

Infection by C. parvum causes inhibition of intestinal epithelial turnover but underlying mechanisms are unclear. This study indicates that parasite Cdg7_FLc_1030 RNA is delivered into host cells during infection, resulting in upregulation and release of DKK1 and, consequently, inhibition of intestinal epithelial cell migration.
Keywords: Cryptosporidium, cryptosporidiosis, intestinal epithelium, parasitic infection, Dkk1, gene transcription, cell migration
Abstract
Intestinal infection by Cryptosporidium is known to cause epithelial cell migration disorder but the underlying mechanisms are unclear. Previous studies demonstrated that a panel of parasite RNA transcripts of low protein-coding potential are delivered into infected epithelial cells. Using multiple models of intestinal cryptosporidiosis, we report here that C. parvum infection induces expression and release of the dickkopf protein 1 (Dkk1) from intestinal epithelial cells. Delivery of parasite Cdg7_FLc_1030 RNA to intestinal epithelial cells triggers transactivation of host Dkk1 gene during C. parvum infection. Release of Dkk1 is involved in C. parvum-induced inhibition of cell migration of epithelial cells, including noninfected bystander cells. Moreover, Dkk1-mediated suppression of host cell migration during C. parvum infection involves inhibition of Cdc42/Par6 signaling. Our data support the hypothesis that attenuation of intestinal epithelial cell migration during Cryptosporidium infection involves parasite Cdg7_FLc_1030 RNA-mediated induction and release of Dkk1 from infected cells.
Cryptosporidium, a genus of protozoa in the phylum Apicomplexa, represents a group of protozoan parasites that can infect humans and many other species of animals [1, 2]. The C. parvum and C. hominis species cause most Cryptosporidium infections in humans, particularly in AIDS patients and in children younger than 2 years old in developing countries [1, 3]. Humans are infected when they ingest Cryptosporidium oocysts. Once ingested, oocysts excyst in the gastrointestinal tract and release infective sporozoites. The sporozoite attaches to a host epithelial cell and forms a vacuole in which the organism remains intracellular but extracytoplasmic. The internalized sporozoite then matures, undergoes asexual and sexual development, and yields oocysts to complete a life cycle within 4–6 days [2]. Cryptosporidium can complete all stages of its development (asexual and sexual) within a single host [2].
The primary infection site of the parasite in humans is the small intestine. The intestinal mucosa is a monolayer of rapidly self-renewing epithelial cells. New functional epithelial cells are produced from stem cells in the crypt base, differentiate, and migrate from the crypt base to the luminal surface, and, eventually, are shed into the lumen after they have reached the tip of the villus; hence, the entire intestinal epithelium is replaced every 2–3 days in mice (3–5 days in humans) [4, 5]. It appears that Cryptosporidium has developed strategies to counteract the rapid turnover of intestinal epithelium to support its intracellular cell cycle. C. parvum infection induces apoptotic resistance in infected epithelial cells during the early stage of infection [6]. We recently observed that C. parvum infection inhibits cell migration of intestinal epithelial cells in culture, including infected cells and noninfected bystander cells [7]. Both the apoptotic resistance in infected cells and attenuation of epithelial cell migration may provide a survival benefit to the parasite cell cycle. However, molecular mechanisms underlying host cell migration inhibition during Cryptosporidium infection are still unclear.
The interactions between Cryptosporidium and intestinal epithelial cells involves exchanges of distinct effector molecules from both sides of the host cell and the parasite at the host-parasite interface [8, 9]. Such exchanges of effector molecules may be involved in parasite invasion and intracellular development [1, 8, 9]. In our previous studies [10], we demonstrated that several C. parvum RNA transcripts of low protein-coding potential are selectively delivered into intestinal epithelial cells during host-parasite interactions and may modulate gene transcription in infected host cells. Specifically, delivery of the parasite Cdg7_FLc_1000 (GenBank ID: FX115830.1) [11] causes trans-suppression of host sphingomyelin phosphodiesterase 3 (SMPD3) gene, resulting in attenuation of cell migration of infected host cells [7]. The dickkopf (Dkk) family encodes secreted proteins and consists of 4 main members in vertebrates (ie, Dkk1,2,3,4) [12]. Dkk1, a secreted protein with 2 cysteine rich regions, is involved in embryonic development [12] and in the regulation of intestinal epithelial cell migration [13]. Induction of Dkk1 was previously demonstrated in human intestinal epithelial cells following C. parvum infection [14]. Here, we report that host delivery of parasite Cdg7_FLc_1030 RNA (GenBank ID: FX115613.1) [11] promotes the transcription of Dkk1 gene in infected intestinal epithelial cells; release of Dkk1 from host cells during C. parvum infection is involved in inhibition of cell migration of epithelial cells, including noninfected bystander cells.
METHODS
C. parvum and Cell Lines
C. parvum oocysts of the Iowa strain were purchased from a commercial source (Bunch Grass Farm, Deary, ID). The human nonmalignant intestinal epithelial cell line (INT; FHs 74 Int, CCL-241) was purchased from ATCC (Manassas, VA). The murine intestinal epithelial cell line (IEC4.1) was a kind gift from Dr. Pingchang Yang (McMaster University, Hamilton, ON, Canada) and cultured as previously reported [7].
Infection Models and Infection Assays
Models of intestinal cryptosporidiosis using cell lines were employed as previously described; infection was with a 1:1 ratio of C. parvum oocysts and host cells [7, 11]. An ex vivo infection model employing enteroids from neonatal mice [15] and a well-developed infection model of cryptosporidiosis in neonatal mice [16, 17] were used for ex vivo and in vivo experiments. At least 5 animals from each group were sacrificed and ileal tissues were obtained for immunohistochemistry and biochemical analyses. Real-time polymerase chain reaction (PCR), immunofluorescence microscopy, and immunohistochemistry were used to assay C. parvum infection as previously reported [18, 19]. Details are described in the Supplementary Materials.
