

This study correlated serial blood transcriptomic profiles of patients with avian influenza A (H7N9) virus infection with clinical data of patients. Biologically significant transcriptomic profiles associated with blood oxygenation and viral load in the lower respiratory tract were defined.
Keywords: H7N9, avian influenza, Pa, o, 2/ F, io, 2, transcriptomic
Abstract
Background
Avian influenza A (H7N9) viruses emerged in China in 2013 and caused zoonotic disease associated with a case-fatality ratio of over 30%. Transcriptional profiles in peripheral blood reflect host responses and can help to elucidate disease pathogenesis.
Methods
We correlated serial blood transcriptomic profiles of patients with avian influenza A (H7N9) virus infection and determined the biological significances from the analysis.
Results
We found that specific gene expression profiles in the blood were strongly correlated with the Pao 2/Fio 2 ratio and viral load in the lower respiratory tract. Cell cycle and leukocyte-related immunity were activated at the acute stage of the infection while T-cell functions and various metabolic processes were associated with the recovery phase of the illness. A transition from systemic innate to adaptive immunity was found.
Conclusions
We developed a novel approach for transcriptomic analysis to identify key host responses that were strongly correlated with specific clinical and virologic parameters in patients with H7N9 infection.
Avian influenza A (H7N9) viruses emerged as a zoonotic respiratory disease in China in early 2013 [1, 2]. From February 2013 to October 2017, 1564 patients with H7N9 disease were reported, 612 of whom died, resulting in an overall case-fatality ratio of around 38% [3]. The infection can rapidly progress to severe pneumonia and acute respiratory distress syndrome. H7N9 virus pathogenesis was caused by multiple factors, including high viral replication, virus tropism for the lower respiratory tract, and dysregulation of the host innate immune responses [4–6]. The measurement of blood oxygen levels reflects the extent of pulmonary dysfunction and is used by clinicians to assess disease severity [7].
Several studies have characterized peripheral blood transcriptional profiles of humans infected with influenza viruses and rhinoviruses, as well as respiratory syncytial viruses (RSV) [8, 9], and determined the relevant biological functions. Two similar studies have recently been carried out in patients with H7N9 virus infection. However, these studies were limited by the small numbers of samples studied (4 and 8 samples) and by a lack of association with detailed clinical parameters including viral load [10, 11].
We aimed to identify host transcriptome responses associated with clinical and virological outcomes in patients with influenza A H7N9 infection in comparison with healthy controls. We adopted a novel strategy for transcriptomic analysis to identify genes that are strongly correlated with the Pao 2/Fio 2 ratio (an indicator that reflects oxygen levels in the blood and lung function) and the viral load at the lower respiratory tract. Our results analyzed the regulation of systemic biological functions and pathways during acute and recovery phases of infection.
MATERIALS AND METHODS
Patients
Eight patients with laboratory-confirmed low pathogenic avian influenza A (H7N9) virus infection admitted for care at the First Affiliated Hospital of Guangzhou Medical University, from 2014 to 2016, were included in this study. The clinical history, physical examination, radiological findings, and hematological, biochemical, and microbiological investigations were recorded. Day 1 of clinical onset was defined as the first day of the appearance of clinical symptoms. The dates of admission, age, and clinical outcomes of these patients are tabulated in Supplementary Table 1; and clinical and immunological features of some of these patients have been reported previously [12]. Approval for the study was obtained from the ethics committee of the First Affiliated Hospital of Guangzhou Medical University (No: 2016–78) and informed consent was obtained from the patients or their family members.
Detection of Virus Infection and Viral Load
Serial throat swabs, bronchoalveolar lavage fluid (BALF), sputum, and endotracheal aspirates were collected, as clinically appropriate, throughout the period of hospitalization. The viral RNA from the samples was extracted and measured as previously described [12].
RNA Purification From Peripheral Whole Blood
The peripheral whole blood samples (3 mL) were collected into Tempus tubes (Applied Biosystems) and the RNA was purified according to manufacturer’s instructions.
Gene Expression Profiling
Gene expression from whole blood samples was determined using Affymetrix Human Gene 2.0 ST following the manufacturer’s instructions. Arrays were scanned by Affymetrix GeneChip Scanner 3000 (Affymetrix, Santa Clara, CA). Command Console Software (Affymetrix, Santa Clara, CA) was used to control the scanner and summarize probe cell intensity data (CEL file generation) with default settings. The raw data were then normalized by Expression Console. All raw and normalized microarray data has been deposited into the NCBI GEO database (Accession number: GSE114466).
Data Analysis
The raw microarray data were normalized using robust multiarray average [13]. Genes with expression that correlated with clinical measurement were identified using significance analysis of microarrays [14], with a false discovery rate of 0.01 and a q value cutoff of 0.01. Calculation of molecular distance to health (MDTH) was adapted from a previously described methodology on a gene-by-gene basis [15]. MDTH outliers were defined as being 3 standard deviations away from the median. Outlier identification was performed iteratively until no further outliers could be identified. Gene ontology enrichment (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways analysis was performed using the web interfaces http://www.geneontology.org and http://www.genome.jp/kegg, respectively [16]. For computing the gene expression change per unit of clinical parameter, linear regression was performed to fit the clinical parameter of interest to the expression level of each gene using linregress function in Python library SciPy. The slope of the linear regression was taken as the gene expression change per unit of clinical parameter.
Statistical Analysis
Wilcoxon rank sum test was employed to compute the P value of the difference in MDTH between week 0 and week 2, 3, and 4. The default function cor.test in R software was employed to compute the P value of the Pearson correlation.
