

Abstract
Antagonists of peripheral type 1 cannabinoid receptors (CB1) may have utility in the treatment of obesity, liver disease, metabolic syndrome and dyslipidemias. We have targeted the purine otenabant (1) analogues for this purpose. The non-tissue selective CB1 antagonist rimonabant (2) was approved as a weight-loss agent in Europe but produced centrally mediated adverse effects in some patients including dysphoria and suicidal ideation leading to its withdrawal. Efforts are now underway to produce compounds with limited brain exposure. While many structure-activity relationship (SAR) studies of 2 have been reported, along with peripheralized compounds, 1 remains relatively less studied. In this report, we pursued analogues of 1 in which the 4-aminopiperidine group was switched to piperazine group to enable a better understanding of SAR to eventually produce compounds with limited brain penetration. To access a binding pocket and modulate physical properties, the piperazine was functionalized with alkyl, heteroalkyl, aryl and heteroaryl groups using a variety of connectors, including amides, sulfonamides, carbamates and ureas. These studies resulted in compounds that are potent antagonists of hCB1 with high selectivity for hCB1 over hCB2. The SAR obtained led to the discovery of 65 (Ki = 4 nM, >10,000-fold selective for hCB1 over hCB2), an orally bioavailable aryl urea with reduced brain penetration, and provides direction for discovering peripherally restricted compounds with good in vitro and in vivo properties.
Keywords: CB1, cannabinoid, peripheral, antagonist, otenabant, purine, CB2, MDCK, blood brain barrier, ADME
Graphical Abstract
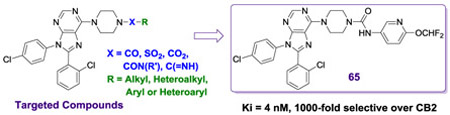
1. INTRODUCTION
Selective modulation of the endocannabinoid (EC) system, comprised of endocannabinoids, receptors, transporters and enzymes, has potential therapeutic applications in a broad range of medical conditions.[1, 2] Cannabinoid receptors CB1 and CB2 are G protein-coupled receptors (GPCRs) whose primary function is to activate G proteins (Gi/o). CB1 receptors are present throughout the body and are highly expressed in the central nervous system (CNS). Selective attenuation of CB1 receptor activation is a validated approach to developing treatments for obesity, diabetes, metabolic syndrome, dyslipidemias and liver diseases. [3, 4]
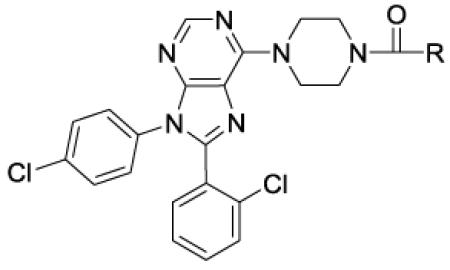
Rimonabant (2, Figure 1), a CNS penetrating CB1 receptor inverse agonist developed by Sanofi-Aventis, was clinically approved for the treatment of obesity in Europe, but had to be withdrawn because of an increase in psychiatric events associated with antagonism of CB1 receptors in the CNS.[5] The adverse side effects seen with 2 precipitated the withdrawal of other CNS-penetrating CB1 receptor antagonists from clinical development, including 1, a compound developed by Pfizer. To avoid side effects associated with CNS activity, efforts are underway to develop compounds that selectively antagonize the CB1 receptors in the periphery.[6, 7] Furthest along among these is TM38837, an analogue of 2, which was tested in humans and demonstrated to have limited brain penetration.[8] Alternative approaches include development of neutral orthosteric antagonists and negative allosteric modulators.[9-11] Recent studies have questioned the usefulness of peripherally restricted CB1 antagonists as pure weight-loss agents.[12-14] However, other studies also indicate that peripherally expressed CB1 receptors may be targeted to treat severe diseases including diabetes and steatohepatitis of the liver.[13, 15] While a peripherally restricted CB1 antagonist with no brain penetration would be considered ideal for such applications, other CB1 antagonists that have limited brain penetration might be also useful for serious disorders with appropriate behavioral monitoring. Analogues of 2 have been extensively explored to date but SAR studies of compounds based on other CB1 scaffolds including the purine 1 have not been extensively reported.[11] Our group has previously reported compounds based on 1 that have limited brain exposure but additional studies are necessary to explore SAR and produce better drug-like molecules.[16, 17] In particular, compound 1 has many characteristics that would enable efficient peripheralization including a topological polar surface area (TPSA) of 102 Å, 3 hydrogen bond donors, and a molecular weight of >500 Da.[18] Studies indicate that intra-molecular H-bonding between the primary amide and the ethyl amine portion of 1 (observed in the X-ray structure) effectively lowers the polarity allowing for penetration into the CNS.[18]
Figure 1.

Examples of clinically tested CB1 antagonists
Identifying a drug-like compound that is both peripherally selective and orally bioavailable is challenging. One approach involves designing compounds with one or more H-bond donors in combination with a polar group and high molecular weight. Analysis of clinical compounds shows that a TPSA of 80-140 Å and a MW of 450-600 Da improve the likelihood of getting the desired profile.[19] Furthermore, incorporation of a mildly basic group into the structure can enhance oral bioavailability by improving solubility in acidic media, such as that of the stomach. Using these strategies, we recently reported the discovery of peripherally selective analogues of 1 and 2 (Figure 2).[17, 20] Compounds related to 1 are particularly attractive because the mildly basic 6-piperidinyl purine core can aid in oral absorption. Our initial efforts to make analogues of 1 with the desired profile resulted in compound 4.[17] This compound has excellent hCB1 potency and good selectivity over hCB2. Although compound 4 is peripherally selective and orally bioavailable, further improvements are needed in both these areas, along with a better understanding of the SAR.
Figure 2.

Peripherally selective CB1 antagonists
Compounds 1 and 4, both contain a 4,4-disubstituted piperidine in the 6-position of the purine core. It was reported that when the much smaller N-methylpiperazine group is in the six position (5 and 6, Figure 3), the compounds are still highly active in a hCB1 binding assay (Ki = 3 nM for 5).[18] Compound 5 has good human liver microsome stability and good solubility in acidic media. Compound 6, a close analogue of compound 5, was reported to have some peripheral selectivity, despite having no hydrogen bond donating or other polar groups. The potential advantages of developing smaller and less lipophilic compounds by replacing the 4-phenylpiperidine group of 4 with a piperazine group attracted our attention. We desired to investigate molecules in which the methyl piperazine is replaced with a piperazine that is functionalized with alkyl, heteroalkyl, aryl or heteroaryl groups connected via an amide, sulfonamide, carbamate, urea, amidine or guanidine (Figure 3). These compounds are reported herein and formed the basis of a thorough SAR understanding which will aid in the design of peripherally restricted hCB1 antagonists.
Figure 3.

Targeted piperazine analogues of 1
Recent crystal structures of hCB1 and docking studies with 1 indicate that such groups would likely be directed toward a binding pocket near the extracellular surface of the membrane that may accommodate polar as well as nonpolar groups.[21, 22] Crystal structures of hCB1 show that the orthosteric binding site is near to the top of the seven transmembrane complex, just below the extracellular membrane. Docking of 2 and 1 into the orthosteric hCB1 binding site showed that the two phenyl rings are in lipophilic pockets in the lower part of the binding site, while the amide is pointed upward to an access channel leading to the extracellular space. A binding pocket leading to the access channel contains polar regions (including residues D104, S123 & H178) and differs significantly from hCB2. By targeting this more polar region near the access channel, we believe it is possible to identify compounds with better physical properties that have good hCB1 potency and excellent selectivity versus hCB2.
2. RESULTS AND DISCUSSION
2.1. Compound synthesis
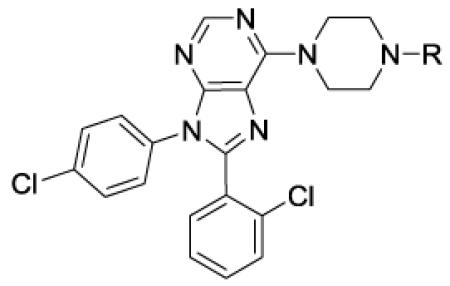
To prepare the targeted compounds, we used the piperazine 8 as a key intermediate. This compound was prepared as shown in Scheme 1. The chloride 7 was prepared as previously reported[18] and then converted to the piperazine 8 in two steps via a nucleophilic reaction with Boc-piperazine, followed by an acid mediated deprotection.
Scheme 1.

Reagents and conditions: (a) BocPiperazine, K2CO3, NMP, 80 °C, 15 h; (b) 6 N HCl, EtOH, 50 °C, 4 h.
Compound 8 was used to prepare sulfonamides, amides, carbamates, ureas, an amidine and a guanidine following the provided procedures, as shown in Scheme 2.
Scheme 2. Reagents and conditions.

(a) RC(=NH)OMe·HCl, NMe3, EtOH, reflux; (b) RNH(C=NH)SMe·HI, i-PrOH, reflux; (c) RSO2Cl, NEt3, DMAP, CH2Cl2, rt; (d) RO2CCl, NEt3, DCE, rt; (e) (1) p-F-PhO2CCl, NEt3, DCE, rt; (2) ROH, NaHMDS, THF, rt; (f) RCOCl, NEt3, DMAP, CH2Cl2, rt; (g) RCO2H, BOP, NEt3, THF, rt; (h) RNCO, THF, rt; (i) (1) p-F-PhO2CCl, NEt3, DCE, rt; (2) RR’NH, NaHMDS, THF, rt.
2.2. In vitro characterization of compounds
All target compounds were evaluated in a FLIPR-based calcium mobilization assay for hCB1 activity (Tables 1-3) as has been described in our previous publications.[17, 20] In general, those that demonstrated apparent antagonist dissociation equilibrium constant (Ke) < 50 nM were further tested for affinity to hCB receptors using radioligand displacement of [3H]CP55940 in purified membrane fractions overexpressing either hCB1 or hCB2. A selection of potent and selective compounds was further tested for potential peripheral selectivity by calculating % apical (A) to basal (B) permeability in a MDCK-mdr1 monolayer permeability assay, which is predictive of brain penetration. [23]
Table 1.
In vitro data for CB1 antagonists – sulfonamides, carbamates & imines
| ||||||
|---|---|---|---|---|---|---|
| # | R | Ke hCB1 (nM) |
Ki hCB1 (nM)a |
Ki hCB2 (nM)a |
Selectivity Ki CB2/CB1 |
MDCK-mdr1 A to B (%)b |
| 2 | Otenabantc | 0.2 | 0.7 | 7700 | 11000 | |
| 5 | Me | 120 | 3d | |||
| 9 | MeSO2 | 3 | 24 | 1700 | 68 | 14 |
| 10 | EtSO2 | 17 | 3 | 980 | 330 | 18 |
| 11 | i-PrSO2 | 1 | 1.4 | 980 | 700 | 11 |
| 12 | c-PrSO2 | 17 | 5 | 2300 | 460 | 4 |
| 13 | n-PrSO2 | 2 | 1.2 | 850 | 710 | 6 |
| 14 | CF3CH2CH2SO2 | 15 | 1.2 | 500 | 420 | |
| 15 | i-BuSO2 | 0.5 | 2 | 2800 | 1400 | 0 |
| 16 | n-BuSO2 | 0.9 | 0.6 | 500 | 830 | 5 |
| 17 | MeOCH2CH2SO2 | 50 | ||||
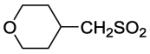
| 18 | c-HexCH2SO2 | 0.6 | 1.7 | 91 | 54 | |
| 19 |
|
4 | 3 | 3000 | 1000 | 9 |
| 20 | MeO2C | 98 | ||||
| 21 | t-BuO2C | 4 | 5 | 3400 | 680 | 3 |
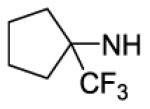
| 22 | MeOCH2CH2O2C | 54 | ||||
| 23 | c-BuO2C | 10 | 3 | 3400 | 1100 | |
| 24 |
|
95 | ||||
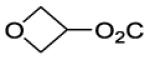
| 25 |
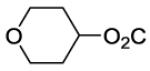
|
105 | ||||
| 26 | c-BuCH2O2C | 12 | 3 | 4600 | 1500 | 0 |
| 27 | 4-F-Ph(HN=)C | 4000 | ||||
| 28 | 4-F-PhNH(HN=)C | 510 | 0.4 | |||
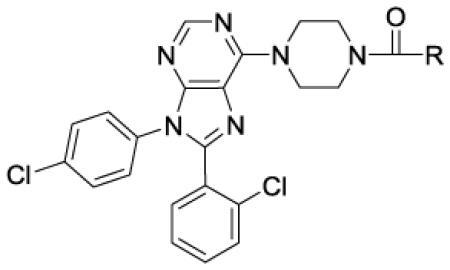
Table 3.