Quantitative Real-Time PCR
For quantitative analysis of mRNA and C. parvum RNA expression, comparative real-time PCR was performed as previous reported [20] using the SYBR Green PCR Master Mix (Applied Biosystems, Carlsbad, CA). Briefly, RNA was extracted using TRI-reagent, treated with DNA-free Kit (Ambion) to remove any remaining DNA. Quantified 500 ng RNA was reverse-transcribed using T100 thermal cyclers (Bio-Rad). Real-time PCR was then performed using 25 ng of template cDNA for each RNA gene of interest. Each sample was run in triplicate. The relative abundance of each RNA was calculated using the ΔΔCt method and normalized to GAPDH or U2 small nuclear RNA (RNU2-1) (a nuclear RNA). The sequences for all the primers are listed in Supplementary Table 1.
siRNAs and Plasmids
Custom-designed siRNA oligos against Cdg7_FLc_1030 and a scrambled siRNA were synthesized by Integrated DNA Technologies (Coralville, IA) and transfected into cells with Lipofectamine RNAimax (Invitrogen). Sequences of siRNAs are: 5′-CGUCAAGGAAUUUACGUAUUU-3′ for Cdg7_FLc_1030 and nonspecific scrambled sequence 5′-UUCUCCGAACGUGUCACGUUU-3′ for the control. The plasmids expressing parasite RNAs were generated by real-time PCR amplification of the cDNA, using RNA from C. parvum sporozoites and cloned into the pcDNA3.1(+) vector, per the manufacturer’s protocol (Invitrogen). The sequences for all the primers are listed in Supplementary Table 1.
Whole Cell Extracts, Western Blot, and Immunofluorescent Staining
Whole cell extracts were prepared using the Mammalian Protein Extraction Reagent (Fisher) supplemented with cocktail protease inhibitors. Cell pellets were incubated in the Mammalian Protein Extraction Reagent, centrifuged at 16100g for 20 minutes and the supernatants were saved as the whole cell extracts, as previously reported [21]. Details for western blot and immunofluorescent staining are described in the Supplementary Materials.
RNA Immunoprecipitation, Chromatin IP, and Chromatin Isolation by RNA Purification Analyses
The formaldehyde cross-linking RNA immunoprecipitation (RIP) was performed as described [22]. For chromatin immunoprecipitation (ChIP) analysis, a commercially available ChIP Assay Kit (Upstate Biotechnologies) was used in accordance with the manufacturer’s instructions. Chromatin isolation by RNA purification (ChIRP) analysis was performed as previously reported [23]. A pool of tiling oligonucleotide probes with affinity specific to the C. parvum Cdg7_FLc_1030 RNA sequences was used and glutaraldehyde cross-linked for chromatin isolation. The sequences for all the primers and probes are listed in Supplementary Table 1 and Table 2. Details are described in the Supplementary Materials.
Cell Migration and MTT Assay
The wound-healing assay was used to analyze cell migration. Cell proliferation assay was carried out using the CellTiter 96 AQueous One Solution Cell Proliferation 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) Assay Kit (Promega Corporation), with details in the Supplementary Materials.
RESULTS
C. parvum Infection Attenuates Intestinal Epithelial Cell Migration in Both Directly Infected Cells and Noninfected Bystander Cells
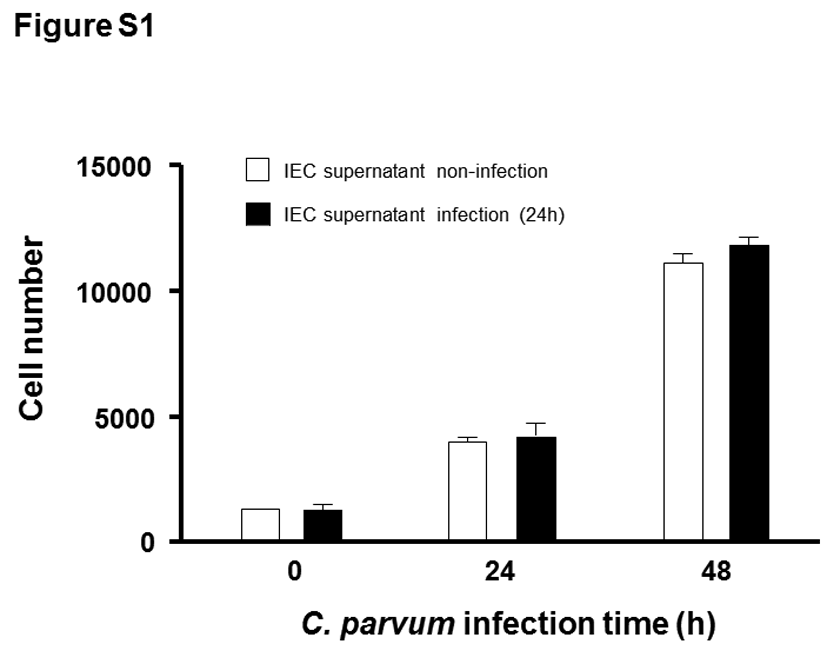
In our previous studies, we demonstrated that C. parvum infection of cultured human intestinal epithelial cells inhibited cell migration and inhibition of migration was not limited to directly infected cells [7]. Inhibition of host cell migration during infection was observed in both directly infected cells and bystander noninfected cells using cultured murine intestinal epithelial cells (IEC4.1 cells) (Figure 1A–1C). To confirm that infection can attenuate cell migration of noninfected bystander cells, we collected the supernatants from IEC4.1 cell cultures after exposure to C. parvum infection for 24 and 48 hours and took a centrifugation approach to remove cell or parasite debris. IEC4.1 cells also showed attenuation of migration after culture with the conditioned media with the supernatants (Figure 1A and 1C). This decrease in cell migration distance was not due to cell death induced by infection [24, 25], as the MTT assay revealed no obvious difference in cell number between the infected cell cultures and the noninfected control (Figure 1D and Supplementary Figure 1). Lack of obvious cell death may reflect the higher infection rate and the fully confluent nature of the cell cultures, as C. parvum-infected cells and confluent cultures are resistant to apoptotic cell death [25].
Figure 1.

