

Abstract
Innate DNA repair mechanisms play a critical role in protecting skin keratinocytes from UV mutagenesis and skin cancer development. We hypothesized that individuals who develop frequent skin cancers may harbor germline defects in DNA repair genes and have increased predisposition to internal malignancies. We enrolled 61 patients with unusually frequent basal cell carcinoma (BCC) development, seen at Stanford Hospital and Clinics from January 2005 until December 2015, for germline analysis of 29 DNA repair genes. In parallel, a case-control retrospective review was performed to interrogate the association of malignancies with frequent BCC development in a large US medical insurance claims database (Truven), which included 13,264 individuals with 6 or more BCCs from 2007 to 2011. 19.7% of the frequent BCC cohort harbored pathogenic mutations in DNA repair genes: APC, BARD1, BRCA1, BRCA2, CDH1, CHEK2, MLH1, MSH2, MSH6, MUTYH, NBN, and PALB2. Individuals with 6 or more BCCs had an increased risk of other malignancies, with a 3.5-fold increase in the frequent BCC cohort and a 3.2-fold increase in the Truven database. Individuals who developed frequent BCCs have an increased prevalence of germline mutations in DNA repair genes and increased malignancy risk. Our data implicate frequent BCC development as an external marker of inherited cancer risk.
Keywords: Dermatology
Keywords: Skin cancer
Individuals who develop frequent basal cell cancers have an increased prevalence of mutations in DNA repair genes and increased malignancy risk.
Introduction
One in five Americans will develop cancer, making it the second leading cause of death in the US. About 5–10 percent of cancers are considered hereditary and occur in individuals genetically predisposed to developing cancer (1). Inherited defects in DNA repair genes have been implicated in cancer susceptibility in colon (2), breast (3, 4), and ovarian (5) cancers. Early identification of cancer-susceptible individuals can aid in risk stratification, screening, and prevention. Unfortunately, there are few clinical signs of DNA repair defects, and universal genetic screening is currently cost prohibitive.
The skin is uniquely dependent on the DNA repair machinery for protection against UV-induced carcinogenesis. When this DNA repair mechanism fails, individuals may develop increased numbers of skin cancers. For example, patients with xeroderma pigmentosum harbor a defect in nucleotide excision repair (NER) genes, resulting in exquisite UV sensitivity and development of numerous skin cancers (6). DNA repair mechanisms beyond NER, including base excision repair and mismatch repair (7), have been also implicated in protection against UV mutagenesis, highlighting a complex, interconnected role of DNA repair mechanisms in the skin.
Basal cell carcinoma (BCC) is a relatively benign skin cancer caused by UV exposure, which develops in over 3 million people in the US annually (8–10). We hypothesized that the constant barrage of UV-induced mutagenesis in the skin may uncover early deficiencies in DNA repair and, accordingly, that frequent BCCs could be a clinical marker of underlying defects in DNA repair. To investigate this, we assessed the prevalence of germline mutations in 29 cancer-susceptibility genes in a rare cohort of individuals who developed unusually frequent BCCs, which represented the top 5% of individuals with BCC seen at Stanford Hospital and Clinics. We further verified the association of frequent BCC and other cancer risk in a large national insurance database.
Results
High-frequency BCC cases.
