

Abstract
Alloreactive T lymphocytes are the primary mediators of immune responses in transplantation, both in the graft-versus-host and host-versus-graft directions. While essentially all clones comprising the human T cell repertoire have been selected on self-peptide presented by self–human leukocyte antigens (self-HLAs), much remains to be understood about the nature of clones capable of responding to allo-HLA molecules. Quantitative tools to study these cells are critical to understand fundamental features of this important response; however, the large size and diversity of the alloreactive T cell repertoire in humans presents a great technical challenge. We have developed a high-throughput T cell receptor (TCR) sequencing approach to characterize the human alloresponse. We present a statistical method to model T cell clonal frequency distribution and quantify repertoire diversity. Using these approaches, we measured the diversity and frequency of distinct alloreactive CD4+ and CD8+ T cell populations in HLA-mismatched responder-stimulator pairs. Our findings indicate that the alloimmune repertoire is highly specific for a given pair of individuals, that most alloreactive clones circulate at low frequencies, and that a high proportion of TCRs is likely able to recognize alloantigens.
Keywords: Immunology, Transplantation
Keywords: Bioinformatics, T cells, T-cell receptor
High-throughput TCR sequencing reveals the allo-specific nature of the alloresponse, the role of HLA in alloreactivity, and the lack of public clones among alloreactive populations.
Introduction
Alloreactive T lymphocytes are the primary mediators of the alloreactive immune response in transplantation, both in the graft-versus-host and host-versus-graft directions. Fundamental questions about alloreactive T cells have long challenged immunologists because of data suggesting considerable size and diversity of alloreactive T cell populations, classically estimated to be 1%–10% of the entire T cell repertoire (1). The early studies that led to these widely quoted values along with many other observations about the alloresponse, however, were performed a decade or more ago (2–12). Furthermore, most studies investigating the T cell alloresponse have been in mice, given the need for transgenic tools and other techniques not feasible in humans (13).
The recent advent of high-throughput T cell receptor (TCR) sequencing (14–16) has led to the emergence of new approaches for studying human alloreactive T cell populations. High-throughput sequencing of the third complementarity-determining region (CDR3) of the TCR β chain, a key region for defining antigen specificity, enables identification of the nucleotide sequence defining each unique T cell clone. Using TCR sequencing combined with a standard in vitro functional assay, the mixed lymphocyte reaction (MLR), we have developed a tool for identifying thousands of alloreactive TCR sequences in humans for any given pair of human leukocyte antigen–mismatched (HLA-mismatched) individuals. We have previously validated the biological relevance of such clones in a small cohort of kidney transplant patients in whom we were able to identify before transplant and then track after transplant the donor-specific T cells for each recipient (17). Analysis of intragraft T cells in intestinal transplant recipients provided further validation of the biological significance of clones identified as alloreactive with this assay; expansions of host-versus-graft clones predominated among recipient TCRs in the grafts during rejection episodes and donor clones identified as graft-versus-host-reactive expanded markedly in association with infiltration of the grafts by recipient antigen-presenting cells (18). We have now applied this approach to pairs of HLA-mismatched healthy adults to quantitatively characterize specific aspects of the human alloimmune response.
While previous studies have primarily focused on functional assays (19) or the pattern of usage of TCR β chains in the alloresponse (20), we set out to measure the size and diversity of CD4+ and CD8+ alloreactive populations at the nucleotide level. We compared repertoires of one individual in response to multiple different potential donors in the context of varying degrees of HLA mismatching. To address these quantitative questions, we have developed a TCR repertoire diversity measurement to focus specifically on the bulk of the clonal distribution, providing information not captured in previous diversity calculations such as clonality and entropy (17, 21). Furthermore, we interrogated circulating T cell populations to evaluate frequency and abundance of alloreactive clones.
One of the central questions in studying T cell populations is how to contextualize results from a single blood sample that captures only a snapshot of the entire repertoire. Based on approaches developed in ecology to study population diversity, different strategies have been proposed to address the unseen species question (14, 22). Here we present a new computational approach to model T cell clonal frequency distribution. Using this model, we seek to extrapolate the circulating frequency of unseen alloreactive clones. We apply this approach to assess the cumulative frequency of alloreactive clones to estimate the proportion of clones with potential for alloreactivity.
Results
Identification and qualitative characterization of human alloreactive T cell populations via high-throughput TCR sequencing.
Using high-throughput TCRβ CDR3 sequencing, we defined circulating (unstimulated sorted T cells) and alloreactive CD4+ and CD8+ T cell populations from healthy adults. We studied 9 distinct HLA-mismatched pairs, 2 with repeat samples obtained 1 year apart (summary statistics included in Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.121256DS1). Individual alloreactive repertoires were identified for individual HLA-mismatched responder-stimulator pairs via our recently published method (17) combining the in vitro carboxyfluorescein succinimidyl ester (CFSE) mixed lymphocyte reaction (MLR) with TCRβ CDR3 sequencing of those T cells that divide in response to the alloantigens of a specific stimulator. Alloreactive populations included thousands of unique CD4+ and CD8+ clones, approximately one-tenth of the number of individual clones found in circulating unstimulated CD4+ and CD8+ samples (Figure 1A). In order to be considered alloreactive, we required 2-fold expansion relative to the frequency of the clone in the unstimulated repertoire to minimize the effects of bystander proliferation and sorting error. An unsupervised clustering analysis (Supplemental Figure 1) confirmed a clear distinction between presumably alloreactive and nonalloreactive clones using the 2-fold-expansion criterion. Importantly, clones failing this fold-expansion requirement were generally low frequency clones in the allostimulated populations (Figure 1B and Supplemental Figure 2).
Figure 1. Defining unstimulated and alloreactive T cell repertoires.

(A) Number of unique productive clones identified in CD4+ and CD8+ unstimulated and alloreactive (2-fold-expansion criterion) populations (mean and standard deviation; n = 11 alloreactive, n = 8 unstimulated). (B) Representative histograms showing the number of clones in CFSElo MLR T cells failing the 2-fold-expansion criterion at each clonal frequency; all samples (n = 9) shown in Supplemental Figure 2.
High diversity of the alloreactive T cell repertoire.
We performed sequencing on CD4+ and CD8+ T cells separately, allowing for key comparisons between the 2 distinct repertoires. Consistent with prior studies (20, 22), the alloreactive populations were not only composed of large numbers of unique clones (Figure 1A), but were also qualitatively diverse in terms of CDR3 amino acid length and Vβ- and Jβ-gene usage. Since the structure of the TCR is determined in part by the number of amino acids that gives rise to the CDR3 region, we compared the distribution of CDR3 amino acid lengths between the unstimulated and alloreactive populations and found no significant difference (Figure 2, A and B, and Supplemental Figure 3). Additionally, both CD4+ and CD8+ alloreactive repertoires reflected marked heterogeneity in V- and J-allele pairing (Figure 2C and Supplemental Figure 4). Although some expanded clones were identified, particularly in the CD8+ samples, alloreactive populations were not dominated by any single Vβ or Jβ family.
Figure 2. Comparison of the unstimulated and alloreactive populations identified via high-throughput T cell receptor sequencing.

(A) CD4 and (B) CD8 representative graphs showing lack of significant difference (Mann-Whitney test) in CDR3 amino acid length distribution between unstimulated and alloreactive repertoires; all samples (n = 9) shown in Supplemental Figure 3. (C) CD4 and CD8 representative Circos plots showing diversity of Vβ and Jβ gene pairing in alloreactive repertoires; all samples (n = 9) shown in Supplemental Figure 4. The thickness of the line between each V-gene (right side of circle) and J-gene (left side of circle) is proportional to the frequency of a given combination.