Inhibition of intestinal epithelial cell migration during Cryptosporidium parvum infection. A, Decreased migration of IEC4.1 cells following C. parvum infection or after incubation with the supernatants from infected IEC4.1 cultures. Cell migration was assessed by measurement of the distance of cell migration after the wound-healing assay. Representative phase images of cell cultures after exposure to C. parvum infection or incubation with addition of the conditioned supernatants for 24 hours are shown. B, Representative dual fluorescent image of cell cultures after exposure to C. parvum infection for 24 hours, showing presence of infected cell (parasite stained in red) and noninfected cells at the migrating edge. Nuclei of cells were stained blue with DAPI (4′,6-diamidino-2-phenylindole). C, Quantitative analysis of the migration distance of IEC4.1 cells following C. parvum infection or after incubation with the supernatants from infected IEC4.1 cultures. D, Quantitative analysis of the cell numbers in the IEC4.1 cultures following C. parvum infection. Data represent means ± SEs from 3 independent experiments. *P < .01 ANOVA versus noninfected control (Ctrl).
C. parvum Infection Induces Expression and Release of Dkk1 from Infected Intestinal Epithelial Cells
Given the inhibitory effect of supernatant from infected cultures on epithelial cell migration, we questioned whether soluble factors in the supernatant released from infected cells are involved in the underlying mechanisms. Genome-wide mRNA array analysis in previous studies revealed significant alterations in gene expression profiles in nonmalignant (INT) and malignant (HCT-8) human intestinal epithelial cells after C. parvum infection in vitro [10, 14, 26]. Genes that are upregulated in host cells include cytokine/chemokine genes and DKK1 gene. There are 4 members of the DKK protein family, that is DKK1–4, conserved in humans and mice [12]. Based on our previous genome-wide array analysis [10], upregulation of DKK1, but not DKK2–4, was shown in INT cells at 48 hours after C. parvum infection (Figure 2A). Similarly, upregulation of DKK1, but not DKK2–4, was observed in INT and IEC4.1 cells after exposure to C. parvum for various periods of time using real-time PCR (Figure 2B). Interestingly, a slight decrease in expression levels of Dkk2 (in INT cells at 48 hours postinfection) and Dkk3 (in IEC4.1 cells at both 24 and 48 hours postinfection) was measured. Using an ex vivo infection model employing enteroids from neonatal mice [15], we detected an increased expression level of Dkk1 RNA in enteroids following C. parvum infection (Figure 2C). An increase in Dkk1 expression was also observed in the intestinal epithelium in infected neonatal mice following oral administration of the parasite (Figure 2C). Different from results with the ex vivo infection model, a higher Dkk1 increase at 48 hours than at 24 hours postinfection was observed in the intestinal epithelium in infected neonatal mice, probably due to the different time course of infection in the 2 models [15]. Moreover, upregulation of Dkk1 at the protein level, but not Dkk2–4, was further confirmed in IEC4.1 cell cultures directly exposed to C. parvum or in supernatants from the cell cultures after exposure to C. parvum infection (Figure 2D).
Figure 2.

Cryptosporidium parvum infection induces expression and release of Dkk1 from infected intestinal epithelial cells. A, Alterations in DKK gene expression profile from genome-wide transcriptome analysis in INT cells following C. parvum infection from our previous study (the GEO database GSE87047) [10]. INT cells were exposed to C. parvum infection for 48 hours, followed by the microarray analysis; noninfected cells were used as the control (Ctrl). The mean value of the expression level for DKK genes is presented as a heatmap and listed in the table. B, Alterations in DKK gene expression in INT cells and IEC4.1 following C. parvum infection. INT and IEC4.1 cells were exposed to C. parvum infection for 24–48 hours, followed by real-time PCR analysis; noninfected cells were used as the control. C, Induction of Dkk1 gene in the enteroids following ex vivo C. parvum infection (Infect.) and in ileum tissues of neonates following in vivo C. parvum infection. Expression levels of Dkk1 gene in enteroids at 24 and 48 hours after ex vivo infection, as well as in the ileum epithelium at 24 and 48 hours after in vivo infection, were measured using real-time PCR. D, Upregulation of Dkk1 at the protein level, but not for Dkk2–4, in IEC4.1 cell cultures directly exposed to C. parvum or in supernatants from the cell cultures after exposure to C. parvum infection. The blotting gels were also stained for protein with silver staining as control. Data represent means ± SEs from 3 independent experiments. *P < .01 ANOVA versus non-infected control.
Dkk1 Released From Infected Epithelial Cells in the Supernatants Is Involved in C. parvum-Induced Inhibition of Cell Migration of the Noninfected Bystander Cells
To explore whether DKK1 released from infected cells is involved in inhibition of epithelial cell migration during C. parvum infection, we used a neutralizing antibody to Dkk1 [27] and measured its effects on cell migration associated with C. parvum infection. The neutralizing antibody to Dkk1 restored cell migration of IEC4.1 cells after incubation with the supernatants of infected cell cultures (Figure 3A and 3B). In accordance, as a positive control, addition of recombinant mouse Dkk1 protein to the culture media inhibited the migration of IEC4.1 cells (Figure 3C).
Figure 3.

Release of Dkk1 in the supernatants is involved in Cryptosporidium parvum-induced inhibition of cell migration of the noninfected bystander cells. A, Representative phase images of IEC4.1 cell cultures after incubation with the supernatants from infected IEC4.1 cultures for 24–48 hours, in the presence or absence of neutralizing anti-Dkk1. Cell migration was assessed by measurement of the distance of cell migration after the wound-healing assay. B, Quantitative analysis of the migration distance of IEC4.1 cells after incubation with the supernatants from infected IEC4.1 cultures, in the presence or absence of neutralizing anti-Dkk1. Proliferation of IEC4.1 cells after incubation with supernatants from infected IEC4.1 cultures, in the presence or absence of the neutralizing anti-Dkk1, was also assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. C, Inhibition of IEC4.1 migration by recombinant mDkk1. The migration distance of IEC4.1 cells, in the presence or absence of the recombinant mouse Dkk1 (mDkk1), was measured and cell proliferation assessed by MTT assay. Data represent means ± SEs from 3 independent experiments. *P < .01 ANOVA versus cells treated with supernatant from noninfected control (Ctrl) or the non-antibody treated Ctrl; #P < .01 ANOVA versus cells after incubation with the supernatants in the absence of anti-Dkk1 treatment.