RESULTS
Demographics of the Patients With H7N9 Virus Infection
Eight patients who were admitted to the hospital from 2013 to 2016 were enrolled in this study. Clinical, virologic, and immunological features of each patient were recorded in detail. Six patients (No. 2, 4, 6, 7, 8, and 9) were discharged and 2 patients (No. 3 and 10) had a fatal outcome. Patients No. 1 and 5 were excluded as no sample was available for the transcriptomic study. All patients had laboratory-confirmed infection with low pathogenic influenza A (H7N9) viruses by reverse transcription polymerase chain reaction using specific HA primers. The demographic and clinical characteristics of the patients during their hospitalization are shown in Supplementary Table 1. Clinical parameters that were used by the clinicians to assist the diagnosis and treatment were recorded on the indicated days. Viral copy numbers were determined from the throat swab (defined as an upper respiratory tract specimen) and from BALF or endotracheal aspirate (defined as lower respiratory tract). Bacteria were identified from the sputum samples in each of the patient to investigate possible bacterial coinfection. Fifteen healthy donors served as controls where we assumed the viral loads in the upper and lower respiratory tracts were zero and their Pao 2/Fio 2 values were 450.
Differential Gene Expression Profile Between H7N9-Infected Patients and Healthy Controls
The gene expression profiles of the total RNA collected from the peripheral blood of the patients, each with multiple time points from the second to fourth week after disease onset (27 samples), were quantified by microarray analysis together with the samples of healthy individuals (15 samples). An overview of the data using principal component analysis showed that the gene expression profiles from the H7N9-infected patients formed a distinct cluster compared to the healthy control group (Figure 1A–1C). The difference can be readily observed in the first 3 components, which together account for >50% of the variance of the data. We next computed the MDTH from each sample to see if it can be used to assess the clinical severity of the H7N9-infected patients (Figure 1D). MDTH is a computational score derived from the full gene set of a blood transcriptomic profile and can provide a summary of overall deviation of gene expression compared to healthy controls. The use of MDTH score has been demonstrated previously to differentiate the severity of the disease in the studies such as pulmonary tuberculosis, septicemic melioidosis, and RSV infection [9, 15, 17]. We found that the MDTH scores derived from all patients’ samples were all higher than the normal controls. When we grouped the samples based on week after disease onset, the MDTH score from each week was also significantly higher than the control (P <2 × 10−10) (Figure 1E).
Figure 1.

Overview of the microarray data. A–C, First 3 components (PC1–3) from principal component analysis of the gene expression profile are plotted. Ellipses represent 0.7 standard deviation from the mean value for the indicated categories (patient and control). D, The molecular distance to health (MDTH) for each sample is shown. E, Samples are categorized by week. The distribution of MDTH for each category is shown as a box plot. ** indicates a significant difference (P value < 2 × 10−5, Wilcoxon rank sum test) compared to the control.
Correlation Between the Blood Transcriptomic Profiling and Clinical Parameters of H7N9-Infected Patients
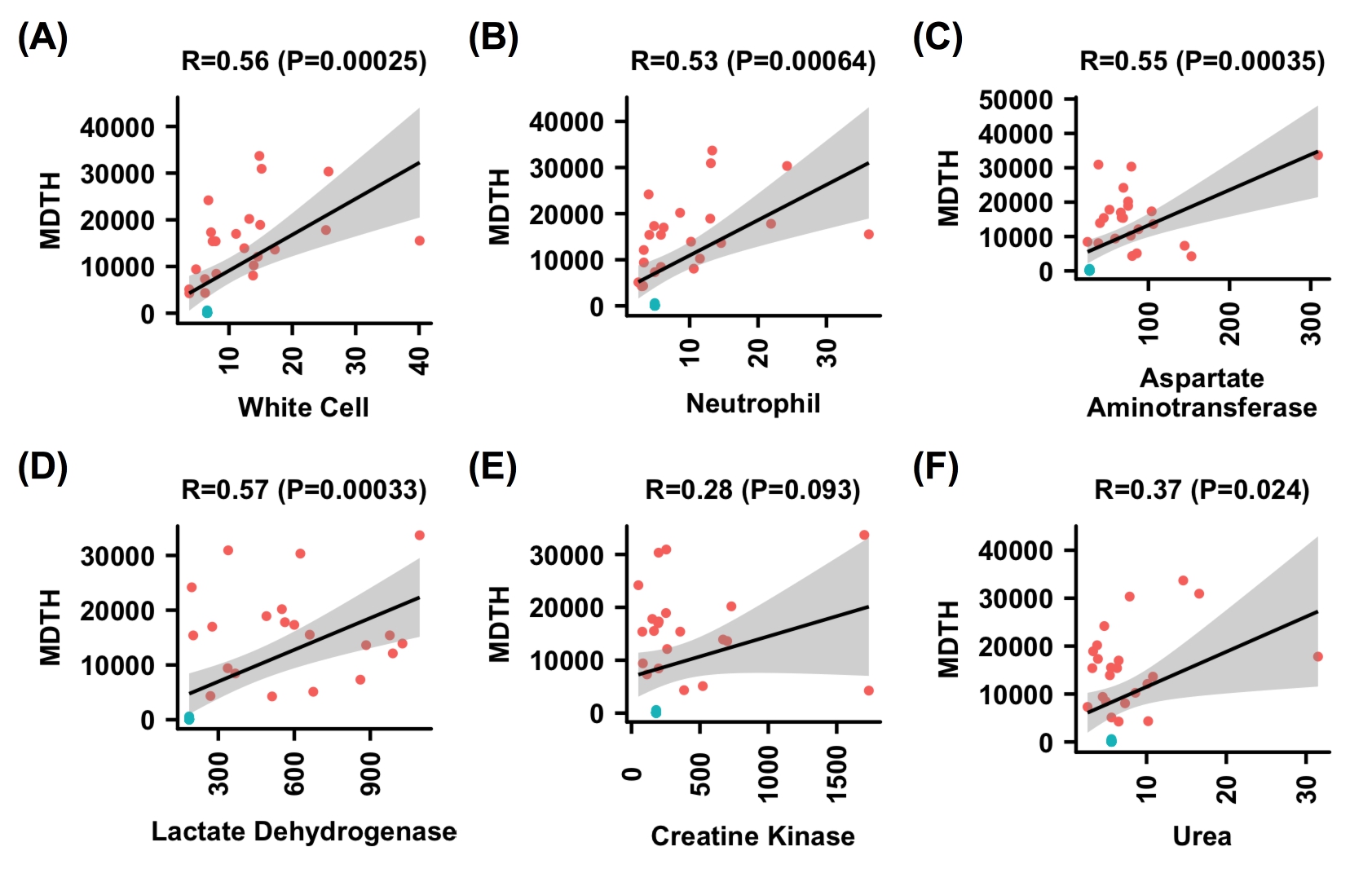
