

Abstract
With more than 150,000 deaths per year in the US alone, lung cancer has the highest number of deaths for any cancer. These poor outcomes reflect a lack of treatment for the most common form of lung cancer, non–small cell lung carcinoma (NSCLC). Lung adenocarcinoma (ADC) is the most prevalent subtype of NSCLC, with the main oncogenic drivers being KRAS and epidermal growth factor receptor (EGFR). Whereas EGFR blockade has led to some success in lung ADC, effective KRAS inhibition is lacking. KRAS-mutant ADCs are characterized by high levels of gel-forming mucin expression, with the highest mucin levels corresponding to worse prognoses. Despite these well-recognized associations, little is known about roles for individual gel-forming mucins in ADC development causatively. We hypothesized that MUC5AC/Muc5ac, a mucin gene known to be commonly expressed in NSCLC, is crucial in KRAS/Kras-driven lung ADC. We found that MUC5AC was a significant determinant of poor prognosis, especially in patients with KRAS-mutant tumors. In addition, by using mice with lung ADC induced chemically with urethane or transgenically by mutant-Kras expression, we observed significantly reduced tumor development in animals lacking Muc5ac compared with controls. Collectively, these results provide strong support for MUC5AC as a potential therapeutic target for lung ADC, a disease with few effective treatments.
Keywords: Oncology, Pulmonology
Keywords: Lung cancer
MUC5AC is linked to KRAS mutant lung adenocarcinoma development and progression, and could provide a novel therapeutic target.
Introduction
Lung cancer is the leading cause of cancer mortality in the US and the world, causing more cancer-related deaths than breast, prostate, and colon cancer — the next most common causes of cancer mortality — combined (1). Lung adenocarcinoma (ADC), a subtype of non–small cell lung carcinoma (NSCLC), is the most prevalent type of lung cancer (1). Even with advances in early disease detection (2), the majority of lung ADC patients still succumb to disease. Whereas recent advancements with receptor tyrosine kinase inhibitors for targets, such as epidermal growth factor receptor (EGFR), have led to successful lung ADC treatments, other proto-oncogenes, such as those encoding RAS GTPases, remain poorly targeted (3). Among RAS family members, KRAS gene mutants are prevalent in human lung ADC, with mutation frequencies of 15%–25% (4, 5). Accordingly, KRAS mutations constitute targets whose potential for improved intervention in lung ADC will be strengthened by determining critical downstream effectors.
ADCs are defined as cancers that begin in secretory cells. Among the numerous markers of lung ADCs, mucins stand out as traits that define tumor origin. Indeed, even in ADC subtypes characterized histochemically as nonmucinous by negative labeling with histochemical dyes such as periodic acid Schiff’s stain, mucin expression is often abundant when tested using more sensitive immunolabeling and gene expression analyses (6). Furthermore, mucinous ADCs are associated with KRAS mutations and worse prognoses for patients (7–12). Nonetheless, aside from the established roles of mucins as markers of tumor type and clinical outcomes, the functional consequences of mucins on lung ADC development are poorly understood.
Mucins can be divided into 2 main families: membrane-bound and secreted. Membrane mucins such as MUC1 and MUC16 are known cancer antigens (KL-6 and CA-125, respectively), and their functions in tumor growth, survival, and invasion are well studied (13, 14). By contrast, the polymeric mucins MUC5AC and MUC5B are abundant in the lungs, and their dysregulated expression and hypersecretion contribute to pathobiological features such as airway obstruction, infection, and tissue remodeling in nonneoplastic disorders of the lung (15–17). In lung ADCs, polymeric mucins are strongly expressed (17, 18). Recent reports have begun to provide clinical associations between mucin expression and lung ADC outcomes (7, 9, 19), including several studies showing that MUC5AC is significantly associated with worsened survival in lung ADC patients (9, 12, 20, 21). These clinical findings are further supported with potential molecular mechanisms identified in gene knockdown studies demonstrating that MUC5AC plays a role in lung ADC cell growth in vitro (22). The effects of complete deficiency have not been tested in vitro or in vivo.
We recently generated lines of Muc5ac and Muc5b gene–KO mice and identified critical but distinct roles for them in the lungs. MUC5B is abundant at baseline, and it is critical for pulmonary host defense under homeostatic conditions. Muc5b gene disruption results in chronic microbial infection and premature mortality (23). By contrast, MUC5AC is present at very low levels at baseline (24), and unlike MUC5B, it is dispensable for airway homeostasis (25). When MUC5AC is induced in models of allergic inflammation, it is required for asthma-like airflow obstruction to occur (26), and its principal adaptive role appears to be to aid in helminth defense (27). Because Muc5ac is a nonessential gene that plays a causative pathophysiological role in mouse lungs, and because human MUC5AC is associated with poor outcomes in lung ADC, we sought to determine the effects of its expression on lung carcinogenesis.
Here, we tested the hypothesis that MUC5AC/Muc5ac expression is associated with the development and progression of KRAS/Kras-driven lung ADC and that MUC5AC/Muc5ac deficiency reduces tumor development. Using 2 independent human cohorts, we found at both mRNA and protein levels that MUC5AC is a significant determinant of poor prognoses in patients with KRAS (but not EGFR) mutant lung ADCs. Furthermore, using 2 unique mouse models, one with constitutive activation of Kras and the other chemically induced with urethane, we also found, for the first time to our knowledge, that Muc5ac plays a functionally causative role in tumor development in vivo. In aggregate, our studies support a role for MUC5AC as an important target in KRAS-driven lung ADCs, which are common yet poorly treated with existing therapies.
Results
MUC5AC expression is associated with ADC compared with squamous cell carcinoma.
On a randomized tissue microarray containing lung ADC (n = 53), adenosquamous carcinoma (ASC) (n = 3), and squamous cell carcinoma (SCC) (n = 21) patient specimens that were labeled immunohistochemically for MUC5AC and evaluated blindly to test for differences in staining, striking differences in MUC5AC protein were observed (Figure 1). Ordinarily, staining for MUC5AC is abundant in normal bronchial, submucosal gland, and proximal bronchiolar epithelia of central airways, but MUC5AC is scant or absent in terminal bronchioles and at bronchoalveolar junctions (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.120941DS1). By contrast, in tissues containing ADC/ASC tumors, which are known to have a bronchoalveolar origin (18), MUC5AC staining was abundant (Figure 1A). Furthermore, although SCC tumors typically arise from transformation of more central tracheobronchial airway regions, staining for MUC5AC was sparse and diffuse in SCCs (Figure 1B). Indeed, blinded evaluation confirmed dramatically higher levels of MUC5AC in ADC/ASC compared with SCC, with histologic scores (H-scores) that were 6.83-fold greater in ADC/ASC relative to SCC (P < 0.0001) (Figure 1C).
Figure 1. MUC5AC production is associated with human lung ADC and poor survival.

(A–C) MUC5AC protein levels were analyzed immunohistochemically using human tissue arrays. Representative images of MUC5AC immunostaining in adenocarcinoma (ADC) (A) and squamous cell carcinoma (SCC) (B) are shown. Scale bars: 200 μm (low magnification) and 10 μm (high magnification). (C) MUC5AC H-scores for ADC (n = 53) and adenosquamous (n = 3) (combined and included with ADC, red circles) were compared with SCC (n = 21, gray circles). MUC5AC levels were significantly higher in ADC tissues. Data are individual values with lines depicting medians ± 95% CIs. Significance was determined by the Wilcoxon rank sum test (61). P < 0.05. (D–F) Effects of MUC5AC expression on survival in ADC and SCC were tested using KM Plotter (http://kmplot.com/analysis/). MUC5AC expression was stratified to define high- (red) and low-expression (black) groups by median transcript levels. Kaplan-Meier survival plot and hazard ratio calculations were compared by multivariate log-rank analyses with sex and stage as covariates; P < 0.05. While sex was not significant, stage was strongly significant for ADC (log-rank P < 1 × 10–14). Significant effects of high MUC5AC gene expression were observed for all human lung ADC (D, n = 534, log-rank P < 0.0001) and for stage I ADC (E, n = 370, log-rank P = 0.001) samples. No differences were observed in SCC (F, n = 315, log-rank P = 0.62). For all groups in D–F, calculated hazard ratios were 2.0 (1.5–2.7) for all ADC, 1.9 (1.3–2.8) for stage I ADC, and 1.1 (0.8–1.5) for SCC.