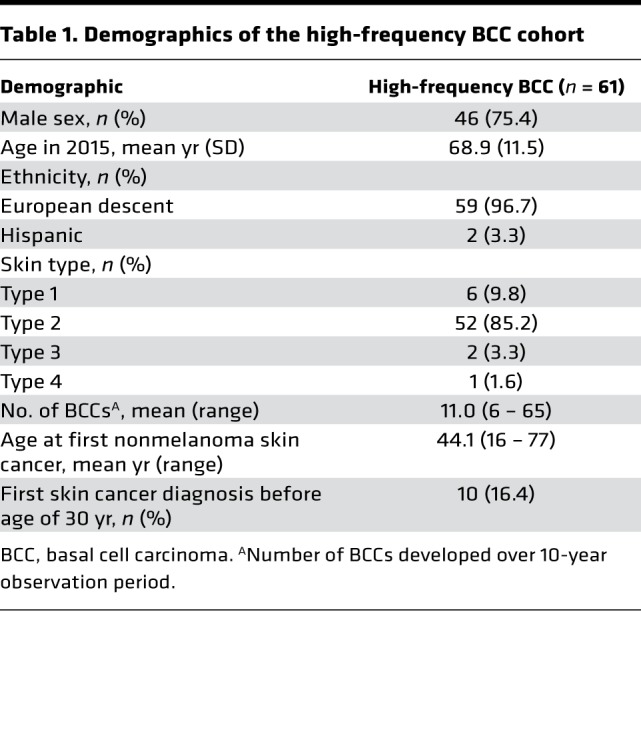
61 (80.3%) of the eligible 76 high-frequency BCC patients were included in the study analysis. The majority of the enrolled cohort was male (n = 46, 75.4%), of European descent (n = 59, 96.7%), and had Fitzpatrick skin type 2 (n = 52, 85.2%) and an average age of 68.9 years in 2016 (Table 1). The number of biopsy-confirmed BCCs during a 10-year observation period ranged from 6 to 65, with an average of 11.0 per patient. The average age at first skin cancer diagnosis was 44.1 years, and 10 (16.4%) subjects developed their first skin cancer before the age of 30 (Table 1).
Table 1. Demographics of the high-frequency BCC cohort.
Germline pathogenic mutations.
Among 61 subjects with high-frequency BCC, a total of 12 (19.7%) subjects harbored 13 reported pathogenic mutations in 12 genes, including APC, BARD1, BRCA1, BRCA2, CDH1, CHEK2, MLH1, MSH2, MSH6, MUTYH, NBN, and PALB2 (Table 2 and Supplemental Figure 2; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.122744DS1). In comparison, 3.0% of non-Finnish European patients in the Exome Aggregation Consortium (ExAC) had pathogenic mutations in these 12 genes (Supplemental Table 3). All 12 genes except APC, CDH1, and MUTYH reached statistically significant enrichment in the high-frequency BCC cohort when compared with the ExAC non-Finnish European population frequency (P < 0.05; Supplemental Table 3). One patient harbored two concurrent pathogenic mutations in BARD1 and MUTYH (Table 2). Among the high-frequency cohort, there was no significant association between the presence of a mutation and BCC frequency (data not shown).
Table 2. Pathogenic mutations identified in the high-frequency BCC cohort.

Cancer risk is increased in the frequent BCC cohort.
21 of 61 (34.4%) patients had a personal history of additional cancers, including 5 invasive melanomas and 5 hematologic, 2 breast, 2 colon, and 5 prostate cancers (Table 3). Compared with the Surveillance, Epidemiology, and End Results–estimated (SEER-estimated) prevalence of any cancer in the 60- to 69-year-old population of European descent, the high-frequency BCC cohort had an increased risk of any cancer with a relative risk (RR) of 3.5 (95% CI: 2.5–4.9) (Table 3). This increased relevant risk was particularly notable for invasive melanoma, potentially due to the additive effect of shared environmental risk factors (RR of 11.9, 95% CI: 5.1–27.6). High-frequency BCC subjects were also more likely to have an increased risk of leukemia and lymphoma, with an RR of 3.5 (95% CI: 1.5–8.0) (P = 0.004); colon cancer, with an RR of 4.5 (95% CI: 1.2–17.7) (P = 0.030), breast cancer, with an RR of 5.6 (95% CI: 1.6–20.5) (P = 0.009), and prostate cancer, with an RR of 4.7 (95% CI: 2.0–10.7) (P < 0.001).
Table 3. Cancer risk in the high-frequency BCC cohort.

Cancer risk in frequent BCC individuals in a large insurance claims data set.