We then investigated if MDTH is associated with specific clinical parameters in the H7N9-infected patients. The correlations of MDTH with the values of different clinical parameters, including Pao 2/Fio 2 ratio, viral loads in the upper (URT) and lower respiratory tract (LRT), procalcitonin (PCT), white cell count, neutrophil count, hemoglobin, aspartate aminotransferase, lactate dehydrogenase, creatine kinase, d-dimer, and urea, which were recorded on the same day as sampling, were examined by Pearson correlation test. We found that the MDTH correlated with most of the parameters except creatine kinase and viral load in the URT (Figure 2 and Supplementary Figure 1). Among all the tested parameters, Pao 2/Fio 2 ratio (Pearson correlation, −0.73; P value 2 × 10−7) and viral load in LRT (Pearson correlation 0.51; P value .0012) were clearly known to associate with the pathogenesis of H7N9 virus during infection. We further aimed to identify genes from the blood transcriptomic profiles that were associated with the Pao 2/Fio 2 ratio and viral load in LRT. We first examined whether there were any genes in the profile that showed a positive or negative correlation to the Pao 2/Fio 2 ratio or viral load in LRT. Positive correlation means the expression of the gene increased when the value of the parameter increased, while negative correlation means the change in gene expression level was opposite to the change in the parameter. From our analysis, we found that there were 4788 and 2189 genes that were positively or negatively correlated with the Pao 2/Fio 2 ratio, respectively. Similarly, 565 and 1845 genes were positively or negatively correlated with the viral load in the LRT, respectively (Figure 3A). During infection, the lung pathology could be caused by diverse factors, including replication of the virus, immune dysregulation, or bacterial coinfection. To specifically determine the host responses that were related to both H7N9 replication and lung dysfunction, we further identified 2 sets of genes with contrasting correlation to both Pao 2/Fio 2 ratio and viral load in LRT : (1) negatively correlated to Pao 2/Fio 2 ratio and positively correlated with viral load in LRT; and (2) positively correlated to Pao 2/Fio 2 ratio and negatively correlated with viral load in LRT (Figure 3B). This was based on the hypothesis that increasing viral load was likely to be associated with impaired lung function, that is decreased Pao 2/Fio 2 ratio. Set 1 represents the genes activated during the acute phase of infection (oxygen level in blood decreased and viral load at the LRT increased) or deactivated during the recovery phase (oxygen level in blood increased and viral load at the LRT decreased). On the contrary, set 2 represents the opposite condition, that is genes deactivated during the acute phase of infection or activated during the recovery phase. From our analysis, 541 and 1803 genes corresponded to sets 1 and 2, respectively.
Figure 2.

Correlation between molecular-distance-to-health (MDTH) and clinical parameters: A, Pao2/Fio2 ratio; B, lower respiratory tract (LRT) titer; C, upper respiratory tract (URT) titer; D, procalcitonin; E, hemoglobin; F, d-dimer. Control samples are blue; patient samples red. Pearson correlation and the corresponding P value are indicated. For controls, Pao2/Fio2 was assumed to be 450; viral titer at the upper and lower respiratory tracts were set as 0. MDTH values of 4 data points, namely patient No. 2 (day 13), patient No. 2 (day 15), Patient No. 3 (day 21), and patient No. 3 (day 28), were identified as outliers (see Materials and Methods) and were excluded from this analysis. Therefore, each plot includes a total of 15 control samples and 23 patient samples.
Figure 3.

Identification of genes with expression levels that correlated with clinical parameters. A, Number of genes identified with expression that showed a negative or positive correlation with clinical parameters of interest. B, Venn diagram showing numbers of genes which were identified from both the gene sets of Pao2/Fio2 ratio and viral titer at the LRT.
