were compared to relevant microarray studies in a database allowing us to assess the involvement of a number of transcription factors. PPARα was found to be the primary molecular target for PFAAs in the mouse liver. Using previously defined predictive gene expression biomarkers, we demonstrated that PFAAs activate CAR in both wild-type and Pparα-null mice and cause feminization of the liver transcriptome through suppression of STAT5B. The suppression of STAT5B could be linked to activation of ERα as there was significant overlap in the genes regulated by PFAAs and an ERα agonist in an ERα-dependent manner. Human PPARγ and ERα were also found to be activated by all four PFAAs in ToxCast trans-activation assays. These results indicate that, in addition to activating PPARα, PFAAs activate other nuclear receptors including CAR, PPARγ and ERα, and may function as suppressors of STAT5B. While these data indicate that the PFAAs mediate a limited number of genomic effects through transcription factors other than PPARα, there is little evidence that these PPARα-independent changes are associated with the key events in the PPARα mode of action for liver cancer. Additional studies would be needed to determine if liver cancer can be induced by PFAAs independently of PPARα.
Supplementary Material
Acknowledgements
We thank Drs. Udayan Apte and Dr. Brian Chorley for their critical review of this manuscript. We would also like to thank members of the NHEERL genomics research core for their support in processing RNA samples for in-house Affymetrix analysis.
The information in this document has been funded by the U.S. Environmental Protection Agency. It has been subjected to review by the National Health and Environmental Research Laboratory and approved for publication. Approval does not signify that the contents necessarily reflect the views and the policies of the Agency, nor does mention of trade names or commercial products constitute endorsement or recommendation for use.
Abbreviations:
- AhR
aryl hydrocarbon receptor
- CAR
constitutive activated receptor
- DEHP
di-(2-ethylhexyl)phthalate
- ERα
estrogen receptor α
- ERE
estrogen response element
- GH
growth hormone
- LXR
liver X receptor
- Nrf2
nuclear factor erythroid-2-related factor 2
- PFAAs
perfluoroalkyl acids
- PPARα
peroxisome proliferator-activated receptor α
- PPARγ
peroxisome proliferator-activated receptor γ
- PFHxS
perfluorohexanesulfonic acid
- PFNA
perfluorononanoic acid
- PFOA
perfluorooctanoic acid
- PFOS
perfluorooctane sulfonate; PFUnDA, perfluoro-n-undecanoic acid
- PPREs
peroxisome proliferator response elements
- PXR
pregnane X receptor
- STAT5B
signal transducer and activator of transcription 5B
- Vtg
vitellogenin
- WY
WY-14,643
Footnotes
Author contributions:
MBR, JR and JCC analyzed the microarray data. KPD performed animal experiments
MBR, CL, and JCC conceived of the study, participated in study design MBR and JCC helped draft manuscript. All authors read and approved the final manuscript.
References
- Abe T, Takahashi M, Kano M, Amaike Y, Ishii C, Maeda K, Kudoh Y, Morishita T, Hosaka T, Sasaki T, Kodama S, Matsuzawa A, Kojima H, Yoshinari K. Activation of nuclear receptor CAR by an environmental pollutant perfluorooctanoic acid. Arch Toxicol. 2016. November 10. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Al-Bader M, Ford C, Al-Ayadhy B and Francis I 2011. Analysis of estrogen receptor isoforms and variants in breast cancer cell lines. Experimental and therapeutic medicine 2, 537–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen CS, Fei C and Gamborg M 2011. Prenatal Exposures to Perfluorinated Chemicals and Anthropometric Measures in Infancy (Vol 172, Pg 1230, 2010). Am J Epidemiol. 173 [DOI] [PubMed] [Google Scholar]
- Apelberg BJ, Witter FR, Herbstman JB, Calafat AM, Halden RU, Needham LL and Goldman LR 2007. Cord serum concentrations of perfluorooctane sulfonate (PFOS) and perfluorooctanoate (PFOA) in relation to weight and size at birth. Environmental health perspectives 115, 1670–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastos Sales L, Kamstra JH, Cenijn PH, van Rijt LS, Hamers T and Legler J 2013. Effects of endocrine disrupting chemicals on in vitro global DNA methylation and adipocyte differentiation. Toxicol In Vitro 27, 1634–1643. [DOI] [PubMed] [Google Scholar]