In vitro data for CB1 antagonists - ureas
| ||||||
|---|---|---|---|---|---|---|
| # | R | Ke hCB1 (nM) |
Ki hCB1 (nM)a |
Ki hCB2 (nM)a |
Selectivity Ki CB2/CB1 |
MDCK-mdr1 A to B (%)b |
| 48 |
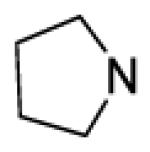
|
66 | 13 | |||
| 49 |
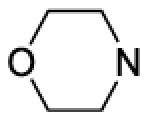
|
670 | ||||
| 50 | i-BuNH | 100 | ||||
| 51 | c-PenNH | 54 | 5 | 740 | 150 | 3 |
| 52 |
|
17 | 24 | 1300 | 54 | |
| 53 | c-HexNH | 41 | 1.4 | 620 | 440 | |
| 54 |
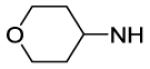
|
2200 | ||||
| 55 |
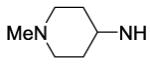
|
4900 | ||||
| 56 | c-HexCH2NH | 35 | 43 | 740 | 17 | |
| 57 |
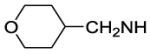
|
5900 | ||||
| 58 |
|
3200 | ||||
| 59 | PhNH | 3 | 6 | 2400 | 400 | 1.0 |
| 60 |
|
8 | 6 | 7100 | 1200 | 1.2 |
| 61 |
|
2 | 6 | 1300 | 220 | 5.0 |
| 62 |
|
10 | 5 | 3200 | 640 | 1.4 |
| 63 |
|
5 | 5 | 4200 | 840 | 0 |
| 64 |
|
2 | 19 | 2000 | 110 | 0 |
| 65 |
|
13 | 4 | >10000 | >1000 | 1.1 |
| 66 |
|
150 | ||||
| 67 |
|
1000 | ||||
| 68 |
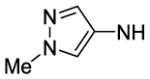
|
1200 | ||||
| 69 |
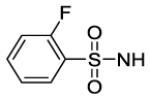
|
420 | 250 | |||
Displacement was measured using [3H]CP55940 in CHO cell membrane preparations overexpressing hCB1 or hCB2 receptors.
% transported from the apical side (A) to the basal side (B).
In Table 1 is presented in vitro data for a set of sulfonamides, carbamates, an amidine and a guanidine. For comparison purposes, the methyl piperazine 5 is included, which has a Ke = 120 nM in the calcium hCB1 assay. In general, the sulfonamide analogues (compounds 9-19) were potent antagonists of hCB1. The methyl sulfonamide 9 has a much improved Ke of 3 nM, compared to 5. The CB2/CB1 selectivity of the methyl sulfonamide 9 in the binding assay was ~68-fold, but larger alkyl groups generally provided better selectivity while maintaining high potency in the calcium hCB1 assay (10-19). The isobutyl sulfonamide 15 is among our most potent and selective compounds, with Ke = 0.5 nM, Ki = 2 nM and CB2/CB1 selectivity of ~1400. Use of ethers to increase TPSA and decrease cLogP resulted in sulfonamides with high selectivity and good, but reduced potency (see compounds 17 vs 16 and 19 vs 18). As with the sulfonamides, alkyl carbamates with good potency and selectivity were also discovered (see compounds 23 and 26), but they are generally less active than the sulfonamides. For example, the methyl sulfonamide 9 has Ke = 3 nM, while the methyl carbamate 20 has Ke = 98 nM). The more polar and basic amidine 27 and guanidine 28 were significantly less active. Several of these compounds were binned by structural features and select compounds were tested in the MDCK-mdr1 assay to predict CNS-permeability. The sulfonamide 15 (TPSA = 84, cLogP = 6.0) and the carbamate 26 (TPSA = 76, cLogP = 6.1), are predicted to be highly peripherally restricted (MDCK-mdr1 < 1%), while other similar and more polar compounds much less so. These mixed results indicated that CNS penetration could be a problem. It is interesting to note that the more polar and basic guanidine 28 (TPSA = 86 Å, cLogP = 7.2) also showed limited permeability (MDCK-mdr1 transport < 1%). Although this compound still has a high cLogP, its basic nature may help mitigate issues such as low solubility that are often associated with nonpolar compounds.
Amides were also targeted as demonstrated in Table 2 and provided some potent and selective antagonists as well. While the methyl amide 29 is similar in potency to the methyl piperazine 5, the sterically similar but electron withdrawing trifluoromethyl amide 30 is over 100-fold more potent in the calcium hCB1 assay (Ke = 0.4 nM). In the MDCK-mdr1 assay, however, 30 performed poorly, indicating that it would readily penetrate the blood-brain barrier (BBB). Larger alkyl amides such as the n-butyl, neopentyl and c-pentylmethyl analogues (31, 35 and 41 respectively) provided exceptionally potent compounds (Ke < 1nM). Of note, the c-pentylmethyl amide 41 is both exceptionally potent (Ke = 0.1 nM, Ki = 0.4 nM, respectively) and selective for hCB1 versus hCB2. As with the sulfonamides, attempts to increase the TPSA and decrease cLogP by use of ethers resulted in compounds of adequate, but reduced potency. For example, the tetrahydrofuran version of 41, compound 42, is still quite potent in the hCB1 binding assay (Ki = 8 nM), but 20-fold less active than 41. More dramatic increases in polarity via the use of an alcohol (see compound 36) or a sulfonyl (see compound 34) resulted in over 1000-fold losses of activity in the calcium hCB1 assay. Good potency can be achieved with a nonpolar basic group, as shown with the piperidin-1-ylmethyl analogue 44 (Ke = 34 nM). A small number of amides were checked in the MDCK-mdr1 assay to predict BBB penetration. Amides 31 (TPSA = 67 Å, cLogP = 6.8) and 40 (TPSA = 86 Å, cLogP = 6.0) are predicted to have low CNS penetration.
Table 2.
In vitro data for CB1 antagonists - amides
| ||||||
|---|---|---|---|---|---|---|
| # | R | Ke hCB1 (nM) |
Ki hCB1 (nM)a |
Ki hCB2 (nM)a |
Selectivity Ki CB2/CB1 |
MDCK-mdr1 A to B (%)b |
| 29 | Me | 120 | ||||
| 30 | CF3 | 0.4 | 1 | 130 | 130 | 500 |
| 31 | n-Bu | 0.6 | 6 | 400 | 67 | 0 |
| 32 | MeOCH2CH2 | 72 | ||||
| 33 | MeCH2OCH2 | 20 | 11 | >10000 | >1000 | |
| 34 | MeSO2CH2CH2 | 3200 | ||||
| 35 | t-BuCH2 | 0.14 | 2 | 5000 | >2500 | |
| 36 |
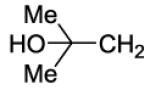
|
200 | ||||
| 37 |
|
410 | ||||
| 38 |
|
2 | 2 | 530 | 265 | |
| 39 |
|
1800 | ||||
| 40 |
|
6 | 5 | 610 | 122 | 0 |
| 41 | c-PenCH2 | 0.1 | 0.4 | >10000 | >25000 | |
| 42 |
|
19 | 8 | 3000 | 375 | |
| 43 |
|
36 | ||||
| 44 |
|
34 | 21 | 5000 | 238 | 5 |
| 45 |
|
370 | ||||
| 46 |
|
740 | ||||
| 47 | Ph(Me)CH | 1.5 | 3 | 560 | 190 | |
Displacement was measured using [3H]CP55940 in CHO cell membrane preparations overexpressing hCB1 or hCB2 receptors.
% transported from the apical side (A) to the basal side (B).
In Table 3 the in vitro data for a set of alkyl and aryl ureas are presented. The alkyl ureas are significantly less potent than the alkyl sulfonamides, amides and carbamates. For example, the i-butyl urea 50 is less potent than the i-butyl sulfonamide 15 (100 nM vs 0.5 nM). Ureas with larger alkyl groups are generally more active and have good selectivity versus hCB2. The cyclohexyl urea 53, for example, has a Ke = 41 nM in the calcium assay, a Ki = 1.4 nM in the binding assay and a hCB2/hCB1 of 440. Increasing the polarity of the large alkyl ureas by employment of ethers or amines (see example 53 vs 54 and 55, respectively) resulted in about a 100-fold loss in activity in the calcium hCB1 assay, making it difficult to attain drug like properties.
Aryl ureas proved to be generally more active, selective and substitution friendly than the alkyl ureas. The phenyl urea 59 (TPSA = 79 Å, cLogP = 6.5), for example, is a potent and selective compound. The 2-pyridyl analogue 60 (TPSA = 92 Å, cLogP = 5.9) is of similar potency, even with an increase of TPSA from 79 to 92 Å. The 6-(difluoromethoxy)pyridin-3-yl analogue 65 has an even higher TPSA of 101 Å and still has good hCB1 potency and exceptional selectivity versus hCB2. Compound 65 and several other aryl ureas were tested in the MDCK-mdr1 assay and generally found to have reduced transport from apical to basal side. A small number of 5-membered heterocyclic analogues were tested and found to have less than satisfactory potency in the calcium hCB1 assay. Finally, the sulfonylurea 69 has a Ki = 250 nM in the hCB1 binding assay. Although this compound is not as potent as desired, it is interesting to note that sulfonyl ureas are acidic and are less likely to cross the BBB. For this reason, the development of more potent sulfonyl ureas is attractive. This avenue will be pursued in future endeavors.
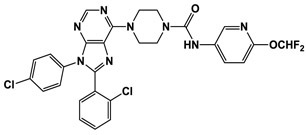
Purine based hCB1 antagonists such as otenabant (1) may be inverse agonists or neutral antagonists. Compound 65, having an excellent in vitro profile and a high TPSA value, was further characterized to establish whether this compound is a neutral antagonist or an inverse agonist of the hCB1 receptor using the calcium mobilization assay. Reference compounds 1 and 2 are known inverse agonists. As indicated in Figure 4, both compounds are inverse agonists of hCB1. Basal signaling through hCB1 was suppressed with increasing concentration of each compound. The EC50’s of 65 and 2 were 590 nM and 586 nM, respectively. Thus, this compound is roughly equipotent to 2 as an inverse agonist of hCB1.
Figure 4. Compound 65 is an inverse agonist of hCB1.

CHO-hCB1 cells were loaded with calcium indicator dye for 60 minutes as described in the “Experimental” section. Cells were then stimulated by various concentrations of 2 as control and 65 and the fluorescence change recorded using a FLIPR Tera (Molecular Devices) instrument. Vehicle only elicited no response. Data is reported as a Mean ± SEM from 3 independent measurements.
2.3. Pharmacokinetic studies
A selection of compounds with the best in vitro results, including low to no permeability in the MDCK-mdr1 transport assay and diverse substituents were tested in rodents to assess their ability to be orally absorbed and kept out of the brain. In general, more polar compounds (higher TPSA values) were favored. Initial pharmacokinetic (pk) studies entailed cassette dosing of rats or mice and measuring the brain and plasma levels at 2 hours after dosing. Subsequently, some compounds were cassette dosed to rats or mice (future efficacy studies in both species were envisioned) in a multiple time point study, thus obtaining plasma and brain maximum concentrations, which were used to calculate and compare brain/plasma levels. In total, 8 different compounds were tested. The main goals were to achieve good plasma levels (>100 ng/mL after an oral dose of 2.5 mg/kg) while keeping the brain to plasma ratio as low as possible, preferably less than 0.05 using the maximum concentrations in the brain and plasma.
In cassette dosing pk experiments, we noted a trend toward better peripheral selectivity with more polar compounds. Of the compounds tested, the dihydrobenzodioxinyl amide 40 and the aryl urea 65, were the most peripherally selective and both have high TPSA values (86 & 101 Å, respectively). In mice, the aryl urea 65 showed the best overall profile. After oral dosing to mice at 2.5 mg/kg in a multiple time point study, good plasma half-life and maximum concentration were observed (6.1 hours and 500 ng/mL, respectively). Compound distribution favored the plasma, with a ratio of the maximum brain to maximum plasma levels of 0.18. Less polar compounds, such as the n-butyl sulfonamide 16 and the n-butyl amide 31 had less favorable brain to plasma ratios, as would be expected (data not shown). For this class of compounds, the MDCK-mdr1 assay was a better predictor of peripheral selectivity for more polar compounds, typically with a cLogP of less than 6.
3. SUMMARY AND CONCLUSIONS
Peripherally restricted CB1 antagonists are of interest to investigate their therapeutic value while minimizing their risk of causing psychiatric disorders. Such compounds could become important tools in treating diabetes, metabolic syndromes, dyslipidemias and liver diseases. In this study, we investigated a series of 1 analogues in which the 4-aminopiperidine group was replaced with a piperazine group. The piperazine was used as a linker to access an additional binding site via functionalization with amides, sulfonamides, carbamates and ureas. Potent hCB1 antagonists with excellent selectivity versus hCB2 were discovered. Positive MDCK-mdr1 results lead us to test several compounds for peripheral restriction in rodents after oral dosing. Varying levels of peripheral restriction were observed, with better results attained with compounds having higher TPSA values. In general, the MDCK-mdr1 assay was more effective at predicting peripheral restriction for compounds of this type that have a CLogP of less than 6. The heteroaryl urea 65 arose as the most interesting compound investigated. It is potent, highly selective and orally bioavailable. While the peripheral restriction observed with 65 is short of our goal, the aryl ureas, as a class of compounds, provide an opportunity to adjust molecular properties to favor peripheral restriction and oral bioavailability, while maintaining a good in vitro profile. Such compounds are currently being optimized for targeting peripheral hCB1 receptors.
4. EXPERIMENTAL
Chemistry General.