Differential Systemic Host Responses at the Acute and Recovery Phase of H7N9 Infection
We further performed GO analysis to investigate the biological and functional processes that were associated with the 2 overlapping gene sets. The genes which were identified from both the gene sets of negative Pao 2/Fio 2 and positive viral load in LRT correlation (set 1) could be clustered into 2 groups of biological processes: cell cycle regulation and leukocyte functions (Table 1). On the other hand, T-cell–related functions and metabolic processes were the top ranks of GO terms in the overlapping gene set with positive Pao 2/Fio 2 and negative viral load at LRT correlation (set 2) (Table 2). Interestingly, few interferon-activated gene expression functions were identified from our data sets, although there was strong evidence of virus shedding in the LRT at the day of sampling. Because a recent study has shown that expression of the gene IFI27 is upregulated in seasonal influenza infection and is a biomarker that discriminates influenza from bacterial infections, we specifically looked its expression in our data set [18]. IFI27 and the related IFI27L1 were two of the interferon pathway-associated genes upregulated in our patients; they were negatively correlated with Pao 2/Fio 2 and positively correlated with higher virus load in the LRT, but were not correlated with PCT, which has been suggested as a biomarker for bacterial infection.
Table 1.
Gene Ontology (GO) Functional Enrichment of Genes With Negative Correlation to Pao2/Fio2 Ratio and Positive Correlation to Viral Load in the Lower Respiratory Tract
| Group | GO Term | GO Biological Process | Fold Enrichment | P Value |
|---|---|---|---|---|
| Leukocyte-related functions | GO:0043299 | Leukocyte degranulation | 5.99 | 8.09E-31 |
| GO:0043312 | Neutrophil degranulation | 6.08 | 2.81E-30 | |
| GO:0002275 | Myeloid cell activation involved in immune response | 5.87 | 2.37E-30 | |
| GO:0002283 | Neutrophil activation involved in immune response | 6.03 | 4.36E-30 | |
| GO:0002446 | Neutrophil mediated immunity | 5.92 | 1.15E-29 | |
| GO:0042119 | Neutrophil activation | 5.93 | 1.04E-29 | |
| GO:0002444 | Myeloid leukocyte mediated immunity | 5.77 | 1.75E-29 | |
| GO:0036230 | Granulocyte activation | 5.89 | 1.59E-29 | |
| GO:0002274 | Myeloid leukocyte activation | 5.42 | 7.14E-29 | |
| GO:0002366 | Leukocyte activation involved in immune response | 5.2 | 1.13E-28 | |
| Cell cycle-related functions | GO:0031536 | Positive regulation of exit from mitosis | 21.92 | .000838 |
| GO:0031577 | Spindle checkpoint | 12.27 | .00000545 | |
| GO:0007094 | Mitotic spindle assembly checkpoint | 11.44 | .0000366 | |
| GO:0071174 | Mitotic spindle checkpoint | 11.44 | .0000366 | |
| GO:0071173 | Spindle assembly checkpoint | 11.44 | .0000366 | |
| GO:0051782 | Negative regulation of cell division | 10.96 | .000891 | |
| GO:0045841 | Negative regulation of mitotic metaphase/anaphase transition | 10.96 | .0000449 | |
| GO:1902100 | Negative regulation of metaphase/anaphase transition of cell cycle | 10.52 | .0000547 | |
| GO:0030071 | Regulation of mitotic metaphase/anaphase transition | 10.12 | .0000000152 | |
| GO:0090307 | Mitotic spindle assembly | 10.05 | .0000000646 |
Top 10 processes were selected from each group.
Table 2.
Gene Ontology (GO) Functional Enrichment of Genes With Positive Correlation to Pao2/Fio2 Ratio and Negative Correlation to Viral Load in the Lower Respiratory Tract
| Group | GO Term | GO Biological Process | Fold Enrichment | P Value |
|---|---|---|---|---|
| Metabolic-related functions | GO:0016070 | RNA metabolic process | 1.68 | 1.13E-28 |
| GO:0090304 | Nucleic acid metabolic process | 1.62 | 1.48E-28 | |
| GO:0044260 | Cellular macromolecule metabolic process | 1.41 | 1.03E-26 | |
| GO:0006139 | Nucleobase-containing compound metabolic process | 1.53 | 2.66E-25 | |
| GO:0046483 | Heterocycle metabolic process | 1.51 | 1.11E-24 | |
| GO:0006725 | Cellular aromatic compound metabolic process | 1.49 | 6.81E-24 | |
| GO:0043170 | Macromolecule metabolic process | 1.33 | 1.71E-23 | |
| GO:0060255 | Regulation of macromolecule metabolic process | 1.4 | 1.77E-23 | |
| GO:0034641 | Cellular nitrogen compound metabolic process | 1.46 | 2.74E-23 | |
| GO:0044237 | Cellular metabolic process | 1.29 | 3.69E-23 | |
| T-cell–related functions | GO:0045061 | Thymic T-cell selection | 6.6 | 0.0000658 |
| GO:0043368 | Positive T-cell selection | 6.23 | 0.0000154 | |
| GO:0045058 | T-cell selection | 5.89 | 0.000000144 | |
| GO:0002293 | Alpha-beta T-cell differentiation involved in immune response | 4.79 | 0.000211 | |
| GO:0002287 | Alpha-beta T-cell activation involved in immune response | 4.79 | 0.000211 | |
| GO:0046632 | Alpha-beta T-cell differentiation | 4.67 | 0.000000944 | |
| GO:0031295 | T-cell costimulation | 4.49 | 0.00000000399 | |
| GO:0042093 | T-helper–cell differentiation | 4.49 | 0.000642 | |
| GO:0002294 | CD4-positive, alpha-beta T-cell differentiation involved in immune response | 4.49 | 0.000642 | |
| GO:0031294 | Lymphocyte costimulation | 4.43 | 0.00000000503 |
Top 10 processes were selected from each group
Our results illustrated the differential systemic host responses in the 2 phases of H7N9 infection. In the acute stage (Pao 2/Fio 2 decreased and viral load increased) the cell cycle regulation and leukocyte functions were activated, while T-cell–related functions and metabolic processes were deactivated. In the recovery stage (Pao 2/Fio 2 increased and viral load decreased) T-cell responses and metabolic processes were dominant but the cell cycle and leukocyte functions were suppressed. Overlapping gene sets were also identified between Pao 2/Fio 2 and either procalcitonin, d-dimer, or hemoglobin using a similar analysis. Interestingly, although differential biological processes were found from these gene sets in the acute phase of infection, T-cell–related functions and metabolic processes were both enriched at the recovery phase (data not shown).