We next sought to measure differences in clonal composition between the alloreactive and unstimulated repertoires, where each clone is defined by its distinct nucleotide sequence. To visualize the clonal diversity for each distinct T cell population, we created abundance plots, shown in Figure 3A and Supplemental Figure 5, key diagrams for illustrating the distribution of clone frequency in the TCR population. In the abundance plot, each point represents the number of individual clones at a given frequency on a logarithmic scale. The left-most portion of the graph covering the low frequency region is limited by the depth of sequencing. In the unstimulated repertoire, large numbers of clones are found at low frequencies, while allostimulation shifts the entire curve to the right due to the expanded nature of the population.
Figure 3. T cell receptor sequencing to quantify diversity of CD4+ and CD8+ unstimulated and alloreactive T cell repertoires.

(A) Representative abundance plots of unstimulated and alloreactive repertoires showing the number of unique clones (clone number) at each frequency within a sample; encircled regions highlight high frequency populations within each sample; plots for all samples (n = 11) are shown in Supplemental Figure 5. (B) Clonality of CD4+ (blue) and CD8+ (red) unstimulated T cells. Each pair of bars represents 1 unstimulated CD4+/CD8+ pair (n = 6). (C) Clonality of CD4+ (blue) and CD8+ (red) alloreactive T cells (2-fold-expansion criterion). Each bar represents 1 alloreactive CD4+/CD8+ pair (n = 9). (D) Box plot (maximum to minimum) comparing fraction of clones accounting for top 20% of reads (R20) in unstimulated and alloreactive populations (samples described in B and C; unpaired t test comparing across alloreactive and unstimulated samples, otherwise paired t test, P = 0.01); tabulated values in Supplemental Table 1. (E) Box plot (maximum to minimum) comparing unstimulated and alloreactive repertoire clonality (samples described in B and C; unpaired t test, P = 0.01); tabulated values in Supplemental Table 1. (F) Representative power law slopes for CD4+ and CD8+ unstimulated sample; solid black line is best-fit line for clone frequency plotted against clone number (number of unique clones or clone count) on the logarithmic scale excluding expanded clones. (G) Box plot (maximum to minimum) comparing slope (S) for unstimulated and alloreactive repertoires (samples described in B and C; unpaired t test comparing across alloreactive and unstimulated comparisons, otherwise paired t test, P = 0.01); tabulated values in Supplemental Table 1.
In an attempt to quantitatively compare CD4+ and CD8+ alloreactive and unstimulated populations, we calculated 2 metrics for the diversity of TCRs. The first is clonality, which is based on Shannon entropy (17, 21), with normalization for sample size. Clonality has a range from 0 to 1, where a clonality of 1 is the least diverse, corresponding to a population consisting of a single clone. Maximum diversity corresponds to a clonality of 0, with all clones present at the same frequency. A striking difference between the unstimulated CD4+ and CD8+ repertoires is that the clonality of CD4+ cells is markedly lower than that of CD8+ cells (Figure 3B), likely due to the increased number of expanded clones in the circulating CD8+ pool (Figure 3A). After allostimulation, however, the clonalities of the expanded CD4+ and CD8+ populations were much more similar. The clonality of alloreactive CD8+ cells was only slightly greater than that of the alloreactive CD4+ cells (Figure 3C).
The second diversity metric is R20. R20 is defined as the fraction of unique clones, in descending order of frequency, that cumulatively account for 20% of the sequenced repertoire: the higher the R20, the less immunodominance there is in a population. The R20 values in Figure 3D show that the unstimulated CD8+ pool includes a dominant population of high frequency clones, whereas unstimulated CD4+ cells have much higher R20s and therefore less immunodominance within the population. The R20 values are markedly reduced for alloreactive compared with unstimulated CD4+ T cells, while a significant trend in the opposite direction was observed for alloreactive compared with unstimulated CD8+ cells, i.e., the R20 value for allostimulated CD8+ cells, while quite low, was greater than that of the unstimulated CD8+ population. When specific dominant clones (frequency > 0.1% in the unstimulated population) were examined in the stimulated versus unstimulated populations, there was very little overlap (Supplemental Table 2), indicating that most immunodominant clones in the alloresponses were not immunodominant in the unstimulated populations.
A quantitative tool for measuring T cell repertoire diversity: power law slopes.
These 2 methods, clonality and R20, are nonparametric metrics for clonal diversity that do not include assumptions of a statistical distribution of clonal abundance. While this nonparametric approach is relatively robust with regard to limitations in sampling, it does not fully utilize the information contained in the shape of the actual distribution. As a result, it has limited ability to distinguish repertoires that are qualitatively different based on biological information. In particular, alloreactive populations are by definition less diverse than their associated unstimulated populations, yet this is not always reflected in the clonality or R20 estimates; while the clonality of alloreactive CD4+ cells is, as expected, significantly increased compared with that of the unstimulated CD4+ cells, such a difference is lacking in the CD8+ cells (Figure 3E); the same issue is seen for R20 (Figure 3D). The increased clonality (and decreased R20) of unstimulated CD8+ cells is apparently driven by a small group of highly abundant or dominant clones, as seen in the abundance distribution (Figure 3A and Supplemental Figure 5). This motivated us to design a new method to measure diversity based on the shape of clonal abundance distribution.
Specifically, we observed that in the abundance plot of clonal size versus frequency (both in log scale), the bulk of the distribution follows a straight line (Figure 3A), implying that the frequencies of clones of a given size are not random, but reflect a constant relationship among the clones that holds for a large proportion of the repertoire. This is known as a power law distribution (23). In a power law distribution, the slope (S), as illustrated in Figure 3F, corresponds to the exponent in the power law and in effect is a measure of population diversity. We therefore present S as a tool for quantifying TCR repertoire diversity: the greater the absolute value of the slope, the greater the diversity.
In a highly diverse population, if one were to sample at random a T cell from this population it would likely be a rare clone with low frequency. As a population approaches maximal diversity (all cells are from distinct and rare clones), the power law slope becomes progressively steeper with fewer and fewer highly abundant clones on the right side of the x axis and higher y values (number of unique clones) on the left side of the distribution. In contrast, a population with relatively fewer rare clones but more abundant clones has a power law slope approaching a horizontal line (slope = 1). These extreme cases illustrate that the greater the slope, the greater the diversity of the repertoire. The slope (Δy/Δx) is indicative of the difference between the number of rare clones on the left side of the distribution and the number of highly abundant clones on the right side. The larger the slope value, the higher the y intercept, and thus the greater the abundance of rare clones circulating at low frequency in the population, consistent with greater diversity. Using S, we can see that the diversity of the unstimulated CD8+ population is indeed much higher than that of allostimulated CD8+ cells, which is biologically reasonable but not evident in the clonality or R20 analyses (Figure 3G). S also captures the increased diversity of unstimulated CD4+ cells compared with unstimulated CD8+ cells and alloreactive CD4+ cells, comparable to results of clonality (Figure 2E).
Furthermore, we performed subsampling analyses to demonstrate that S is robust with regard to changes in the number of clones sampled above a certain minimum (Supplemental Figure 6A). While the x intercept migrates with variations in the number of cells sequenced, the slope itself remains reasonably constant (Supplemental Figure 6B) except for the ultralow range (<104 cells). S focuses on the majority of clones in a population, but as a diversity tool must be used in combination with other approaches that analyze the top expanded clones in a population (highlighted in a distinct color in Figure 3F) to fully describe the entire sequenced repertoire. Additionally, it is a tool that is only relevant in a population that indeed follows a power law distribution.