Purity and characterization of compounds were established by a combination of MS, NMR, HPLC and TLC analytical techniques described below. NMR spectra were recorded on a Bruker Avance DPX-300 (300 MHz) spectrometer and were determined in chloroform-d (7.26 ppm) or methanol-d4 (3.31 ppm) with tetramethylsilane (0.00 ppm) or solvent peaks as the internal reference unless otherwise noted. Chemical shifts are reported in ppm relative to the solvent signal and coupling constant (J) values are reported in hertz (Hz). Thin-layer chromatography (TLC) was performed on EMD precoated silica gel 60 F254 plates. TLC spots were visualized with UV light or I2 detection. Low-resolution mass spectra were obtained using a single quadrupole PE Sciex API 150EX (ESI). Unless stated otherwise, all test compounds were at least 95% pure as determined by HPLC. HPLC method: an Agilent-Varian system equipped with Prostar 210 pumps, a Prostar 335 Diode UV detector and a Phenomenex Synergi 4 μm Hydro RP 80A C18 250 × 4.6 mm column using a 20-minute gradient elution of 5-95% solvent B at 1 mL/min followed by 5 minutes at 95% solvent B (solvent A, water with 0.1% TFA; solvent B, acetonitrile with 0.1% TFA and 5% water; absorbance monitored at 220 and 280 nm).
General Procedure A: Sulfonamides of 8 from Sulfonyl Chlorides.
To a solution of 8 (0.2 mmol, 1 equiv) in CH2Cl2 (2 ml) was added DMAP (0.02 mmol, 0.1 equiv), followed by NEt3 (0.4 mmol, 2 equiv). The sulfonyl chloride (0.24 mmol, 1.2 equiv) was added and the reaction stirred at rt under nitrogen for 16 hours. The mixture was concentrated and then chromatographed on a 4-gram silica gel column using a 0-100% gradient of EtOAc in hexanes to provide the purified sulfonamide.
General Procedure B: Carbamates of 8 from Carbamoyl Chlorides.
To an ice-cold solution of 8 (0.2 mmol, 1 equiv) in DCE (1 mL) was added NEt3 (0.24 mmol, 1.2 equiv), followed by slow addition of the chloroformate (0.24 mmol, 1.2 equiv). The ice bath was removed and stirring continued for 2 hours. Ethyl acetate (3 mL) was added, followed by saturated NaHCO3 solution (0.8 mL) and water (0.4 mL). After 10 minutes, the aqueous layer was removed. Celite (600 mg) was added to the organic layer and the solvent evaporated. Flash chromatography using silica gel with an EtOAc/hexanes gradient provided the purified carbamate.
General Procedure C: Carbamates of 8 from Alcohols.
To an ice-cold solution of the alcohol (0.3 mmol, 1.5 equiv) in THF (1 mL) was slowly added NaHMDS (1 M/THF, 0.24 mL, 1.2 equiv). After 10 minutes, the 4-fluorophenyl carbamate of 8 (0.2 mmol; prepared using general procedure B) was added. After 15 minutes, the ice bath was removed, and the mixture stirred at rt for 15 hours. Ethyl acetate (3 mL) was added, followed by brine (1 mL) and 2 N NaOH (0.4 mL). After 10 minutes, the aqueous layer was removed, and the organic layer was washed with 0.8 M NaHCO3 solution (2×1 mL). Celite (600 mg) was added to the organic layer and the solvent evaporated. Flash chromatography using silica gel with an EtOAc/hexanes gradient provided the purified carbamate.
General Procedure D: Amides of 8 from Acid Chlorides.
To a solution of 8 (0.2 mmol, 1 equiv) in CH2Cl2 (2 ml) was added DMAP (0.02 mmol, 0.1 equiv), followed by NEt3 (0.4 mmol, 2 equiv). The acid chloride (0.24 mmol, 1.2 equiv) was added and the reaction stirred at rt under nitrogen for 16 hours. The mixture was concentrated and then chromatographed on a 4-gram silica gel column using a 0-100% gradient of EtOAc in hexanes to provide the purified amide.
General Procedure E: Amides of 8 from Carboxylic Acids.
To a solution of 8 (0.2 mmol, 1 equiv) in CH2Cl2 (2 ml) were added NEt3 (0.6 mmol, 3 equiv), N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU, 0.26 mmol, 1.3 equiv) and the appropriate carboxylic acid (1.25 equiv). The reaction was stirred overnight and then concentrated. The crude material was purified by silica gel column chromatography using a gradient of 0–100% EtOAc/hexanes.
General Procedure F: Ureas of 8 from Isocyanates.
To a solution of 8 (0.2 mmol, 1 equiv) in THF (1 mL) was added the isocyanate (0.24 mmol, 1.2 equiv), followed by NEt3 (0.24 mmol, 1.2 equiv). The mixture was stirred at rt for 15 hours. Water (0.4 mL) was added, followed by ethyl acetate (3 mL) and then saturated NaHCO3 solution (0.8 mL). After 10 minutes, the aqueous layer was removed. Celite (600 mg) was added to the organic layer and the solvent was evaporated. Flash chromatography using silica gel with an EtOAc/hexanes gradient provided the purified urea.
General Procedure G: Ureas of 8 from Amines.
To an ice-cold solution of 8 (0.2 mmol, 1 equiv) in CH2Cl2 (1 mL) was added NaHCO3 (0.6 mmol, 3 equiv), followed by saturated NaHCO3 solution (0.2 mL). Triphosgene (0.2 mmol, 1 equiv) was added and after 10 minutes, the ice bath was removed and the mixture was stirred at rt for 1 hour (gas evolution). Saturated NaHCO3 solution (0.8 mL) and water (0.4 mL) were added. After 10 minutes, the aqueous layer was removed and the organic layer dried with sodium sulfate (20 minutes). The mixture was filtered, toluene (0.5 ml) was added and the solvent evaporated. THF (1 mL) was added followed by the amine (0.4 mmol, 2 equiv) and then NEt3 (0.5 mmol, 2.5 equiv). The mixture was stirred at rt for 15 hours. Ethyl acetate (3 mL) was added, followed by saturated NaHCO3 solution (0.8 mL) and water (0.4 mL). After 10 minutes, the aqueous layer was removed. Celite (600 mg) was added to the organic layer and the solvent was evaporated. Flash chromatography using silica gel with an EtOAc/hexanes gradient provided the purified urea.
General Procedure H: Ureas of 8 from Amines.
To an ice-cold solution of the amine (0.4 mmol, 2 equiv) in CH2Cl2 (1 mL) was added NaHCO3 (0.6 mmol, 3 equiv), followed by saturated NaHCO3 solution (0.6 mL). Triphosgene (0.2 mmol, 1 equiv) was added and after 10 minutes, the ice bath was removed and the mixture was stirred at rt for 1 hour (gas evolution). Saturated NaHCO3 solution (0.8 mL) and water (0.4 mL) were added. After 10 minutes, the aqueous layer was removed and the organic layer dried with sodium sulfate (20 minutes). The mixture was filtered, toluene (0.5 ml) was added and most of the solvent evaporated. THF (1 mL) was added followed by 8 (0.2 mmol, 1 equiv) and then NEt3 (0.4 mmol, 2 equiv). The mixture was stirred at rt for 15 hours. Ethyl acetate (3 mL) was added, followed by saturated NaHCO3 solution (0.8 mL) and water (0.4 mL). After 10 minutes, the aqueous layer was removed. Celite (600 mg) was added to the organic layer and the solvent was evaporated. Flash chromatography using silica gel with an EtOAc/hexanes gradient provided the purified urea.
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-6-(piperazin-1-yl)-9H-purine (8).
To a heterogeneous mixture of 21 (1.5 g, 2.8 mmol) and ethanol (6 mL) was added 6 N HCl (3 mL). The mixture was stirred at rt for 10 minutes and then at 50 °C for 4 hours (homogeneous). The mixture was cooled in an ice bath. Chloroform (15 mL) was added, followed by brine (6 mL). 6 N NaOH (3.2 mL) was added slowly. After 5 minutes, the bath was removed. After 10 minutes, the organic layer was removed and the aqueous layer was saturated with sodium chloride. The aqueous layer was extracted with chloroform (2×6 mL). The combined organic layers was dried (sodium sulfate for 20 minutes), filtered and evaporated. Toluene (3 mL) was added and the solvent evaporated. The resulting thick residue was dissolved in methylene chloride (3 mL) and evaporated, providing 1.2 g (100%) of a tan amorphous solid. 1H NMR (300 MHz, CDCl3) δ 8.39 (s, 1H), 7.47-7.59 (m, 1H), 7.30-7.43 (m, 5H), 7.20 (d, J = 8.7 Hz, 2H), 4.36 (br s, 4H), 2.93-3.16 (m, 4H), 1.83 (br s, 1H). MS (m/z) 425.2 (M+1). HPLC = 93% at 11.38 minutes.
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-6-[4-(methylsulfonyl)piperazin-1-yl]-9H-purine (9).
The title compound was prepared by the general procedure A to provide 10.1 mg (20%) of a solid, mp 159-161 °C. 1H NMR (300 MHz, CDCl3) δ 8.41 (s, 1H), 7.47-7.56 (m, 1H), 7.30-7.46 (m, 5H), 7.15-7.24 (m, 2H), 4.52 (br s, 4H), 3.31-3.48 (m, 4H), 2.81 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 153.7, 153.1, 152.5, 146.4, 134.5, 134.1, 132.8, 132.4, 131.6, 130.1, 129.5, 129.4, 127.9, 127.0, 119.8, 98.4, 45.9, 44.9, 34.5. MS (m/z) 503.3 (M+1). HPLC = 95% at 20.17 minutes.
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-6-[4-(ethylsulfonyl)piperazin-1-yl]-9H-purine (10).
The title compound was prepared by the general procedure A to provide 7.0 mg (14%) of a solid, mp 175-176 °C. 1H NMR (300 MHz, CDCl3) δ 8.41 (s, 1H), 7.46-7.55 (m, 1H), 7.30-7.45 (m, 5H), 7.14-7.23 (m, 2H), 4.48 (br s, 4H), 3.37-3.56 (m, 4H), 2.99 (q, J = 7.4 Hz, 2H), 1.80 (br s, 1H), 1.40 (t, J = 7.4 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 153.7, 153.0, 152.4, 146.3, 134.5, 134.2, 132.8, 132.4, 131.6, 130.1, 129.5, 129.4, 128.0, 127.0, 119.8, 98.4, 45.9, 45.4, 44.0. MS (m/z) 517.4 (M+1). HPLC = 96% at 20.71 minutes.
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-6-{4-[(1-methylethyl)sulfonyl]piperazin-1-yl}-9H-purine (11).
The title compound was prepared by the general procedure A to provide 3.9 mg (7%) of a solid, mp 207-209 °C. 1H NMR (300 MHz, CDCl3) δ 8.41 (s, 1H), 7.47-7.56 (m, 1H), 7.31-7.45 (m, 6H), 7.16-7.23 (m, 2H), 4.46 (br s, 4H), 3.50-3.61 (m, 4H), 3.23 (quin, J = 6.8 Hz, 1H), 1.82 (br s, 2H), 1.37 (d, J = 6.8 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 153.7, 153.0, 152.7, 146.3, 134.5, 134.2, 132.9, 132.4, 131.6, 130.1, 129.4, 129.4, 128.0, 127.0, 119.8, 53.4, 46.5, 45.8, 16.8. MS (m/z) 531.3 (M+1). HPLC = 90% at 21.28 minutes.
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-6-[4-(cyclopropylsulfonyl)piperazin-1-yl]-9H-purine (12).
The title compound was prepared by the general procedure A to provide 17.8 mg (34%) of a solid, mp 202-204 °C. 1H NMR (300 MHz, CDCl3) δ 8.41 (s, 1H), 7.48-7.54 (m, 1H), 7.30-7.46 (m, 5H), 7.14-7.24 (m, 2H), 4.50 (br s, 4H), 3.38-3.56 (m, 4H), 2.27 (tt, J = 4.8, 8.0 Hz, 1H), 1.93 (br s, 1H), 1.15-1.22 (m, 2H), 0.93-1.04 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 153.7, 153.1, 152.4, 146.3, 134.5, 134.1, 132.8, 132.4, 131.6, 130.1, 129.4, 129.4, 127.9, 127.0, 119.8, 46.3, 45.0, 25.5, 4.4. MS (m/z) 529.5 (M+1). HPLC = 99% at 21.07 minutes.
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-6-[4-(propylsulfonyl)piperazin-1-yl]-9H-purine (13).
The itle compound was prepared by the general procedure A to provide 6.1 mg (11%) of a solid, mp 211-212 °C. 1H NMR (300 MHz, CDCl3) δ 8.41 (s, 1H), 7.47-7.55 (m, 1H), 7.30-7.45 (m, 5H), 7.14-7.23 (m, 2H), 4.49 (br s, 4H), 3.37-3.53 (m, 4H), 2.84-2.98 (m, 2H), 1.79-1.98 (m, 3H), 1.06 (t, J = 7.4 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 153.7, 153.1, 152.4, 146.3, 134.5, 134.2, 132.8, 132.4, 131.6, 130.1, 129.4, 129.4, 128.0, 127.0, 119.8, 51.1, 45.9, 45.3, 16.8, 13.2. MS (m/z) 531.3 (M+1). HPLC = 97% at 21.47 minutes.
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-6-{4-[(3,3,3-trifluoropropyl)sulfonyl]piperazin-1-yl}-9H-purine (14).