We then performed KEGG pathway analysis from the 2 overlapping gene sets between Pao 2/Fio 2 and viral load in LRT to identify the pathways that involve products of these genes (Figure 4). Interestingly, pathways related to systemic lupus erythematosus and alcoholism were strongly enriched (Figure 4A). The cell cycle pathway was consistently identified from the overlapping gene set of negative Pao 2/Fio 2 and positive viral load in LRT (set 1) (Figure 4C). On the other hand, the pathways related to the different immune responses, especially those associated with adaptive immunity, were significantly enriched in the overlapping gene set of positive Pao 2/Fio 2 and negative viral load at LRT (set 2) (Figure 4B and 4D).
Figure 4.


Top 20 Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways for each of the gene sets. Negative log-transformed adjusted P values for each of the top 20 KEGG pathways are plotted for (A) the overlapping gene set with negative Pao2/Fio2 and positive viral load at LRT correlation; and (B) the overlapping gene set with positive Pao2/Fio2 and negative viral load at LRT correlation. Red lines indicate an adjusted P value cutoff of 0.05. Bars are arranged in a descending order of negative log-transformed adjusted P values. A higher negative log-transformed adjusted P value indicates greater change in the corresponding pathway. Schematic gene pathways of (C) cell cycle and (D) T-cell receptor signaling with genes identified from our analysis marked with yellow.
Finally, we identified genes from sets 1 and 2 that were significantly sensitive to the change in Pao 2/Fio 2 ratio and viral load in LRT. A linear regression analysis was performed to compute the change in gene expression level per unit of the 2 parameters. Genes with R values higher than 0.4 and P value lower than .05 were considered as significant. The selected genes that were related to our identified biological processes are shown in Table 3. In the gene group represented by the acute stage of infection, CD177 and MMP9 were found to be the most sensitive genes to those parameters. Consistent with the results from GO and KEGG analysis, a group of T-cell–related genes, including TRAV12-2, TRAJ17, CD28, TRAT1, and THEMIS, were mainly activated during the improvement of Pao 2/Fio 2 ratio and viral load in LRT. Interestingly, genes related to natural killer cell-mediated cytotoxicity, including KLRB1, KLRF1, KLRD1, KLRC3, KLRG1, and KLRC1, were identified, which was in line with the results of KEGG analysis.
Table 3.
Sensitivity of Genes to Pao2/Fio2 and Viral Load in the Lower Respiratory Tract (LRT)
| Gene | Gene Description | Change per Unit of Pao2/Fio2 | Pearson Correlation R | P Value | Change per Unit of Viral Load in LRT | Pearson Correlation R | P Value |
|---|---|---|---|---|---|---|---|
| Genes with negative correlation to Pao2/Fio2 and positive correlation to viral load in LRT (set 1) | |||||||
| CD177 | CD177 molecule | −0.0134 | −0.81 | 9.67E-31 | 0.9987 | 0.74 | 1.78E-22 |
| MMP9 | Matrix metallopeptidase 9 | −0.0120 | −0.72 | 8.74E-08 | 0.8881 | 0.64 | 6.98E-06 |
| ALPL | Alkaline phosphatase, liver/ bone/kidney | −0.0088 | −0.60 | 2.43E-05 | 0.6733 | 0.56 | 1.62E-04 |
| IL1R2 | Interleukin 1 receptor, type II | −0.0092 | −0.70 | 2.94E-07 | 0.6154 | 0.56 | 1.28E-04 |
| MGAM2 | Maltase-glucoamylase 2 (putative) | −0.0094 | −0.63 | 1.34E-65 | 0.5873 | 0.47 | 3.49E-33 |
| TNFAIP6 | Tumor necrosis factor, alpha-induced protein 6 | −0.0072 | −0.65 | 3.55E-06 | 0.5779 | 0.62 | 1.30E-05 |
| MMP8 | Matrix metallopeptidase 8 | −0.0105 | −0.73 | 3.87E-08 | 0.5013 | 0.44 | 4.04E-03 |
| ARG1 | Arginase 1 | −0.0087 | −0.77 | 3.51E-09 | 0.4739 | 0.52 | 5.40E-04 |
| CXCR1 | Chemokine (C-X-C motif) receptor 1 | −0.0054 | −0.49 | 1.13E-03 | 0.4281 | 0.46 | 2.38E-03 |
| MMP25 | Matrix metallopeptidase 25 | −0.0052 | −0.59 | 3.34E-05 | 0.3748 | 0.52 | 5.30E-04 |
| Genes with positive correlation to Pao2/Fio2 and negative correlation to viral load in LRT (set 2) | |||||||
| KLRB1 | Killer cell lectin-like receptor subfamily B, member 1 | 0.0088 | 0.85 | 9.02E-13 | −0.5407 | −0.64 | 7.98E-06 |
| KLRF1 | Killer cell lectin-like receptor subfamily F, member 1 | 0.0089 | 0.83 | 7.60E-12 | −0.4472 | −0.51 | 6.77E-04 |
| TRAV12-2 | T-cell receptor alpha variable 12-2 | 0.0062 | 0.66 | 1.74E-06 | −0.3893 | −0.51 | 6.84E-04 |
| KLRD1 | Killer cell lectin-like receptor subfamily D, member 1 | 0.0070 | 0.86 | 3.52E-13 | −0.3847 | −0.58 | 7.99E-05 |
| KLRC3 | Killer cell lectin-like receptor subfamily C, member 3 | 0.0073 | 0.80 | 2.95E-10 | −0.3836 | −0.50 | 7.83E-04 |
| TRAJ17 | T-cell receptor alpha joining 17 | 0.0060 | 0.71 | 1.14E-07 | −0.3760 | −0.55 | 2.12E-04 |
| KLRG1 | Killer cell lectin-like receptor subfamily G, member 1 | 0.0064 | 0.82 | 4.01E-11 | −0.3714 | −0.58 | 6.25E-05 |