We hypothesized that if, indeed, a subset of individuals with high frequency of BCCs harbored genetic susceptibility to internal cancers, we would be able to detect increased cancer incidence in individuals with numerous BCCs in larger clinical data sets. We therefore investigated the prevalence of internal malignancy using the Truven MarketScan, a large medical insurance claims database, encompassing over 250 million individuals seen in the US annually. The BCC cohort identified in the Truven MarketScan database consisted of 111,562 subjects with 1 BCC, 13,264 subjects with 6 or more BCCs, and 2,920 subjects with 12 or more BCCs. The BCC cohort had an average age of 48.7, 50.1, and 50.0 years at first enrollment, respectively (Supplemental Table 4). The majority of the BCC subjects were between 40 and 59 years of age and were male (Supplemental Table 4). After adjustment for age and sex, subjects with 1, 6 or more, and 12 or more BCCs had an increased risk of any cancer compared with the control cohort, at an odds ratio of 1.61 (95%CI: 1.58–1.65), 3.12 (95%CI: 2.98–3.26), and 4.15 (95%CI: 3.79–4.53), respectively (Table 4 and Supplemental Table 5). The odds ratio for malignancy showed an upward trend with increasing number of BCCs. Consistent with the enrolled cohort, the Truven BCC cohort with 6 or more BCCs and 12 or more BCCs had an increased risk of colon, melanoma, and hematologic malignancies (non-Hodgkin lymphoma, Hodgkin’s lymphoma, leukemia) compared with the control group (Table 4). The comparison of age at first diagnosis with any cancer among the control group and cohort with 1 BCC and 6 or more BCCs highlighted a slight decrease in age of onset of 54.6 years, 53.7 years, and 54.0 years, respectively (P < 0.001) (Supplemental Table 6). The limited decrease in age between each BCC group may be a reflection of the relatively younger age of the Truven cohort and shorter duration of longitudinal follow-up from their enrollment time. These data confirm that individuals who develop numerous BCCs are at increased risk of developing internal malignancies.
Table 4. Increased cancer risk associated with frequent BCC in Truven MarketScan data.

Discussion
Our study identified frequent BCC development as a clinical sign of inherited cancer risk. Individuals who develop frequent BCCs have a higher prevalence of inherited mutations in DNA repair genes and are at increased risk of developing internal organ malignancies.
A total of 12 (19.7%) subjects from the high-frequency BCC cohort carried a pathogenic mutation in 13 cancer-susceptibility genes. This prevalence of inherited mutations is even higher than those reported in individuals with life-threatening malignancies, including metastatic prostate (11) (11.8%), primary ovarian (5) (18%), and stage I–III breast cancer (3, 4) (10.7%, 14.6%). All 12 genes with pathogenic mutations were involved in DNA repair pathway, including base excision repair (MUTYH), mismatch excision repair (MSH1, MSH2, MSH6), homologous recombination (BRCA1, BRCA2, NBN, PALB2), and DNA damage signaling and transduction (APC, BARD1, CDH1, CHEK2) (12). Increased frequency of mutations in the DNA repair pathways may reflect the greater susceptibility of these individuals to the environmental contribution of UV damage. Consistent with this, prior studies have identified polymorphisms in DNA repair genes associated with increased risk of skin cancer development (13), suggesting a broader role for DNA repair in BCC.
Over 85% of individuals in the US develop their first skin cancer after 55 years of age (14). The high-frequency BCC cohort developed their first BCC at an earlier age, averaging 44.1 years. In addition, 16.4% (n = 10) of the high-frequency BCC cohort developed their first skin cancer before the age of 30 compared with 1.3% of general population developing incident cases before age 35 (14). The earlier onset of first skin cancer may suggest a higher contribution from genetic susceptibility in addition to increased environmental risk factors.
The frequent BCC cohort also harbored an increased risk of cutaneous and noncutaneous malignancies. Indeed, 34.4% had a history of additional cancers. A similar enrichment was observed in a large independent US claims data set (Truven), which included 13,264 registered individuals with 6 or more BCCs. The increased malignancy risk was observed in melanoma, breast, colon, and prostate cancer. The highest increase in risk was observed in other skin cancers, specifically melanoma and squamous cell carcinoma, likely due to shared UV exposure; this is consistent with previous reports of high coincidences between melanoma and keratinocyte carcinoma (15). While previous studies have not investigated malignancy risk in individuals who develop frequent BCCs, one longitudinal study of 190,000 individuals in Denmark identified an increased risk of internal malignancy after the diagnosis of one or more BCC (16). Interestingly, patients receiving a diagnosis of BCC at a younger age had higher risk for developing subsequent internal organ cancers, including breast cancer and lymphoma, highlighting a potential shared underlying genetic susceptibility for BCC and internal malignancy.