MUC5AC expression is associated with reduced overall survival in lung ADC patients.
To test whether clinical outcomes correlated with differences in MUC5AC levels like those described above, MUC5AC mRNA expression in ADC vs. SCC was analyzed using http://kmplot.com/analysis/ to perform a meta-analysis on 1,541 patients (27, 28). Using global gene expression and mortality metadata stratified by median MUC5AC mRNA expression and corrected for sex and cancer stage, we observed a significant association with high MUC5AC expression and reduced overall survival (OS). In ADC patients (n = 534), there was a hazard ratio of 2.0 (range 1.5–2.7) that supports a greater mortality risk in those with MUC5AC levels above the overall ADC median (Figure 1D). Further, stage I ADC patients (n = 371) also showed worsened OS when stratified for high vs. low MUC5AC expression (Figure 1E; hazard ratio of 1.9, range 1.3–2.8; log-rank P = 0.001). By contrast, in SCC patients (n = 525), differences in mortality risk relative to MUC5AC expression were not observed (Figure 1F). Collectively, these data suggest a significant role for MUC5AC involvement, specifically in lung ADC compared with SCC, and the findings provide additional evidence to support roles for MUC5AC at both early and late stages of lung ADC. These data also suggest that there may be a significant association between differential expression of MUC5AC and prognoses in these diseases. We thus sought to explore these associations causatively in mouse models of lung cancer.
Muc5ac deficiency reduces tumor burden in urethane-induced carcinogenesis.
Urethane is a known carcinogen that elicits lung ADC following i.p. injection. After 10 weeks of treatment, urethane significantly increased Muc5ac mRNA (Figure 2A), histochemically detectable mucin (Figure 2B), and Muc5ac protein levels (Figure 2C) in the lungs of WT BALB/cJ mice. Having established an association between urethane exposure and Muc5ac levels, a functional role for MUC5AC in early-stage lung ADC was investigated using Muc5ac-null mice on a BALB/cJ background (Muc5ac–/–). At 10 weeks following the initiation of tumorigenesis with urethane, lung tumor multiplicity was significantly decreased in the Muc5ac–/– compared with WT Muc5ac+/+ mice. Muc5ac–/– animals showed reduced tumor numbers on lung surfaces (Figure 2D) and significantly lower overall tumor area within lung tissues (Figure 2E), thereby providing functional evidence of Muc5ac involvement in the development of lung ADC. In addition, the protective effect of Muc5ac gene deficiency persisted in mice examined 20 weeks after urethane exposure, supporting a significant role for MUC5AC in lung ADC progression (Figure 2, D–G). This causative relationship was explored further by testing the relationships between MUC5AC gene expression and molecular markers of lung ADC in human patients.
Figure 2. Muc5ac is elevated in urethane-induced tumors, and Muc5ac-deficiency leads to reduced tumor burden.

(A) Muc5ac mRNA expression is significantly elevated in response to urethane in WT mice at 10 weeks after urethane (n = 5 mice per group). *P < 0.05 between saline (Sal) and urethane (Ure) treatments determined by 2-tailed t test (n = 5 mice for each treatment group). (B) Periodic acid fluorescent Schiff’s (PAFS) positive mucin staining (red) indicates increased mucins in urethane-treated WT lungs. (C) Mucin-positive tumors contain immunohistochemically detectable Muc5ac in WT lungs. (D) Surface tumor counts in Muc5ac-sufficient (Muc5ac+/+) and -deficient (Muc5ac–/–) mice 10 and 20 weeks following the first urethane injection. Significant differences were observed between genotypes at both time points (10 weeks, n = 6 Muc5ac+/+, n = 8 Muc5ac–/– mice; 20 weeks, n = 14 Muc5ac+/+, n = 19 Muc5ac–/– mice). (E) Tumor areas within tissues were significantly lower in Muc5ac–/– mice compared with Muc5ac+/+ mice (10 weeks, n = 4 Muc5ac+/+, n = 4 Muc5ac–/– mice; 20 weeks, n = 10 per genotype). Data are mean ± SEM in A, D, and E. P < 0.05, determined by 2-tailed t test. (F and G) Lung histopathology of Muc5ac+/+ and Muc5ac–/– mice. Arrowheads identify tumors. Scale bar: 100 μm in B and C, 1 mm in F and G.
MUC5AC as a potential target for KRAS mutant lung tumors.
KRAS gene mutations are among the most prevalent in lung ADC in humans, and urethane-induced lung carcinogenesis is primarily associated with Kras mutations in mice (29, 30). Therefore, the links between KRAS and MUC5AC expression and lung ADC were investigated. An independent cohort of human lung ADC patients was analyzed to interrogate the links between MUC5AC gene expression, KRAS gene mutation status, and clinical outcomes. MUC5AC mRNA expression was measured in surgically resected lung ADC samples from the Profiling of Resistance patterns and Oncogenic Signaling Pathways in Evaluation of Cancers of the Thorax (PROSPECT) cohort, comprising 150 patients who did not receive neoadjuvant therapy and whose detailed prognostic, genetic, and gene expression data have been collected (31). Patients were stratified based on median MUC5AC mRNA expression using above- and below-median levels of expression to dichotomize high- and low-expressing populations, as was done for KM-plot meta-analyses, above (Figure 1E). In this independent population, greater-than-median MUC5AC expression was also significantly associated with worsened OS (Figure 3A, P = 0.003), and it was likewise associated with decreased disease-free survival (Figure 3B, P = 0.05). Furthermore, when PROSPECT study patients were further divided into subgroups based on KRAS gene mutation status, the effects of high MUC5AC expression on survival were even more apparent. In KRAS-mutant lung ADC patients, there was a trend for association between MUC5AC expression and worsened OS (Figure 3C, P = 0.07), and there was a significant association between MUC5AC expression and worsened disease-free survival (Figure 3D, P = 0.02). Lastly, we also evaluated EGFR gene mutation status and MUC5AC expression for any associations; none were identified (data not shown). In aggregate, these data demonstrate that MUC5AC expression is associated with more aggressive clinical phenotypes in lung ADC, including a tight link between MUC5AC expression levels and outcomes in patients with mutations in the KRAS proto-oncogene. Having thus demonstrated that KRAS-driven lung ADC and MUC5AC expression in lung ADC patients were associated, this relationship was interrogated in mice to test for causation.
Figure 3. Muc5ac expression worsens both human and mouse lung cancer with KRAS mutations.

(A–D) Human ADCs (A and B; n = 152) and KRAS-mutant ADCs (C and D; n = 39) from the PROSPECT Cohort were stratified based on Muc5ac median mRNA expression (high, red; low, black). Patient subgroups were then analyzed for differences in overall and disease-free survival using the Kaplan-Meier method for estimation of survival probability and the log-rank test; P < 0.05. (E–G) KRAS mutant lung cancer was modeled in mice (CC-LR). Muc5ac gene expression from CC-LR mice was compared with control littermates by qPCR (n = 3 mice per genotype). (F) Periodic acid fluorescent Schiff’s (PAFS) positive mucin staining (red) indicates increased mucins in CC-LR mice lungs. (G) Mucin-positive tumors contain immunohistochemically detectable Muc5ac. Data are mean ± SEM in E. P < 0.05 by 2-tailed t test. Scale bar: 50 μm in F and 200 μm in G.
Lack of Muc5ac suppresses Kras-driven lung cancer.
A transgenic mouse model (CC-LR) was previously developed by our group (32) by targeting mutant Kras expression to mouse bronchiolar epithelia (see Methods). CC-LR mice harbor a glycine to aspartic acid mutation that is commonly found in human KRAS and that results in rapid development of ADC in Kras mutant transgene–expressing mice (33). As was also observed in the urethane model above, Muc5ac gene expression (Figure 3E), mucin glycoprotein (Figure 3F), and MUC5AC protein (Figure 3G) were induced in CC-LR mice.