Delivery of Parasite Cdg7_FLc_1030 RNA to Infected Epithelial Cells Triggers Transactivation of Host Dkk1 Gene During C. parvum Infection
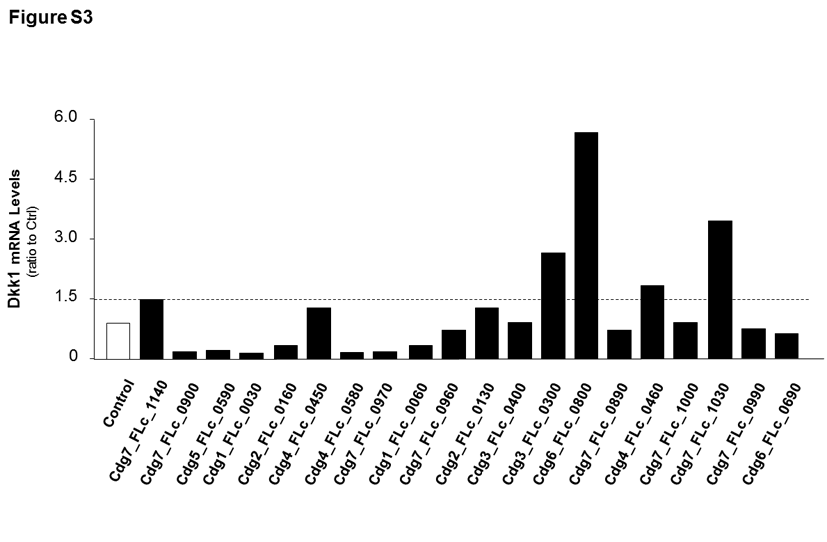
Our previous studies demonstrate that a panel of C. parvum RNA transcripts is selectively delivered into epithelial cells during host cell invasion and may modulate gene transcription in infected cells [10]. We generated constructs expressing each of these C. parvum RNAs and transfection of each of these constructs resulted in significant expression of the corresponding parasite RNA in IEC4.1 cells (Supplementary Figure 2). Expression levels of Dkk1 were then measured, by real-time PCR, in IEC4.1 cells after transfection of these constructs. Interestingly, a high level of Dkk1 RNA was detected in cells following transfection of parasite Cdg7_FLc_1030, an RNA transcript from the chromatin 7 of low protein-coding potential (GenBank ID: FX115613.1) [21, 22] (Figure 4A and 4B). No significant alteration in Dkk1 expression was detected in cells transfected with the other parasite RNA transcripts (Supplementary Figure 3). We then questioned whether nuclear delivery of Cdg7_FLc_1030 causes Dkk1 transcription. Because conventional genetic tools are very difficult, if not impossible, to modify C. parvum genes [1, 28], we developed a method to treat cells with an siRNA to Cdg7_FLc_1030 for 4 hours and then exposed them to C. parvum. The increase in Cdg7_FLc_1030 RNA level in cells induced by C. parvum infection was significantly suppressed by pretreatment with the siRNA to Cdg7_FLc_1030 (Figure 4C). Accordingly, upregulation of Dkk1 RNA expression induced by C. parvum infection was at least partially inhibited through pretreatment of the siRNA to Cdg7_FLc_1030 (Figure 4C).
Figure 4.

Induction of the Dkk1 gene in epithelial cells following Cryptosporidium parvum infection is associated with delivery of Cdg7_FLc_1030 into the host cells. A, Chromatic location and sequence of the Cdg7_FLc_1030 gene in C. parvum. B, Expression of Full-Cdg7_FLc_1030, but not other parasite RNAs, resulted in induction of Dkk1 expression in IEC4.1 cells. IEC4.1 was transfected with the constructs expressing various full-lengths of parasite RNAs for 48 hours. Cells transfected with the empty vector were used as the control. Whole-cell extracts were collected and expression levels of corresponding parasite RNAs and Dkk1 were measured with real-time PCR. C, Inhibition of Cdg7_FLc_1030 in host cells by the siRNA treatment attenuated the induction of Dkk1 following C. parvum infection. IEC4.1 cells were treated with an siRNA to Cdg7_FLc_1030 for 12 hours and then exposed to C. parvum infection for additional 24 hours. A nonspecific scrambled siRNA was used as the control (Ctrl). Contents of Cdg7_FLc_1030 and Dkk1 cells were quantified by real-time PCR. Data represent means ± SEs from 3 independent experiments. *P < .01 ANOVA versus noninfected cells treated with the control siRNA or empty vector controls; #P < .01 ANOVA versus infected cells treated with the control siRNA.
To explore how Cdg7_FLc_1030 may promote Dkk1 expression in the host cells, we measured the enrichment of transcriptional activity markers within the promoter region of the Dkk1 gene locus in cells following infection. Histone modifications, such as H3K4 and H3K36 methylations, are generally associated with gene transcriptional activation [20]. Increased enrichment of H3K4me1 and H3K36me3 was detected in the Dkk1 gene locus in infected cells using ChIP analysis with anti-H3K4me1 or anti-H3K36me3 and the PCR primer sets designed to cover the various promoter regions of the Dkk1 gene locus (Figure 5A). In addition, increased enrichment of the RNA polymerase II (Pol II) was also detected in the Dkk1 gene locus in infected cells using ChIP analysis with anti-Pol II and the same PCR primer sets (Figure 5A). To test whether Cdg7_FLc_1030 is assembled into the Pol II complex in the infected cells, we preformed RIP analysis of infected cells. A significant amount of Cdg7_FLc_1030, but not the control RNU-2 RNA, was detected in the immunoprecipitates from infected cells using anti-Pol II (Figure 5B). To define whether Cdg7_FLc_1030 is physically recruited to the Dkk1 gene locus in infected cells, we used a pool of biotinylated tiling oligonucleotide probes specific to Cdg7_FLc_1030 for ChIRP analysis. Recruitment of Cdg7_FLc_1030 was detected within the promoter region of the Dkk1 gene locus in cells following infection (Figure 5C). To further explore how Cdg7_FLc_1030 may activate transcription of the Dkk1 gene, we generated various luciferase reporter vectors encompassing the various upstream regions of the transcription start site of the Dkk1 gene (Figure 5D). IEC4.1 cells were transfected with the luciferase reporter plasmids, followed by exposure to C. parvum infection. No significant increase in luciferase activity associated with the Dkk1 promoter regions was detected in cells following infection (Figure 5D). In contrast, a marked increase in luciferase activity associated with the human interleukin 8 luciferase reporter plasmid, as a positive control for C. parvum-induced transcription of host genes [29], was measured in infected cells (Figure 5D). Because the luciferase reporter assay cannot mimic the chromatin-remodeling–mediated regulation of gene transcription, we speculate that Cdg7_FLc_1030 may activate transcription of the Dkk1 gene through modulation of chromatin-remodeling–associated histone modifications.