- Beggs KM, McGreal SR, McCarthy A, Gunewardena S, Lampe JN, Lau C, Apte U. The role of hepatocyte nuclear factor 4-alpha in perfluorooctanoic acid- and perfluorooctanesulfonic acid-induced hepatocellular dysfunction. Toxicol Appl Pharmacol. 2016. August 1;304:18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benninghoff AD, Bisson WH, Koch DC, Ehresman DJ, Kolluri SK and Williams DE 2011. Estrogen-like activity of perfluoroalkyl acids in vivo and interaction with human and rainbow trout estrogen receptors in vitro. Toxicological sciences : an official journal of the Society of Toxicology 120, 42–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benninghoff AD, Orner GA, Buchner CH, Hendricks JD, Duffy AM and Williams DE 2012. Promotion of hepatocarcinogenesis by perfluoroalkyl acids in rainbow trout. Toxicological sciences : an official journal of the Society of Toxicology 125, 69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijland S, Rensen PC, Pieterman EJ, Maas AC, van der Hoorn JW, van Erk MJ, Havekes LM, Willems van Dijk K, Chang SC, Ehresman DJ, Butenhoff JL and Princen HM 2011. Perfluoroalkyl sulfonates cause alkyl chain length-dependent hepatic steatosis and hypolipidemia mainly by impairing lipoprotein production in APOE*3-Leiden CETP mice. Toxicological sciences : an official journal of the Society of Toxicology 123, 290–303. [DOI] [PubMed] [Google Scholar]
- Bjork JA, Butenhoff JL and Wallace KB 2011. Multiplicity of nuclear receptor activation by PFOA and PFOS in primary human and rodent hepatocytes. Toxicology 288, 8–17. [DOI] [PubMed] [Google Scholar]
- Boulanger B, Vargo JD, Schnoor JL and Hornbuckle KC 2005. Evaluation of perfluorooctane surfactants in a wastewater treatment system and in a commercial surface protection product. Environ Sci Technol 39, 5524–5530. [DOI] [PubMed] [Google Scholar]
- Buhrke T, Kruger E, Pevny S, Rossler M, Bitter K and Lampen A 2015. Perfluorooctanoic acid (PFOA) affects distinct molecular signalling pathways in human primary hepatocytes. Toxicology 333, 53–62. [DOI] [PubMed] [Google Scholar]
- Butenhoff JL, Chang SC, Ehresman DJ and York RG 2009. Evaluation of potential reproductive and developmental toxicity of potassium perfluorohexanesulfonate in Sprague Dawley rats. Reprod Toxicol 27, 331–341. [DOI] [PubMed] [Google Scholar]
- Cheng X and Klaassen CD 2008. Critical role of PPAR-alpha in perfluorooctanoic acid- and perfluorodecanoic acid-induced downregulation of Oatp uptake transporters in mouse livers. Toxicological sciences : an official journal of the Society of Toxicology 106, 37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HY, Jung JY, Park H, Yang JY, Jung S, An JH, Cho SW, Kim SW, Kim SY, Kim JE, Park YJ and Shin CS 2014. In vivo deletion of CAR resulted in high bone mass phenotypes in male mice. Journal of cellular physiology 229, 561–571. [DOI] [PubMed] [Google Scholar]
- Cook JC, Murray SM, Frame SR and Hurtt ME 1992. Induction of Leydig cell adenomas by ammonium perfluorooctanoate: a possible endocrine-related mechanism. Toxicology and applied pharmacology 113, 209–217. [DOI] [PubMed] [Google Scholar]
- Corton JC, Cunningham ML, Hummer BT, Lau C, Meek B, Peters JM, Popp JA, Rhomberg L, Seed J and Klaunig JE 2014. Mode of action framework analysis for receptor-mediated toxicity: The peroxisome proliferator-activated receptor alpha (PPARalpha) as a case study. Crit Rev Toxicol 44, 1–49. [DOI] [PubMed] [Google Scholar]
- Costa G, Sartori S and Consonni D 2009. Thirty years of medical surveillance in perfluooctanoic acid production workers. J Occup Environ Med 51, 364–372. [DOI] [PubMed] [Google Scholar]
- Dannan GA, Porubek DJ, Nelson SD, Waxman DJ and Guengerich FP 1986. 17 beta-estradiol 2- and 4-hydroxylation catalyzed by rat hepatic cytochrome P-450: roles of individual forms, inductive effects, developmental patterns, and alterations by gonadectomy and hormone replacement. Endocrinology 118, 1952–1960. [DOI] [PubMed] [Google Scholar]
- Das KP, Wood CR, Lin MT, Starkov AA, Lau C, Wallace KB, Corton JC, Abbott BD. Perfluoroalkyl acids-induced liver steatosis: Effects on genes controlling lipid homeostasis. Toxicology. 2017. March 1;378:37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das KP, Grey BE, Rosen MB, Wood CR, Tatum-Gibbs KR, Zehr RD, Strynar MJ, Lindstrom AB and Lau C 2015. Developmental toxicity of perfluorononanoic acid in mice. Reprod Toxicol 51, 133–144. [DOI] [PubMed] [Google Scholar]
- de Cock M and van de Bor M 2014. Obesogenic effects of endocrine disruptors, what do we know from animal and human studies? Environ Int 70, 15–24. [DOI] [PubMed] [Google Scholar]
- DeLuca JG, Doebber TW, Kelly LJ, Kemp RK, Molon-Noblot S, Sahoo SP, Ventre J, Wu MS, Peters JM, Gonzalez FJ, Moller DE. Evidence for peroxisome proliferator-activated receptor (PPAR)alpha-independent peroxisome proliferation: effects of PPARgamma/delta-specific agonists in PPARalpha-null mice. Mol Pharmacol. 2000. September;58(3):470–6. [DOI] [PubMed] [Google Scholar]
- DeWitt JC, Shnyra A, Badr MZ, Loveless SE, Hoban D, Frame SR, Cunard R, Anderson SE, Meade BJ, Peden-Adams MM, Luebke RW and Luster MI 2009. Immunotoxicity of perfluorooctanoic acid and perfluorooctane sulfonate and the role of peroxisome proliferator-activated receptor alpha. Crit Rev Toxicol 39, 76–94. [DOI] [PubMed] [Google Scholar]