To test the promoting role of MUC5AC in lung cancer, we crossed CC-LR mice with Muc5ac-null mice to generate a Kras mutant mouse lacking Muc5ac (CC-LR/Muc5ac−/− mice). The lack of MUC5AC in this triple transgenic line significantly inhibited lung cancer promotion in CC-LR mice compared with age- and sex-matched control CC-LR mice (CC-LR/Muc5ac+/+) with intact MUC5AC (Figure 4, A and B). Surface tumor numbers decreased by 2.5-fold (Figure 4B), and histologic tumor area also decreased robustly (~1.9-fold) (Supplemental Figure 2A). Tumor suppression caused by MUC5AC absence was further confirmed by reduction in the amount of the mutant form of KRAS (KrasG12D) protein in Western blot analysis of whole lung homogenate from CC-LR/Muc5ac–/– mice compared with age- and sex-matched CC-LR/Muc5ac+/+ mice (Supplemental Figure 2B).
Figure 4. Lack of Muc5ac suppresses Kras-induced lung tumorigenesis.

(A) Histopathological appearance of lung tissue and (B) lung surface tumor number in CC-LR/Muc5ac+/+ and CC-LR/Muc5ac–/– mice at 14 weeks age (n = 12 CC-LR/Muc5ac+/+; n = 26 CC-LR/Muc5ac–/– mice). Ki-67 (C and D) and ERG (E and F) positive cells in lung tissue of CC-LR/Muc5ac+/+ and CC-LR/Muc5ac–/– mice at the age of 14 weeks (n = 3 per genotype). Data in B, D, and F are mean ± SEM. P < 0.05 by 2-tailed t test. Scale bars: 200 μm in A and 50 μm in C and E.
Further histopathologic examination of the lung sections from CC-LR/Muc5ac–/– mice showed that most lung tumors remained at early stages of development. Reduced tumor cell proliferation in CC-LR/Muc5ac–/– mice compared with CC-LR/Muc5ac+/+ controls was indicated by the presence of fewer Ki67-expressing cells (Figure 4, C and D), and reduced tumor angiogenesis was detected by decreased presence of the markers ERG (34, 35) and CD31 (Figures 4, E and F, and Supplemental Figure 3). Lastly, the CC-LR/Muc5ac–/– tumors had significantly less adjacent inflammatory cell infiltrates compared with CC-LR/Muc5ac+/+ mice (Figure 4A and Figure 5, A–C). The links between diminishments in MUC5AC- and KRAS-dependent carcinogenesis and tumor-associated inflammation suggest that a potential mechanism through which MUC5AC promotes lung ADC occurs through effects of MUC5AC on pulmonary inflammatory responses in CC-LR mice.
Figure 5. Lack of Muc5ac suppresses protumor lung inflammation and STAT3 pathway activation.

(A) Total macrophages, (B) lymphocytes, and (C) neutrophils, as well as levels of IL-6 (D), IL-17 (E), and KC (F) in lung lavage fluid of CC-LR/Muc5ac+/+ and CC-LR/Muc5ac–/– mice at the age of 14 weeks are shown (n = 6 for lavage cellular differentials and n = 3 for cytokine analysis). (G and H) Representative photomicrograph (G) and quantitative analysis (H) of pSTAT3-positive cells in lung tissue of CC-LR/Muc5ac+/+ and CC-LR/Muc5ac–/– mice at the age of 14 weeks (n = 4 mice per group). Data are mean ± SEM. P < 0.05 by 2-tailed t test. Scale bar: 25 µm.
Muc5ac-deficiency inhibits lung inflammation and STAT3 activation.
Having previously shown that lung tumor promotion in Kras mutant tumors is associated with inflammation and activation of the STAT3 pathway (36), we compared inflammatory cell populations in the lungs of CC-LR/Muc5ac–/– mice with age- and sex-matched CC-LR/Muc5ac+/+ mice. The absence of Muc5ac changed the BALF inflammatory cell profile of CC-LR/Muc5ac+/+ mice with a predominant effect on the macrophage population (Figure 5A). To further verify the potential role of MUC5AC on Kras-associated tumor inflammation, the levels of proinflammatory cytokines with known effects in mutant Kras-driven lung tumorigenesis were measured (36, 37). Muc5ac deficiency was associated with significant reductions in the levels of cytokines IL-6 (Figure 5D) and IL-17 (Figure 5E), as well as chemokine KC (Figure 5F), which we previously demonstrated to have important roles in mutant KRAS-driven lung tumorigenesis (36, 37). Immunohistochemical analysis of lung tumor tissues for pSTAT3, the main IL-6–responsive transcription factor, also revealed a significant suppression of STAT3 pathway activation (Figures 5, G and H). One caveat with this observed reduction in inflammation in CC-LR/Muc5ac–/– mice is that reduced tumor burden is often associated with reduced inflammation.
Discussion
ADC is a subtype of lung cancer that is prevalent, progressive, and lethal, with 5-year survival rates that rapidly fall below 60% as disease progresses beyond stage I (38). Activating KRAS mutations are found in around 30% of lung cancer overall (39), and they exist in the majority of mucin-overexpressing ADCs (8, 40–43). Moreover, it has been recently shown that histologic patterns of mucin in lung ADC are associated with KRAS mutations (9, 10). Whole-exome sequencing also demonstrated significant increases in MUC5AC copy number in 11% of the ADC patients tested, further supporting studies on MUC5AC (44). The studies presented here investigated the mucin MUC5AC as a common marker in lung ADC and its role in human disease and in mouse models. Using independent cohorts of human lung cancer patients, we determined that (a) MUC5AC protein is a specific marker of ADC, (b) high MUC5AC mRNA expression was significantly associated with reduced OS, and (c) the detrimental effect of high MUC5AC expression on patient survival is linked to KRAS gene mutations. These association data linking MUC5AC to the development and progression of lung ADC strongly suggest that MUC5AC is a key promoter of carcinogenic progression that may provide a target for therapeutic intervention for a disease whose only effective treatment is surgery. This is further strengthened by our findings in 2 different mouse models — chemical and genetic — both of which are dependent on activating Kras gene mutations (29, 30, 32) and, as we show in this study, on the expression of the Muc5ac gene. Our finding that MUC5AC is an effector of KRAS-driven lung cancer in mice may be of additional importance, given the challenges associated with inhibiting KRAS in human patients. Here, we found that MUC5AC is a significant predictor of poor survival and prognosis in human KRAS-mutant lung ADC, and it has a significant role in promoting Kras-mutant lung cancer in mice.
In lung ADC cell lines in vitro, MUC5AC gene expression is controlled by EGFR- and HIF-1–dependent pathways (45), both of which are important factors in lung carcinogenesis. However, several studies have concluded that EGFR mutations did not associate with MUC5AC protein expression in lung ADCs (20, 46–49). This discrepancy could reflect limitations of in vitro tests in both cell lines and primary cultures. It could also reflect heterogeneous mechanisms that regulate the localization of MUC5AC overexpression in lung ADC, since its expression is in regions of the distal lung where it is not normally localized (Supplemental Figure 1). The tumor-promoting effects of MUC5AC in terminal bronchiolar and bronchoalveolar regions could be mediated through extracellular or intracellular mechanisms, such as interactions between integrin β4 and MUC5AC (40). A gene regulatory network for mucinous ADC was also identified where transcription factors SPDEF and FOXA3 induced MUC5AC expression in human mucinous lung cancer cells with mutant KRAS (9). Additionally, the bronchoalveolar junction is the location of the stem cells for ADC (bronchioalveolar stem cells [BASCs] or bronchoalveolar stem cells) (50). Taken together, these findings point to potential pathways where future studies will focus on suppressing MUC5AC gene expression. Such interventions could be beneficial not only for slowing lung cancer progression, but also for potentially enhancing cancer prevention.