Taken together, our diversity analyses support the highly diverse nature of both for CD4+ and CD8+ alloreactive populations without a dominant CDR3 length or Vβ/Jβ family, most accurately quantified with the diversity measurement S.
The human alloreactive repertoire is highly allospecific.
We compared the alloreactive repertoire for 1 responder to 2 different stimulators, hypothesizing that the pool of clones reacting to 1 specific HLA-mismatched stimulator would be highly distinct from that responding to another, HLA-disparate, stimulator. For 3 individual responders (designated R1, R2, R3), we compared the alloreactive repertoire generated in response to 3 pairs of different stimulators (designated S1–S5) with extensive HLA mismatching compared with the responder. Scatter plots showing the number of individual clones shared between the 2 distinct alloreactive repertoires confirms their disparate nature for both CD4+ and CD8+ T cell repertoires (Figure 4A). A standard quantitative measure of repertoire overlap is Jensen-Shannon divergence (JSD) (24, 25), a tool that accounts for both clone number and frequency and is normalized on a scale of 0 to 1: a JSD of 1 indicates that all clones in 2 populations are distinct. A small group of high frequency shared clones was detected in the CD8+ repertoires of 2 of the 3 pairs (red in Figure 4A). We computed the corresponding JSD and found an associated decrease between repertoires among top clones ranked by frequency (Figure 4B). Notably, the stimulators in these 2 pairs (S3 and S4; S1 and S5) shared 2 of 6 class I HLA alleles that were not shared by the responder. Furthermore, when the 2 stimulators shared no HLA-A, -B, or -C alleles (S1 and S2), the identified alloreactive repertoires were nearly entirely distinct (Figure 4B). These findings not only support the highly allospecific nature of the alloreactive repertoire for each responder-stimulator pair, but also suggest that some clones shared across repertoires may arise from shared HLA alleles between stimulators.
Figure 4. Allospecificity of the alloreactive repertoire and the role of HLA.

(A) Scatter plots showing lack of overlap between alloreactive clones for 2 distinct alloreactive repertoires generated by 1 responder (R) paired with 2 different stimulators (S) (2-fold-expansion criterion). (B) Jensen-Shannon divergence (JSD) quantitatively compares T cell receptor (TCR) repertoire overlap between the distinct alloreactive populations in A, taking into account clone frequency (JSD 0 = complete overlap; JSD 1 = complete divergence). HLA matches between stimulators shown for class I; for class II, 0 or 1 of 4 (HLA-DR, -DQ) (2 fold-expansion criterion). (C) Schematic highlighting that different analytic strategies for investigating role of HLA-disparities in the alloresponse: comparisons can be between different stimulators (S) or between responder (R) and stimulator (S). (D) Illustrative example of 2 alloreactive repertoires, one HLA mismatched (red) and the other haploidentical (blue), showing power law abundance with a best-fit line used for slope calculation. Alloreactive populations were obtained from a kidney transplant subject. (E) Comparison of CD4 and CD8 slope measurement of alloreactive repertoires for 4 kidney transplant subjects each in response to 2 distinct stimulators, one unrelated HLA-mismatched (left) and one related haploidentical with 2 or more class I (HLA-A, -B) and class II (HLA-DR, -DQ) matches (right). Dashed line connects stimulator pairs for the same subject; Wilcoxon’s test for statistical comparison, P = 0.05). Supplemental Figure 7A shows individual slope plots for each subject (n = 4); raw data included in Supplemental Table 3 along including clonality and R20.
Relationship between alloreactive repertoire diversity and HLA disparity.
We hypothesized that the extent of HLA allele matching between the responder and stimulator (schematized in Figure 4C) would affect the diversity of the alloresponse. To compare overall repertoire diversity, we used S to measure differences between the alloreactive repertoire generated in response to different degrees of HLA matching in a small group of pre–kidney transplant samples from patients who received haploidentical related donor transplants. We were able to compare S between the haploidentical donor stimulators and unrelated fully HLA-mismatched stimulators (Figure 4D and Supplemental Figure 7A). In the setting of greater HLA disparity between responder and stimulator, we identified a greater repertoire diversity as defined by a greater slope (S) (Figure 4E) and by higher R20 and lower clonality (Supplemental Table 3). When the HLA disparity was less marked between the responder-stimulator pairs (2 unrelated stimulators sharing 0–1 allele in class I [HLA-A, -B, -C] and/or 0–2 alleles in class II [HLA-DR, -DQ]), S was similar between the 2 samples for both CD4+ and CD8+ cells (Supplemental Figure 7B).
Lack of evidence for dominant antiviral reactivity in the alloreactive repertoire.
Many human virus-reactive clones have been shown to have cross-reactive alloreactivity (26). We therefore interrogated our alloreactive repertoires for known public (identified in 2 or more individuals) clones cross-reactive to viral antigens and alloantigens. Literature review yielded previously identified public clones reactive to EBV (n = 25), CMV (n =12), HSV (n = 3), and influenza A (n = 8) (Supplemental Table 4). We then looked for those clones in our unstimulated and alloreactive repertoires (without fold-expansion criteria), hypothesizing that some of the dominant clones failing fold-expansion criteria were cross-reactive to viral antigens. We found 24 clones matching the same amino acid sequence of the CDR3 region of the public clones in our alloreactive populations, but only 7 clones with the same V and J gene and 10 associated with the same HLA type previously associated with the public clone (Supplemental Table 5). A majority of those clones were CD8+ and all were associated with EBV or influenza A. None of the clones were present at high frequency, either in the unstimulated repertoire (Supplemental Table 6) or alloreactive repertoire (Supplemental Table 5). We also looked for the public clones in the alloreactive repertoire of the pre–kidney transplant subjects and again few public virus-reactive clones were identified (Supplemental Table 5). Of the 7 likely virus-reactive clones identified in both the unstimulated and CFSElo populations in MLR, 5 did not meet the 2-fold-expansion criterion, perhaps because these clones were bystanders rather than truly alloreactive; furthermore, the frequency of these clones in the unstimulated population was less than 0.01%. Overall, our results do not support a role for virus-reactive clones playing a major role in the identified alloreactive repertoires.
Alloreactive T cells are primarily low frequency clones in circulation.
Our high-throughput sequencing approach enabled investigation of the baseline circulating frequency of those clones. Here, we introduced a minimum-frequency threshold of 10–5 in MLR for identifying alloreactive clones to help ensure that those clones meeting the 2-fold-expansion criterion were not simply included because their frequency in the unstimulated repertoire was undetectable. Strikingly, we found that the circulating frequencies of most alloreactive clones were so low in the unstimulated CD4+ and CD8+ populations that most were not detected via deep sequencing (Figure 5A and Supplemental Figure 8). Histograms highlighting the alloreactive clones detected within the unstimulated repertoire show the presence of only a few clones at frequencies greater than 10–3, mostly among CD8+ cells, while the overwhelming majority of alloreactive clones seem to circulate at very low frequency.
Figure 5. Frequency of alloreactive clones in circulation.

(A) Representative histograms for 3 healthy adults showing frequency distribution of alloreactive clones (blue CD4+, red CD8+) within the unstimulated population (gray). Undetected alloreactive clones (2-fold-expansion criterion, minimum-frequency threshold 10–5 within stimulated) clones are plotted to the left of the y axis. Histograms for all subjects (n = 9) shown in Supplemental Figure 8. (B) Sum frequency of CD4+ and CD8+ alloreactive clones detected in the corresponding unstimulated CD4+ and CD8+ populations (light blue and light red, respectively) and the total estimated frequency of alloreactive clones: sum frequency of detected clones with added sum frequency of unseen alloreactive clones (dark blue, dark red). Unseen frequencies of alloreactive clones calculated via the statistical model schematized in C and further described in the supplemental methods (n = 9 alloreactive, n = 6 unstimulated, individual values included in Table 1). (C) Schematic illustrating statistical model used to estimate average frequency of alloreactive clones not detected within the circulating population. (D) Cumulative frequency of detected (solid fill bars) and undetected (unfilled bars) alloreactive clones for 1 responder to 2 distinct stimulators (n = 3, corresponding to sample pairs shown in Figure 3A; “A,” “B,” and “C” on the x axis refer to each different responder).