- Du G, Hu J, Huang H, Qin Y, Han X, Wu D, Song L, Xia Y and Wang X 2013. Perfluorooctane sulfonate (PFOS) affects hormone receptor activity, steroidogenesis, and expression of endocrine-related genes in vitro and in vivo. Environ Toxicol Chem 32, 353–360. [DOI] [PubMed] [Google Scholar]
- Eagon PK, Chandar N, Epley MJ, Elm MS, Brady EP and Rao KN 1994. Di(2-ethylhexyl)phthalate-induced changes in liver estrogen metabolism and hyperplasia. International journal of cancer 58, 736–743. [DOI] [PubMed] [Google Scholar]
- Elcombe CR, Peffer RC, Wolf DC, Bailey J, Bars R, Bell D, Cattley RC, Ferguson SS, Geter D, Goetz A, Goodman JI, Hester S, Jacobs A, Omiecinski CJ, Schoeny R, Xie W, Lake BG. Mode of action and human relevance analysis for nuclear receptor-mediated liver toxicity: A case study with phenobarbital as a model constitutive androstane receptor (CAR) activator. Crit Rev Toxicol. 2014. January;44(1):64–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elcombe CR, Elcombe BM, Foster JR, Chang SC, Ehresman DJ and Butenhoff JL 2012. Hepatocellular hypertrophy and cell proliferation in Sprague-Dawley rats from dietary exposure to potassium perfluorooctanesulfonate results from increased expression of xenosensor nuclear receptors PPARalpha and CAR/PXR. Toxicology 293, 16–29. [DOI] [PubMed] [Google Scholar]
- Elcombe CR, Elcombe BM, Foster JR, Farrar DG, Jung R, Chang SC, Kennedy GL and Butenhoff JL 2010. Hepatocellular hypertrophy and cell proliferation in Sprague-Dawley rats following dietary exposure to ammonium perfluorooctanoate occurs through increased activation of the xenosensor nuclear receptors PPARalpha and CAR/PXR. Arch Toxicol 84, 787–798. [DOI] [PubMed] [Google Scholar]
- Ellis DA, Martin JW, De Silva AO, Mabury SA, Hurley MD, Sulbaek Andersen MP and Wallington TJ 2004. Degradation of fluorotelomer alcohols: a likely atmospheric source of perfluorinated carboxylic acids. Environ Sci Technol 38, 3316–3321. [DOI] [PubMed] [Google Scholar]
- Eriksen KT, Raaschou-Nielsen O, McLaughlin JK, Lipworth L, Tjonneland A, Overvad K and Sorensen M 2013. Association between plasma PFOA and PFOS levels and total cholesterol in a middle-aged Danish population. PloS one 8, e56969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksen KT, Raaschou-Nielsen O, Sorensen M, Roursgaard M, Loft S and Moller P 2010. Genotoxic potential of the perfluorinated chemicals PFOA, PFOS, PFBS, PFNA and PFHxA in human HepG2 cells. Mutat Res 700, 39–43. [DOI] [PubMed] [Google Scholar]
- Fei C, McLaughlin JK, Lipworth L and Olsen J 2009. Maternal levels of perfluorinated chemicals and subfecundity. Hum Reprod 24, 1200–1205. [DOI] [PubMed] [Google Scholar]
- Fei C, McLaughlin JK, Tarone RE and Olsen J 2007. Perfluorinated chemicals and fetal growth: a study within the Danish National Birth Cohort. Environmental health perspectives 115, 1677–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng YX, Fang XM, Shi ZM, Xu MQ and Dai JY 2010. Effects of PFNA exposure on expression of junction-associated molecules and secretory function in rat Sertoli cells. Reproductive Toxicology 30, 429–437. [DOI] [PubMed] [Google Scholar]
- Fisher M, Arbuckle TE, Wade M and Haines DA 2013. Do perfluoroalkyl substances affect metabolic function and plasma lipids?--Analysis of the 2007–2009, Canadian Health Measures Survey (CHMS) Cycle 1. Environ Res 121, 95–103. [DOI] [PubMed] [Google Scholar]
- Fitz-Simon N, Fletcher T, Luster MI, Steenland K, Calafat AM, Kato K and Armstrong B 2013. Reductions in serum lipids with a 4-year decline in serum perfluorooctanoic acid and perfluorooctanesulfonic acid. Epidemiology 24, 569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisbee SJ, Shankar A, Knox SS, Steenland K, Savitz DA, Fletcher T and Ducatman AM 2010. Perfluorooctanoic acid, perfluorooctanesulfonate, and serum lipids in children and adolescents: results from the C8 Health Project. Arch Pediatr Adolesc Med 164, 860–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo V, Leonardi G, Brayne C, Armstrong B and Fletcher T 2013. Serum perfluoroalkyl acids concentrations and memory impairment in a large cross-sectional study. BMJ Open 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger SD, Xiao J, Ducatman A, Frisbee S, Innes K and Shankar A 2014. The association between PFOA, PFOS and serum lipid levels in adolescents. Chemosphere 98, 78–83. [DOI] [PubMed] [Google Scholar]
- Gonzalez FJ and Shah YM 2008. PPARalpha: mechanism of species differences and hepatocarcinogenesis of peroxisome proliferators. Toxicology 246, 2–8. [DOI] [PubMed] [Google Scholar]
- Griesbacher T, Pommer V, Schuligoi R, Tiran B, Peskar BA. Anti-inflammatory actions of perfluorooctanoic acid and peroxisome proliferator-activated receptors (PPAR) alpha and gamma in experimental acute pancreatitis. Int Immunopharmacol. 2008. February;8(2):325–9. [DOI] [PubMed] [Google Scholar]