| CD28 | CD28 molecule | 0.0061 | 0.79 | 5.55E-10 | −0.3632 | −0.57 | 1.05E-04 |
| TRAT1 | T-cell receptor associated transmembrane adaptor 1 | 0.0053 | 0.76 | 7.45E-09 | −0.3428 | −0.59 | 5.60E-05 |
| CD3G | CD3g molecule, gamma (CD3- TCR complex) | 0.0053 | 0.74 | 2.57E-08 | −0.3421 | −0.58 | 7.12E-05 |
DISCUSSION
Traditionally, biological functions were determined from blood transcriptomic analysis using the whole gene set identified from the sample. The results were then correlated with “time” and the changes were characterized during the disease progression. However, we believed that phenotypic measurements (ie, clinical parameters) are a more reliable assessment for the condition of a patient because one could expect that progression of a disease is likely to vary among individuals. In respiratory infection, the Pao 2/Fio 2 ratio measured in the blood and the viral load in the respiratory tract are both useful quantitative parameters to determine disease severity [12, 19, 20]. One technical advantage of our approach is that samples do not need to be collected at fixed time points after admission, which is anyway not correlated with the time after onset of disease. Instead, the use of clinical parameters can enable a correlation analysis by pulling together all time points from all individuals. Two patients had a fatal outcome and both of them required extracorporeal membrane oxygenation. However, we did not find genes or MDTH that were associated with, or predictive of, this outcome. Indeed, the disease outcome of a patient can be affected by multiple factors, including the clinical practice, response to the treatment, secondary infections, or unexpected complications. The transcriptomic host responses associated with a fatal outcome need to be identified by a larger cohort with similar sample collection criteria. Our analysis, therefore, has the limitation that it does not provide predictive host responses that are associated with an adverse outcome. Instead, our approach analyzed and identified gene expression associated with 2 key clinical parameters: Pao 2/Fio 2 and viral load in the LRT, which are closely related to disease severity but may not invariably be linked to disease outcome (recovered patients can also have low Pao 2/Fio 2 value and high viral load at some stages of their illness). We found that MDTH correlated well with viral load at the LRT but not in the URT, which is understandable for a virus such as H7N9 that causes a primary viral pneumonia. Because dysregulation of proinflammatory cytokines has been shown to be associated with the pathogenesis of avian influenza H7N9 in humans and experimental animal models, it would be interesting to examine possible correlations between cytokine responses in plasma and gene expression levels (as well as MDTH) in blood in future studies [12, 21–24].
Our data showed that the activities of neutrophil during the acute phase of infection were associated with the viral infection. Neutrophils are involved in various lung diseases [25, 26]. A neutrophil response has long been considered a marker of bacterial infection [27, 28]. However, rapid recruitment of neutrophils to the lung was seen in naive mice after nasal inoculation with avian influenza viruses [29, 30]. These results suggest that infection with avian influenza viruses can also lead to recruitment and activation of neutrophils in the lung. Our study found that CD177 was the most sensitive to both the Pao 2/Fio 2 ratio and the viral load in LRT. CD177 (NB-1) is a glycophosphatidylinositol-anchored protein, which is responsible for mediating the migration, degranulation, and superoxide generation by neutrophils during inflammation [31, 32]. Activation of matrix metalloproteinase (MMP) 8, 9, and 25 were also found during the acute phase of H7N9 infection. A case-control study indicated that mRNA levels of MMP8 and MMP9 in peripheral blood mononuclear cells increased in children with LRT infection compared to healthy subjects [33]. It has been shown that MMP9 plays a role in regulating phagocytosis and reactive oxygen species production in neutrophils [34, 35]. It is also interesting that cell cycle-related processes were strongly associated with the change in viral load in the lower lung during H7N9 infection. A previous transcriptomic study also noted cell cycle perturbations in the peripheral blood specimens collected from patients with severe seasonal influenza infection [36]. Although cell cycle pathways were also found to be activated in other studies, their significance was masked by other biological processes such as bacteria-related or immunological pathways when the data were analyzed without association with specific clinical parameters, as has been done in this study [8, 10]. Our results suggest that activation of cell cycle processes maybe a hallmark of severe influenza virus infection.