An approach to estimate the total frequency of unseen alloreactive clones.
The cumulative frequency of CD4+ and CD8+ alloreactive clones detected in circulation accounted for 0.83% ± 0.73% and 0.76% ± 0.75% of all clones, respectively (mean ± standard deviation) (Figure 5B and Table 1). While fold-expansion and minimum-frequency criteria affected this result to varying degrees (Supplemental Figure 9), we sought an analytic strategy that could include the greatest number of clones most likely to be alloreactive.
Table 1. Extrapolation of total frequency of unseen alloreactive clones estimated via parametric statistical model.

Since this total frequency calculation only includes the very small percentage of alloreactive clones detected in the unstimulated population in a given experiment, frequencies of the majority of alloreactive clones in unstimulated circulating populations were below the threshold of detection achieved with current deep sequencing platforms. To estimate the total frequency of undetected clones for a given experiment, we developed a statistical model, schematized in Figure 5C and described in detail in the Methods section, that describes the frequency distribution of alloreactive clones detected within the circulating repertoire by a power law. The slope of the power law is then used to extrapolate the average frequency of undetected clones (Table 1 and Supplemental Figure 10) under the assumption that the clonal frequency of undetected clones continues to obey a power law. Multiplying this estimated average clone frequency by the number of undetected clones, we calculated an additional frequency of 1.5% ± 0.69% for CD4+ and 0.53% ± 0.31% for CD8+ cells (Figure 5B). We refer to this method as parametric because it utilizes a fitted power law distribution for clonal abundance.
This approach allowed us to examine the combined total frequency of alloreactive clones in an individual that responded to 2 different allogeneic stimulators. Because these repertoires shared so few overlapping clones, when we assessed the sum frequency of both detected and undetected alloreactive clones, the total frequency of all alloreactive clones to 2 different stimulators was nearly twice that of the allostimulated population in response to 1 stimulator alone (Figure 5D).
Thus, our model enabled the estimation of the total frequency of both detected and undetected alloreactive clones within the unstimulated repertoires. Applying this strategy, we observed that a substantial fraction of the circulating TCR repertoire (0.5%–6%) responded to just 2 different allogeneic stimulators.
Discussion
Observations about the potent immune phenomenon that mediates both allograft rejection and graft-versus-host disease emerged primarily from rodent and other nonhuman animal studies prior to the high-throughput sequencing era. Our work seeks to build on the previous findings about the alloresponse in humans via analyses of alloimmune T cells using a sequencing approach. Our results help to quantitatively define central features of the human T cell alloreactive repertoire. We provide fundamental information about the diversity and frequency of CD4+ and CD8+ alloreactive clones and in so doing have developed a tool for measuring T cell population diversity that captures distinct aspects of the repertoire not highlighted by existing methods. Here, we also provide insight into the role of HLA disparity in alloreactivity and investigate public virus-reactive TCRs within the alloresponse. Furthermore, we propose a statistical model to address the unseen species question to estimate the frequency of alloreactive clones not detected in a given experiment. Taken together, the results of our study of the human alloresponse suggest that many, if not all, T cell clones have the potential to be alloreactive.
Alloreactive clones could not be differentiated from the unstimulated repertoire on the basis of fundamental features such as CDR3 length or Vβ/Jβ-gene usage. These results are consistent with previous observations about the lack of dominant CDR3 length or Vβ/Jβ-gene usage within alloreactive T cell populations (27, 28). While this diversity is part of what makes the alloresponse difficult to target in the clinical setting, it suggests that there may not be anything distinct about the structure of an alloreactive TCR that inherently denotes a given clone as having alloreactive potential. Similarly, we identified minimal overlap between alloreactive TCRs and known public virus-reactive clones implicated in the response to pathogens, suggesting that alloreactive clones are not distinguished by a particular prior immunologic role. The ontogeny of alloreactive T cells has been somewhat enigmatic (29). Our current understanding reflects the inherent recognition of MHC/peptide by TCR structures and the flexibility in the configuration of TCR interactions with alternative ligands (30–34). Consequently, T cells have the potential to interact with more than one HLA/peptide pair, a phenomenon known as degeneracy. It is this capacity for cross-reactivity that may explain the high probability that a given T cell clone will have alloreactivity. Indeed, the T cell clones in any given individual are selected on self-antigens and not alloantigens in the thymus and thus by definition alloreactivity can be thought of as an incidental function arising from the degeneracy of the TCR.
The great potential for alloreactivity across the T cell repertoire is reflected in the vast diversity of the alloreactive population. Because this study focused on healthy adults without known prior exposure to alloantigens, the frequency distribution of alloreactive clones ranged from rare, presumably capturing naive T cells, to highly abundant, presumably capturing cross-reactive memory clones (Figure 3A). It is this diversity that necessitated the creation of a tool to make accurate quantitative comparisons capturing the abundance of these clones. As the number of TCR sequencing studies continues to grow, it is essential that we have the necessary tools to quantitatively compare key aspects of T cell populations. The available methods for measuring TCR diversity, such as clonality, Simpson’s index, and entropy, emerged predominantly from the ecology literature. While these tools are valuable for comparing overall population diversity, they are each affected by the presence of expanded/dominant clones and do not describe the nature of the physical distribution of a population.
Prior studies have highlighted the greater diversity of CD4+ populations compared with CD8+ cells (21, 35). Our results show a similar pattern in peripheral blood T cells (Figure 3B), which is likely due to a small number of highly dominant clones in the CD8+ circulating population as captured by the R20 analysis. These few abundant clones thereby crowd out the lower frequency clones and thus lead to a functionally smaller CD8+ T cell compartment. These clonal expansions create a statistical challenge when analyzing repertoire size and diversity. A clear example of this is the failure of clonality or R20 to accurately capture the increased diversity of the unstimulated CD8+ pool compared with alloreactive clones. We therefore developed a diversity measurement, S, that emphasizes the bulk of the TCR population rather than the expanded, highest frequency clones (Figure 3F) and thereby does distinguish the diversity of the unstimulated CD8+ repertoire from that of alloreactive CD8+ cells.
S is a diversity measure that focuses on the majority of clones within a population, highlights fundamental features of the T cell repertoire not identified via existing diversity calculations, and is robust with regard to variations in sample size. Furthermore, a major advantage of S as a measure of T cell repertoire diversity is that it describes the physical distribution of a population such that it can be recreated and used for statistical modeling, which we apply in our approach for estimating the frequency of unseen clones. S emerges from the confirmation that the power law is the best model to describe the distribution for the bulk of TCR clones (22, 25). The steepness of S reflects the difference in the number of rare clones on the left side of the distribution in an abundance plot compared with the number that are highly frequent on the right. As S increases, so does the proportion of clones within a population that are low frequency or rare, consistent with greater diversity. Using S, we are able to assess the relationship between HLA mismatching and alloresponse diversity, obtaining results supporting the notion that a greater number of potential alloantigens in the HLA-mismatched setting results in a more diverse alloreactive repertoire. However, our sample size was limited and further work will be necessary to discern how the extent and structure of matched class I and class II alleles affects both CD4+ and CD8+ alloreactive repertoire diversity.