Figure 5.

Host delivery of Cdg7_FLc_1030 triggers transcription of the Dkk1 gene in epithelial cells. A, Levels of the transcription activity markers, H3K4me1 and H3K36me3, as well as enrichment of Pol II, associated with the Dkk1 gene locus in IEC4.1 cells following Cryptosporidium parvum infection. Cells were exposed to C. parvum infection for 24 hours, followed by chromatin immunoprecipitation (ChIP) analysis using anti-H3K4me1, anti-H3K36me3, or anti-Pol II, respectively, and the PCR primer sets as designed. Increased enrichment of H3K4me1 and H3K36me3, as well as Pol II, was detected in the Dkk1 gene locus in cells following infection. B, Assembly of Cdg7_FLc_1030 to the Pol II complex in cells following C. parvum infection. IEC4.1 were exposed to C. parvum infection for 24 hours or transfected with Full-Cdg7_FLc_1030 for 24 hours, followed by RNA immunoprecipitation (RIP) analysis using anti-Pol II and PCR primers for Cdg7_FLc_1030. Assembly of the U2 RNA was also measured as control (Ctrl). C, Recruitment of Cdg7_FLc_1030 to the Dkk1 gene locus in IEC4.1 cells following C. parvum infection. Cells were exposed to C. parvum infection for 24 hours, following by chromatin isolation by RNA purification (ChIRP) analysis using a pool of probes specific to Cdg7_FLc_1030 and the PCR primer sets as designed. Increased recruitment of Cdg7_FLc_1030 was detected in the Dkk1 gene locus in cells following infection. D, C. parvum infection did not promote the luciferase activity associated with the transcription of the Dkk1-promoter luciferase reporter constructs. Various regions of Dkk1 promoter were cloned into the luciferase reporter constructs and the interleukin 8 luciferase reporter (IL8-Luc) plasmid containing human IL8 promoter was used for the positive control. IEC4.1 cells were transfected with the constructs for 12 hours and followed by exposure to C. parvum infection for 24 hours. Cells transfected with the empty vector were used as the control. The luciferase activities of the cells were then measured and normalized to β-gal. Data represent means ± SEs from 3 independent experiments. *P < .01 ANOVA versus noninfected or empty vector controls.
DKK1-Mediated Suppression of Host Cell Migration during C. parvum Infection Involves Inhibition of Cdc42/Par6 Signaling
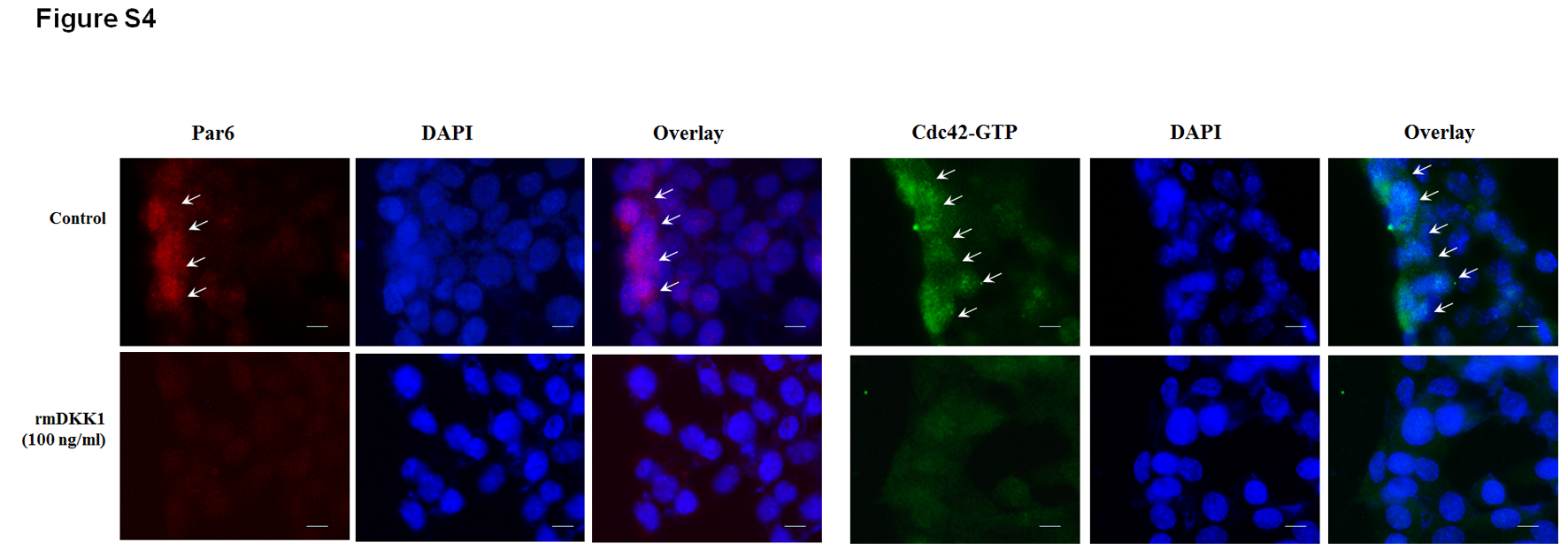
Previous studies demonstrated that Dkk1 mediates cell migration through inhibition of Cdc42/Par6 signaling [13]. To define whether Cdc42/Par6 signaling is involved in C. parvum-induced inhibition of intestinal epithelial cell migration, we performed immunostaining of Cdc42-active and Par6 in IEC4.1 cell cultures upon wound healing after incubation with supernatants from infected cell cultures. An increased staining of Cdc42-active and Par6 was detected in cells along the migrating edge after wounding when they were cultured with normal control media (Figure 6A and 6B). In contrast, we detected a weakened staining of both Cdc42-active and Par6 in cells along the migrating edge upon wounding in the cell cultures after incubation with the conditioned supernatants (Figure 6A and 6B). Addition of neutralizing anti-Dkk1 to the culture media restored the staining level of Cdc42-active and Par6 in cells along the migrating edge (Figure 6A and 6B). Decreased staining of Cdc42-active and Par6 along the migrating edge was also detected in cells following culture with addition of the recombinant mouse Dkk1 (Supplementary Figure 4).