The title compound was prepared by the general procedure A to provide 51.4 mg (44%) of a solid, mp 209-210 °C. 1H NMR (300 MHz, CDCl3) δ 8.42 (s, 1H), 7.50 (dd, J = 0.8, 7.0 Hz, 1H), 7.30-7.45 (m, 5H), 7.12-7.24 (m, 2H), 4.51 (br s, 4H), 3.37-3.58 (m, 4H), 3.03-3.20 (m, 2H), 2.53-2.78 (m, 2H). 19F NMR (282 MHz, CDCl3) δ −66.14. 13C NMR (75 MHz, CDCl3) δ 153.6, 153.1, 152.5, 146.5, 134.5, 134.2, 132.8, 132.4, 131.6, 130.1, 129.5, 127.9, 127.0, 119.9, 45.9, 28.8, 28.4. MS (m/z) 585.3 (M+1). HPLC = 99% at 21.83 minutes.
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-6-{4-[(2-methylpropyl)sulfonyl]piperazin-1-yl}-9H-purine (15).
The title compound was prepared by the general procedure A to provide 15.0 mg (27%) of a solid, mp 196-197 °C. 1H NMR (300 MHz, CDCl3) δ 8.41 (s, 1H), 7.46-7.54 (m, 1H), 7.30-7.45 (m, 5H), 7.14-7.23 (m, 2H), 4.49 (br s, 4H), 3.31-3.53 (m, 4H), 2.77 (d, J = 6.6 Hz, 2H), 2.32 (qd, J = 6.7, 13.4 Hz, 1H), 1.96 (br s, 1H), 1.12 (d, J = 6.7 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 153.7, 153.0, 152.4, 146.3, 134.5, 134.2, 132.8, 132.4, 131.6, 130.1, 129.5, 129.4, 128.0, 127.0, 119.8, 56.2, 45.8, 45.2, 24.4, 22.7. MS (m/z) 545.5 (M+1). HPLC = 97% at 22.24 minutes.
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-6-[4-(butylsulfonyl)piperazin-1-yl]-9H-purine (16).
The title compound was prepared by the general procedure A to provide 9.5 mg (17%) of a solid, mp 182-184 °C. 1H NMR (300 MHz, CDCl3) δ 8.41 (s, 1H), 7.47-7.56 (m, 1H), 7.30-7.45 (m, 5H), 7.13-7.24 (m, 2H), 4.49 (br s, 4H), 3.35-3.53 (m, 4H), 2.86-3.01 (m, 2H), 1.93 (br s, 1H), 1.73-1.88 (m, 2H), 1.45 (qd, J = 7.4, 14.9 Hz, 2H), 0.95 (t, J = 7.4 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 153.7, 153.0, 152.4, 146.3, 134.5, 134.2, 132.8, 132.4, 131.6, 130.1, 129.4, 129.4, 128.0, 127.0, 119.8, 49.1, 45.9, 45.3, 25.0, 21.7, 13.5. MS (m/z) 545.4 (M+1). HPLC = 96% at 22.27 minutes.
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-6-{4-[(2-methoxyethyl)sulfonyl]piperazin-1-yl}-9H-purine (17).
The title compound was prepared by the general procedure A to provide 68.6 mg (63%) of a solid, mp 180-183 °C. 1H NMR (300 MHz, CDCl3) δ 8.41 (s, 1H), 7.47-7.56 (m, 1H), 7.30-7.45 (m, 5H), 7.15-7.24 (m, 2H), 4.48 (br s, 4H), 3.76 (t, J = 5.8 Hz, 2H), 3.40-3.51 (m, 4H), 3.35 (s, 3H), 3.22 (t, J = 5.8 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 153.8, 153.1, 152.4, 146.3, 134.4, 134.2, 132.9, 132.4, 131.6, 130.1, 129.4, 128.0, 127.0, 119.8, 77.5, 77.0, 76.6, 66.1, 58.8, 50.2, 45.5, 45.3. MS (m/z) 547.3 (M+1). HPLC = 99% at 20.39 minutes.
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-6-{4-[(cyclohexylmethyl)sulfonyl]piperazin-1-yl}-9H-purine (18).
The title compound was prepared by the general procedure A to provide 53.9 mg (46%) of a solid, mp 195-196 °C. 1H NMR (300 MHz, CDCl3) δ 8.41 (s, 1H), 7.46-7.54 (m, 1H), 7.30-7.45 (m, 5H), 7.15-7.23 (m, 2H), 4.49 (br s, 4H), 3.41 (t, J = 4.9 Hz, 4H), 2.76 (d, J = 6.1 Hz, 2H), 1.96 (d, J = 11.9 Hz, 3H), 1.60-1.79 (m, 3H), 0.97-1.43 (m, 5H). 13C NMR (75 MHz, CDCl3) δ 153.7, 153.1, 146.3, 134.2, 132.9, 132.4, 131.6, 130.1, 129.4, 127.9, 127.0, 119.8, 72.9, 55.0, 45.8, 33.2, 25.8, 25.8, 12.0. MS (m/z) 585.4 (M+1). HPLC = >99% at 24.05 minutes.
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-6-{4-[(tetrahydro-2H-pyran-4-ylmethyl)sulfonyl]piperazin-1-yl}-9H-purine (19).
The title compound was prepared by the general procedure A to provide 83.6 mg (71%) of a solid, mp 195-196 °C. 1H NMR (300 MHz, CDCl3) δ 8.41 (s, 1H), 7.46-7.55 (m, 1H), 7.31-7.46 (m, 5H), 7.15-7.24 (m, 2H), 4.50 (br s, 4H), 3.96 (dd, J = 3.1, 11.5 Hz, 2H), 3.31-3.54 (m, 6H), 2.80 (d, J = 6.6 Hz, 2H), 2.17-2.40 (m, 1H), 1.87 (dd, J = 1.8, 13.0 Hz, 2H), 1.34-1.58 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 153.7, 153.1, 152.4, 146.4, 134.4, 134.1, 132.8, 132.4, 131.6, 130.1, 129.4, 129.4, 127.9, 127.0, 119.8, 67.4, 54.3, 45.8, 45.1, 32.8, 30.7. MS (m/z) 587.4 (M+1). HPLC = 99% at 20.89 minutes.
Methyl 4-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperazine-1-carboxylate (20).
The title compound was prepared by the general procedure B to provide 68.3 mg (57%) of a solid, mp 198-201 °C. 1H NMR (300 MHz, CDCl3) δ 8.40 (s, 1H), 7.48-7.55 (m, 1H), 7.30-7.44 (m, 5H), 7.20 (d, J = 8.7 Hz, 2H), 4.37 (br s, 4H), 3.75 (s, 3H), 3.56-3.71 (m, 4H). MS (m/z) 483.3 (M+1). HPLC = >99% at 20.92 minutes.
tert-Butyl 4-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperazine-1-carboxylate (21).
To a solution of 7 (1.13 g, 3 mmol) and Boc-piperazine (670 mg, 1.2 equiv) in NMP (6 mL) was added potassium carbonate (1.2 g, 3 equiv). The heterogeneous mixture was stirred at rt for 10 minutes and then heated at 80 °C for 15 hours (turned dark brown). Ethyl acetate (30 mL) was added at rt, followed by brine (12 mL) and water (6 mL). The aqueous layer was removed and the organic layer was washed with brine/water (3/1, 2×12 mL). Celite (6 g) was added to the organic layer and the solvent evaporated. Purification by flash chromatography provided 1.57 g (99%) of an off white crystalline solid, mp 209-210 °C. Rf = 0.33 (30% EtOAc/hexanes; blue w UV). 1H NMR (300 MHz, CDCl3) δ 8.40 (s, 1H), 7.47-7.56 (m, 1H), 7.30-7.43 (m, 5H), 7.20 (d, J = 8.8 Hz, 2H), 4.35 (br s, 4H), 3.52-3.67 (m, 4H), 1.49 (s, 9H). MS (m/z) 525.5 (M+1). HPLC = 98% at 23.95 minutes.
2-Methoxyethyl 4-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperazine-1-carboxylate (22).
The title compound was prepared by the general procedure B to provide 84 mg (100%) of a white amorphous solid, mp 137-138 °C. Rf = 0.31 (1% MeOH/60% EtOAc/hexanes; blue with UV). 1H NMR (300 MHz, CDCl3) δ 8.40 (s, 1H), 7.51 (d, J = 6.6 Hz, 1H), 7.30-7.45 (m, 5H), 7.20 (d, J = 8.7 Hz, 2H), 4.38 (br s, 4H), 4.29 (t, J = 6.6 Hz, 2H), 3.56-3.78 (m, 6H), 3.40 (s, 3H). MS (m/z) 527.4 (M+1). HPLC = >99% at 20.31 minutes.
Cyclobutyl 4-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperazine-1-carboxylate (23).
The title compound was prepared by the general procedure C to provide 78 mg (100%) of a white amorphous solid, mp 174-175 °C. Rf = 0.23 (40% EtOAc/hexanes; blue with UV). 1H NMR (300 MHz, CDCl3) δ 8.40 (s, 1H), 7.51 (d, J = 6.8 Hz, 1H), 7.30-7.43 (m, 5H), 7.20 (d, J = 6.7 Hz, 2H), 4.90-5.08 (m, 1H), 4.37 (br s, 4H), 3.55-3.74 (m, 4H), 2.27-2.49 (m, 2H), 1.99-2.20 (m, 2H), 1.55-1.80 (m, 2H). MS (m/z) 523.2 (M+1). HPLC = >99% at 23.03 minutes.
Oxetan-3-yl 4-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperazine-1-carboxylate (24).
The title compound was prepared by the general procedure C to provide 80 mg (100%) of a white amorphous solid, mp 170-171 °C. Rf = 0.28 (1% MeOH/60% EtOAc/hexanes; blue with UV). 1H NMR (300 MHz, CDCl3) δ 8.41 (s, 1H), 7.51 (d, J = 6.8 Hz, 1H), 7.30-7.46 (m, 5H), 7.20 (d, J = 6.7 Hz, 2H), 5.45 (quin, J = 5.8 Hz, 1H), 4.52-4.94 (m, 4H), 4.40 (br s, 4H), 3.68 (br s, 4H). MS (m/z) 525.5 (M+1). HPLC = >99% at 20.15 minutes.
Oxan-4-yl 4-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperazine-1-carboxylate (25).
The title compound was prepared by the general procedure C to provide 66 mg (80%) of a white amorphous solid, mp 180-181 °C. Rf = 0.40 (1% MeOH/60% EtOAc/hexanes; blue with UV). 1H NMR (300 MHz, CDCl3) δ 8.41 (s, 1H), 7.51 (d, J = 7.0 Hz, 1H), 7.30-7.45 (m, 5H), 7.20 (d, J = 6.7 Hz, 2H), 4.84-5.00 (m, 1H), 4.38 (br s, 4H), 3.83-4.02 (m, 2H), 3.48-3.75 (m, 6H), 1.91-2.05 (m, 2H), 1.63-1.79 (m, 2H). MS (m/z) 553.3 (M+1). HPLC = >99% at 21.25 minutes.
Cyclobutylmethyl 4-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperazine-1-carboxylate (26).
The title compound was prepared by the general procedure C to provide 59 mg (74%) of a white crystalline solid, mp 129-131 °C. Rf = 0.24 (30% EtOAc/hexanes; blue with UV). 1H NMR (300 MHz, CDCl3) δ 8.40 (s, 1H), 7.51 (d, J = 6.6 Hz, 1H), 7.30-7.46 (m, 5H), 7.20 (d, J = 6.7 Hz, 2H), 4.37 (br s, 4H), 4.10 (d, J = 6.6 Hz, 2H), 3.55-3.75 (m, 4H), 2.52-2.73 (m, 1H), 1.73-2.13 (m, 6H). MS (m/z) 537.3 (M+1). HPLC = >99% at 24.16 minutes.
1-{4-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperazin-1-yl}-1-(4-fluorophenyl) methanimine (27).
A mixture of 8 (0.2 mmol, 99.6 mg), 4-fluorobenzamidic acid methyl ester hydrochloride salt{Connolly, 2004 #4} (47 mg, 1.25 equiv) and trimethylamine (0.28 mL, 10 equiv) in ethanol (1 mL) was heated at reflux overnight. The crude product was concentrated and chromatographed on a 12 g silica gel column using a 0-100% EtOAc/hexanes gradient, thus providing 33 mg (30%) of a colorless solid. 1H NMR (300 MHz, CDCl3) δ 8.38 (s, 1H), 7.49-7.83 (m, 3H), 7.29-7.47 (m, 10H), 7.15-7.24 (m, 2H), 5.67 (br s, 2H), 4.10 (s, 2H), 3.25 (t, J = 11.4 Hz, 1H), 3.01 (t, J = 12.4 Hz, 2H), 2.14 (d, J = 10.9 Hz, 2H), 1.93 (s, 1H), 1.66-1.88 (m, 2H), 0.07-0.98 (m, 3H). 13C NMR (75 MHz, CDCl3) δ 153.9, 153.2, 152.2, 145.3, 140.5, 134.3, 134.2, 133.2, 132.5, 131.3, 130.0, 129.8, 129.3, 128.4, 128.0, 126.9, 119.7, 55.2, 54.2, 45.6, 36.9, 30.7. MS (m/z) 546.4 (M+1). HPLC = 97% at 19.10 and 19.36 min (E & Z isomers of the imine).
4-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-N-(4-fluorophenyl)piperazine-1-carboximidamide (28).