The question of how many clones are indeed alloreactive has been longstanding. Original estimates were all impressively large and classically cited as 1%–10% of the entire T cell repertoire. While techniques and species varied, many estimates arose from in vitro functional assays in rodents (4, 6, 7, 36–38), though one of the earliest studies was based on a the classical MLR with human cells (39) and another was performed in chickens (5). While transgenic approaches enabled more sophisticated approaches in rodents (12), functional assays, with their many limitations, have been the main approach in humans (9). With the development of the CFSE-MLR, new analytic techniques emerged to quantify precursor frequencies of alloreactive cells based on the dilution of CFSE (11, 19). A major limitation of this approach is that the quantification occurs at the end of the multiday assay and thereby the denominator excludes the large fraction of clones that die each day in the MLR due to lack of antigenic stimulation. An advantage of our approach is that we are able to look at the original circulating frequency of alloreactive clones by interrogating the unstimulated population. Although our estimates are biased for identifying high frequency and thereby likely memory clones in the unstimulated circulating pool due to the limited depth of sequencing and number of clones sequenced, they are overall consistent with previous estimates based on functional readouts alone.
It was striking that most alloreactive clones identified in MLRs were not detected in our unstimulated T cell populations from the same sample. This again supports the notion that alloreactive clones, though highly abundant, are not specifically expanded within the circulating T cell pool. It was therefore necessary to develop a statistical model to incorporate such clones into our frequency estimates. The only studies that we are aware of that estimate TCR repertoire size from a high-throughput sequencing approach are the nonparametric method proposed by Robins et al. in 2009 (14) and the parametric method proposed by Laydon et al. in 2015 (22). Most nonparametric approaches rely on accurate quantification of rare clones, which are the least well sampled and the most prone to error during sequencing. Thus, deriving good estimates from these methods requires a large sample size of several million clones that is not available in a typical study. The parametric method in this study does not presume to know a true repertoire size, but instead utilizes measurable quantities: power law slopes and clones that are not shared between the unstimulated and alloreactive populations. Because we base alloreactive clone identification on only a single MLR experiment for each pair, we recognize that our results may underestimate the total frequency of alloreactive clones that could potentially be identified via multiple MLRs. Importantly, our statistical approach to extrapolate the circulating frequency of a population of unseen clones has a broad range of applications for TCR sequencing analysis in any setting in which only a subgroup of clones is identified within a circulating T cell population.
Notably, a sequencing approach enabled demonstration of the allospecificity of a given repertoire of clones for one responder paired with a unique stimulator. One interesting trend that emerged is the sharing of CD8+ clones when stimulators shared HLA class I alleles. Further structural analysis will be needed to investigate whether shared HLA/peptide complexes presented by the stimulators are the alloimmune response targets of the responder T cells. Furthermore, because the repertoire identified for each responder-stimulator pair was distinct, we were able to quantify the cumulative frequency of alloreactive clones for 1 adult donor against 2 separate stimulators. These total frequencies were nearly twice that of each individual alloreactive repertoire. If one were to continue to add together the cumulative frequency of alloreactive clones for one responder to many different stimulators of all possible HLA mismatches, one could imagine that even greater than 10% if not all T cell clones are potentially alloreactive.
One of the greatest challenges in this study is setting the definition of alloreactive clones without having an in vivo test to know if all clones proliferating in an MLR are indeed alloreactive. Although previous studies performed using our TCR sequencing tool to study alloreactive clones in transplant recipients support the biological relevance of the clones identified (17, 18), setting definitions such as fold-expansion criteria and minimum-frequency thresholds are hardly straightforward. We believe a fold-expansion criterion is critical to remove highly dominant clones in the circulating pool that likely proliferated by bystander affect and/or were found in the CFSElo pool due to sorting error. Reassuringly, this approach is supported by our unsupervised clustering analysis, in which we are able to separate the alloreactive and nonalloreactive clones using a method analogous to gating in flow cytometry. While the majority of clones failing our fold-expansion criteria are low frequency, some large clones are inevitably excluded (Figure 1B and Supplemental Figure 1), biasing against considering circulating immunodominant clones as alloreactive. Similarly, although we have algorithms to correct for CD4 and CD8 sorting error, there is still variability in sorting purity as well as in the sorting gates themselves based on the pattern of CFSE dilution. Furthermore, while both naive and memory cells proliferate in an MLR (19), it is not certain whether their frequency distribution remains constant throughout the MLR, making it possible to sequence more of one subgroup than another. Similarly, other subpopulations of T cells, such as T regulatory cells, may not proliferate as readily as other subsets in an MLR. Other variables to consider include CMV/EBV exposure history (40–42) and subject age (24), which likely also play a role in the balance of naive and memory cells and nature of the alloresponse.
Methods
Study design
The aim of this study was to quantitatively characterize the human alloreactive T cell repertoire using high-throughput TCR sequencing. Laboratory and statistical investigations were performed on HLA-typed (allele-level resolution, either via sequence-based typing or sequence-specific oligonucleotides by One Lamda) healthy adult peripheral blood mononuclear cells (HLA results included in Supplemental Table 7). Additional analyses were performed on TCR sequencing results from 4 of 5 combined kidney bone-marrow transplant (CKBMT) subjects from the ITN036ST study as described in Morris et al. (17); patient samples were provided by the Immune Tolerance Network. There was no randomization or blinding.
Identifying the circulating and alloreactive T cell repertoires
High-throughput TCR sequencing (ImmunoSeq, Adaptive Biotechnologies) of the responder T cells dividing in response to HLA-mismatched stimulators in a CFSE (CellTrace CFSE Proliferation Kit, Molecular Probes, catalog C34554) MLR was performed as described previously (17). Briefly, responder cells from healthy HLA-typed adults labeled with CFSE dye and violet-labeled irradiated stimulator cells (BD Horizon Violet Proliferation Dye 450, catalog 562158) were cocultured for 6 days. Deep sequencing was performed on genomic DNA extracted from CFSElo and therefore divided responder T cells isolated via FACS. Sequencing was also performed on unstimulated responder T cells, enabling comparison between the unstimulated and alloreactive T cell populations.
TCR repertoire sequencing and statistical analyses
TCR repertoire sequencing and quality control.
CD4+ and CD8+ sorted T cells were sent to Adaptive Biotechnologies for β-chain profiling according to protocols and standards for sequencing and error correction that comprise their immunoSEQ Platform (24, 43, 44). In summary, the Adaptive deep sequencing protocol extracts approximately 1,200 ng of T cell DNA. PCR amplification of the CDR3 region is performed using specialized primers that anneal to the V and J recombination cassettes with minimal crossover. Unique molecular identifiers are added during library preparation to track template numbers. After sequencing, a computational pipeline identifies CDR3 nucleotide regions and V and J cassettes. Clonal copy numbers are corrected for sequencing and PCR error based on known error rates and clonal frequencies.
Datasets were downloaded from Adaptive servers and further filtered to correct for CD4+/CD8+ sorting error using template frequencies from individual clones. A minimum 2-fold difference was required between CD4+ and CD8+ subsets, as previously described in Thome et al. (24) (Supplemental Figure 11), removing 0.6% of the total clonal population on average, with a maximum of 1.6% consistent with proportions for sorting error.
Defining frequency thresholds for alloreactive populations.