The extracellular space in respiratory tissues is sustained by the defensive functions of an effective mucociliary apparatus. The normal functions of airway mucus include trapping and eliminating inhaled particles, recruited inflammatory cells, and dead resident cells and debris from the lungs (16, 45). While an effective mucus barrier is critical for protecting the lungs from injury, excessive mucin production — specifically overproduction of MUC5AC (15, 16) — in chronic obstructive pulmonary disease (COPD) can impair mucociliary clearance and contributes to COPD pathogenesis (16). COPD is a risk factor for lung cancer (51, 52), and lung ADC with mucin overexpression has higher malignancy potential and poorer prognosis than other types of lung cancer (7, 9, 12, 19–21, 40). It is not clear whether a mucociliary defect tied to lung ADC can be explained directly by poor mucociliary functions in remodeled cancerous tissues. These should be explored in future studies.
In addition to extracellular effects of MUC5AC overexpression, excessive or mislocalized expression of MUC5AC could also drive cell intrinsic pathologies. ADCs derived from nonrespiratory epithelia, including those of the breast, prostate, and pancreas, commonly overexpress mucins (14). These tissues are not exposed to the same particulate loads as the airways, which are exposed to 8,000–12,000 liters of air containing hundreds of billions of particles daily (53). A lack of clear environmental stimuli suggests that intrinsic factors may mediate detrimental effects of mucin overexpression in nonrespiratory ADC cell autonomously. Polymeric mucins, including MUC5AC, are extraordinarily large molecules whose synthesis requires tight regulation of gene expression in the nucleus, complex processing to facilitate disulfide-mediated polymer assembly in the ER, specific programming for the elaboration of O-linked glycans in the Golgi, and coordinated regulation of the secretory and postsecretory fates of mature mucin glycoproteins. Accordingly, there are numerous proteins whose expression parallels mucins, and whose functions are essential for protein homeostasis (proteostasis). Indeed, mucin overexpression or misfolding can have dramatic effects on cellular, organ, and host responses that result in inflammation, tissue remodeling, and disease pathogenicity (54). Along these lines, altered epithelial polarity and migration of lung ADC cell lines have been directly linked to MUC5AC expression in vitro (40). Future studies testing the downstream cellular effects of MUC5AC/Muc5ac expression may identify mechanisms in mouse and human lung ADCs that are potential targets for novel treatments.
Targeted therapies for lung ADCs are elusive. There are over a dozen primary oncogenic drivers that could be considered as therapeutic targets for NSCLC. Two targets whose prevalence of mutations accounts for 25%–45% of disease are EGFR and KRAS (4, 55, 56). While EGFR and some other rare targets have seen successful treatments develop, the more frequently mutated oncogene KRAS has remained a challenge. Options for direct inhibition of KRAS, such as antisense RNA and small molecule inhibitors, have been tested with very little success (57–60). Therefore, targeting downstream effectors of KRAS presents an innovative alternative approach to inhibit the detrimental effects of this pathway in lung ADC. The data presented here show significant associations of MUC5AC with human lung ADC and patient outcomes, and they further demonstrate in animal models that MUC5AC plays a causative role in lung ADC. Since Muc5ac is a nonessential gene in the airways whose expression is linked to pathophysiological obstruction (25), our finding puts MUC5AC as an alternative potential therapeutic target downstream to KRAS, as well as independently of KRAS, due to the reduction of tumor development observed in the presence of Kras mutations. Thus, targeting MUC5AC could be used in combination with a conventional cytotoxic drug or for improvement of newly available immunotherapeutic modalities (e.g., immune checkpoint blockade) (61, 62).
In summary, the studies reported here show, for the first time to our knowledge, that the polymeric mucin MUC5AC plays a significant role in the development and pathogenesis of KRAS-induced lung ADC. Given the lack of efficacious targeting and the poor outcomes for patients, therapies directed at downstream effectors such as MUC5AC could be beneficial. One approach may be to exploit transcriptional networks that regulate MUC5AC gene expression, such as HIF, Notch, or Nkx2-1 (24, 63–67). In addition, manipulating pathways involved in mucin glycoprotein biosynthesis and secretion, or developing efficacious mucolytic treatments that allow for better elimination of MUC5AC from the distal airways, could also prove beneficial. Along these lines, it will be crucial for such therapies to inhibit the detrimental effects of mucus dysfunction while preserving, or even potentially promoting, the beneficial effects of mucus on lung homeostasis and airway defense.
Methods
MUC5AC immunostaining in NSCLC patients.
Tissue microarrays were obtained through approval from the University of Colorado Cancer Center SPORE in Lung Cancer Tissue Bank. Tumor stage and histopathology was performed by a board certified pathologist (D.T. Merrick). The clinical characteristics of the tissues used in this study are presented in Table 1. Tissues were immunolabeled with biotinylated mouse monoclonal anti-MUC5AC antibody 45M1 (Thermo Fisher Scientific), using streptavidin-linked HRP (Thermo Fisher Scientific) and 3,3’-diaminobenzadene (47) (Vector Laboratories) to stain MUC5B brown. Antibodies were diluted 1:200 in 0.1% Tween 20/PBS and incubated on tissues for 1 hour at room temperature. Nuclei blue were counterstained with Meyer’s hematoxylin. All chemicals were obtained from MilliporeSigma, unless otherwise indicated.
Table 1. Patient demographics for tissue arrays.
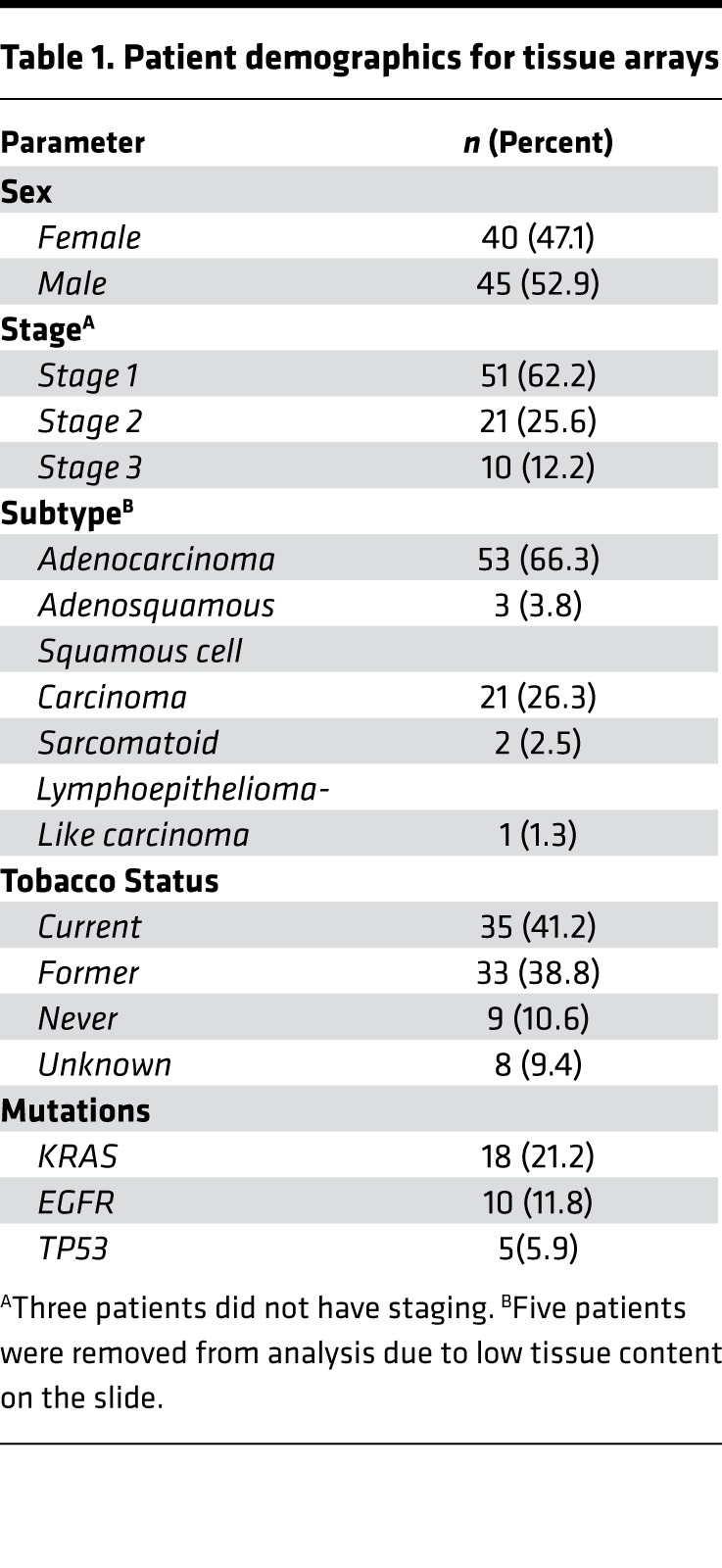
Once these tissue microarrays were stained for MUC5AC, 4 reviewers (A.K. Bauer, C.M. Evans, D.T. Merrick, P.R. Mann) determined H-scores for all samples in a blinded manner. H-scores were determined by the percentage of staining using a 1–3 scoring method with the following formula ([1 × (% cells 1+)] + [2 × (% cells 2+)] + [3 × (% cells 3+)]) (68, 69). D.T. Merrick, a board-certified pathologist, reviewed any slide discrepancies. The distributions of MUC5AC H-scores were compared between SCC and ADC/ASC samples using a Wilcoxon rank sum test.