Our findings implicate frequent BCC development as an external clinical marker of DNA repair deficiency and cancer susceptibility. Similar to cancer-prone phenotypes in other malignancies (12), the frequent BCC population carries increased germline mutations for internal malignancy. The study has two key limitations. First, the genetic analysis is limited by small sample size of frequent BCC cohort (n = 61) in a single institution. Second, claims databases such as Truven MarketScan highlight the association between frequent BCC and malignancy but lack detailed clinical and demographic data, such as race and genetic testing results. Further studies are needed to determine if our findings are generalizable in independent cohorts.
Given the overall banal nature of BCC and the earlier onset of disease compared with internal malignancies, frequent BCCs may serve as a low-risk clinical marker to identify a subset of individuals at highest risk of internal malignancies. These patients may benefit from clinical screening for family history of malignancy and multigene cancer-susceptibility panel testing. However, a better understanding of how multigene panel testing can be incorporated in clinical care is warranted before a widespread implementation. In addition, a further characterization of gene-environment interaction in patients with frequent BCCs is needed to identify risk factors that may be independently associated with increased malignancy.
Methods
Study population recruitment.
In a retrospective cohort study using pathology medical records, we identified 2,407 subjects with new biopsy-confirmed BCC seen at Stanford Hospital and Clinics between 2005 and 2015. Of the 2,407 subjects, 65.6% (n = 1,578) had a single BCC, 29.6% (n = 712) had 2 to 5 BCCs, 4.8% (n = 117) had 6 or more BCCs (Figure 1A). We defined frequent BCC development as 6 or more BCCs in the 10-year time frame, representing the top 5% of the total patients with BCC and termed this cohort the high-frequency BCC cohort (Figure 1B). Basal cell nevus syndrome patients were excluded from analysis based on chart review. Of the 76 patients eligible for study recruitment, 63 patients enrolled, and saliva was collected for sequencing. After excluding two subjects with positive PTCH1 mutations, a final cohort of 61 subjects was included for study analysis (Supplemental Figures 1 and 3).
Figure 1. Distribution of individuals with multiple BCCs at Stanford Hospital and Clinics.

(A) Waterfall plot depicting the distribution of individuals by BCC number ordered by increasing number of BCC count from 2005 to 2015 (range 1–65 BCCs). The cohort consists of 2,407 individuals, with the top 5% having 6 or more BCCs. (B) Subject with history of multiple BCCs showing 6 biopsy-confirmed BCCs on the back.
Sequencing assay.
2 ml of saliva sample was collected from each enrolled subject using the OGR-500 collection kit (DNA Genotek). Germline DNA was extracted and subjected to targeted capture of all coding sequences using the Fulgent Genetics Focus Cancer panel array (Fulgent Diagnostics). 29 genes were analyzed for mutations, including APC, ATM, AXIN2, BARD1, BMPR1A, BRCA1, BRCA2, BRIP1, CDH1, CHEK2, EPCAM, HOXB13, MLH1, MRE11, MSH2, MSH6, MUTYH, NBN, PALB2, PMS2, POLD1, POLE, PTEN, RAD50, RAD51C, RAD51D, SMAD4, STK11, and TP53. The panel included 22 genes involved in DNA repair (17) (Supplemental Table 1). The mean coverage across all genes was 53.8×. Identified pathogenic mutations were confirmed by Sanger sequencing.
Bioinformatics analysis and variant classification.
Paired end reads were aligned to the hg19 reference human genome using BWA-MEM (18) (version 0.7.15; http://bio-bwa.sourceforge.net/), and duplicates were marked with Picard (version 2.4.1; http://broadinstitute.github.io/picard/). Germline variations were called using GATK HaplotypeCaller (19) according to the Best Practice guideline (20) (version 3.7; https://broadinstitute.org/gatk). Annotations were performed using ANNOVAR (21) (version 2015 Jun17). Population allele frequencies were obtained from ExAC (22) (version 0.3.1, March 14, 2016; http://exac.broadinstitute.org/), with the reference population being of non-Finnish European descent. Pathogenic mutations were determined according to American College of Medical Genetics and Genomics and Association for Molecular Pathology consensus criteria guidelines using ClinVar (23) (version June 29 2015; http://www.ncbi.nlm.nih.gov/clinvar/) and HGMD Professional (24) (version 2016.4; http://www.hgmd.cf.ac.uk/). Variants in DNA mismatch repair genes were evaluated using the variant classification determined by the International Society of Gastrointestinal Hereditary Tumors (InSiGHT; http://www.insight-group.org/), and BRCA1 and BRCA2 variants were similarly evaluated using Breast Cancer Information Core (https://research.nhgri.nih.gov/bic/). The variants were additionally filtered for less than 1% population frequency to exclude common polymorphisms with low penetrance.