- Houde M, Martin JW, Letcher RJ, Solomon KR and Muir DC 2006. Biological monitoring of polyfluoroalkyl substances: A review. Environ Sci Technol 40, 3463–3473. [DOI] [PubMed] [Google Scholar]
- Huang W, Zhang J, Washington M, Liu J, Parant JM, Lozano G and Moore DD 2005. Xenobiotic stress induces hepatomegaly and liver tumors via the nuclear receptor constitutive androstane receptor. Mol Endocrinol 19, 1646–1653. [DOI] [PubMed] [Google Scholar]
- Ishibashi H, Ishida H, Matsuoka M, Tominaga N and Arizono K 2007. Estrogenic effects of fluorotelomer alcohols for human estrogen receptor isoforms alpha and beta in vitro. Biological & pharmaceutical bulletin 30, 1358–1359. [DOI] [PubMed] [Google Scholar]
- Ishii T, Nishimura K and Nishimura M 2006. Administration of xenobiotics with anti-estrogenic effects results in mRNA induction of adult male-specific cytochrome P450 isozymes in the livers of adult female rats. Journal of pharmacological sciences 101, 250–255. [DOI] [PubMed] [Google Scholar]
- Jensen TK, Andersen LB, Kyhl HB, Nielsen F, Christesen HT and Grandjean P 2015. Association between perfluorinated compound exposure and miscarriage in Danish pregnant women. PloS one 10, e0123496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson N, Eriksson P and Viberg H 2009. Neonatal exposure to PFOS and PFOA in mice results in changes in proteins which are important for neuronal growth and synaptogenesis in the developing brain. Toxicological sciences : an official journal of the Society of Toxicology 108, 412–418. [DOI] [PubMed] [Google Scholar]
- Johansson N, Fredriksson A and Eriksson P 2008. Neonatal exposure to perfluorooctane sulfonate (PFOS) and perfluorooctanoic acid (PFOA) causes neurobehavioural defects in adult mice. Neurotoxicology 29, 160–169. [DOI] [PubMed] [Google Scholar]
- Judson RS, Houck KA, Kavlock RJ, Knudsen TB, Martin MT, Mortensen HM, Reif DM, Rotroff DM, Shah I, Richard AM, Dix DJ. 2010. In vitro Screening of Environmental Chemicals for Targeted Testing Prioritization: The ToxCast Project. Environ Health Perspect 118(4): 485–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato K, Basden BJ, Needham LL and Calafat AM 2011. Improved selectivity for the analysis of maternal serum and cord serum for polyfluoroalkyl chemicals. J Chromatogr A 1218, 2133–2137. [DOI] [PubMed] [Google Scholar]
- Kavlock R, Chandler K, Houck K, Hunter S, Judson R, Kleinstreuer N, Knudsen T, Martin M, Padilla S, Reif D, Richard A, Rotroff D, Sipes N and Dix D. 2012. Update on EPA’s ToxCast program: providing high throughput decision support tools for chemical risk management. Chem Res Toxicol 25(7): 1287–1302. [DOI] [PubMed] [Google Scholar]
- Kersten S 2014. Physiological regulation of lipoprotein lipase. Biochim Biophys Acta 1841, 919–933. [DOI] [PubMed] [Google Scholar]
- Khalil N, Chen A, Lee M, Czerwinski SA, Ebert JR, DeWitt JC and Kannan K 2015. Association of Perfluoroalkyl Substances, Bone Mineral Density, and Osteoporosis in the U.S. Population in NHANES 2009–2010. Environmental health perspectives. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjeldsen LS and Bonefeld-Jorgensen EC 2013. Perfluorinated compounds affect the function of sex hormone receptors. Environmental science and pollution research international 20, 8031–8044. [DOI] [PubMed] [Google Scholar]
- Klaunig JE, Babich MA, Baetcke KP, Cook JC, Corton JC, David RM, DeLuca JG, Lai DY, McKee RH, Peters JM, Roberts RA, Fenner-Crisp PA. PPARalpha agonist-induced rodent tumors: modes of action and human relevance. Crit Rev Toxicol. 2003;33(6):655–780. [DOI] [PubMed] [Google Scholar]
- Kudo N and Kawashima Y 1997. Fish oil-feeding prevents perfluorooctanoic acid-induced fatty liver in mice. Toxicology and applied pharmacology 145, 285–293. [DOI] [PubMed] [Google Scholar]
- Kudo N and Kawashima Y 2003. Induction of triglyceride accumulation in the liver of rats by perfluorinated fatty acids with different carbon chain lengths: comparison with induction of peroxisomal beta-oxidation. Biological & pharmaceutical bulletin 26, 47–51. [DOI] [PubMed] [Google Scholar]
- Kudo N, Mizuguchi H, Yamamoto A and Kawashima Y 1999. Alterations by perfluorooctanoic acid of glycerolipid metabolism in rat liver. Chem Biol Interact 118, 69–83. [DOI] [PubMed] [Google Scholar]
- Kupershmidt I, Su QJ, Grewal A, Sundaresh S, Halperin I, Flynn J, Shekar M, Wang H, Park J, Cui W, Wall GD, Wisotzkey R, Alag S, Akhtari S and Ronaghi M 2010. Ontology-based meta-analysis of global collections of high-throughput public data. PloS one 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau C, Anitole K, Hodes C, Lai D, Pfahles-Hutchens A and Seed J 2007. Perfluoroalkyl acids: a review of monitoring and toxicological findings. Toxicological sciences : an official journal of the Society of Toxicology 99, 366–394. [DOI] [PubMed] [Google Scholar]