Interferons and interferon-activated pathways are important to control viral infection. A clinical study in seasonal influenza virus infections showed that activation of these pathways was reduced dramatically at around day 6 after disease onset [8]. Our study could not identify significant activation of interferon related pathways (with the exception of IFI27) even though we could still clearly observe virus shedding in the lower lung. Thus, our findings are compatible with interferon pathways being activated at early stages of infection but being downregulated fairly quickly. However, it should be noted that we did not have specimens available for transcriptomic analysis in the first week of illness. Specimens collected earlier in the course of illness will be needed in order to test this hypothesis. Interestingly, a previous blood transcriptomic study from 4 H7N9 patients (with 1 sample from each patient) showed a GO enrichment of “Suppression by virus of host interferon receptor activity (GO:0039511)” [10]. However, that study did not provide details on day of illness in relation to sampling and data on severity of illness were lacking. Thus we could not make a direct comparison with our data. While studies using animals such as mice and macaques all showed that H7N9 could effectively trigger the interferon pathways in the lung during the early stage of infection [37–39], it is also possible that interferon activities were only localized in the lung of the patients during the H7N9 infection.
A recent study has shown that the expression of the gene IFI27 is upregulated in seasonal influenza infection and is a biomarker that discriminates influenza from bacterial infections [18]. We therefore specifically looked for the expression of IFI27 in our data set. Expression of IFI27 and the related IFI27L1 were two of the interferon pathway-associated genes that were upregulated in our cohort of H7N9 infected patients. It was negatively correlated with Pao 2/Fio 2 (gene expression increases with lower lung oxygenation) and a positively correlated with higher virus load in the LRT. Interestingly, the expression of IFI27 was not correlated with PCT. ILI27 has been reported to be expressed primarily by plasmacytoid dendritic cells in response to viral infection [18].
It has been shown that patients with H7N9 disease who were discharged earlier from hospital had better T-cell and natural killer (NK) cell recruitment and antibody induction [11]. Our transcriptomic analysis supported their findings in that T-cell processes including selection, differentiation, and stimulation were activated during the recovery phase of H7N9 infection. Other than T-cells–specific genes, a group of genes from the killer cell lectin-like receptor subfamily, which are expressed on the surface of T and NK cells, were also activated during the recovery stage. These proteins have been shown to regulate diverging functions of T, NK, and γδT cells during infection. For example, previous study have showed that Vγ9Vδ2 T cells, which express KLRG1, can effective kill influenza virus-infected cells [40].
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
{kind=link}
Notes
Acknowledgment. We would like to thank all the clinical and research staff who contributed to this study.
Financial support. This work was supported by the National Natural Science Foundation of China (grant number 81761128014); Science Research Project of the Guangdong Province (grant number 2016A050503047); Municipal Science and Technology Bureau Foundation of Guangzhou (grant number 2014Y2-00031); Research Grants Council of the Hong Kong Special Administrative Region, China, through the Theme Based Research Scheme (reference T11-705/14N); National Institutes of Health (grant number R56 AI127371); and a Croucher Foundation Fellowship (N.C.W.).
Potential conflicts of interest. All authors: No reported conflicts of interest. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1. Gao R, Cao B, Hu Y, et al. Human infection with a novel avian-origin influenza A (H7N9) virus. N Engl J Med 2013; 368:1888–97. [DOI] [PubMed] [Google Scholar]
- 2. World Health Organization. Disease outbreak news. Human infection with avian influenza A(H7N9) virus-China. http://www.who.int/csr/don/22-february-2017-ah7n9-china/en/. Accessed 22 February 2017. [Google Scholar]
- 3. World Health Organization. Influenza monthly risk assessment summary, 2016. http://www.who.int/influenza/human_animal_interface/Influenza_Summary_IRA_HA_interface_10_30_2017.pdf?ua=1. Accessed 27 November 2017. [Google Scholar]
- 4. Chan MC, Chan RW, Chan LL, et al. Tropism and innate host responses of a novel avian influenza A H7N9 virus: an analysis of ex-vivo and in-vitro cultures of the human respiratory tract. Lancet Respir Med 2013; 1:534–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Husain M. Avian influenza A (H7N9) virus infection in humans: epidemiology, evolution, and pathogenesis. Infect Genet Evol 2014; 28:304–12. [DOI] [PubMed] [Google Scholar]
- 6. Zhou J, Wang D, Gao R, et al. Biological features of novel avian influenza A (H7N9) virus. Nature 2013; 499:500–3. [DOI] [PubMed] [Google Scholar]
- 7. Mandell LA, Wunderink RG, Anzueto A, et al. ; Infectious Diseases Society of America; American Thoracic Society Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis 2007; 44:S27–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhai Y, Franco LM, Atmar RL, et al. Host transcriptional response to influenza and other acute respiratory viral infections–a prospective cohort study. PLoS Pathog 2015; 11:e1004869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mejias A, Dimo B, Suarez NM, et al. Whole blood gene expression profiles to assess pathogenesis and disease severity in infants with respiratory syncytial virus infection. PLoS Med 2013; 10:e1001549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mei B, Ding X, Xu HZ, Wang MT. Global gene expression changes in human peripheral blood after H7N9 infection. Gene 2014; 551:255–60. [DOI] [PubMed] [Google Scholar]