Figure 6.

The Cdc42-Par6 signaling pathway is involved in DKK1-mediated suppression of host cell migration during Cryptosporidium parvum infection. IEC4.1 cells were incubated with the supernatants from infected IEC4.1 cultures for 24 hours, in the presence or absence of the neutralizing anti-Dkk1. Wound healing was applied to the cell cultured as the cell migration assay, followed by immunostaining with anti-Par6 or anti-Cdc42-GTP (Cdc42-active), respectively. Representative images showing the staining of Par6 (A) and Cdc42-GTP (B) in cells along the migrating edge (indicated by arrowheads) after wounding are shown. Data represent means ± SEs from 3 independent experiments. Bar = 20 µm.
DISCUSSION
In this study, we report that host delivery of a parasite Cdg7_FLc_1030 RNA transcript into infected intestinal epithelial cells during C. parvum infection activates transcription of the Dkk1 gene and increases release of Dkk1 protein from host epithelial cells. Consequently, induction and release of Dkk1 inhibits cell migration of both infected cells and noninfected bystander cells. Coupled with our previous study on the trans-suppression of the Smpd3 gene through Cdg7_FLc_1000-mediated epigenetic suppression to inhibit cell migration in infected cells [7], our data support the hypothesis that delivery of parasite RNA transcripts into infected host cells during Cryptosporidium infection causes inhibition of epithelial cell migration of both infected and noninfected bystander cell populations, contributing to disturbances of intestinal epithelial homeostasis following Cryptosporidium infection (Figure 7).
Figure 7.

Delivery of parasite RNA transcripts into the nuclei of infected host epithelial cells following cryptosporidial infection activates transcription of the Dkk1 gene (via delivery of Cdg7_FLc_1030) and suppresses the transcription of Smpd3 gene (via delivery of Cdg7_FLc_1000) [7], resulting in the inhibition of epithelial cell migration of both infected host cells and noninfected bystander cells, a process that may benefit the parasite intracellular cell cycle in host cells.
Consistent with data from previous studies [10, 14], we detected induction of Dkk1, but not other members of the Dkk family, in intestinal epithelial cells after C. parvum infection, using in vitro, ex vivo, and in vivo infection models. Interestingly, tissue damage itself can cause an upregulation of Dkk-1 and production of Dkk-1 is a normal response to manage tissue remodeling in response to injury [30–32]. Increased amounts of Dkk1 have also been reported in chronic inflammatory diseases such as various types of cancers, rheumatoid arthritis, and lupus [33–35]. Here, our data indicate that C. parvum infection of intestinal epithelial cells triggers Dkk1 transcription via host delivery of Cdg7_FLc_1030, a parasite RNA transcript of low protein potential [11]. This induction of Dkk1 through host delivery of Cdg7_FLc_1030 appears to be specific as expression of other parasite RNA transcripts, even though they were also delivered into infected host cells, failed to induce Dkk1 transcription. Moreover, Dkk1 induction was not detected in intestinal epithelial cells following infection by Escherichia coli or Aeromonas caviae [36, 37], suggesting that this is not a general intestinal epithelial cell response to pathogen infection or inflammatory stimulation.
Mechanistically, transactivation of the Dkk1 gene in intestinal epithelial cells after C. parvum infection is associated with an increased methylation of H3K4 and H3K36 in its gene promoter. We speculate that Cdg7_FLc_1030 may activate transcription of the Dkk1 gene through modulation of chromatin-remodeling–associated histone modifications. Consistent with the mechanism of transactivation for most genes in mammalian cells, the enrichment of Pol II to the Dkk1 gene locus was also detected in cells following infection. It is still unclear how Cdg7_FLc_1030 RNA may trigger transcription of the Dkk1 gene. Increasing evidence supports the hypothesis that RNA molecules in mammalian cells can function as scaffold molecules to affect gene transcription through their interactions with various RNA-binding components in the chromatin-remodeling complexes [38, 39]. As such, Cdg7_FLc_1030 may modulate the recruitment of those chromatin-remodeling complexes to the Dkk1 gene locus. In our previous studies, we demonstrated that nuclear delivery of the parasite Cdg7_FLc_1000 transcript (GeneBank ID: FX115830.1) [11] causes trans-suppression of host SMPD3 gene [7]. Cdg7_FLc_1000 can interact with the positive regulatory domain zinc finger protein 1, an RNA-binding protein with a role in the regulation of histone methylation [40, 41], and, consequently, causes trans-suppression of the SMPD3 gene [7]. Another potential mechanism is that Cdg7_FLc_1030 may trigger Dkk1 transcription through direct binding to a specific DNA motif in its promoter regions, as RNAs may interact directly with DNA molecules to form a triple-helical structure [38]. However, C. parvum infection failed to trigger luciferase activity in intestinal epithelial cells that were transfected with plasmids expressing the promoter region-(1/2/3) of the Dkk1 gene locus.