A mixture of 8 (0.2 mmol, 100 mg) and 4-fluorophenyl S-methyl thiourea hydroiodide salt{Rasmussen, 1988 #5} (78 mg, 1.25 equiv) in isopropanol (1 mL) was heated at reflux overnight. The crude product was concentrated and chromatographed on a 12-g silica gel column using a 0-100% EtOAc/hexanes gradient, thus providing 4.1 mg (4%) of a colorless solid. 1H NMR (300 MHz, CDCl3) δ 8.33 (s, 1H), 7.45-7.55 (m, 1H), 7.28-7.43 (m, 10H), 7.12-7.25 (m, 5H), 7.08 (d, J = 6.9 Hz, 2H), 5.63 (br s, 2H), 4.26-4.40 (m, 1H), 3.57-3.77 (m, 1H), 3.21-3.53 (m, 3H), 3.05 (t, J = 12.4 Hz, 2H), 2.67 (dd, J = 3.3, 13.8 Hz, 1H), 2.44 (dd, J = 9.8, 13.8 Hz, 1H), 2.11-2.29 (m, 2H), 1.68-1.92 (m, 2H), 0.06-0.99 (m, 2H). MS (m/z) 561.2 (M+1). HPLC = 96% at 17.37 min.
6-(4-Acetylpiperazin-1-yl)-8-(2-chlorophenyl)-9-(4-chlorophenyl)-9H-purine (29).
The title compound was prepared by the general procedure D to provide 91.5 mg (78%) of a solid, mp 189-190 °C. 1H NMR (300 MHz, CDCl3) δ 8.41 (s, 1H), 7.48-7.56 (m, 1H), 7.31-7.45 (m, 5H), 7.21 (d, J = 8.8 Hz, 2H), 4.24-4.60 (m, 4H), 3.56-3.87 (m, 4H), 2.16 (s, 3H). MS (m/z) 467.4 (M+1). HPLC = 97% at 19.77 minutes.
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-6-[4-(trifluoroacetyl)piperazin-1-yl]-9H-purine (30).
The title compound was prepared by the general procedure D to provide 11.2 mg (21%) of a solid, mp 210-211 °C. 1H NMR (300 MHz, CDCl3) δ 8.43 (s, 1H), 7.47-7.55 (m, 1H), 7.30-7.43 (m, 5H), 7.15-7.24 (m, 2H), 4.46 (d, J = 5.4 Hz, 4H), 3.71-3.95 (m, 4H), 1.96 (br s, 1H). 19F NMR (282 MHz, CDCl3) δ −68.79. MS (m/z) 521.4 (M+1). HPLC = >99% at 22.15 minutes.
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-6-(4-pentanoylpiperazin-1-yl)-9H-purine (31).
The title compound was prepared by the general procedure D to provide 77.8 mg (61%) of a solid, mp 114-116 °C. 1H NMR (300 MHz, CDCl3) δ 8.41 (s, 1H), 7.47-7.56 (m, 1H), 7.30-7.45 (m, 5H), 7.15-7.25 (m, 2H), 4.19-4.71 (m, J = 19.7 Hz, 4H), 3.57-3.89 (m, 4H), 2.34-2.45 (m, 2H), 1.66 (quin, J = 7.6 Hz, 2H), 1.39 (qd, J = 7.4, 14.9 Hz, 2H), 0.94 (t, J = 7.4 Hz, 3H). MS (m/z) 509.4 (M+1). HPLC = 99% at 22.05 minutes.
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-6-[4-(3-methoxypropanoyl)piperazin-1-yl]-9H-purine (32).
The title compound was prepared by the general procedure E to provide 9.3 mg (9%) of a colorless film. 1H NMR (300 MHz, CDCl3) δ 8.41 (s, 1H), 7.45-7.59 (m, 1H), 7.28-7.44 (m, 6H), 7.20 (d, J = 8.7 Hz, 2H), 4.39 (d, J = 14.2 Hz, 4H), 3.58-3.91 (m, 6H), 3.29-3.47 (m, 3H), 2.56-2.80 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 169.3, 153.9, 153.1, 152.4, 146.2, 134.4, 134.2, 132.9, 132.4, 131.5, 130.1, 129.5, 129.4, 128.0, 127.0, 119.8, 68.8, 58.9, 45.8, 41.7, 33.6. MS (m/z) 511.1 (M+1). HPLC = 96% at 19.53 minutes.
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-6-[4-(ethoxyacetyl)piperazin-1-yl]-9H-purine (33).
The title compound was prepared by the general procedure E to provide 73 mg (71%) of a solid, mp 176-178 °C. 1H NMR (300 MHz, CDCl3) δ 8.41 (s, 1H), 7.45-7.57 (m, 1H), 7.29-7.45 (m, 5H), 7.20 (d, J = 8.7 Hz, 2H), 4.40 (br s, 4H), 4.21 (s, 2H), 3.66-3.87 (m, 4H), 3.60 (q, J = 7.0 Hz, 2H), 1.26 (t, J = 7.1 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 168.3, 153.9, 153.1, 152.4, 146.2, 134.4, 134.2, 132.9, 132.4, 131.5, 130.1, 129.5, 129.4, 128.0, 127.0, 119.9, 70.4, 66.9, 45.2, 42.0, 15.1. MS (m/z) 511.2 (M+1). HPLC = 99% at 19.83 minutes.
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-6-{4-[3-(methylsulfonyl)propanoyl]piperazin-1-yl}-9H-purine (34).
The title compound was prepared by the general procedure E to provide 88.4 mg (79%) of a solid, mp 129-132 °C. 1H NMR (300 MHz, CDCl3) δ 8.41 (s, 1H), 7.45-7.57 (m, 1H), 7.29-7.45 (m, 5H), 7.21 (d, J = 8.7 Hz, 2H), 4.41 (d, J = 17.2 Hz, 4H), 3.60-3.92 (m, 4H), 3.48 (t, J = 7.2 Hz, 2H), 3.23 (q, J = 7.2 Hz, 2H), 3.00 (s, 3H), 1.36 (t, J = 7.4 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 167.8, 153.8, 153.1, 152.4, 146.4, 134.4, 134.1, 132.9, 132.5, 131.6, 130.1, 129.4, 129.4, 128.0, 127.0, 119.9, 50.4, 47.5, 45.5, 42.1, 41.7, 26.1. MS (m/z) 559.2 (M+1). HPLC = 95% at 18.19 minutes.
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-6-[4-(3,3-dimethylbutanoyl)piperazin-1-yl]-9H-purine (35).
The title compound was prepared by the general procedure E to provide 77.4 mg (74%) of a solid, mp 194-195 °C. 1H NMR (300 MHz, CDCl3) δ 8.41 (s, 1H), 7.47-7.56 (m, 1H), 7.30-7.46 (m, 5H), 7.14-7.26 (m, 2H), 4.16-4.68 (m, 4H), 3.76-3.92 (m, 2H), 3.64-3.75 (m, 2H), 2.33 (s, 2H), 1.08 (s, 9H). 13C NMR (75 MHz, CDCl3) δ 170.7, 153.9, 153.1, 152.4, 146.2, 134.4, 134.2, 132.9, 132.4, 131.5, 130.1, 129.5, 129.4, 128.0, 127.0, 119.8, 46.7, 44.8, 41.5, 38.6, 31.5, 30.1. MS (m/z) 523.5 (M+1). HPLC = >99% at 22.43 minutes.
4-{4-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]piperazin-1-yl}-2-methyl-4-oxobutan-2-ol (36).
The title compound was prepared by the general procedure E to provide 40.7 mg (39%) of a solid, mp 192-193 °C. 1H NMR (300 MHz, CDCl3) δ 8.41 (s, 1H), 7.50 (d, J = 7.1 Hz, 1H), 7.30-7.45 (m, 5H), 7.13-7.24 (m, 2H), 4.40 (br s, 4H), 3.74-3.90 (m, 2H), 3.57-3.71 (m, 2H), 2.51 (s, 2H), 1.32 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 171.5, 153.8, 153.0, 152.3, 146.4, 134.5, 134.2, 132.8, 132.4, 131.6, 130.1, 129.5, 129.4, 128.0, 127.0, 119.9, 69.3, 45.6, 43.1, 41.4, 29.6, 29.5. MS (m/z) 525.5 (M+1). HPLC = 98% at 19.33 minutes.
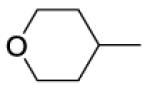
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-6-[4-(tetrahydro-2H-pyran-4-ylcarbonyl)piperazin-1-yl]-9H-purine (37).
The title compound was prepared by the general procedure E to provide 73 mg (68%) of a solid, mp 71-72 °C. 1H NMR (300 MHz, CDCl3) δ 8.41 (s, 1H), 7.51 (d, J = 7.1 Hz, 1H), 7.29-7.44 (m, 5H), 7.20 (d, J = 8.7 Hz, 2H), 4.16-4.67 (m, 4H), 3.89-4.13 (m, 3H), 3.60-3.89 (m, 4H), 3.37-3.56 (m, 3H), 3.13-3.32 (m, 2H), 2.67-2.87 (m, 1H), 1.73-2.03 (m, 3H), 1.65 (d, J = 12.1 Hz, 2H), 1.32-1.45 (m, 3H). 13C NMR (75 MHz, CDCl3) δ 173.2, 153.8, 153.0, 152.3, 146.3, 134.5, 134.2, 132.8, 132.4, 131.6, 130.1, 129.4, 129.4, 128.0, 127.0, 119.8, 67.2, 67.1, 47.6, 39.7, 37.7, 29.1, 28.6. MS (m/z) 537.3 (M+1). HPLC = 99% at 19.77 minutes.
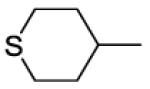
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-6-[4-(tetrahydro-2H-thiopyran-4-ylcarbonyl)piperazin-1-yl]-9H-purine (38).
The title compound was prepared by the general procedure E to provide 65.3 mg (59%) of a solid, mp 127-129 °C. 1H NMR (300 MHz, CDCl3) δ 8.41 (s, 1H), 7.51 (d, J = 6.9 Hz, 1H), 7.30-7.45 (m, 5H), 7.20 (d, J = 8.6 Hz, 2H), 4.04-4.74 (m, 4H), 3.47-3.94 (m, 4H), 2.51-2.76 (m, 5H), 1.89-2.15 (m, 4H). 13C NMR (75 MHz, CDCl3) δ 173.5, 153.8, 153.1, 152.4, 146.2, 134.4, 134.2, 132.8, 132.4, 131.6, 130.1, 129.4, 128.0, 127.0, 119.8, 45.5, 41.7, 40.1, 30.3, 28.0. MS (m/z) 553.3 (M+1). HPLC = 94% at 20.84 minutes.
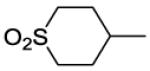
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-6-{4-[(1,1-dioxidotetrahydro-2H-thiopyran-4-yl)carbonyl]piperazin-1-yl}-9H-purine (39).
The title compound was prepared by the general procedure E to provide 116.9 mg (100%) of a solid, mp 106-108 °C. 1H NMR (300 MHz, CDCl3) δ 8.41 (s, 1H), 7.48-7.56 (m, 1H), 7.31-7.43 (m, 5H), 7.15-7.23 (m, 2H), 6.68 (br s, 1H), 4.40 (d, J = 11.8 Hz, 4H), 3.61-3.90 (m, 4H), 3.30 (br s, 2H), 2.92-3.14 (m, 3H), 2.31-2.48 (m, 2H), 2.12-2.31 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 172.0, 153.7, 153.0, 152.3, 146.3, 134.4, 134.0, 132.8, 132.5, 131.6, 130.0, 129.4, 129.3, 128.0, 127.1, 119.8, 49.8, 47.6, 45.7, 41.9, 35.7, 27.1. MS (m/z) 585.3 (M+1). HPLC = 98% at 18.32 minutes.
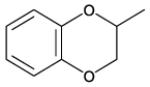
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-6-[4-(2,3-dihydro-1,4-benzodioxin-2-ylcarbonyl)piperazin-1-yl]-9H-purine (40).
The title compound was prepared by the general procedure E to provide 28.2 mg (19%) of a solid, mp 135-136 °C. 1H NMR (300 MHz, CDCl3) δ 8.43 (s, 1H), 7.47-7.57 (m, 1H), 7.31-7.45 (m, 5H), 7.16-7.24 (m, 2H), 6.80-6.98 (m, 4H), 4.89 (dd, J = 2.5, 8.0 Hz, 1H), 4.60-4.82 (m, 2H), 4.54 (dd, J = 2.5, 12.0 Hz, 1H), 4.37 (dd, J = 8.0, 12.0 Hz, 2H), 4.01 (d, J = 12.2 Hz, 2H), 3.58-3.82 (m, 2H). MS (m/z) 587.2 (M+1). HPLC = >99% at 22.65 minutes.
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-6-[4-(cyclopentylacetyl)piperazin-1-yl]-9H-purine (41).
The title compound was prepared by the general procedure D to provide 67.9 mg (63%) of a solid, mp 133-134 °C. 1H NMR (300 MHz, CDCl3) δ 8.41 (s, 1H), 7.45-7.56 (m, 1H), 7.29-7.45 (m, 5H), 7.20 (d, J = 8.8 Hz, 2H), 4.38 (d, J = 16.5 Hz, 4H), 3.55-3.92 (m, 4H), 2.42 (d, J = 7.2 Hz, 2H), 2.27 (td, J = 7.6, 15.1 Hz, 1H), 1.88 (dd, J = 5.6, 10.4 Hz, 2H), 1.45-1.72 (m, 4H), 1.08-1.31 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 171.6, 153.9, 153.1, 152.4, 146.2, 134.4, 134.2, 132.9, 132.4, 131.5, 130.1, 129.5, 129.4, 128.0, 127.0, 119.9, 45.5, 41.6, 39.3, 36.8, 32.8, 25.0. MS (m/z) 535.5 (M+1). HPLC = >99% at 23.01 minutes.