Meta-analysis for comparison between MUC5AC expression in NSCLC subtypes and overall patient survival.
The program kmplot.org (http://kmplot.com/lung; 2015 version; Feb 1, 2017–March 1, 2017) was used to evaluate MUC5AC mRNA expression (217187_at) to the NSCLC subtypes, ADC (n = 534 patients) and SCC (n = 524 patients), for OS, similar to Bauer et al., 2017 (70). This database uses publicly available data sets (Gene Expression Omnibus [GEO]; Cancer Biomedical Informatics Grid [caBIG]; The Cancer Genome Atlas [TCGA]) from several human Affymetrix microarray platforms because these arrays have 22,277 probe sets in common (27). Briefly, the raw CEL files were MAS5 normalized using the Affy Bioconductor library (27, 28). Data were then analyzed using kmplot.org, which uses R (Bioconductor package within kmplot.org) and the database PostgreSQL to integrate the gene expression and clinical data (27, 28). We performed a multivariate COX regression analysis for OS with the covariates stage and sex, as in Győrffy et al., 2013 (27), based on the clinical information available. For the stage I analysis with ADC patients (n = 371 patients), we performed a univariate analysis. Kaplan Meier survival plots and log-rank P values were then calculated in kmplot.com and graphed in GraphPad stratifying by the median values for the MUC5AC gene expression as the threshold for high and low expression.
Urethane-induced carcinogenesis.
Five- to 6-week-old male Muc5ac-deficient mice (Muc5ac–/–) were generated from a breeding colony at the University of Colorado animal facility. This line was previously backcrossed >10 generations onto a congenic BALB/cJ strain background using strain-specific microsatellite marker–assisted analysis. Age-matched WT mice (BALB/cJ) were purchased from the Jackson Laboratory and allowed to acclimate for 1 week prior to studies. Animals were housed in a virus- and antigen-free room. Mice were fed irradiated mouse chow (Teklad, 2920X) and water ad libitum and housed in cages that were humidity- and temperature-controlled.
Mice were injected i.p. with either urethane dissolved in saline (1 mg per g body weight; MilliporeSigma) or with vehicle (saline) weekly for 7 weeks. Urethane and saline solutions were sterilized by passage through 0.2-μm filters (Thermo Fisher Scientific). Mice were euthanized 10 or 20 weeks after the first urethane injection by CO2 exposure, followed by exsanguination. The left lung lobe was then fixed for 48 hours using methacarn (60% methanol, 30% chloroform, 10% glacial acetic acid), a highly permeable noncrosslinking fixative (71, 72). Tumors were then enumerated and sized as described previously (32, 73–75).
MUC5AC expression in a mutant KRAS human cohort.
MUC5AC mRNA expression was determined by array analysis (Illumina v3) of surgically resected lung ADC from 150 patients that did not receive neoadjuvant therapy. This cohort was obtained from the PROSPECT study, developed in 2006 at MD Anderson Cancer Center (31). MUC5AC mRNA expression from these patients was log2 transformed, and median expression was computed. We then dichotomized lung ADC patients based on median MUC5AC mRNA expression in the manner previously performed (76). Patients with relatively low expression displayed lower than the median MUC5AC levels, whereas patients with relatively high expression exhibited greater than the median MUC5AC expression levels. Of these patients, those with KRAS mutations (n = 39 of 150) were then stratified based on MUC5AC expression in this same set of patients.
Mutant Kras mouse model.
CCSPCre/LSL-KrasG12D mice (CC-LR) were generated by crossing mice harboring an activating Kras allele (Lox-Stop-Lox-KrasG12D) with mice containing Cre recombinase inserted into the Scgb1a1 club cell secretory protein (CCSP), also called Secregatlobin-1a1, locus as previously described (32). Muc5ac–/– mice were developed on congenic C57BL/6J and BALB/cJ lineages as previously described (26). C57BL/6J congenic Muc5ac–/– mice were crossed to CC-LR mice and are referred to as CC-LR/Muc5ac–/–. Both male and female CC-LR/Muc5ac–/– were used for these studies, and littermate CC-LR/Muc5ac+/+ mice were used as controls.
At 14 weeks of age, mice were anesthetized and sacrificed by CO2 inhalation or i.p. injection of tribromoethanol (MilliporeSigma), and the lungs were infused with 10% buffered formalin (MilliporeSigma). The sections were then stained with H&E. The H&E-stained slides were examined by a pathologist blinded to genotype and treatment, and proliferative lesions of the lungs were evaluated in accordance with the recommendations of the Mouse Models of Human Cancer Consortium (77).
Immunostaining.
IHC staining was performed and evaluated for expression of pSTAT3 (Tyr705) (1:250; Cell Signaling Technology, 91125), Ki-67 (1:200; Abcam, ab16667), CD31 (1:50, BD Biosciences, 550274), ERG (ETS-related gene; 1:250; Abcam, Ab92513), rabbit anti–mouse MUC5AC (78), and mouse anti–human MUC5AC (biotinylated 45M1; 1:500; Abcam, ab212636). Heat-induced antigen retrieval was performed using 10 mmol/l of citrate buffer (pH 6.0) in a pressure cooker for 20 minutes. Immunoreactivity for IHC was detected using biotinylated IgG secondary antibodies specific for each primary antibody, followed by incubation with ABC kit (Vector Laboratories), and stained with DAB. Slides were counter-stained with Harris hematoxylin. Images were obtained at 4×, 20×, and 40× magnification by an Olympus BX60 microscope with Image-Pro Plus (version 4.5.1.22) or on an Olympus BX63 microscope with cellSens (version 1.18). The numbers of labeled positive cells for each marker were quantitated as a fraction of total tumor nuclei per high power field (40×) in at least 10 fields from 3–5 mice of each group. Results were expressed as percentage of positive cells ± SEM.
For fluorescent labeling of mucin, tissues from both urethane-treated and CC-LR mice were stained using a periodic acid fluorescent Schiff (PAFS) staining procedure as described previously (79). Briefly, tissues were oxidized in 1% periodic acid (10 minutes), rinsed, treated with acriflavine fluorescent Schiff’s reagent (0.5% acriflavine HCl, 1% sodium metabisulfite, 0.01 N HCl) for 20 minutes, rinsed in double deionized H2O, and rinsed 2 × 5 minutes in acid alcohol (0.1 N HCl in 70% ethanol). Slides were then analyzed with fluorescence microscopy, with mucin granules showing red fluorescence and nuclei and cytoplasm showing green fluorescence (79).
As mucin labeling controls, lung tissues from urethane-treated Muc5ac–/– mice, saline challenged WT and Muc5ac–/– mice, and allergen challenged WT and Muc5ac–/– mice were used (Supplemental Figure 4).
Assessment of lung tumor burden and inflammation in CC-LR and urethane model.