Dkk1 was originally identified in the regulation of head formation of Xenopus laevis and is known to inhibit the canonical Wnt signaling pathway [42, 43]. Dkk1 is an antagonistic inhibitor of the Wnt signaling pathway that acts by isolating the Lrp5/6 receptors so that it cannot aid in activating the Wnt signaling pathway [44]. This inhibition plays a key role in heart, head, and forelimb development during anterior morphogenesis of the embryo [45]. A polarized localization of active Cdc42 and Par6 was demonstrated in the leading edge of migrating cells in cultured intestinal epithelial cells [13]. Dkk1 was reported to disrupt the polarized localization of active Cdc42 and Par6 in the leading edge of migrating cells, resulting in disturbance of cell migration [13]. In our experimental setting, increased staining of active Cdc42 and Par6 was observed in the migrating cells along the wounded edge. Our data also implicate the involvement of Cdc42/Par6 signaling in the attenuation of intestinal epithelial cell migration associated with release of Dkk1 during C. parvum infection. Dkk1 is a well-characterized Wnt signaling inhibitor and acts as an antagonist of the canonical Wnt pathway by binding to the Wnt receptor [46, 47]. Given the critical role of Wnt signaling in regulating intestinal epithelial cell migration [48, 49], future studies should investigate the potential role of Wnt signaling associated with Dkk1 release in inhibition of cell migration following infection. Cdg7_FLc_1030-mediated cell migration through induction and release of Dkk1 may be critical to the parasite intracellular cell cycle during intestinal infection, which merits in vivo investigation using cell-type specific Dkk1 knockout mice. Moreover, targeting Dkk1 may be of relevance to the development of therapeutic strategies for intestinal cryptosporidiosis.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Notes
Acknowledgments. We thank Drs Quanghui Zhao (Northwest A&F University, China) and Shibin Ma (Creighton University) for helpful and stimulating discussions, and Barbara L. Bittner (Creighton University) for her assistance in writing the manuscript.
Financial Support. This work was supported by the National Institutes of Health (grant numbers AI116323 and AI136877 to X.-M. C.); Nebraska Department of Health and Human Services (grant number LB595 Cancer and Smoking Disease Research Program Development Grant to X.-M. C.); National Center for Research Resources (grant number G20RR024001); China Scholarship Council (to Z. M.); and National Natural Science Foundation of China (grant number 31372194 to Z. M.).
Potential conflicts of interest. All authors: No reported conflicts of interest. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1. Striepen B. Parasitic infections: time to tackle cryptosporidiosis. Nature 2013; 503:189–91. [DOI] [PubMed] [Google Scholar]
- 2. O’Donoghue PJ. Cryptosporidium and cryptosporidiosis in man and animals. Int J Parasitol 1995; 25:139–95. [DOI] [PubMed] [Google Scholar]
- 3. Checkley W, White AC Jr, Jaganath D et al. A review of the global burden, novel diagnostics, therapeutics, and vaccine targets for Cryptosporidium. Lancet Infect Dis 2015; 15:85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barker N. Adult intestinal stem cells: critical drivers of epithelial homeostasis and regeneration. Nat Rev Mol Cell Biol 2014; 15:19–33. [DOI] [PubMed] [Google Scholar]
- 5. Creamer B, Shorter RG, Bamforth J. The turnover and shedding of epithelial cells. I. The turnover in the gastro-intestinal tract. Gut 1961; 2:110−8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen XM, Keithly JS, Paya CV, LaRusso NF. Cryptosporidiosis. N Engl J Med 2002; 346:1723–31. [DOI] [PubMed] [Google Scholar]
- 7. Ming ZP, Gong AY, Wang Y et al. Involvement of Cryptosporidium parvum Cdg7_FLc_1000 RNA in the attenuation of intestinal epithelial cell migration via trans-suppression of host cell SMPD3 gene. J Infect Dis 2018; 217:122–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sateriale A, Striepen B. Beg, borrow and steal: three aspects of horizontal gene transfer in the protozoan parasite, Cryptosporidium parvum. PLoS Pathog 2016; 12:e1005429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sibley LD. Intracellular parasite invasion strategies. Science 2004; 304:248–53. [DOI] [PubMed] [Google Scholar]
- 10. Wang Y, Gong AY, Ma S et al. Delivery of parasite RNA transcripts into infected epithelial cells during Cryptosporidium infection and its potential impact on host gene transcription. J Infect Dis 2017; 215:636–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yamagishi J, Wakaguri H, Sugano S et al. Construction and analysis of full-length cDNA library of Cryptosporidium parvum. Parasitol Int 2011; 60:199–202. [DOI] [PubMed] [Google Scholar]
- 12. Mukhopadhyay M, Shtrom S, Rodriguez-Esteban C et al. Dickkopf1 is required for embryonic head induction and limb morphogenesis in the mouse. Dev Cell 2001; 1:423–34. [DOI] [PubMed] [Google Scholar]
- 13. Koch S, Capaldo CT, Samarin S et al. Dkk-1 inhibits intestinal epithelial cell migration by attenuating directional polarization of leading edge cells. Mol Biol Cell 2009; 20:4816–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Deng M, Lancto CA, Abrahamsen MS. Cryptosporidium parvum regulation of human epithelial cell gene expression. Int J Parasitol 2004; 34:73–82. [DOI] [PubMed] [Google Scholar]
- 15. Zhang XT, Gong AY, Wang Y et al. Cryptosporidium parvum infection attenuates the ex vivo propagation of murine intestinal enteroids. Physiol Rep 2016; 4:e13060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kapel N, Benhamou Y, Buraud M, Magne D, Opolon P, Gobert JG. Kinetics of mucosal ileal gamma-interferon response during cryptosporidiosis in immunocompetent neonatal mice. Parasitol Res 1996; 82:664–7. [DOI] [PubMed] [Google Scholar]
- 17. Lacroix S, Mancassola R, Naciri M, Laurent F. Cryptosporidium parvum-specific mucosal immune response in C57BL/6 neonatal and gamma interferon-deficient mice: role of tumor necrosis factor alpha in protection. Infect Immun 2001; 69:1635–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sasahara T, Maruyama H, Aoki M et al. Apoptosis of intestinal crypt epithelium after Cryptosporidium parvum infection. J Infect Chemother 2003; 9:278–81. [DOI] [PubMed] [Google Scholar]