Alloreactive populations were further filtered to account for bystander clones that may be erroneously present due to the analog nature of fluorescence-based quantification. To establish an appropriate threshold, sample frequencies of alloreactive and corresponding unstimulated samples were visualized on a scatter plot. A 2D kernel density estimation approach was used to separate the data into clusters of distinct T cell populations, using all clones with frequency greater than 5 × 10–5, since low frequency clones were not separable. Different linear cutoff thresholds from 1- to 10-fold change were compared visually for their ability to recreate the cluster pattern (Supplemental Figure 2). Additionally, for each threshold, a sum frequency was calculated for unstimulated clones also present in the alloreactive population as a way to assess the stringency of the cutoff criteria (Supplemental Figure 9.) The 2-fold cutoff was most robust across samples, providing a conservative filter for alloreactivity while still retaining the majority of clones by frequency. Finally, clones with frequency less than 1 × 10–5 in the alloreactive population were removed for some analyses, corresponding to the minimum possible frequency for a proliferated clone in a sample containing 2 × 105 sequenced cells.
Statistical measures of repertoire diversity.
There are many ways of defining the diversity of a population, with each method providing a different representation of the number of species present (richness) and of their relative frequencies (evenness). Shannon entropy weighs both of these aspects of diversity equally, providing an intuitive measure whereby the maximum value is determined by the total size of the repertoire, and entropy values decreases with increasing inequality of frequencies as a result of clonal expansion. The Shannon entropy in a population of N clones with nucleotide frequencies pi is defined by:
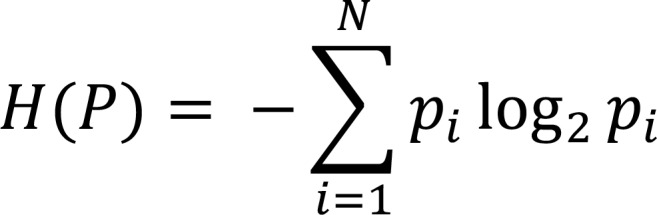 Equation 1.
Equation 1.
A related measure, which we call clonality (17, 21), can be obtained from the normalized sample entropy, defining maximum diversity at 0, when all clones are equally represented, and minimal diversity at 1, corresponding to a population that consists of a single clone:
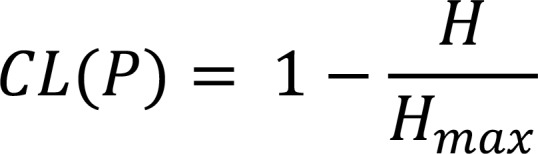 Equation 2
Equation 2
is the maximum attainable entropy, with all clones sampled at the same frequency, 1/N. To perform these calculations, we used template frequency as a proxy for the cell frequency of each clone in the sample, as described in TCR repertoire sequencing and quality control.
The R20 score measures diversity for the top 20% of the population, indicating the extent to which this population is expanded relative to the bottom 80%. R20 is obtained by first sorting clonal frequencies in decreasing order, then starting with the highest frequency clone and going in decreasing order to compute the fraction of all clones included in the top 20% of templates (T):
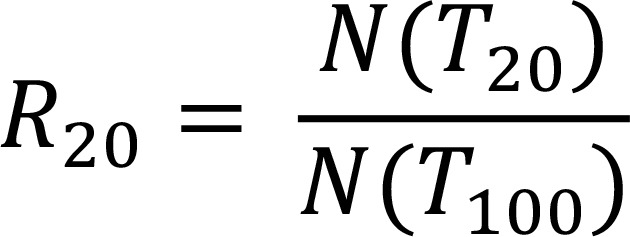 Equation 3,
Equation 3,
where N(T) defines the number of clones that account for T templates.
Modeling the frequency distribution of the TCR repertoire
T cell repertoires have consistently been seen to follow a long-tailed distribution, with 2 major subpopulations: a bulk component into which most clones fall and a smaller component consisting of high frequency clones. The bulk component is linear on the log-log scale, and is well described by a power law (23, 45, 46), defined by y = Kx–S. Taking a logarithm produces the equation of a line log y = −S log(x) + log(K), with exponent S as the slope and log(K) as the intercept.
Data were separated into a power law component and an expanded portion using as a cutoff the second smallest unique expanded clone by frequency. In this study a unique expanded clone is defined as any clone with a unique cell count in the population and no other sampled clones having the same cell count. Linear fits of template counts were calculated for all samples using a maximum likelihood, least squares fit, as computed by the “lm” function in R, applied to the log-transformed data. This fitting procedure produced a value for slope and intercept (K).
While the lowest frequency unique expanded clone was sufficient for most of the data, adding the second-smallest clone improved the accuracy of the slope estimate for several cases where the data were undersampled and the smallest such clone observed was erroneous. Large expansions sometimes created a second unique expanded clone with much higher frequency than the first, and therefore the 2 clones considered were required to be near to one another, with the best results achieved when the log10 distance was less than 1.5. If the distance was larger, only the single lowest frequency clone was used. In rare cases where there were one or fewer total unique expanded clones, either due to a lack of significant expansion or the result of small sample size, the entire dataset was used to perform the fit.
Quantifying repertoire divergence
Interpopulation differences were determined by the JSD (47), which has previously been applied to TCR repertoire analysis (24, 25). Rather than quantifying unevenness within a population, the JSD provides a measure of dissimilarity between the labeled frequencies of 2 populations. The use of this measure assumes populations initially belong to the same starting pool — observed frequencies of each clone may change, and even be zero, but the comprehensive biological set of clones does not. Alloreactive populations from the same healthy control can therefore be compared by CDR3 nucleotide sequence.
Measuring the sum frequency of alloreactive clones
Initial estimates of alloreactivity were obtained from the frequencies of unstimulated clones observed in both the unstimulated and alloreactive populations. Additional frequencies for unobserved alloreactive clones were inferred using a parametric method.
Parametric method for estimating frequency of unseen clones
Most clones found in the alloreactive TCR population are not observed in the unstimulated population due to the large repertoire diversity relative to sample size. The parametric method uses estimated power law slopes to measure an average frequency for such missing clones.
In this method, power law parameters, S and K, are obtained as described above to fit the subset of unstimulated clones captured in the alloreactive population. An unseen clone is represented by a template count of zero in the power law, which cannot be identified on a log-log scale. However, when these counts are converted into frequencies, the power law can be extrapolated to lower frequency values. A new y intercept is defined by the number of clones, N, observed only in the alloreactive population. A vertical line is extended down from this y intercept to define the x intercept, x*, which represents the average frequency of an unseen clone.
The x intercept can be obtained mathematically from the definition of the slope of a line, with values transformed on the log scale: x* = exp([log(N) – log(K)]/–S). To test the model, we downloaded reference datasets from Adaptive’s public repository (https://clients.adaptivebiotech.com/pub/healthy-adult-time-course-TCRB) containing sequenced TCRB Time Course data taken from healthy human peripheral blood mononucleocyte samples. Three subjects were selected, each with 3 samples taken from the same time point. These datasets contained approximately 300,000 clones on average, with 20 million reads per sample (Supplemental Table 8). After converting the reads into template counts, ranging from 3 × 105 to 9 × 105 templates, the data were subsampled into 2 populations, one with 150,000 templates and a second with 45,000, representing the typical numbers of sequenced unstimulated and alloreactive samples in our experiments (Supplemental Figure 13A). The parametric method was applied to predict unseen species frequencies from CDR3 overlap (Supplemental Figure 13B). Subsampling and prediction were performed 10 times for each of the 9 samples. Each predicted frequency was compared to the true unseen frequency of clones in the alloreactive population (true frequency – prediction) that was not present in the overlap. Several examples are shown in Supplemental Figure 13C; results varied slightly based on the initial number of templates in the original sample, and likely the quality of the read fitting, but all fell within a margin of ±3% with only one sample deviating to +4.8% (Supplemental Figure 13D). Most of the results were very slight overestimates of the true value.