Truven MarketScan data set selection.
The Truven Health MarketScan database represents claims data from approximately 100 commercial plans, representing over 250 million individuals in all US census regions from 2007 onward. The study met the criteria for Institutional Review Board human subjects determination waiver, as no identifiable protected health information is contained in the data set. Subjects ages 18 years or older with a diagnosis of BCC between 2007 and 2015 were identified. A diagnosis of each BCC event was based on a combination of ICD-9 CM codes and CPT codes associated with biopsy, destruction, excision, or Mohs (Supplemental Table 2). All included subjects were enrolled for a minimum of 3 years between 2012 and 2015. Prior history of skin cancer was identified by similar combination of ICD-9 CM codes and CPT codes from 2007 to 2011. Subjects with 1–5 BCCs had no history of prior skin cancer between 2007 and 2011, while subjects with 6 or more BCCs were allowed to have a history of prior skin cancer. We selected 5 age- and sex-matched controls for each BCC case. Solid-organ malignancies, including breast, lung, stomach, colon, pancreatic, liver, bladder, renal/urinary, prostate, cancer, uterine, ovarian, and hematologic and other cutaneous cancers, were also determined from ICD-9 codes (Supplemental Table 2).
Statistics.
Demographic characteristics were summarized with descriptive statistics, including mean, range, and SDs. Comparisons between each BCC groups (0, 1, and ≤6 BCCs) were made using the χ2 test (categorical variables) and the 2-tailed Student’s t test (continuous variables). A P value less than 0.05 was considered significant. Logistic regression was applied to estimate the associations between BCC groups and controls for each cancer for Truven data set analyses. RR, its standard error, and 95% confidence intervals were calculated according to Altman (25). All analyses were performed using SAS Institute Inc. (version 9.4). Data analyses were performed by HGC and SL.
Study approval.
The present study in humans was reviewed and approved by the Stanford Institutional Review Board. Subjects provided informed consent prior to their participation in the study, and written consent was received from participants prior to inclusion in the study (IRB-36263). Written informed consent was provided for use of the patient photo that appears in the manuscript.
Author contributions
KYS designed research. HGC, KYK, IB, SA, ALSC, AEO, EE, JYT, and KYS acquired data. HGC, KYK, SL, JYT, and KYS analyzed data. HGC and KYS wrote the manuscript.
Supplementary Material
Acknowledgments
This study was supported by PellePharm Inc. (to KYS); the Dermatology Foundation (to KYS); the Stanford Society of Physician Scholars (to KYK and HGC); the American Skin Association (to HGC and KYS); and the National Cancer Institute and National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under awards K23 CA211793 (to KYS) and AR0AR46786 (to AEO). This work was presented at the Society for Investigative Dermatology meeting in Portland, Oregon, USA, on April 28, 2017. Portions of this work have appeared in abstract form (26).
Version 1. 08/09/2018
Electronic publication
Footnotes
Conflict of interest: JYT is a cofounder and director of PellePharm.
Reference information: JCI Insight. 2018;3(15):e122744. https://doi.org/10.1172/jci.insight.122744.
Contributor Information
Karen Y. Kuo, Email: karenyan@stanford.edu.
Shufeng Li, Email: shufeng@stanford.edu.
Irene Bailey, Email: baileyhi@stanford.edu.
Anthony E. Oro, Email: oro@stanford.edu.
Kavita Y. Sarin, Email: ksarin@stanford.edu.
References
- 1. Hereditary Cancer Syndromes. National Cancer Institute. https://www.cancer.gov/about-cancer/causes-prevention/genetics Accessed August 4, 2018.