- 11. Wang Z, Wan Y, Qiu C, et al. Recovery from severe H7N9 disease is associated with diverse response mechanisms dominated by CD8⁺ T cells. Nat Commun 2015; 6:6833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang ZF, Mok CK, Liu XQ, et al. Clinical, virological and immunological features from patients infected with re-emergent avian-origin human H7N9 influenza disease of varying severity in Guangdong province. PLoS One 2015; 10:e0117846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res 2003; 31:e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Efron B, Tibshirani R. Empirical Bayes methods and false discovery rates for microarrays. Genet Epidemiol 2002; 23:70–86. [DOI] [PubMed] [Google Scholar]
- 15. Pankla R, Buddhisa S, Berry M, et al. Genomic transcriptional profiling identifies a candidate blood biomarker signature for the diagnosis of septicemic melioidosis. Genome Biol 2009; 10:R127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gene Ontology Consortium. Gene Ontology Consortium: going forward. Nucleic Acids Res 2015; 43:D1049–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Berry MP, Graham CM, McNab FW, et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 2010; 466:973–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tang BM, Shojaei M, Parnell GP, et al. A novel immune biomarker IFI27 discriminates between influenza and bacteria in patients with suspected respiratory infection. Eur Respir J 2017; 49:pii:1602098. [DOI] [PubMed] [Google Scholar]
- 19. Shi SJ, Li H, Liu M, et al. Mortality prediction to hospitalized patients with influenza pneumonia: PO2 /FiO2 combined lymphocyte count is the answer. Clin Respir J 2017; 11:352–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cao B, Huang Y, She DY, et al. Diagnosis and treatment of community-acquired pneumonia in adults: 2016 clinical practice guidelines by the Chinese Thoracic Society, Chinese Medical Association. Clin Respir J 2018; 12:1320–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Belser JA, Gustin KM, Pearce MB, et al. Pathogenesis and transmission of avian influenza A (H7N9) virus in ferrets and mice. Nature 2013; 501:556–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhu H, Wang D, Kelvin DJ, et al. Infectivity, transmission, and pathology of human-isolated H7N9 influenza virus in ferrets and pigs. Science 2013; 341:183–6. [DOI] [PubMed] [Google Scholar]
- 23. Watanabe T, Kiso M, Fukuyama S, et al. Characterization of H7N9 influenza A viruses isolated from humans. Nature 2013; 501:551–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mok CK, Lee HH, Chan MC, et al. Pathogenicity of the novel A/H7N9 influenza virus in mice. MBio 2013; 4:pii:e00362-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Camp JV, Jonsson CB. A role for neutrophils in viral respiratory disease. Front Immunol 2017; 8:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Williams AE, Chambers RC. The mercurial nature of neutrophils: still an enigma in ARDS?Am J Physiol Lung Cell Mol Physiol 2014; 306:L217–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Craig A, Mai J, Cai S, Jeyaseelan S. Neutrophil recruitment to the lungs during bacterial pneumonia. Infect Immun 2009; 77:568–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mizgerd JP. Molecular mechanisms of neutrophil recruitment elicited by bacteria in the lungs. Semin Immunol 2002; 14:123–32. [DOI] [PubMed] [Google Scholar]
- 29. Xu L, Bao L, Deng W, et al. The mouse and ferret models for studying the novel avian-origin human influenza A (H7N9) virus. Virol J 2013; 10:253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang C, Lee HH, Yang ZF, Mok CK, Zhang Z. PB2-Q591K mutation determines the pathogenicity of avian H9N2 influenza viruses for mammalian species. PLoS One 2016; 11:e0162163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Goldschmeding R, van Dalen CM, Faber N, et al. Further characterization of the NB 1 antigen as a variably expressed 56-62 kD GPI-linked glycoprotein of plasma membranes and specific granules of neutrophils. Br J Haematol 1992; 81:336–45. [DOI] [PubMed] [Google Scholar]
- 32. Demaret J, Venet F, Plassais J, et al. Identification of CD177 as the most dysregulated parameter in a microarray study of purified neutrophils from septic shock patients. Immunol Lett 2016; 178:122–30. [DOI] [PubMed] [Google Scholar]
- 33. Brand KH, Ahout IM, de Groot R, Warris A, Ferwerda G, Hermans PW. Use of MMP-8 and MMP-9 to assess disease severity in children with viral lower respiratory tract infections. J Med Virol 2012; 84:1471–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hong JS, Greenlee KJ, Pitchumani R, et al. Dual protective mechanisms of matrix metalloproteinases 2 and 9 in immune defense against Streptococcus pneumoniae. J Immunol 2011; 186:6427–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Warner RL, Beltran L, Younkin EM, et al. Role of stromelysin 1 and gelatinase B in experimental acute lung injury. Am J Respir Cell Mol Biol 2001; 24:537–44. [DOI] [PubMed] [Google Scholar]
- 36. Parnell G, McLean A, Booth D, Huang S, Nalos M, Tang B. Aberrant cell cycle and apoptotic changes characterise severe influenza A infection–a meta-analysis of genomic signatures in circulating leukocytes. PLoS One 2011; 6:e17186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. de Wit E, Rasmussen AL, Feldmann F, et al. Influenza virus A/Anhui/1/2013 (H7N9) replicates efficiently in the upper and lower respiratory tracts of cynomolgus macaques. mBio 2014; 5:e01331–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Morrison J, Josset L, Tchitchek N, et al. H7N9 and other pathogenic avian influenza viruses elicit a three-pronged transcriptomic signature that is reminiscent of 1918 influenza virus and is associated with lethal outcome in mice. J Virol 2014; 88:10556–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Baas T, Baskin CR, Diamond DL, et al. Integrated molecular signature of disease: analysis of influenza virus-infected macaques through functional genomics and proteomics. J Virol 2006; 80:10813–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tu W, Zheng J, Liu Y, et al. The aminobisphosphonate pamidronate controls influenza pathogenesis by expanding a gammadelta T cell population in humanized mice. J Exp Med 2011; 208:1511–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.