To further assess the reliability of this method, we looked at a pair of biological replicates of alloreactive samples from our dataset, together with the single unstimulated sample from which both derived (Supplemental Figure 14, A and B). The predicted unseen frequencies of the 2 samples were added together, taking an average for the portion of clones present in both. This result was compared to the frequency prediction from a single sample made by combining the two (Supplemental Figure 14C). Adding the 2 replicates individually produced an average unseen clone frequency of 0.0166, compared with 0.0151 from the combined sample, an overestimate of of 0.15% demonstrating the precision of this approach.
Statistics
Unpaired t tests were used for comparisons across alloreactive and unstimulated samples. Paired t tests were used for analyses of paired CD4+/CD8+ samples. Significance was defined as a P value of 0.05 except where specifically noted in Figure 3, D–G, where the P value threshold was set at 0.01 based on Bonferroni’s correction to adjust for multiple comparisons. Wilcoxon’s test was used for analysis of S in paired CD4+/CD8+ samples (Figure 4E). Statistical tests were performed with GraphPad Prism. All TCR sequencing analyses were performed in R.
Study approval
The study protocols were approved by the Columbia University Medical Center and Massachusetts General Hospital Institutional Review Boards (Boston, Massachusetts, USA). All subjects provided informed consent prior to their participation in the study.
Author contributions
SDW designed and performed experiments, analyzed and interpreted data, and wrote the manuscript. BG developed analytic tools, analyzed and interpreted data, and wrote the manuscript. TS, SPL, BS, HM, and JZ processed samples, performed experiments, and analyzed and interpreted data. AO analyzed data. SY participated in sample procurement. RW supervised analysis and edited the manuscript. MS designed and oversaw laboratory studies, interpreted the data, and wrote the manuscript. YS oversaw computational analyses, interpreted the data, and wrote the manuscript.
Supplementary Material
Acknowledgments
We thank Nicole Casio for assistance with submission. We thank the Immune Tolerance Network for providing patient peripheral blood mononuclear cell samples. The work was supported in part by NIH grants P01AI106697-01 (to B. Grinshpun and Y. Shen), R21AR064473 (to Y. Shen), and NIH National Institute of Allergy and Infectious Diseases (NIAID) grants P01 AI106697-06 and UM1 AI109565 (both to M. Sykes). S. DeWolf was supported by an American Society of Hematology HONORS award. Research reported in this publication was performed in the CCTI Flow Cytometry Core, supported in part by the Office of the Director, NIH, under awards S10RR027050 and S10OD020056. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Code for TCR analysis is available at https://github.com/ShenLab/Repertoire/tree/master/Alloreactive
Version 1. 08/09/2018
Electronic publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: JCI Insight. 2018;3(15):e121256. https://doi.org/10.1172/jci.insight.121256.
Contributor Information
Susan DeWolf, Email: sed7009@nyp.org.
Boris Grinshpun, Email: bgrinshpun@gmail.com.
Thomas Savage, Email: tms2157@cumc.columbia.edu.
Sai Ping Lau, Email: saipinglau@gmail.com.
Aleksandar Obradovic, Email: azo2104@cumc.columbia.edu.
Brittany Shonts, Email: bas2203@cumc.columbia.edu.
Suxiao Yang, Email: sy2495@cumc.columbia.edu.
Heather Morris, Email: hm2334@cumc.columbia.edu.
Julien Zuber, Email: julien.zuber@aphp.fr.
Megan Sykes, Email: megan.sykes@columbia.edu.
Yufeng Shen, Email: ys2411@cumc.columbia.edu.
References
- 1.Sherman LA, Chattopadhyay S. The molecular basis of allorecognition. Annu Rev Immunol. 1993;11:385–402. doi: 10.1146/annurev.iy.11.040193.002125. [DOI] [PubMed] [Google Scholar]
- 2.Wilson DB. Quantitative studies on the mixed lymphocyte interaction in rats. I. Conditions and parameters of response. J Exp Med. 1967;126(4):625–654. doi: 10.1084/jem.126.4.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wilson DB, Silvers WK, Nowell PC. Quantitative studies on the mixed lymphocyte interaction in rats. II. Relationship of the proliferative response to the immunologic status of the donors. J Exp Med. 1967;126(4):655–665. doi: 10.1084/jem.126.4.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilson DB, Blyth JL, Nowell PC. Quantitative studies on the mixed lymphocyte interaction in rats. 3. Kinetics of the response. J Exp Med. 1968;128(5):1157–1181. doi: 10.1084/jem.128.5.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nisbet NW, Simonsen M, Zaleski M. The frequency of antigen-sensitive cells in tissue transplantation. A commentary on clonal selection. J Exp Med. 1969;129(3):459–467. doi: 10.1084/jem.129.3.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ford WL, Atkins RC. The proportion of lymphocytes capable of recognizing strong transplantation antigens in vivo. Adv Exp Med Biol. 1973;29(0):255–262. doi: 10.1007/978-1-4615-9017-0_37. [DOI] [PubMed] [Google Scholar]
- 7.Lindahl KF, Wilson DB. Histocompatibility antigen-activated cytotoxic T lymphocytes. II. Estimates of the frequency and specificity of precursors. J Exp Med. 1977;145(3):508–522. doi: 10.1084/jem.145.3.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Oers MH, Pinkster J, Zeijlemaker WP. Quantification of antigen-reactive cells among human T lymphocytes. Eur J Immunol. 1978;8(7):477–484. doi: 10.1002/eji.1830080706. [DOI] [PubMed] [Google Scholar]
- 9.Sharrock CE, Man S, Wanachiwanawin W, Batchelor JR. Analysis of the alloreactive T cell repertoire in man. I. Differences in precursor frequency for cytotoxic T cell responses against allogeneic MHC molecules in unrelated individuals. Transplantation. 1987;43(5):699–703. doi: 10.1097/00007890-198705000-00019. [DOI] [PubMed] [Google Scholar]
- 10.Orosz CG, Adams PW, Ferguson RM. Frequency of human alloantigen-reactive T lymphocytes. II. Method for limiting dilution analysis of alloantigen-reactive helper T cells in human peripheral blood. Transplantation. 1987;43(5):718–724. [PubMed] [Google Scholar]
- 11.Noorchashm H, et al. A direct method for the calculation of alloreactive CD4+ T cell precursor frequency. Transplantation. 1999;67(9):1281–1284. doi: 10.1097/00007890-199905150-00015. [DOI] [PubMed] [Google Scholar]
- 12.Suchin EJ, Langmuir PB, Palmer E, Sayegh MH, Wells AD, Turka LA. Quantifying the frequency of alloreactive T cells in vivo: new answers to an old question. J Immunol. 2001;166(2):973–981. doi: 10.4049/jimmunol.166.2.973. [DOI] [PubMed] [Google Scholar]
- 13.DeWolf S, Shen Y, Sykes M. A new window into the human alloresponse. Transplantation. 2016;100(8):1639–1649. doi: 10.1097/TP.0000000000001064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robins HS, et al. Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood. 2009;114(19):4099–4107. doi: 10.1182/blood-2009-04-217604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robins H, et al. Ultra-sensitive detection of rare T cell clones. J Immunol Methods. 2012;375(1–2):14–19. doi: 10.1016/j.jim.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carlson CS, et al. Using synthetic templates to design an unbiased multiplex PCR assay. Nat Commun. 2013;4:2680. doi: 10.1038/ncomms3680. [DOI] [PubMed] [Google Scholar]