- 2.Pearlman R, et al. Prevalence and spectrum of germline cancer susceptibility gene mutations among patients with early-onset colorectal cancer. JAMA Oncol. 2017;3(4):464–471. doi: 10.1001/jamaoncol.2016.5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Couch FJ, et al. Inherited mutations in 17 breast cancer susceptibility genes among a large triple-negative breast cancer cohort unselected for family history of breast cancer. J Clin Oncol. 2015;33(4):304–311. doi: 10.1200/JCO.2014.57.1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tung N, et al. Frequency of germline mutations in 25 cancer susceptibility genes in a sequential series of patients with breast cancer. J Clin Oncol. 2016;34(13):1460–1468. doi: 10.1200/JCO.2015.65.0747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Norquist BM, et al. Inherited mutations in women with ovarian carcinoma. JAMA Oncol. 2016;2(4):482–490. doi: 10.1001/jamaoncol.2015.5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kraemer KH, DiGiovanna JJ. Xeroderma Pigmentosum. In: Adam MP, et al. eds. GeneReviews (R). Seattle, Washington, USA: University of Washington; 1993. https://www.ncbi.nlm.nih.gov/books/NBK1116/ Accessed July 30, 2018.
- 7.Young LC, Hays JB, Tron VA, Andrew SE. DNA mismatch repair proteins: potential guardians against genomic instability and tumorigenesis induced by ultraviolet photoproducts. J Invest Dermatol. 2003;121(3):435–440. doi: 10.1046/j.1523-1747.2003.12450.x. [DOI] [PubMed] [Google Scholar]
- 8.van Dam RM, et al. Risk factors for basal cell carcinoma of the skin in men: results from the health professionals follow-up study. Am J Epidemiol. 1999;150(5):459–468. doi: 10.1093/oxfordjournals.aje.a010034. [DOI] [PubMed] [Google Scholar]
- 9.Chuang TY, Popescu A, Su WP, Chute CG. Basal cell carcinoma. A population-based incidence study in Rochester, Minnesota. J Am Acad Dermatol. 1990;22(3):413–417. doi: 10.1016/0190-9622(90)70056-N. [DOI] [PubMed] [Google Scholar]
- 10.Rogers HW, et al. Incidence estimate of nonmelanoma skin cancer in the United States, 2006. Arch Dermatol. 2010;146(3):283–287. doi: 10.1001/archdermatol.2010.19. [DOI] [PubMed] [Google Scholar]
- 11.Pritchard CC, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med. 2016;375(5):443–453. doi: 10.1056/NEJMoa1603144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ruczinski I, et al. A population-based study of DNA repair gene variants in relation to non-melanoma skin cancer as a marker of a cancer-prone phenotype. Carcinogenesis. 2012;33(9):1692–1698. doi: 10.1093/carcin/bgs170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin Y, et al. Association study of genetic variation in DNA repair pathway genes and risk of basal cell carcinoma. Int J Cancer. 2017;141(5):952–957. doi: 10.1002/ijc.30786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dacosta Byfield S, Chen D, Yim YM, Reyes C. Age distribution of patients with advanced non-melanoma skin cancer in the United States. Arch Dermatol Res. 2013;305(9):845–850. doi: 10.1007/s00403-013-1357-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu S, Cho E, Li WQ, Qureshi AA. History of keratinocyte carcinoma and risk of melanoma: a prospective cohort study. J Natl Cancer Inst. 2017;109(4):djw268. doi: 10.1093/jnci/djw268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frisch M, Hjalgrim H, Olsen JH, Melbye M. Risk for subsequent cancer after diagnosis of basal-cell carcinoma. A population-based, epidemiologic study. Ann Intern Med. 1996;125(10):815–821. doi: 10.7326/0003-4819-125-10-199611150-00005. [DOI] [PubMed] [Google Scholar]
- 17.Wood RD, Mitchell M, Sgouros J, Lindahl T. Human DNA repair genes. Science. 2001;291(5507):1284–1289. doi: 10.1126/science.1056154. [DOI] [PubMed] [Google Scholar]
- 18. Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM [q-bioGN]. Cornell University Library. https://arxiv.org/abs/1303.3997 Accessed July 24, 2018.
- 19.McKenna A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van der Auwera GA, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:11.10.1–11.1033. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lek M, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Landrum MJ, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42(Database issue):D980–D985. doi: 10.1093/nar/gkt1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Richards S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Altman D. Practical statistics for medical research. London, UK: Chapman and Hall; 1991. [Google Scholar]
- 26.Cho H, et al. Frequent basal cell cancer development is a clinical marker for inherited cancer susceptibility. J Invest Dermatol. 2018;138(5):S28. doi: 10.1172/jci.insight.122744. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.