In some mice, lung-surface tumor numbers were counted. Then, lungs were prepared for histological analysis. H&E sections were prepared from 3–5 animals per group. Five randomly selected microscopic fields from peripheral and central regions of the lungs were photographed, and the percentage of the lung field occupied by tumors was measured by overlaying of these images on a dotted grid as previously described (32). In other mice, lung-surface tumor numbers were also counted, and then bronchoalveolar lavage fluid (BALF) was obtained by sequentially instilling and collecting 2 aliquots of 1 ml PBS through a tracheostomy cannula (32). The lungs were snap frozen and stored for future RNA and protein analysis. Total leukocyte count was determined using a hemacytometer, and cell populations were determined by cytocentrifugation followed by Wright–Giemsa (W&G; MilliporeSigma) staining. The remaining BALF was centrifuged at 1,250 g for 10 minutes, and supernatants were collected and stored at –80°C for further analysis.
Cytokines/chemokine measurement.
The levels of selected cytokines in the BALF were assessed using the MCYTOMAG-70 K assay (MilliporeSigma) according to the manufacturer’s instructions. Data were collected using a Luminex 100 (Luminex Corporation). Standard curves were generated using a 5-parameter logistic curve fitting equation (StarStation V 2.0; Applied Cytometry Systems). Each sample reading was interpolated from the appropriate standard curve.
Quantitative PCR analysis.
Total RNA was isolated from whole lung according to the TRIzol reagent protocol (Thermo Fisher Scientific) and purified by E.Z.N.A. total RNA kit I (OMEGA) at MD Anderson and the Nucleospin RNA II kit (Macherey-Nagel) at the University of Colorado. Reverse-transcription PCR (RT-PCR) was performed using the qScript cDNA SuperMix (Quanta Biosciences) at MD Anderson and using MMLV reverse transcriptase (Invitrogen) and oligo-dT (Invitrogen) at the University of Colorado, following previous protocols (80). Quantitative PCR (qPCR) was performed according to a standard protocol using Muc5ac and Hprt-specific primers (Mm01276718_m1 and Hs99999901_s1, respectively) in a TaqMan Master Mix (Thermo Fisher Scientific). Products were amplified in 3 technical replicates on 96-well plates and measured on an ABI Viia 7 PCR system (ABI). The expression of individual genes was calculated and normalized with the ΔΔCt method.
Statistics.
Summary statistics for cell counts, and cytokine levels in BALF, mRNA expression, and nuclear staining of Ki-67, ERG, and pSTAT3 in tumors were computed within treatment groups, and unpaired 2-tailed t tests with adjustment for unequal variances and nonnormal distributions was performed to examine the differences between the mean cell counts of the groups. For tumor counts and tumor areas, comparisons of groups were made using unpaired 2-tailed Student’s t test, and presented as mean ± SEM. Differences were considered significant for P < 0.05. Assessment of disease-free survival was performed using Cox proportional hazard regression analysis, and the Kaplan Meier method for estimation of survival probability.
Study approval.
All animal use at University of Colorado was conducted in facilities accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care, approved by the University of Colorado IACUC, and following the Helsinki convention for the use and care of animals, including pathogen free conditions. All animal use at MD Anderson was also conducted in facilities accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care, and mice were housed in specific pathogen-free conditions and handled in accordance with the IACUC of MD Anderson Cancer Center.
All human tissues used at the University of Colorado were approved by the IRB at the University of Colorado as exempt because the samples only included the use of existing pathologic specimens that were publicly available and are identified to the investigator only by a Tissue Bank identification number. These studies are in compliance with the Helsinki Declaration, including the additional amendments. For the samples from MD Anderson, patient data is publicly available from the PROSPECT cohort (University of Texas MD Anderson Cancer Center) (43); these patient samples were deidentified and approved by the MD Anderson IRB.
Author contributions
AKB, CME, and SJM designed the study; AKB, MU, VLR, AMC, AQH, NK, ZA, NMH, MM, MSC, DTM, KV, PRM, and SJM designed, performed, and analyzed experiments; CE, IIW, HK, AVB, and BFD contributed reagents and analytic tools; HK, MSC, XL, AEB, and SJM analyzed data; AKB, CME, HK, and SJM wrote the manuscript; and SJM supervised and conceptualized the study.
Supplementary Material
Acknowledgments
This research was funded in part by NIH grants P50CA058187 (AKB, CME), R01HL080396, R01HL130938 (CME), P50CA058187 (Colorado Lung SPORE Tissue Biobank and Biomarker Core), and P30CA046934 (University of Colorado Cancer Center Tissue Biobanking and Histology Shared Resource); by American Cancer Society grant RSG-10-162-01LIB (AKB); and by the MD Anderson Cancer Center University Cancer Foundation Institutional Research Grant and Lung Cancer Moonshot Programs (SJM).
Version 1. 08/09/2018
Electronic publication
Funding Statement
This was pilot funding from the Lung cancer spore grant.
R01 funding (Dr. Evans).
Research Scholars grant (AKB).
University of Colorado Cancer Center Tissue Biobanking and Histology Shared Resource
Seed fund from Lung Cancer Moonshot Program, UT MD Anderson Cancer Center
University Cancer Foundation via the IRG program (SJM)
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: JCI Insight. 2018;3(15):e120941. https://doi.org/10.1172/jci.insight.120941.
Contributor Information
Alison K. Bauer, Email: alison.bauer@ucdenver.edu.
Misha Umer, Email: misha.umer@gmail.com.
Anna Q. Harder, Email: annaqharder@gmail.com.
Nasim Khosravi, Email: khosravinassim1@gmail.com.
Zoulikha Azzegagh, Email: zazzegagh@mdanderson.org.
Camille Ehre, Email: camille_ehre@med.unc.edu.
Maedeh Mohebnasab, Email: m_mohebnasab@yahoo.com.
Mauricio S. Caetano, Email: mau.s.caetano@gmail.com.
Adrie van Bokhoven, Email: adrie.vanbokhoven@ucdenver.edu.
Humam Kadara, Email: hk94@aub.edu.lb.
Kalpana Velmurugan, Email: kalpana.velmurugan@ucdenver.edu.
Xian Lu, Email: xian.lu@kp.org.
Christopher M. Evans, Email: christopher.evans@ucdenver.edu.