- 17.Morris H, et al. Tracking donor-reactive T cells: Evidence for clonal deletion in tolerant kidney transplant patients. Sci Transl Med. 2015;7(272):272ra10. doi: 10.1126/scitranslmed.3010760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zuber J, et al. Bidirectional intragraft alloreactivity drives the repopulation of human intestinal allografts and correlates with clinical outcome. Sci Immunol. 2016;1(4):eaah3732. doi: 10.1126/sciimmunol.aah3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Macedo C, et al. Contribution of naïve and memory T-cell populations to the human alloimmune response. Am J Transplant. 2009;9(9):2057–2066. doi: 10.1111/j.1600-6143.2009.02742.x. [DOI] [PubMed] [Google Scholar]
- 20.Sebille F, et al. Direct recognition of foreign MHC determinants by naive T cells mobilizes specific Vbeta families without skewing of the complementarity-determining region 3 length distribution. J Immunol. 2001;167(6):3082–3088. doi: 10.4049/jimmunol.167.6.3082. [DOI] [PubMed] [Google Scholar]
- 21.Thome JJ, et al. Spatial map of human T cell compartmentalization and maintenance over decades of life. Cell. 2014;159(4):814–828. doi: 10.1016/j.cell.2014.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laydon DJ, Bangham CR, Asquith B. Estimating T-cell repertoire diversity: limitations of classical estimators and a new approach. Philos Trans R Soc Lond, B, Biol Sci. 2015;370(1675) doi: 10.1098/rstb.2014.0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Desponds J, Mora T, Walczak AM. Fluctuating fitness shapes the clone-size distribution of immune repertoires. Proc Natl Acad Sci USA. 2016;113(2):274–279. doi: 10.1073/pnas.1512977112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thome JJ, et al. Longterm maintenance of human naive T cells through in situ homeostasis in lymphoid tissue sites. Sci Immunol. 2016;1(6):eaah6506. doi: 10.1126/sciimmunol.aah6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sims JS, et al. Diversity and divergence of the glioma-infiltrating T-cell receptor repertoire. Proc Natl Acad Sci USA. 2016;113(25):E3529–E3537. doi: 10.1073/pnas.1601012113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Amir AL, et al. Allo-HLA reactivity of virus-specific memory T cells is common. Blood. 2010;115(15):3146–3157. doi: 10.1182/blood-2009-07-234906. [DOI] [PubMed] [Google Scholar]
- 27.Guillet M, Sebille F, Soulillou J. TCR usage in naive and committed alloreactive cells: implications for the understanding of TCR biases in transplantation. Curr Opin Immunol. 2001;13(5):566–571. doi: 10.1016/S0952-7915(00)00260-0. [DOI] [PubMed] [Google Scholar]
- 28.Morris GP, Ni PP, Allen PM. Alloreactivity is limited by the endogenous peptide repertoire. Proc Natl Acad Sci USA. 2011;108(9):3695–3700. doi: 10.1073/pnas.1017015108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DeWolf S, Sykes M. Alloimmune T cells in transplantation. J Clin Invest. 2017;127(7):2473–2481. doi: 10.1172/JCI90595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mason D. A very high level of crossreactivity is an essential feature of the T-cell receptor. Immunol Today. 1998;19(9):395–404. doi: 10.1016/S0167-5699(98)01299-7. [DOI] [PubMed] [Google Scholar]
- 31.Reiser JB, et al. CDR3 loop flexibility contributes to the degeneracy of TCR recognition. Nat Immunol. 2003;4(3):241–247. doi: 10.1038/ni891. [DOI] [PubMed] [Google Scholar]
- 32.Archbold JK, et al. Alloreactivity between disparate cognate and allogeneic pMHC-I complexes is the result of highly focused, peptide-dependent structural mimicry. J Biol Chem. 2006;281(45):34324–34332. doi: 10.1074/jbc.M606755200. [DOI] [PubMed] [Google Scholar]
- 33.Colf LA, et al. How a single T cell receptor recognizes both self and foreign MHC. Cell. 2007;129(1):135–146. doi: 10.1016/j.cell.2007.01.048. [DOI] [PubMed] [Google Scholar]
- 34.Gras S, Kjer-Nielsen L, Chen Z, Rossjohn J, McCluskey J. The structural bases of direct T-cell allorecognition: implications for T-cell-mediated transplant rejection. Immunol Cell Biol. 2011;89(3):388–395. doi: 10.1038/icb.2010.150. [DOI] [PubMed] [Google Scholar]
- 35.Curran SA, et al. Nucleotide sequencing of psoriatic arthritis tissue before and during methotrexate administration reveals a complex inflammatory T cell infiltrate with very few clones exhibiting features that suggest they drive the inflammatory process by recognizing autoantigens. J Immunol. 2004;172(3):1935–1944. doi: 10.4049/jimmunol.172.3.1935. [DOI] [PubMed] [Google Scholar]
- 36.Atkins RC, Ford WL. Early cellular events in a systemic graft-vs.-host reaction. I. The migration of responding and nonresponding donor lymphocytes. J Exp Med. 1975;141(3):664–680. doi: 10.1084/jem.141.3.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ford WL, Simmonds SJ, Atkins RC. Early cellular events in a systemic graft-vs.-host reaction. II. Autoradiographic estimates of the frequency of donor lymphocytes which respond to each Ag-B-determined antigenic complex. J Exp Med. 1975;141(3):681–696. doi: 10.1084/jem.141.3.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ryser JE, MacDonald HR. Limiting dilution analysis of alloantigen-reactive T lymphocytes. I. Comparison of precursor frequencies for proliferative and cytolytic responses. J Immunol. 1979;122(5):1691–1696. [PubMed] [Google Scholar]
- 39.Bach F, Hirschhorn K, Schreibman RR, Ripps C. Immunological responses of human lymphocytes in vitro. Ann N Y Acad Sci. 1964;120:299–302. doi: 10.1111/j.1749-6632.1964.tb34728.x. [DOI] [PubMed] [Google Scholar]
- 40.Catalina MD, Sullivan JL, Bak KR, Luzuriaga K. Differential evolution and stability of epitope-specific CD8(+) T cell responses in EBV infection. J Immunol. 2001;167(8):4450–4457. doi: 10.4049/jimmunol.167.8.4450. [DOI] [PubMed] [Google Scholar]
- 41.Khan N, et al. Cytomegalovirus seropositivity drives the CD8 T cell repertoire toward greater clonality in healthy elderly individuals. J Immunol. 2002;169(4):1984–1992. doi: 10.4049/jimmunol.169.4.1984. [DOI] [PubMed] [Google Scholar]
- 42.Torti N, Oxenius A. T cell memory in the context of persistent herpes viral infections. Viruses. 2012;4(7):1116–1143. doi: 10.3390/v4071116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hsu M, et al. TCR sequencing can identify and track glioma-infiltrating T cells after DC vaccination. Cancer Immunol Res. 2016;4(5):412–418. doi: 10.1158/2326-6066.CIR-15-0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robins H. Immunosequencing: applications of immune repertoire deep sequencing. Curr Opin Immunol. 2013;25(5):646–652. doi: 10.1016/j.coi.2013.09.017. [DOI] [PubMed] [Google Scholar]
- 45. West GB, Brown JH, Enquist BJ. The origin of universal scaling laws in biology. In: Brown JH, ed. Scaling in Biology. Oxford, UK: Oxford University Press; 2000:87–112. [Google Scholar]
- 46.Bolkhovskaya OV, Zorin DY, Ivanchenko MV. Assessing T cell clonal size distribution: a non-parametric approach. PLoS ONE. 2014;9(9):e108658. doi: 10.1371/journal.pone.0108658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin J. Divergence measures based on the Shannon entropy. IEEE Trans Inform Theory. 1991;37:145–151. doi: 10.1109/18.61115. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.