- Le Bouter S, Rodriguez M, Guigal-Stephan N, Courtade-Gaïani S et al. Coordinate Transcriptomic and Metabolomic Effects of the Insulin Sensitizer Rosiglitazone on Fundamental Metabolic Pathways in Liver, Soleus Muscle, and Adipose Tissue in Diabetic db/db Mice. PPAR Res 2010;2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis RC, Johns LE and Meeker JD 2015. Serum Biomarkers of Exposure to Perfluoroalkyl Substances in Relation to Serum Testosterone and Measures of Thyroid Function among Adults and Adolescents from NHANES 2011–2012. Int J Environ Res Public Health 12, 6098–6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindeman B, Maass C, Duale N, Gutzkow KB, Brunborg G and Andreassen A 2011. Effects of per- and polyfluorinated compounds on adult rat testicular cells following in vitro exposure. Reprod Toxicol. [DOI] [PubMed] [Google Scholar]
- Liu C, Du Y and Zhou B 2007. Evaluation of estrogenic activities and mechanism of action of perfluorinated chemicals determined by vitellogenin induction in primary cultured tilapia hepatocytes. Aquatic toxicology 85, 267–277. [DOI] [PubMed] [Google Scholar]
- Lopez-Espinosa MJ, Fletcher T, Armstrong B, Genser B, Dhatariya K, Mondal D, Ducatman A and Leonardi G 2011. Association of Perfluorooctanoic Acid (PFOA) and Perfluorooctane Sulfonate (PFOS) with Age of Puberty among Children Living near a Chemical Plant. Environ Sci Technol. [DOI] [PubMed] [Google Scholar]
- Louis GM, Chen Z, Schisterman EF, Kim S, Sweeney AM, Sundaram R, Lynch CD, Gore-Langton RE and Barr DB 2015. Perfluorochemicals and human semen quality: the LIFE study. Environmental health perspectives 123, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv Z, Li G, Li Y, Ying C, Chen J, Chen T, Wei J, Lin Y, Jiang Y, Wang Y, Shu B, Xu B and Xu S 2013. Glucose and lipid homeostasis in adult rat is impaired by early-life exposure to perfluorooctane sulfonate. Environmental toxicology 28, 532–542. [DOI] [PubMed] [Google Scholar]
- Maher JM, Aleksunes LM, Dieter MZ, Tanaka Y, Peters JM, Manautou JE and Klaassen CD 2008. Nrf2- and PPAR alpha-mediated regulation of hepatic Mrp transporters after exposure to perfluorooctanoic acid and perfluorodecanoic acid. Toxicological sciences : an official journal of the Society of Toxicology 106, 319–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloney EK and Waxman DJ 1999. trans-Activation of PPARalpha and PPARgamma by structurally diverse environmental chemicals. Toxicology and applied pharmacology 161, 209–218. [DOI] [PubMed] [Google Scholar]
- Maras M, Vanparys C, Muylle F, Robbens J, Berger U, Barber JL, Blust R and De Coen W 2006. Estrogen-like properties of fluorotelomer alcohols as revealed by mcf-7 breast cancer cell proliferation. Environmental health perspectives 114, 100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgensen UB, Grandjean P, Heilmann C, Nielsen F, Weihe P and Budtz-Jorgensen E 2015. Structural equation modeling of immunotoxicity associated with exposure to perfluorinated alkylates. Environ Health 14, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson JW, Hatch EE and Webster TF 2010. Exposure to polyfluoroalkyl chemicals and cholesterol, body weight, and insulin resistance in the general U.S. population. Environmental health perspectives 118, 197–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen GW and Zobel LR 2007. Assessment of lipid, hepatic, and thyroid parameters with serum perfluorooctanoate (PFOA) concentrations in fluorochemical production workers. Int Arch Occup Environ Health 81, 231–246. [DOI] [PubMed] [Google Scholar]
- Olsen GW, Burris JM, Ehresman DJ, Froehlich JW, Seacat AM, Butenhoff JL and Zobel LR 2007. Half-life of serum elimination of perfluorooctanesulfonate,perfluorohexanesulfonate, and perfluorooctanoate in retired fluorochemical production workers. Environmental health perspectives 115, 1298–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen GW, Butenhoff JL and Zobel LR 2009. Perfluoroalkyl chemicals and human fetal development: an epidemiologic review with clinical and toxicological perspectives. Reprod Toxicol 27, 212–230. [DOI] [PubMed] [Google Scholar]
- Olsen GW, Ellefson ME, Mair DC, Church TR, Goldberg CL, Herron RM, Medhdizadehkashi Z, Nobiletti JB, Rios JA, Reagen WK and Zobel LR 2011. Analysis of a Homologous Series of Perfluorocarboxylates from American Red Cross Adult Blood Donors, 2000–2001 and 2006. Environ Sci Technol. [DOI] [PubMed] [Google Scholar]
- Onishchenko N, Fischer C, Wan Ibrahim WN, Negri S, Spulber S, Cottica D and Ceccatelli S 2011. Prenatal exposure to PFOS or PFOA alters motor function in mice in a sex-related manner. Neurotox Res 19, 452–461. [DOI] [PubMed] [Google Scholar]