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-6-[4-(tetrahydrofuran-3-ylacetyl)piperazin-1-yl]-9H-purine (42).
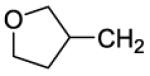
The title compound was prepared by the general procedure E to provide 74.5 mg (69%) of a solid, mp 105-106 °C. 1H NMR (300 MHz, CDCl3) δ 8.41 (s, 1H), 7.46-7.57 (m, 1H), 7.30-7.45 (m, 5H), 7.20 (d, J = 8.7 Hz, 2H), 4.37 (br s, 4H), 3.59-4.06 (m, 8H), 3.36-3.54 (m, 1H), 3.23 (q, J = 7.4 Hz, 1H), 2.67-2.83 (m, 1H), 2.51 (dd, J = 3.7, 7.2 Hz, 2H), 2.17 (t, J = 6.3 Hz, 1H), 1.48-1.70 (m, 1H), 1.37 (t, J = 7.4 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 170.6, 153.8, 153.0, 152.3, 146.3, 134.4, 134.1, 132.8, 132.4, 131.6, 130.1, 129.4, 129.4, 128.0, 127.0, 119.8, 73.2, 67.6, 47.5, 45.5, 41.7, 37.5, 37.1, 35.5, 32.2. MS (m/z) 537.3 (M+1). HPLC = 93% at 19.88 minutes.
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-6-[4-(tetrahydro-2H-pyran-4-ylacetyl)piperazin-1-yl]-9H-purine (43).
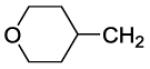
The title compound was prepared by the general procedure E to provide 103.9 mg (94%) of a solid, mp 73-75 °C. 1H NMR (300 MHz, CDCl3) δ 8.41 (s, 1H), 7.46-7.59 (m, 1H), 7.30-7.45 (m, 5H), 7.15-7.25 (m, 2H), 4.39 (d, J = 18.6 Hz, 4H), 3.95 (dd, J = 3.2, 11.3 Hz, 2H), 3.60-3.87 (m, 4H), 3.43 (dt, J = 1.7, 11.7 Hz, 2H), 3.09-3.32 (m, 2H), 2.33 (d, J = 6.9 Hz, 2H), 2.13 (ddd, J = 3.6, 7.3, 14.4 Hz, 1H), 1.71 (dd, J = 1.6, 12.8 Hz, 2H), 1.26-1.46 (m, 5H). 13C NMR (75 MHz, CDCl3) δ 170.4, 153.8, 153.0, 152.3, 146.2, 134.4, 134.1, 132.9, 132.4, 131.6, 130.0, 129.4, 128.0, 127.0, 119.8, 67.8, 47.5, 45.7, 41.6, 40.0, 33.1, 32.2, 8.4. MS (m/z) 551.2 (M+1). HPLC = 90% at 20.48 minutes.
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-6-[4-(piperidin-1-ylacetyl)piperazin-1-yl]-9H-purine (44).
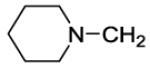
The title compound was prepared by the general procedure E to provide 6.1 mg (6%) of a colorless film. 1H NMR (300 MHz, CDCl3) δ 8.41 (s, 1H), 7.47-7.56 (m, 1H), 7.31-7.45 (m, 5H), 7.15-7.24 (m, 2H), 4.22-4.60 (m, 4H), 3.80 (td, J = 5.1, 15.2 Hz, 4H), 3.24 (s, 2H), 2.49 (br s, 4H), 1.71 (br s, 4H), 1.60 (td, J = 5.5, 10.7 Hz, 4H), 1.45 (d, J = 5.0 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 170.6, 153.9, 153.1, 152.3, 146.1, 134.4, 134.3, 132.4, 131.5, 130.1, 129.4, 128.0, 127.0, 119.8, 103.2, 96.8, 82.6, 62.5, 54.5, 50.7, 45.8, 42.1, 25.9, 23.8. MS (m/z) 550.4 (M+1). HPLC = 99% at 18.05 minutes.
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-6-[4-(morpholin-4-ylacetyl)piperazin-1-yl]-9H-purine (45).
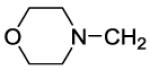
The title compound was prepared by the general procedure E to provide 52.1 mg (47%) of a solid, mp 110-111 °C. 1H NMR (300 MHz, CDCl3) δ Shift 8.41 (s, 1H), 7.47-7.57 (m, 1H), 7.30-7.46 (m, 5H), 7.15-7.25 (m, 2H), 4.21-4.66 (m, 4H), 3.68-3.88 (m, 8H), 3.25 (s, 2H), 2.45-2.64 (m, 4H). 13C NMR (75 MHz, CDCl3) δ 168.0, 153.9, 153.1, 152.4, 146.2, 134.4, 134.2, 132.9, 132.4, 131.6, 130.1, 129.4, 129.4, 128.0, 127.0, 119.8, 66.8, 61.8, 53.5, 45.7, 41.9. MS (m/z) 552.5 (M+1). HPLC = >99% at 16.97 minutes.
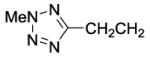
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-6-{4-[3-(2-methyl-2H-tetrazol-5-yl)propanoyl]piperazin-1-yl}-9H-purine (46).
The title compound was prepared by the general procedure E to provide 103.6 mg (92%) of a solid, mp 125 °C (Dec.). 1H NMR (300 MHz, CDCl3) δ 8.40 (s, 1H), 7.47-7.56 (m, 1H), 7.31-7.45 (m, 5H), 7.16-7.26 (m, 2H), 4.58 (t, J = 6.2 Hz, 2H), 4.19-4.52 (m, 4H), 3.69-3.84 (m, 2H), 3.54-3.67 (m, 2H), 2.67 (s, 3H), 2.04 (s, 2H). 13C NMR (75 MHz, CDCl3) δ 167.5, 153.7, 153.0, 152.4, 146.3, 134.4, 134.1, 132.8, 132.4, 131.6, 130.0, 129.4, 129.4, 128.0, 127.0, 119.8, 45.3, 45.1, 44.8, 42.6, 41.8, 32.5. MS (m/z) 563.4 (M+1). HPLC = >99% at 18.81 minutes.
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-6-[4-(2-phenylpropanoyl)piperazin-1-yl]-9H-purine (47).
The title compound was prepared by the general procedure D to provide 68.7 mg (62%) of a solid, mp 107-109 °C. 1H NMR (300 MHz, CDCl3) δ 8.35 (s, 1H), 7.44-7.53 (m, 1H), 7.24-7.40 (m, 10H), 7.11-7.20 (m, 2H), 4.21-4.76 (m, 2H), 3.85-4.08 (m, 3H), 3.44-3.71 (m, 4H), 1.48 (d, J = 6.9 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 172.3, 153.8, 153.0, 152.3, 146.1, 141.9, 134.4, 134.2, 132.9, 132.4, 131.5, 130.1, 129.4, 129.4, 129.1, 128.0, 127.2, 127.0, 126.9, 119.7, 77.5, 77.1, 76.6, 43.5, 20.7. MS (m/z) 557.6 (M+1). HPLC = 99% at 22.52 minutes.
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-6-{4-[(pyrrolidin-1-yl)carbonyl]piperazin-1-yl}-9H-purine (48).
A mixture of the 4-fluorophenylcarbamate of 8 (85 mg, 0.15 mmol, prepared from 8 by general procedure B), pyrrolidine (0.038 mL, 3 equiv) and NMP (0.4 mL) was heated at 80-85 for 20 hours. Ethyl acetate (3 mL) was added, followed by brine (0.6 mL) and 2 N NaOH (0.3 mL). After 10 minutes, the aqueous layer was removed and the organic layer was washed with 0.8 M NaHCO3 (2×1 mL). Celite (600 mg) was added to the organic layer and the solvent evaporated. Purification by flash chromatography provided 40 mg (51%) of a white crystalline solid, mp 200-201 °C. Rf = 0.17 (1% MeOH/60% EtOAc/hexanes; blue with UV). 1H NMR (300 MHz, CDCl3) δ 8.40 (s, 1H), 7.52 (d, J = 6.4 Hz, 1H), 7.15-7.45 (m, 7H), 4.39 (br s, 2H), 3.30-3.57 (m, 10H), 1.74-1.97 (m, 4H). MS (m/z) 522.2 (M+1). HPLC = >99% at 21.25 minutes.
8-(2-Chlorophenyl)-9-(4-chlorophenyl)-6-{4-[(morpholin-4-yl)carbonyl]piperazin-1-yl}-9H-purine (49).
To an ice-cold solution of morpholine (0.026 mL, 2 equiv) in THF (0.8 mL) was added dropwise n-butyl lithium (1.6 M/hexanes, 0.12 mL, 1.2 equiv). After 5 minutes, the 4-fluorophenylcarbamate of 8 (85 mg, 0.15 mmol, prepared from 8 by general procedure B) was added. After 5 minutes, the ice bath was removed and the mixture was stirred at rt for 1 hour. Saturated NaHCO3 solution (0.8 mL), ethyl acetate (3 mL) and water (0.4 mL) were added. After 10 minutes, the aqueous layer was removed. Celite (600 mg) was added to the organic layer and the solvent evaporated. Purification by flash chromatography provided 25 mg (31%) of a white crystalline solid, mp 181-182 °C. Rf = 0.14 (1% MeOH/60% EtOAc/hexanes; blue with UV). 1H NMR (300 MHz, CDCl3) δ 8.40 (s, 1H), 7.51 (d, J = 6.0 Hz, 1H), 7.30 - 7.44 (m, 4H), 7.20 (d, J = 8.6 Hz, 2H), 4.39 (br s, 4H), 3.71 (br s, 4H), 3.45 (br s, 4H), 3.32 (br s, 4H). MS (m/z) 538.1 (M+1). HPLC = >99% at 19.76 minutes.
4-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-N-(2-methylpropyl)piperazine-1-carboxamide (50).
The title compound was prepared by the general procedure G to provide 57 mg (54%) of a white crystalline solid, mp 222-223 °C. Rf = 0.24 (2% MeOH/60% EtOAc/Hexanes; blue with UV). 1H NMR (300 MHz, CDCl3) δ 8.40 (s, 1H), 7.51 (d, J = 6.8 Hz, 1H), 7.30-7.45 (m, 5H), 7.20 (d, J = 8.7 Hz, 2 H), 4.56 (t, J = 5.3 Hz, 1H), 4.40 (br s, 4H), 3.48-3.67 (m, 4H), 3.10 (dd, J = 6.3, 6.3 Hz, 2H), 1.71-1.92 (m, 1H), 0.93 (d, J = 6.8 Hz, 6H). MS (m/z) 524.3 (M+1). HPLC = >99% at 20.35 minutes.
4-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-N-cyclopentylpiperazine-1-carboxamide (51).
The title compound was prepared by the general procedure F to provide 51 mg (63%) of a white crystalline solid, mp 248-250 °C. Rf = 0.22 (1% MeOH/60% EtOAc/hexanes; blue with UV). 1H NMR (300 MHz, CDCl3) δ 8.40 (s, 1H), 7.51 (d, J = 6.8 Hz, 1H), 7.30-7.44 (m, 5H), 7.20 (d, J = 8.7 Hz, 2H), 4.39 (d, J = 6.2 Hz, 4H), 4.04-4.23 (m, 1H), 3.42-3.63 (m, 4H), 1.84-2.13 (m, 2H), 1.52-1.77 (m, 5H), 1.30-1.45 (m, 2H). MS (m/z) 536.2 (M+1). HPLC = >99% at 20.85 minutes.
4-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-N-[1-(trifluoromethyl)cyclopentyl]-piperazine-1-carboxamide (52).
The title compound was prepared by the general procedure H to provide 36 mg (50%) of an off white crystalline solid, mp 202-204 °C. Rf = 0.47 (2% MeOH/60% EtOAc/hexanes; blue with UV). 1H NMR (300 MHz, CDCl3) δ 8.40 (s, 1H), 7.51 (d, J = 6.8 Hz, 1H), 7.31-7.43 (m, 5H), 7.20 (d, J = 8.7 Hz, 2H), 4.50 (s, 1H), 4.40 (br s, 4H), 3.46-3.65 (m, 4H), 2.20-2.35 (m, 2H), 2.01-2.20 (m, 2H), 1.67-1.97 (m, 4H). MS (m/z) 604.4 (M+1). HPLC = 99% at 21.64 minutes.
4-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-N-cyclohexylpiperazine-1-carboxamide (53).
The title compound was prepared by the general procedure G to provide 43 mg (65%) of a white crystalline solid, mp 228-229 °C. Rf = 0.31 (2% MeOH)/60%EtOAc/hexanes; blue with UV). 1H NMR (300 MHz, CDCl3) δ 8.40 (s, 1H), 7.51 (d, J = 6.8 Hz, 1H), 7.30-7.44 (m, 5H), 7.20 (d, J = 8.7 Hz, 2H), 4.23-4.53 (m, 5H), 3.61-3.79 (m, 1H), 3.46-3.61 (m, 4H), 1.89-2.07 (m, 2H), 1.65-1.81 (m, 2H), 1.29-1.50 (m, 2H), 1.02-1.29 (m, 4H). MS (m/z) 550.4 (M+1). HPLC = >99% at 21.49 minutes.