Seyed Javad Moghaddam, Email: smoghadd@mdanderson.org.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.National Lung Screening Trial Research Team, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med. 2011;365(5):395–409. doi: 10.1056/NEJMoa1102873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mayekar MK, Bivona TG. Current Landscape of Targeted Therapy in Lung Cancer. Clin Pharmacol Ther. 2017;102(5):757–764. doi: 10.1002/cpt.810. [DOI] [PubMed] [Google Scholar]
- 4.Riely GJ, et al. Frequency and distinctive spectrum of KRAS mutations in never smokers with lung adenocarcinoma. Clin Cancer Res. 2008;14(18):5731–5734. doi: 10.1158/1078-0432.CCR-08-0646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun Y, et al. Lung adenocarcinoma from East Asian never-smokers is a disease largely defined by targetable oncogenic mutant kinases. J Clin Oncol. 2010;28(30):4616–4620. doi: 10.1200/JCO.2010.29.6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McGuckin MA, Thornton DJ. Detection and quantitation of mucins using chemical, lectin, and antibody methods. Methods Mol Biol. 2000;125:45–55. doi: 10.1385/1-59259-048-9:045. [DOI] [PubMed] [Google Scholar]
- 7.Awaya H, Takeshima Y, Yamasaki M, Inai K. Expression of MUC1, MUC2, MUC5AC, and MUC6 in atypical adenomatous hyperplasia, bronchioloalveolar carcinoma, adenocarcinoma with mixed subtypes, and mucinous bronchioloalveolar carcinoma of the lung. Am J Clin Pathol. 2004;121(5):644–653. doi: 10.1309/U4WGE9EBFJN6CM8R. [DOI] [PubMed] [Google Scholar]
- 8.Duruisseaux M, et al. The impact of intracytoplasmic mucin in lung adenocarcinoma with pneumonic radiological presentation. Lung Cancer. 2014;83(3):334–340. doi: 10.1016/j.lungcan.2013.12.016. [DOI] [PubMed] [Google Scholar]
- 9.Guo M, et al. Gene signature driving invasive mucinous adenocarcinoma of the lung. EMBO Mol Med. 2017;9(4):462–481. doi: 10.15252/emmm.201606711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kadota K, et al. Associations between mutations and histologic patterns of mucin in lung adenocarcinoma: invasive mucinous pattern and extracellular mucin are associated with KRAS mutation. Am J Surg Pathol. 2014;38(8):1118–1127. doi: 10.1097/PAS.0000000000000246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mansuet-Lupo A, et al. The new histologic classification of lung primary adenocarcinoma subtypes is a reliable prognostic marker and identifies tumors with different mutation status: the experience of a French cohort. Chest. 2014;146(3):633–643. doi: 10.1378/chest.13-2499. [DOI] [PubMed] [Google Scholar]
- 12.Kim YK, et al. MUC5AC and MUC5B enhance the characterization of mucinous adenocarcinomas of the lung and predict poor prognosis. Histopathology. 2015;67(4):520–528. doi: 10.1111/his.12693. [DOI] [PubMed] [Google Scholar]
- 13.Hollingsworth MA, Swanson BJ. Mucins in cancer: protection and control of the cell surface. Nat Rev Cancer. 2004;4(1):45–60. doi: 10.1038/nrc1251. [DOI] [PubMed] [Google Scholar]
- 14.Kufe DW. Mucins in cancer: function, prognosis and therapy. Nat Rev Cancer. 2009;9(12):874–885. doi: 10.1038/nrc2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Evans CM, et al. Idiopathic Pulmonary Fibrosis: A Genetic Disease That Involves Mucociliary Dysfunction of the Peripheral Airways. Physiol Rev. 2016;96(4):1567–1591. doi: 10.1152/physrev.00004.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fahy JV, Dickey BF. Airway mucus function and dysfunction. N Engl J Med. 2010;363(23):2233–2247. doi: 10.1056/NEJMra0910061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu M, Wang DC, Wang X, Zhang Y. Correlation between mucin biology and tumor heterogeneity in lung cancer. Semin Cell Dev Biol. 2017;64:73–78. doi: 10.1016/j.semcdb.2016.08.027. [DOI] [PubMed] [Google Scholar]
- 18.Travis WD, et al. International association for the study of lung cancer/american thoracic society/european respiratory society international multidisciplinary classification of lung adenocarcinoma. J Thorac Oncol. 2011;6(2):244–285. doi: 10.1097/JTO.0b013e318206a221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nishiumi N, et al. Use of 11p15 mucins as prognostic factors in small adenocarcinoma of the lung. Clin Cancer Res. 2003;9(15):5616–5619. [PubMed] [Google Scholar]
- 20.Sumiyoshi S, et al. Non-terminal respiratory unit type lung adenocarcinoma has three distinct subtypes and is associated with poor prognosis. Lung Cancer. 2014;84(3):281–288. doi: 10.1016/j.lungcan.2014.03.013. [DOI] [PubMed] [Google Scholar]
- 21.Yu CJ, Shih JY, Lee YC, Shun CT, Yuan A, Yang PC. Sialyl Lewis antigens: association with MUC5AC protein and correlation with post-operative recurrence of non-small cell lung cancer. Lung Cancer. 2005;47(1):59–67. doi: 10.1016/j.lungcan.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 22.Lakshmanan I, et al. MUC5AC interactions with integrin β4 enhances the migration of lung cancer cells through FAK signaling. Oncogene. 2016;35(31):4112–4121. doi: 10.1038/onc.2015.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roy MG, et al. Muc5b is required for airway defence. Nature. 2014;505(7483):412–416. doi: 10.1038/nature12807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Young HW, et al. Central role of Muc5ac expression in mucous metaplasia and its regulation by conserved 5’ elements. Am J Respir Cell Mol Biol. 2007;37(3):273–290. doi: 10.1165/rcmb.2005-0460OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Evans CM, et al. The polymeric mucin Muc5ac is required for allergic airway hyperreactivity. Nat Commun. 2015;6:6281. doi: 10.1038/ncomms7281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hasnain SZ, et al. Muc5ac: a critical component mediating the rejection of enteric nematodes. J Exp Med. 2011;208(5):893–900. doi: 10.1084/jem.20102057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Győrffy B, Surowiak P, Budczies J, Lánczky A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One. 2013;8(12):e82241. doi: 10.1371/journal.pone.0082241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szász AM, et al. Cross-validation of survival associated biomarkers in gastric cancer using transcriptomic data of 1,065 patients. Oncotarget. 2016;7(31):49322–49333. doi: 10.18632/oncotarget.10337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin L, et al. Additional evidence that the K-ras protooncogene is a candidate for the major mouse pulmonary adenoma susceptibility (Pas-1) gene. Exp Lung Res. 1998;24(4):481–497. doi: 10.3109/01902149809087382. [DOI] [PubMed] [Google Scholar]
- 30.Zerbe LK, et al. Inhibition by erlotinib of primary lung adenocarcinoma at an early stage in male mice. Cancer Chemother Pharmacol. 2008;62(4):605–620. doi: 10.1007/s00280-007-0644-z. [DOI] [PubMed] [Google Scholar]
- 31.Cardnell RJ, et al. An Integrated Molecular Analysis of Lung Adenocarcinomas Identifies Potential Therapeutic Targets among TTF1-Negative Tumors, Including DNA Repair Proteins and Nrf2. Clin Cancer Res. 2015;21(15):3480–3491. doi: 10.1158/1078-0432.CCR-14-3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moghaddam SJ, et al. Promotion of lung carcinogenesis by chronic obstructive pulmonary disease-like airway inflammation in a K-ras-induced mouse model. Am J Respir Cell Mol Biol. 2009;40(4):443–453. doi: 10.1165/rcmb.2008-0198OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jackson EL, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15(24):3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McLaughlin F, Ludbrook VJ, Cox J, von Carlowitz I, Brown S, Randi AM. Combined genomic and antisense analysis reveals that the transcription factor Erg is implicated in endothelial cell differentiation. Blood. 2001;98(12):3332–3339. doi: 10.1182/blood.V98.12.3332. [DOI] [PubMed] [Google Scholar]
- 35.Shah AV, Birdsey GM, Randi AM. Regulation of endothelial homeostasis, vascular development and angiogenesis by the transcription factor ERG. Vascul Pharmacol. 2016;86:3–13. doi: 10.1016/j.vph.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Caetano MS, et al. IL6 Blockade Reprograms the Lung Tumor Microenvironment to Limit the Development and Progression of K-ras-Mutant Lung Cancer. Cancer Res. 2016;76(11):3189–3199. doi: 10.1158/0008-5472.CAN-15-2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chang SH, et al. T helper 17 cells play a critical pathogenic role in lung cancer. Proc Natl Acad Sci USA. 2014;111(15):5664–5669. doi: 10.1073/pnas.1319051111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goldstraw P, et al. The IASLC Lung Cancer Staging Project: Proposals for Revision of the TNM Stage Groupings in the Forthcoming (Eighth) Edition of the TNM Classification for Lung Cancer. J Thorac Oncol. 2016;11(1):39–51. doi: 10.1016/j.jtho.2015.09.009. [DOI] [PubMed] [Google Scholar]
- 39.Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359(13):1367–1380. doi: 10.1056/NEJMra0802714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lakshmanan I, et al. Mucins in lung cancer: diagnostic, prognostic, and therapeutic implications. J Thorac Oncol. 2015;10(1):19–27. doi: 10.1097/JTO.0000000000000404. [DOI] [PubMed] [Google Scholar]