- Oshida K, Vasani N, Jones C, Moore T, Hester S, Nesnow S, Auerbach S, Geter DR, Aleksunes LM, Thomas RS, Applegate D, Klaassen CD and Corton JC 2015b. Identification of chemical modulators of the constitutive activated receptor (CAR) in a gene expression compendium. Nucl Recept Signal 13, e002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshida K, Vasani N, Thomas RS, Applegate D, Gonzalez FJ, Aleksunes LM, Klaassen CD and Corton JC 2015a. Screening a mouse liver gene expression compendium identifies modulators of the aryl hydrocarbon receptor (AhR). Toxicology 336, 99–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshida K, Vasani N, Thomas RS, Applegate D, Rosen M, Abbott B, Lau C, Guo G, Aleksunes LM, Klaassen C and Corton JC 2015c. Identification of modulators of the nuclear receptor peroxisome proliferator-activated receptor alpha (PPARalpha) in a mouse liver gene expression compendium. PloS one 10, e0112655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshida K, Vasani N, Waxman DJ and Corton JC 2016a. Disruption of STAT5b-Regulated Sexual Dimorphism of the Liver Transcriptome by Diverse Factors Is a Common Event. PloS one 11, e0148308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshida K, Waxman DJ and Corton JC 2016b. Chemical and Hormonal Effects on STAT5b-Dependent Sexual Dimorphism of the Liver Transcriptome . PloS one 11, e0150284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedram A, Razandi M, O’Mahony F, Harvey H, Harvey BJ and Levin ER 2013. Estrogen reduces lipid content in the liver exclusively from membrane receptor signaling. Sci Signal 6, ra36. [DOI] [PubMed] [Google Scholar]
- Rakhshandehroo M, Sanderson LM, Matilainen M, Stienstra R, Carlberg C, de Groot PJ, Muller M and Kersten S 2007. Comprehensive analysis of PPARalpha-dependent regulation of hepatic lipid metabolism by expression profiling. PPAR Res 2007, 26839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren H, Aleksunes LM, Wood C, Vallanat B, George MH, Klaassen CD, Corton JC. Characterization of peroxisome proliferator-activated receptor alpha--independent effects of PPARalpha activators in the rodent liver: di-(2-ethylhexyl) phthalate also activates the constitutive-activated receptor. Toxicol Sci. 2010. January;113(1):45–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren H, Vallanat B, Nelson DM, Yeung LW, Guruge KS, Lam PK, Lehman-McKeeman LD and Corton JC 2009. Evidence for the involvement of xenobiotic-responsive nuclear receptors in transcriptional effects upon perfluoroalkyl acid exposure in diverse species. Reprod Toxicol 27, 266–277. [DOI] [PubMed] [Google Scholar]
- Rockwell CE, Turley AE, Cheng X, Fields PE and Klaassen CD 2013. Acute Immunotoxic Effects of Perfluorononanoic Acid (PFNA) in C57BL/6 Mice. Clinical & experimental pharmacology Suppl 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers JM, Ellis-Hutchings RG, Grey BE, Zucker RM, Norwood J Jr., Grace CE, Gordon CJ and Lau C 2014. Elevated blood pressure in offspring of rats exposed to diverse chemicals during pregnancy. Toxicological sciences : an official journal of the Society of Toxicology 137, 436–446. [DOI] [PubMed] [Google Scholar]
- Rosen MB, Abbott BD, Wolf DC, Corton JC, Wood CR, Schmid JE, Das KP, Zehr RD, Blair ET and Lau C 2008a. Gene profiling in the livers of wild-type and PPARalpha-null mice exposed to perfluorooctanoic acid. Toxicologic pathology 36, 592–607. [DOI] [PubMed] [Google Scholar]
- Rosen MB, Lee JS, Ren H, Vallanat B, Liu J, Waalkes MP, Abbott BD, Lau C and Corton JC 2008b. Toxicogenomic dissection of the perfluorooctanoic acid transcript profile in mouse liver: evidence for the involvement of nuclear receptors PPAR alpha and CAR. Toxicological sciences : an official journal of the Society of Toxicology 103, 46–56. [DOI] [PubMed] [Google Scholar]
- Rosen MB, Schmid JR, Corton JC, Zehr RD, Das KP, Abbott BD and Lau C 2010. Gene Expression Profiling in Wild-Type and PPARalpha-Null Mice Exposed to Perfluorooctane Sulfonate Reveals PPARalpha-Independent Effects. PPAR Res 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakr CJ, Kreckmann KH, Green JW, Gillies PJ, Reynolds JL and Leonard RC 2007a. Cross-sectional study of lipids and liver enzymes related to a serum biomarker of exposure (ammonium perfluorooctanoate or APFO) as part of a general health survey in a cohort of occupationally exposed workers. J Occup Environ Med 49, 1086–1096. [DOI] [PubMed] [Google Scholar]
- Sakr CJ, Leonard RC, Kreckmann KH, Slade MD and Cullen MR 2007b. Longitudinal study of serum lipids and liver enzymes in workers with occupational exposure to ammonium perfluorooctanoate. J Occup Environ Med 49, 872–879. [DOI] [PubMed] [Google Scholar]