4-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-N-(oxan-4-yl)piperazine-1-carboxamide (54).
The title compound was prepared by the general procedure G to provide 45 mg (58%) of a white amorphous solid, mp 255-257 °C. Rf = 0.23 (4% MeOH/EtOAc; blue with UV). 1H NMR (300 MHz, CDCl3) δ 8.41 (s, 1H), 7.50 (d, J = 6.8 Hz, 1H), 7.30-7.45 (m, 5H), 7.20 (d, J = 8.5 Hz, 2H), 4.23-4.55 (m, 5H), 3.81-4.07 (m, 3H), 3.34-3.69 (m, 6H), 1.96 (d, J = 11.1 Hz, 2H), 1.37-1.55 (m, 2H). MS (m/z) 552.4 (M+1). HPLC = >99% at 19.04 minutes.
4-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-N-[(1-methylpiperidin-4-yl)methyl]piperazine-1-carboxamide (55).
The title compound was prepared by the general procedure G to provide 19 mg (28%) of a white amorphous solid, mp 128-130 °C. Rf = 0.09 (10% (20% NH4OH/MeOH)/EtOAc; blue with UV). 1H NMR (300 MHz, CDCl3) δ 8.40 (s, 1H), 7.50 (d, J = 6.8 Hz, 1H), 7.30-7.45 (m, 5H), 7.20 (d, J = 8.5 Hz, 2H), 4.25-4.55 (m, 5H), 3.62-3.85 (m, 1H), 3.55 (br s, 4H), 2.72-2.86 (m, 2H), 2.28 (s, 3H), 1.90-2.20 (m, 4H), 1.37-1.56 (m, 2H). MS (m/z) 565.2 (M+1). HPLC = >99% at 16.77 minutes.
4-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-N-(cyclohexylmethyl)piperazine-1-carboxamide (56).
The title compound was prepared by the general procedure G to provide 44 mg (65%) of a white amorphous solid, mp 105-107 °C. Rf = 0.31 (2% MeOH)/60%EtOAc/hexanes; blue with UV). 1H NMR (300 MHz, CDCl3) δ 8.40 (s, 1H), 7.51 (d, J = 6.6 Hz, 1H), 7.29-7.45 (m, 5H), 7.20 (d, J = 8.5 Hz, 2H), 4.61 (t, J = 5.5 Hz, 1H), 4.33-4.45 (m, 4H), 3.56 (br s, 4H), 3.11 (dd, J = 6.0, 6.0 Hz, 2H), 1.66-1.80 (m, 4H), 1.42-1.54 (m, 1H), 1.08-1.34 (m, 4H), 0.82-1.02 (m, 2H). MS (m/z) 564.2 (M+1). HPLC = >99% at 22.32 minutes.
4-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-N-(oxan-4-ylmethyl)piperazine-1-carboxamide (57).
The title compound was prepared by the general procedure G to provide 56 mg (82%) of a white amorphous solid, mp 114-116 °C. Rf = 0.22 (4% MeOH/EtOAc; blue with UV). 1H NMR (300 MHz, CDCl3) δ 8.40 (s, 1H), 7.51 (d, J = 6.8 Hz, 1H), 7.30-7.46 (m, 5H), 7.20 (d, J = 8.7 Hz, 2H), 4.69 (t, J = 5.4 Hz, 1H), 4.40 (br s, 4 H), 3.98 (dd, J = 11.1, 3.2 Hz, 2H), 3.49-3.68 (m, 4H), 3.38 (t, J = 11.2 Hz, 2H), 3.17 (dd, J = 6.1, 6.1 Hz, 2H), 1.60-1.70 (m, 2H), 1.22-1.43 (m, 3H). MS (m/z) 566.0 (M+1). HPLC = >99% at 19.29 minutes.
4-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-N-[(1-methylpiperidin-4-yl)methyl]piperazine-1-carboxamide (58).
The title compound was prepared by the general procedure G to provide 40 mg (58%) of a white amorphous solid, mp 100-102 °C. Rf = 0.06 (10% (20% NH4OH/MeOH)/EtOAc; blue with UV). 1H NMR (300 MHz, CDCl3) δ 8.40 (s, 1H), 7.51 (d, J = 6.8 Hz, 1H), 7.31-7.46 (m, 5H), 7.20 (d, J = 8.5 Hz, 2H), 4.63 (t, J = 5.5 Hz, 1H), 4.39 (br s, 4H), 3.46-3.63 (m, 4H), 3.13-3.27 (m, 2H), 2.75-2.96 (m, 2H), 2.26 (s, 3H), 1.84-2.01 (m, 2H), 1.64-1.79 (m, 2H), 1.43-1.62 (m, 1H), 1.18-1.41 (m, 2H). MS (m/z) 579.4 (M+1). HPLC = >99% at 16.96 minutes.
4-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-N-phenylpiperazine-1-carboxamide (59).
The title compound was prepared by the general procedure G to provide 67 mg (100%) of an off white amorphous solid, mp 121-123 °C. Rf = 0.43 (2% MeOH/60% EtOAc/hexanes; blue with UV). 1H NMR (300 MHz, CDCl3) δ 8.42 (s, 1H), 7.51 (d, J = 6.8 Hz, 1H), 7.23-7.44 (m, 9H), 7.20 (d, J = 8.5 Hz, 2H), 6.96-7.11 (m, 1H), 6.61 (s, 1H), 4.43 (br s, 4H), 3.67 (br s, 4H). MS (m/z) 544.3 (M+1). HPLC = >99% at 20.36 minutes.
4-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-N-(pyridin-2-yl)piperazine-1-carboxamide (60).
The title compound was prepared by the general procedure G to provide 65 mg (99%) of an off white amorphous solid, mp 120-122 °C. Rf = 0.25 (2% MeOH/60% EtOAc/hexanes; blue with UV). 1H NMR (300 MHz, CDCl3) δ 8.42 (d, J = 3.8 Hz, 1H), 8.21 (br s, 1H), 8.04 (d, J = 8.3 Hz, 1H), 7.67 (t, J = 7.4 Hz, 1H), 7.52 (d, J = 6.4 Hz, 1H), 7.29-7.45 (m, 5H), 7.20 (d, J = 8.3 Hz, 2H), 6.89-7.03 (m, 1H), 6.40-6.70 (m, 1H), 4.45 (br s, 4H), 3.74 (br s, 2H), 3.50 (br s, 2H). MS (m/z) 545.7 (M+1). HPLC = >97% at 18.15 minutes.
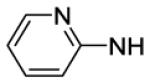
4-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-N-(2-fluorophenyl)piperazine-1-carboxamide (61).
The title compound was prepared by the general procedure G to provide 15 mg (22%) of an off white amorphous solid, mp 214-215 °C. Rf = 0.52 (2% MeOH/60% EtOAc/hexanes; blue with UV). 1H NMR (300 MHz, CDCl3) δ 8.42 (s, 1H), 8.00-8.19 (m, 1H), 7.52 (d, J = 6.8 Hz, 1H), 7.30-7.45 (m, 5H), 7.20 (d, J = 8.5 Hz, 2H), 6.89-7.16 (m, 3H), 6.58-6.75 (m, 1H), 4.47 (br s, 4H), 3.60-3.86 (m, 4H). MS (m/z) 562.2 (M+1). HPLC = 95% at 20.97 minutes.
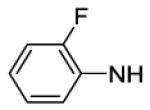
4-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-N-(2,4-difluorophenyl)piperazine-1-carboxamide (62).
The title compound was prepared by the general procedure H to provide 15 mg (22%) of an off white crystalline solid, mp 220-221 °C. Rf = 0.48 (2% MeOH/60% EtOAc/hexanes; blue with UV). 1H NMR (300 MHz, CDCl3) δ 8.42 (s, 1H), 7.92-8.11 (m, 1H), 7.52 (d, J = 6.6 Hz, 1H), 7.30-7.47 (m, 5H), 7.21 (d, J = 8.3 Hz, 2H), 6.80-6.95 (m, 2H), 6.51 (s, 1H), 4.47 (br s, 4H), 3.71 (br s, 4H). MS (m/z) 580.4 (M+1). HPLC = >98% at 20.53 minutes.
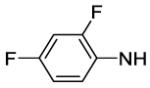
4-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-N-(3,4-difluorophenyl)piperazine-1-carboxamide (63).
The title compound was prepared by the general procedure G to provide 39 mg (56%) an off white amorphous solid, mp 205-207 °C. Rf = 0.46 (2% MeOH/60% EtOAc/hexanes; blue with UV). 1H NMR (300 MHz, CDCl3) δ 8.42 (s, 1H), 7.51 (d, J = 6.6 Hz, 1H), 7.30-7.46 (m, 6H), 7.20 (d, J = 8.5 Hz, 2H), 6.89-7.12 (m, 2H), 6.58 (s, 1H), 4.44 (br s, 4H), 3.68 (br s, 4H). MS (m/z) 580.4 (M+1). HPLC = >98% at 21.28 minutes.
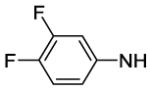
4-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-N-(4-fluorophenyl)piperazine-1-carboxamide (64).
The title compound was prepared by the general procedure G to provide 71 mg (100%) of an off white amorphous solid, mp 234-235 °C. Rf = 0.42 (2% MeOH/60% EtOAc/hexanes; blue with UV). 1H NMR (300 MHz, CDCl3) δ 8.42 (s, 1H), 7.51 (d, J = 6.22 Hz, 1H), 7.24-7.45 (m, 7H), 7.20 (d, J =8.1 Hz, 2H), 6.90-7.05 (m, 2H), 6.61 (s, 1H), 4.43 (br s, 4H), 3.67 (br s, 4H). MS (m/z) 561.9 (M+1). HPLC = >99% at 20.44 minutes.
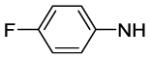
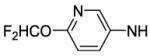
4-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-N-[6-(difluoromethoxy)pyridin-3-yl]piperazine-1-carboxamide (65).
The title compound was prepared by the general procedure G to provide 71 mg (100%) of an off white amorphous solid, mp 118-120 °C. Rf = 0.28 (2% MeOH/60% EtOAc/hexanes; blue with UV). 1H NMR (300 MHz, CDCl3) δ 8.42 (s, 1H), 8.07 (d, J = 2.3 Hz, 1H), 7.94 (dd, J = 8.8, 2.5 Hz, 1H), 7.51 (d, J = 7.0 Hz, 1H), 7.30-7.44 (m, 6H), 7.20 (d, J = 8.5 Hz, 2H), 6.87 (d, J = 8.8 Hz, 1H), 6.57 (s, 1H), 4.46 (br s, 4H), 3.60-3.85 (m, 4H). MS (m/z) 611.9 (M+1). HPLC = >99% at 20.59 minutes.
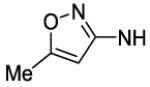
4-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-N-(5-methyl-1,2-oxazol-3-yl)piperazine-1-carboxamide (66).
The title compound was prepared by the general procedure G to provide 48 mg (73%) of a white crystalline solid, mp 230-231 °C. Rf = 0.52 (40% EtOAc/Hexanes; blue with UV). 1H NMR (300 MHz, CDCl3) δ 8.42 (s, 1H), 7.50 (d, J = 7.0 Hz, 1H), 7.31-7.46 (m, 6H), 7.20 (d, J = 8.7 Hz, 2H), 4.45 (br s, 4H), 3.91 (br s, 2H), 3.83 (br s, 2H), 1.59 (s, 3H). MS (m/z) no TIC (M+1). HPLC = >98% at 21.63 minutes.
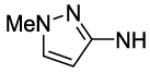
4-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-N-(1-methyl-1H-pyrazol-3-yl)piperazine-1-carboxamide (67).
The title compound was prepared by the general procedure G to provide 48 mg (73%) of an off white crystalline solid, mp 231-233 °C. Rf = 0.31 (4% MeOH/80% EtOAc/Hexanes; blue with UV). 1H NMR (300 MHz, CDCl3) δ 8.41 (br s, 1H), 7.52 (d, J = 6.8 Hz, 1H), 7.15-7.47 (m, 7H), 6.54 (s, 1H), 4.42 (br s, 4H), 3.77 (s, 3H), 3.67 (br s, 4H). MS (m/z) 549.3 (M+1). HPLC = >99% at 18.77 minutes.
4-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-N-(1-methyl-1H-pyrazol-4-yl)piperazine-1-carboxamide (68).
The title compound was prepared by the general procedure G to provide 55 mg (84%) of a tan crystalline solid, mp 213-215 °C. Rf = 0.23 (6% MeOH/80% EtOAc/Hexanes; blue with UV). 1H NMR (300 MHz, CDCl3) δ 8.42 (s, 1H), 7.72 (s, 1H), 7.51 (d, J = 6.8 Hz, 1H), 7.30-7.46 (m, 6H), 7.20 (d, J = 8.5 Hz, 2 ), 6.34 (s, 1H), 4.44 (br s, 4H), 3.85 (s, 3H), 3.53-3.75 (m, 4H). MS (m/z) 549.9 (M+1). HPLC = >99% at 18.93 minutes.
4-[8-(2-Chlorophenyl)-9-(4-chlorophenyl)-9H-purin-6-yl]-N-[(2-fluorobenzene)sulfonyl]piperazine-1-carboxamide (69).