- 41.Qu Y, et al. Prognostic analysis of primary mucin-producing adenocarcinoma of the lung: a comprehensive retrospective study. Tumour Biol. 2016;37(1):887–896. doi: 10.1007/s13277-015-3869-1. [DOI] [PubMed] [Google Scholar]
- 42.Righi L, et al. Retrospective Multicenter Study Investigating the Role of Targeted Next-Generation Sequencing of Selected Cancer Genes in Mucinous Adenocarcinoma of the Lung. J Thorac Oncol. 2016;11(4):504–515. doi: 10.1016/j.jtho.2016.01.004. [DOI] [PubMed] [Google Scholar]
- 43.Skoulidis F, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015;5(8):860–877. doi: 10.1158/2159-8290.CD-14-1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kadara H, et al. Whole-exome sequencing and immune profiling of early-stage lung adenocarcinoma with fully annotated clinical follow-up. Ann Oncol. 2017;28(1):75–82. doi: 10.1093/annonc/mdw436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Evans CM, Kim K, Tuvim MJ, Dickey BF. Mucus hypersecretion in asthma: causes and effects. Curr Opin Pulm Med. 2009;15(1):4–11. doi: 10.1097/MCP.0b013e32831da8d3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gray T, Koo JS, Nettesheim P. Regulation of mucous differentiation and mucin gene expression in the tracheobronchial epithelium. Toxicology. 2001;160(1-3):35–46. doi: 10.1016/S0300-483X(00)00455-8. [DOI] [PubMed] [Google Scholar]
- 47.Takeyama K, et al. Epidermal growth factor system regulates mucin production in airways. Proc Natl Acad Sci USA. 1999;96(6):3081–3086. doi: 10.1073/pnas.96.6.3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhen G, et al. IL-13 and epidermal growth factor receptor have critical but distinct roles in epithelial cell mucin production. Am J Respir Cell Mol Biol. 2007;36(2):244–253. doi: 10.1165/rcmb.2006-0180OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wakata K, et al. A favourable prognostic marker for EGFR mutant non-small cell lung cancer: immunohistochemical analysis of MUC5B. BMJ Open. 2015;5(7):e008366. doi: 10.1136/bmjopen-2015-008366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim CF, et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121(6):823–835. doi: 10.1016/j.cell.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 51.de-Torres JP, et al. Lung cancer in patients with chronic obstructive pulmonary disease. Development and validation of the COPD Lung Cancer Screening Score. Am J Respir Crit Care Med. 2015;191(3):285–291. doi: 10.1164/rccm.201407-1210OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Takiguchi Y, Sekine I, Iwasawa S, Kurimoto R, Tatsumi K. Chronic obstructive pulmonary disease as a risk factor for lung cancer. World J Clin Oncol. 2014;5(4):660–666. doi: 10.5306/wjco.v5.i4.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.O’Connell S, Au-Yeung HK, Gregory CJ, Matthews IP. Outdoor and indoor respirable air particulate concentrations in differing urban traffic microenvironments. J Toxicol Environ Health Part A. 2008;71(16):1069–1072. doi: 10.1080/15287390802112000. [DOI] [PubMed] [Google Scholar]
- 54.Wang M, Kaufman RJ. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat Rev Cancer. 2014;14(9):581–597. doi: 10.1038/nrc3800. [DOI] [PubMed] [Google Scholar]
- 55.Lynch TJ, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 56.Pao W, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA. 2004;101(36):13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gysin S, Salt M, Young A, McCormick F. Therapeutic strategies for targeting ras proteins. Genes Cancer. 2011;2(3):359–372. doi: 10.1177/1947601911412376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang Y, Mukhopadhyay T, Donehower LA, Georges RN, Roth JA. Retroviral vector-mediated transduction of K-ras antisense RNA into human lung cancer cells inhibits expression of the malignant phenotype. Hum Gene Ther. 1993;4(4):451–460. doi: 10.1089/hum.1993.4.4-451. [DOI] [PubMed] [Google Scholar]
- 59.Maurer T, et al. Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc Natl Acad Sci USA. 2012;109(14):5299–5304. doi: 10.1073/pnas.1116510109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ostrem JM, Shokat KM. Direct small-molecule inhibitors of KRAS: from structural insights to mechanism-based design. Nat Rev Drug Discov. 2016;15(11):771–785. doi: 10.1038/nrd.2016.139. [DOI] [PubMed] [Google Scholar]
- 61.Garon EB, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372(21):2018–2028. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 62.Rizvi NA, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maeda Y, et al. Airway epithelial transcription factor NK2 homeobox 1 inhibits mucous cell metaplasia and Th2 inflammation. Am J Respir Crit Care Med. 2011;184(4):421–429. doi: 10.1164/rccm.201101-0106OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maeda Y, et al. Kras(G12D) and Nkx2-1 haploinsufficiency induce mucinous adenocarcinoma of the lung. J Clin Invest. 2012;122(12):4388–4400. doi: 10.1172/JCI64048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Milewski D, et al. FOXM1 activates AGR2 and causes progression of lung adenomas into invasive mucinous adenocarcinomas. PLoS Genet. 2017;13(12):e1007097. doi: 10.1371/journal.pgen.1007097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Polosukhin VV, et al. Hypoxia-inducible factor-1 signalling promotes goblet cell hyperplasia in airway epithelium. J Pathol. 2011;224(2):203–211. doi: 10.1002/path.2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tsao PN, et al. Notch signaling prevents mucous metaplasia in mouse conducting airways during postnatal development. Development. 2011;138(16):3533–3543. doi: 10.1242/dev.063727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hirsch FR, et al. Epidermal growth factor receptor immunohistochemistry: comparison of antibodies and cutoff points to predict benefit from gefitinib in a phase 3 placebo-controlled study in advanced nonsmall-cell lung cancer. Cancer. 2008;112(5):1114–1121. doi: 10.1002/cncr.23282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hirsch FR, et al. Epidermal growth factor receptor in non-small-cell lung carcinomas: correlation between gene copy number and protein expression and impact on prognosis. J Clin Oncol. 2003;21(20):3798–3807. doi: 10.1200/JCO.2003.11.069. [DOI] [PubMed] [Google Scholar]
- 70.Bauer AK, Upham BL, Rondini EA, Tennis MA, Velmuragan K, Wiese D. Toll-like receptor expression in human non-small cell lung carcinoma: potential prognostic indicators of disease. Oncotarget. 2017;8(54):91860–91875. doi: 10.18632/oncotarget.19463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci USA. 2008;105(39):15064–15069. doi: 10.1073/pnas.0803124105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Puchtler H, Waldrop FS, Meloan SN, Terry MS, Conner HM. Methacarn (methanol-Carnoy) fixation. Practical and theoretical considerations. Histochemie. 1970;21(2):97–116. doi: 10.1007/BF00306176. [DOI] [PubMed] [Google Scholar]
- 73.Bauer AK, et al. Toll-like receptor 4 in butylated hydroxytoluene-induced mouse pulmonary inflammation and tumorigenesis. J Natl Cancer Inst. 2005;97(23):1778–1781. doi: 10.1093/jnci/dji403. [DOI] [PubMed] [Google Scholar]
- 74.Bauer AK, et al. Transcriptomic analysis of pathways regulated by toll-like receptor 4 in a murine model of chronic pulmonary inflammation and carcinogenesis. Mol Cancer. 2009;8:107. doi: 10.1186/1476-4598-8-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bauer AK, Rondini EA. Review paper: the role of inflammation in mouse pulmonary neoplasia. Vet Pathol. 2009;46(3):369–390. doi: 10.1354/vp.08-VP-0217-B-REV. [DOI] [PubMed] [Google Scholar]
- 76.Behrens C, et al. EZH2 protein expression associates with the early pathogenesis, tumor progression, and prognosis of non-small cell lung carcinoma. Clin Cancer Res. 2013;19(23):6556–6565. doi: 10.1158/1078-0432.CCR-12-3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nikitin AY, et al. Classification of proliferative pulmonary lesions of the mouse: recommendations of the mouse models of human cancers consortium. Cancer Res. 2004;64(7):2307–2316. doi: 10.1158/0008-5472.CAN-03-3376. [DOI] [PubMed] [Google Scholar]
- 78.Ehre C, et al. Overexpressing mouse model demonstrates the protective role of Muc5ac in the lungs. Proc Natl Acad Sci USA. 2012;109(41):16528–16533. doi: 10.1073/pnas.1206552109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Evans CM, et al. Mucin is produced by clara cells in the proximal airways of antigen-challenged mice. Am J Respir Cell Mol Biol. 2004;31(4):382–394. doi: 10.1165/rcmb.2004-0060OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Osgood RS, Upham BL, Bushel PR, Velmurugan K, Xiong KN, Bauer AK. Secondhand Smoke-Prevalent Polycyclic Aromatic Hydrocarbon Binary Mixture-Induced Specific Mitogenic and Pro-inflammatory Cell Signaling Events in Lung Epithelial Cells. Toxicol Sci. 2017;157(1):156–171. doi: 10.1093/toxsci/kfx027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.