- Savitz DA 2007. Guest editorial: biomarkers of perfluorinated chemicals and birth weight. Environmental health perspectives 115, A528–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharmach E, Buhrke T, Lichtenstein D and Lampen A 2012. Perfluorooctanoic acid affects the activity of the hepatocyte nuclear factor 4 alpha (HNF4alpha). Toxicol Lett 212, 106–112. [DOI] [PubMed] [Google Scholar]
- Schupp M, Cristancho AG, Lefterova MI, Hanniman EA, Briggs ER, Steger DJ, Qatanani M, Curtin JC, Schug J, Ochsner SA, McKenna NJ, Lazar MA. Re-expression of GATA2 cooperates with peroxisome proliferator-activated receptor-gamma depletion to revert the adipocyte phenotype. J Biol Chem. 2009. April 3;284(14):9458–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z, Feng Y, Wang J, Zhang H, Ding L and Dai J 2010. Perfluorododecanoic acid-induced steroidogenic inhibition is associated with steroidogenic acute regulatory protein and reactive oxygen species in cAMP-stimulated Leydig cells. Toxicological sciences : an official journal of the Society of Toxicology 114, 285–294. [DOI] [PubMed] [Google Scholar]
- Shipley JM and Waxman DJ 2004. Simultaneous, bidirectional inhibitory crosstalk between PPAR and STAT5b. Toxicology and applied pharmacology 199, 275–284. [DOI] [PubMed] [Google Scholar]
- Stauber AJ, Brown-Borg H, Liu J, Waalkes MP, Laughter A, Staben RA, Coley JC, Swanson C, Voss KA, Kopchick JJ and Corton JC 2005. Constitutive expression of peroxisome proliferator-activated receptor alpha-regulated genes in dwarf mice. Mol Pharmacol 67, 681–694. [DOI] [PubMed] [Google Scholar]
- Steenland K, Tinker S, Frisbee S, Ducatman A and Vaccarino V 2009. Association of perfluorooctanoic acid and perfluorooctane sulfonate with serum lipids among adults living near a chemical plant. Am J Epidemiol 170, 1268–1278. [DOI] [PubMed] [Google Scholar]
- Steenland K, Tinker S, Shankar A and Ducatman A 2010. Association of perfluorooctanoic acid (PFOA) and perfluorooctane sulfonate (PFOS) with uric acid among adults with elevated community exposure to PFOA. Environmental health perspectives 118, 229–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenland K, Zhao L, Winquist A and Parks C 2013. Ulcerative colitis and perfluorooctanoic acid (PFOA) in a highly exposed population of community residents and workers in the mid-Ohio valley. Environmental health perspectives 121, 900–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takacs ML and Abbott BD 2007. Activation of mouse and human peroxisome proliferator-activated receptors (alpha, beta/delta, gamma) by perfluorooctanoic acid and perfluorooctane sulfonate. Toxicological sciences : an official journal of the Society of Toxicology 95, 108–117. [DOI] [PubMed] [Google Scholar]
- Tan Y, Muise ES, Dai H, Raubertas R, Wong KK, Thompson GM, Wood HB, Meinke PT, Lum PY, Thompson JR, Berger JP. Novel transcriptome profiling analyses demonstrate that selective peroxisome proliferator-activated receptor γ (PPARγ) modulators display attenuated and selective gene regulatory activity in comparison with PPARγ full agonists. Mol Pharmacol. 2012. July;82(1):68–79. [DOI] [PubMed] [Google Scholar]
- Taxvig C, Dreisig K, Boberg J, Nellemann C, Schelde AB, Pedersen D, Boergesen M, Mandrup S and Vinggaard AM 2012. Differential effects of environmental chemicals and food contaminants on adipogenesis, biomarker release and PPARgamma activation. Molecular and cellular endocrinology 361, 106–115. [DOI] [PubMed] [Google Scholar]
- Thomas M, Bayha C, Klein K, Müller S, Weiss TS, Schwab M, Zanger UM. The truncated splice variant of peroxisome proliferator-activated receptor alpha, PPARα-tr, autonomously regulates proliferative and pro-inflammatory genes. BMC Cancer. 2015. June 30;15:488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson JP, Hunter JM, Lempiainen H, Muller A, Terranova R, Moggs JG and Meehan RR 2013. Dynamic changes in 5-hydroxymethylation signatures underpin early and late events in drug exposed liver. Nucleic acids research 41, 5639–5654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilton SC, Orner GA, Benninghoff AD, Carpenter HM, Hendricks JD, Pereira CB and Williams DE 2008. Genomic profiling reveals an alternate mechanism for hepatic tumor promotion by perfluorooctanoic acid in rainbow trout. Environmental health perspectives 116, 1047–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udy GB, Towers RP, Snell RG, Wilkins RJ, Park SH, Ram PA, Waxman DJ and Davey HW 1997. Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. Proc Natl Acad Sci U S A 94, 7239–7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Heuvel JP, Thompson JT, Frame SR and Gillies PJ 2006. Differential activation of nuclear receptors by perfluorinated fatty acid analogs and natural fatty acids: a comparison of human, mouse, and rat peroxisome proliferator-activated receptor-alpha, -beta, and -gamma, liver X receptor-beta, and retinoid X receptor-alpha. Toxicological sciences : an official journal of the Society of Toxicology 92, 476–489. [DOI] [PubMed] [Google Scholar]