To an ice-cold solution of 2-fluorobenzenesulfonamide (42 mg, 2 equiv) in THF (1 mL), 1 M NaHMDS (0.26 mL, 2.2 equiv) was added drop-wise (precipitate formed). After 10 minutes, the 4-nitrophenylcarbamate of 8 (71 mg, 0.12 mmol, prepared from 8 by general procedure B) was added (turned yellow). After 10 minutes, the mixture was stirred at rt of 1 hour. DMF (0.8 mL) was added and the mixture heated at 60 °C for 15 hours. Brine (0.8 mL) was added, followed by 2 N HCl (0.18 mL, 3 equiv). Ethyl acetate (3 mL) was added and after 10 minutes, the aqueous layer was removed. The organic layer was washed with brine (2×0.5 mL). Celite (600 mg) was added to the organic layer and the solvent was evaporated. Purification by flash chromatography provided 53 mg (71%) of a white crystalline solid, mp 209-211 °C. Rf = 0.24 (10% MeOH/70% EtOAc/Hexanes; blue with UV). 1H NMR (300 MHz, CDCl3) δ 8.34 (s, 1H), 7.88-8.07 (m, 1H), 7.49 (d, J = 6.6 Hz, 1H), 7.27-7.44 (m, 6H), 7.07-7.25 (m, 4 H), 4.27 (br s, 4H), 3.66 (br s, 4H). MS (m/z) 626.5 (M+1), 624.6 (M-1). HPLC = 98%, 58% at 11.72 min & 40% at 15.12 min.
Calcium mobilization and radioligand displacement assays
Each compound was biologically characterized using a functional fluorescent hCB1 activated Gαq16-coupled intracellular calcium mobilization assay in CHO-K1 cells, as has been described in our previous publications and apparent affinity (Ke) values were determined.[17, 20] Briefly, CHO-K1 cells were engineered to co-express human CB1 and Gαq16. Activation of CB1 by an agonist then leads to generation of inositol phospahatase 3 (IP3) and activation of IP3 receptors, which leads to mobilization of intracellular calcium. Calcium flux was monitored in a 96-well format using the fluorescent dye Calcein-4 AM in an automated plate reader (Flexstation, Molecular Devices). The antagonism of a test compound was measured by its ability to shift the concentration response curve of the synthetic CB1 agonist CP55940 rightwards using the equation:
Ke = [Ligand]/[DR-1] where DR is the EC50 ratio of CP55940 in the presence or absence of a test agent.
For some assays, cells were loaded with Calcein-4 AM as described below and directly stimulated with various concentrations of a test agent for 90 seconds. Decrease in basal fluorescence was used in these assays to calculate EC50 values.
Further characterization of select compounds was performed using radioligand displacement of [3H]CP55940 and equilibrium dissociation constant (Ki) values were determined as described previously.[17, 20] Selectivity of these compounds at hCB1 versus hCB2 was also determined by obtaining Ki values at either receptor in membranes of CHO cells over-expressing either receptor. Data reported are average values from 3-6 measurements typically with <30% standard error.
MDCK-mdr1 permeability assays
MDCK-mdr1 cells obtained from the Netherlands Cancer Institute were grown on Transwell type filters (Corning) for 4 days to confluence in DMEM/F12 media containing 10% fetal bovine serum and antibiotics. Compounds were added to the apical side at a concentration of 10 μM in a transport buffer comprising of 1X Hank’s balanced salt solution, 25 mM D-glucose and buffered with HEPES to pH 7.4. Samples were incubated for 1 h at 37 °C and carefully collected from both the apical and basal side of the filters. Compounds selected for MDCK-mdr1 cell assays were infused on an Applied Biosystems API-4000 mass spectrometer to optimize for analysis using multiple reaction monitoring (MRM), as previously described.[25] The chromatography was conducted with an Agilent 1100 binary pump with a flow rate of 0.5 mL/min. Mobile phase solvents were 0.1% formic acid in water (A) and 0.1% formic acid in methanol (B). The solvent conditions were 10% B for 1 minute, followed by a gradient to 95% B over 5 minutes. Data reported are average values from 2-3 measurements.
Pharmacokinetic testing
Male and female C57BL6 mice were procured from Jackson Laboratories at 8 weeks of age. Animals were dosed with compounds in a vehicle comprised of 100% corn oil, 90% corn oil and 10% DMSO, or 1% NMP and 0.3% Tween 80 in 0.5% sodium carboxymethylcellulose (medium viscosity; deionized water). Animals were sacrificed at various time-points and samples were removed. Pharmacokinetic analyses were performed as has been described in our previous publications using Phoenix WinNonlin (Certara).[17]
Table 4.
Mouse PK Data for Compound 65
| Structure | PO Dose mg/kg |
Half-Lifea hours | Plasma Tmax hours |
Max. Plasma Conc. ng/mL |
Max. Brain/ Plasma |
|---|---|---|---|---|---|
|
2.5 | 6.1 | 4.0 | 500 | 0.18 |
Time points were: 1, 2, 4, 8 & 24 hours post-dosing.
Acknowledgments
We express our gratitude to the NIDA drug supply program for providing radiolabeled probes and to Dr. Brian Thomas for supplying the CB1 cells. We thank Keith Warner, Taylor Rosa, Aliah Hackney, Elaine Gay and Melody Markley for excellent technical help. This research was funded by research grants AA022235 and DK100414 to RM from NIH.
Abbreviations Used
- BBB
Blood-Brain Barrier
- CB1
Cannabinoid Receptor 1
- CB2
Cannabinoid Receptor 2
- CHO-K1
Chinese Hamster Ovary Cells
- CNS
Central Nervous System
- EtOH
ethanol
- EtOAc
ethyl acetate
- Ke
apparent affinity constant
- MDCK-mdr1
Madin-Darby canine kidney cells transfected with the human MDR1 gene
- IP3
Inositol Phospahatase 3
- MRM
Multiple Reaction Monitoring
- rt
room temperature
- TEA
triethylamine
- TFA
trifluoroacetic acid
Footnotes
Conflict of interest statement
Authors do not have any known conflicts of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Janero DR, Makriyannis A. Cannabinoid receptor antagonists: pharmacological opportunities, clinical experience, and translational prognosis. Expert Opin Emerg Drugs. 2009; 14: 43–65. [DOI] [PubMed] [Google Scholar]
- 2.Porter AC, Felder CC. The endocannabinoid nervous system: unique opportunities for therapeutic intervention. Pharmacol Ther 2001; 90: 45–60. [DOI] [PubMed] [Google Scholar]
- 3.Pacher P, Batkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev 2006; 58: 389–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pacher P, Kunos G. Modulating the endocannabinoid system in human health and disease--successes and failures. FEBS J 2013; 280: 1918–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Christensen R, Kristensen PK, Bartels EM, Bliddal H, Astrup A. Efficacy and safety of the weight-loss drug rimonabant: a meta-analysis of randomised trials. Lancet. 2007; 370: 1706–1713. [DOI] [PubMed] [Google Scholar]
- 6.Chorvat RJ. Peripherally restricted CB1 receptor blockers. Bioorg Med Chem Lett 2013; 23: 4751–4760. [DOI] [PubMed] [Google Scholar]
- 7.Chang CP, Wu CH, Song JS, Chou MC, Wong YC, Lin Y, Yeh TK, Sadani AA, Ou MH, Chen KH, Chen PH, Kuo PC, Tseng CT, Chang KH, Tseng SL, Chao YS, Hung MS, Shia KS. Discovery of 1-(2,4-dichlorophenyl)-N-(piperidin-1-yl)-4-((pyrrolidine-1-sulfonamido)methyl)-5-(5-((4-(trifluoromethyl)phenyl)ethynyl)thiophene-2-yl)-1H-pyrazole-3-carboxamide as a novel peripherally restricted cannabinoid-1 receptor antagonist with significant weight-loss efficacy in diet-induced obese mice. J Med Chem 2013; 56: 9920–9933. [DOI] [PubMed] [Google Scholar]
- 8.Klumpers LE, Fridberg M, de Kam ML, Little PB, Jensen NO, Kleinloog HD, Elling CE, van Gerven JM. Peripheral selectivity of the novel cannabinoid receptor antagonist TM38837 in healthy subjects. Br J Clin Pharmacol 2013; 76: 846–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Navarro HA, Howard JL, Pollard GT, Carroll FI. Positive allosteric modulation of the human cannabinoid (CB(1)) receptor by RTI-371, a selective inhibitor of the dopamine transporter. Br J Pharmacol 2009; electronic, PMID: 19226282, see attached publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ross RA. Allosterism and cannabinoid CB(1) receptors: the shape of things to come. Trends Pharmacol Sci 2007; 28: 567–572. [DOI] [PubMed] [Google Scholar]
- 11.Sharma MK, Murumkar PR, Barmade MA, Giridhar R, Yadav MR. A comprehensive patents review on cannabinoid 1 receptor antagonists as antiobesity agents. Expert Opin Ther Pat 2015; 25: 1093–1116. [DOI] [PubMed] [Google Scholar]
- 12.Varga B, Kassai F, Szabo G, Kovacs P, Fischer J, Gyertyan I. Pharmacological comparison of traditional and non-traditional cannabinoid receptor 1 blockers in rodent models in vivo. Pharmacol Biochem Behav 2017; 159: 24–35. [DOI] [PubMed] [Google Scholar]
- 13.Matthews JM, McNally JJ, Connolly PJ, Xia M, Zhu B, Black S, Chen C, Hou C, Liang Y, Tang Y, Macielag MJ. Tetrahydroindazole derivatives as potent and peripherally selective cannabinoid-1 (CB1) receptor inverse agonists. Bioorg Med Chem Lett 2016; 26: 5346–5349. [DOI] [PubMed] [Google Scholar]
- 14.Rover S, Andjelkovic M, Benardeau A, Chaput E, Guba W, Hebeisen P, Mohr S, Nettekoven M, Obst U, Richter WF, Ullmer C, Waldmeier P, Wright MB. 6-Alkoxy-5-aryl-3-pyridinecarboxamides, a new series of bioavailable cannabinoid receptor type 1 (CB1) antagonists including peripherally selective compounds. J Med Chem 2013; 56: 9874–9896. [DOI] [PubMed] [Google Scholar]
- 15.Tam J, Liu J, Mukhopadhyay B, Cinar R, Godlewski G, Kunos G. Endocannabinoids in liver disease. Hepatology. 2011; 53: 346–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fulp A, Bortoff K, Zhang Y, Snyder R, Fennell T, Marusich JA, Wiley JL, Seltzman H, Maitra R. Peripherally selective diphenyl purine antagonist of the CB1 receptor. J Med Chem 2013; 56: 8066–8072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fulp A, Bortoff K, Zhang Y, Seltzman H, Mathews J, Snyder R, Fennell T, Maitra R. Diphenyl purine derivatives as peripherally selective cannabinoid receptor 1 antagonists. J Med Chem 2012; 55: 10022–10032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Griffith DA, Hadcock JR, Black SC, Iredale PA, Carpino PA, DaSilva-Jardine P, Day R, DiBrino J, Dow RL, Landis MS, O'Connor RE, Scott DO. Discovery of 1-[9-(4-chlorophenyl)-8-(2-chlorophenyl)-9H-purin-6-yl]-4-ethylaminopiperidine-4- carboxylic acid amide hydrochloride (CP-945,598), a novel, potent, and selective cannabinoid type 1 receptor antagonist. J Med Chem 2009; 52: 234–237. [DOI] [PubMed] [Google Scholar]
- 19.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews. 2001; 46: 3–26. [DOI] [PubMed] [Google Scholar]
- 20.Fulp A, Bortoff K, Seltzman H, Zhang Y, Mathews J, Snyder R, Fennell T, Maitra R. Design and synthesis of cannabinoid receptor 1 antagonists for peripheral selectivity. J Med Chem 2012; 55: 2820–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shao Z, Yin J, Chapman K, Grzemska M, Clark L, Wang J, Rosenbaum DM. High-resolution crystal structure of the human CB1 cannabinoid receptor. Nature. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hua T, Vemuri K, Pu M, Qu L, Han GW, Wu Y, Zhao S, Shui W, Li S, Korde A, Laprairie RB, Stahl EL, Ho JH, Zvonok N, Zhou H, Kufareva I, Wu B, Zhao Q, Hanson MA, Bohn LM, Makriyannis A, Stevens RC, Liu ZJ. Crystal Structure of the Human Cannabinoid Receptor CB1. Cell. 2016; 167: 750–762 e714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Q, Rager JD, Weinstein K, Kardos PS, Dobson GL, Li J, Hidalgo IJ. Evaluation of the MDR-MDCK cell line as a permeability screen for the blood-brain barrier. Int J Pharm 2005; 288: 349–359. [DOI] [PubMed] [Google Scholar]
- 24.Hadcock JR, Griffith DA, Iredale PA, Carpino PA, Dow RL, Black SC, O'Connor R, Gautreau D, Lizano JS, Ward K, Hargrove DM, Kelly-Sullivan D, Scott DO. In vitro and in vivo pharmacology of CP-945,598, a potent and selective cannabinoid CB(1) receptor antagonist for the management of obesity. Biochem Biophys Res Commun 2010; 394: 366–371. [DOI] [PubMed] [Google Scholar]
- 25.Fulp A, Bortoff K, Zhang Y, Seltzman H, Snyder R, Maitra R. Towards rational design of cannabinoid receptor 1 (CB1) antagonists for peripheral selectivity. Bioorg Med Chem Lett 2011; 21: 5711–5714. [DOI] [PMC free article] [PubMed] [Google Scholar]