- 19. Zhou R, Gong AY, Eischeid AN, Chen XM. miR-27b targets KSRP to coordinate TLR4-mediated epithelial defense against Cryptosporidium parvum infection. PLoS Pathog 2012; 8:e1002702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dong X, Weng Z. The correlation between histone modifications and gene expression. Epigenomics 2013; 5:113–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Abmayr SM, Yao T, Parmely T, Workman JL. Preparation of nuclear and cytoplasmic extracts from mammalian cells. Curr Protoc Mol Biol 2006; 35:12.3.1–12.3.10 [DOI] [PubMed] [Google Scholar]
- 22. Niranjanakumari S, Lasda E, Brazas R, Garcia-Blanco MA. Reversible cross-linking combined with immunoprecipitation to study RNA-protein interactions in vivo. Methods 2002; 26:182–90. [DOI] [PubMed] [Google Scholar]
- 23. Chu C, Qu K, Zhong FL, Artandi SE, Chang HY. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol Cell 2011; 44:667–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu J, Deng M, Lancto CA, Abrahamsen MS, Rutherford MS, Enomoto S. Biphasic modulation of apoptotic pathways in Cryptosporidium parvum-infected human intestinal epithelial cells. Infect Immun 2009; 77:837–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen XM, Levine SA, Splinter PL et al. Cryptosporidium parvum activates nuclear factor kappaB in biliary epithelia preventing epithelial cell apoptosis. Gastroenterology 2001; 120:1774–83. [DOI] [PubMed] [Google Scholar]
- 26. Yang YL, Serrano MG, Sheoran AS, Manque PA, Buck GA, Widmer G. Over-expression and localization of a host protein on the membrane of Cryptosporidium parvum infected epithelial cells. Mol Biochem Parasitol 2009; 168:95–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pozzi S, Fulciniti M, Yan H et al. In vivo and in vitro effects of a novel anti-Dkk1 neutralizing antibody in multiple myeloma. Bone 2013; 53:487–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vinayak S, Pawlowic MC, Sateriale A et al. Genetic modification of the diarrhoeal pathogen Cryptosporidium parvum. Nature 2015; 523:477–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhou R, Hu G, Liu J, Gong AY, Drescher KM, Chen XM. NF-kappaB p65-dependent transactivation of miRNA genes following Cryptosporidium parvum infection stimulates epithelial cell immune responses. PLoS Pathog 2009; 5:e1000681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Price RM, Tulsyan N, Dermody JJ, Schwalb M, Soteropoulos P, Castronuovo JJ Jr. Gene expression after crush injury of human saphenous vein: using microarrays to define the transcriptional profile. J Am Coll Surg 2004; 199:411–8. [DOI] [PubMed] [Google Scholar]
- 31. Li X, Grisanti M, Fan W et al. Dickkopf-1 regulates bone formation in young growing rodents and upon traumatic injury. J Bone Miner Res 2011; 26:2610–21. [DOI] [PubMed] [Google Scholar]
- 32. Koch S, Nava P, Addis C et al. The Wnt antagonist Dkk1 regulates intestinal epithelial homeostasis and wound repair. Gastroenterology 2011; 141:259–68, 268.e1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Diarra D, Stolina M, Polzer K et al. Dickkopf-1 is a master regulator of joint remodeling. Nat Med 2007; 13:156–63. [DOI] [PubMed] [Google Scholar]
- 34. Sato N, Yamabuki T, Takano A et al. Wnt inhibitor Dickkopf-1 as a target for passive cancer immunotherapy. Cancer Res 2010; 70:5326–36. [DOI] [PubMed] [Google Scholar]
- 35. Wang XD, Huang XF, Yan QR, Bao CD. Aberrant activation of the WNT/β-catenin signaling pathway in lupus nephritis. PLoS One 2014; 9:e84852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ukena SN, Westendorf AM, Hansen W et al. The host response to the probiotic Escherichia coli strain Nissle 1917: specific up-regulation of the proinflammatory chemokine MCP-1. BMC Med Genet 2005; 6:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hayes SL, Lye DJ, McKinstry CA, Vesper SJ. Aeromonas caviae strain induces Th1 cytokine response in mouse intestinal tract. Can J Microbiol 2010; 56:27–31. [DOI] [PubMed] [Google Scholar]
- 38. Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell 2013; 154:26–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Guttman M, Donaghey J, Carey BW et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 2011; 477:295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gyory I, Wu J, Fejér G, Seto E, Wright KL. PRDI-BF1 recruits the histone H3 methyltransferase G9a in transcriptional silencing. Nat Immunol 2004; 5:299–308. [DOI] [PubMed] [Google Scholar]
- 41. Shin HM, Kapoor V, Guan T, Kaech SM, Welsh RM, Berg LJ. Epigenetic modifications induced by blimp-1 regulate CD8⁺ T cell memory progression during acute virus infection. Immunity 2013; 39:661–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cruciat CM, Niehrs C. Secreted and transmembrane wnt inhibitors and activators. Cold Spring Harb Perspect Biol 2013; 5:a015081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Glinka A, Wu W, Delius H, Monaghan AP, Blumenstock C, Niehrs C. Dickkopf-1 is a member of a new family of secreted proteins and functions in head induction. Nature 1998; 391:357–62. [DOI] [PubMed] [Google Scholar]
- 44. Lewis SL, Khoo PL, De Young RA et al. Dkk1 and Wnt3 interact to control head morphogenesis in the mouse. Development 2008; 135:1791–801. [DOI] [PubMed] [Google Scholar]
- 45. Schneider VA, Mercola M. Wnt antagonism initiates cardiogenesis in Xenopus laevis. Genes Dev 2001; 15: 304–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Glinka A, Wu W, Delius H, Monaghan AP, Blumenstock C, Niehrs C. Dickkopf-1 is a member of a new family of secreted proteins and functions in head induction. Nature 1998; 391:357–62. [DOI] [PubMed] [Google Scholar]
- 47. Krupnik VE, Sharp JD, Jiang C et al. Functional and structural diversity of the human Dickkopf gene family. Gene 1999; 238:301–13. [DOI] [PubMed] [Google Scholar]
- 48. de Lau W, Barker N, Clevers H. WNT signaling in the normal intestine and colorectal cancer. Front Biosci 2007; 12:471–91. [DOI] [PubMed] [Google Scholar]
- 49. Ouko L, Ziegler TR, Gu LH, Eisenberg LM, Yang VW. Wnt11 signaling promotes proliferation, transformation, and migration of IEC6 intestinal epithelial cells. J Biol Chem 2004; 279:26707–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.