- Velez MP, Arbuckle TE and Fraser WD 2015. Maternal exposure to perfluorinated chemicals and reduced fecundity: the MIREC study. Hum Reprod 30, 701–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Ibrahim WN, Tofighi R, Onishchenko N, Rebellato P, Bose R, Uhlén P, Ceccatelli S. Perfluorooctane sulfonate induces neuronal and oligodendrocytic differentiation in neural stem cells and alters the expression of PPARγ in vitro and in vivo. Toxicol Appl Pharmacol. 2013. May 15;269(1):51–60. [DOI] [PubMed] [Google Scholar]
- Watkins AM, Wood CR, Lin MT and Abbott BD 2015. The effects of perfluorinated chemicals on adipocyte differentiation in vitro. Molecular and cellular endocrinology 400, 90–101. [DOI] [PubMed] [Google Scholar]
- Waxman DJ 1988. Interactions of hepatic cytochromes P-450 with steroid hormones. Regioselectivity and stereospecificity of steroid metabolism and hormonal regulation of rat P-450 enzyme expression. Biochemical pharmacology 37, 71–84. [DOI] [PubMed] [Google Scholar]
- Wei S, Chen LQ, Taniyasu S, So MK, Murphy MB, Yamashita N, Yeung LW and Lam PK 2007. Distribution of perfluorinated compounds in surface seawaters between Asia and Antarctica. Mar Pollut Bull 54, 1813–1818. [DOI] [PubMed] [Google Scholar]
- White SS, Stanko JP, Kato K, Calafat AM, Hines EP and Fenton SE 2011. Gestational and chronic low-dose PFOA exposures and mammary gland growth and differentiation in three generations of CD-1 mice. Environmental health perspectives 119, 1070–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winquist A and Steenland K 2014. Modeled PFOA Exposure and Coronary Artery Disease, Hypertension, and High Cholesterol in Community and Worker Cohorts. Environmental health perspectives 122, 1299–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf CJ, Schmid JE, Lau C and Abbott BD 2012. Activation of mouse and human peroxisome proliferator-activated receptor-alpha (PPARalpha) by perfluoroalkyl acids (PFAAs): Further investigation of C4-C12 compounds. Reprod Toxicol 33, 546–551. [DOI] [PubMed] [Google Scholar]
- Wolf CJ, Takacs ML, Schmid JE, Lau C and Abbott BD 2008a. Activation of mouse and human peroxisome proliferator-activated receptor alpha by perfluoroalkyl acids of different functional groups and chain lengths. Toxicological sciences : an official journal of the Society of Toxicology 106, 162–171. [DOI] [PubMed] [Google Scholar]
- Wolf CJ, Zehr RD, Schmid JE, Lau C and Abbott BD 2010. Developmental effects of perfluorononanoic Acid in the mouse are dependent on peroxisome proliferator-activated receptor-alpha. PPAR Res 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf DC, Moore T, Abbott BD, Rosen MB, Das KP, Zehr RD, Lindstrom AB, Strynar MJ and Lau C 2008b. Comparative hepatic effects of perfluorooctanoic acid and WY 14,643 in PPAR-alpha knockout and wild-type mice. Toxicologic pathology 36, 632–639. [DOI] [PubMed] [Google Scholar]
- Yamamoto J, Yamane T, Oishi Y and Kobayashi-Hattori K 2015. Perfluorooctanoic acid binds to peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation in 3T3-L1 adipocytes. Biosci Biotechnol Biochem 79, 636–639. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Moore R, Goldsworthy TL, Negishi M and Maronpot RR 2004. The orphan nuclear receptor constitutive active/androstane receptor is essential for liver tumor promotion by phenobarbital in mice. Cancer research 64, 7197–7200. [DOI] [PubMed] [Google Scholar]
- Yamashita N, Kannan K, Taniyasu S, Horii Y, Petrick G and Gamo T 2005. A global survey of perfluorinated acids in oceans. Mar Pollut Bull 51, 658–668. [DOI] [PubMed] [Google Scholar]
- Yang B, Zou W, Hu Z, Liu F, Zhou L, Yang S, Kuang H, Wu L, Wei J, Wang J, Zou T and Zhang D 2014. Involvement of oxidative stress and inflammation in liver injury caused by perfluorooctanoic acid exposure in mice. Biomed Res Int 2014, 409837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao PL, Ehresman DJ, Rae JM, Chang SC, Frame SR, Butenhoff JL, Kennedy GL, Peters JM. Comparative in vivo and in vitro analysis of possible estrogenic effects of perfluorooctanoic acid. Toxicology. 2014. December 4;326:62–73. [DOI] [PubMed] [Google Scholar]
- Yao X and Zhong L 2005. Genotoxic risk and oxidative DNA damage in HepG2 cells exposed to perfluorooctanoic acid. Mutat Res 587, 38–44. [DOI] [PubMed] [Google Scholar]
- Yao Y, Volchek K, Brown CE, Robinson A and Obal T 2014. Comparative study on adsorption of perfluorooctane sulfonate (PFOS) and perfluorooctanoate (PFOA) by different adsorbents in water. Water Sci Technol 70, 1983–19 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.