

Abstract
T-cell dysfunction contributes to tumor immune escape in patients with cancer and is particularly severe amidst glioblastoma (GBM). Among other defects, T-cell lymphopenia is characteristic, yet often attributed to treatment. We reveal that even treatment-naïve patients and mice with GBM can harbor AIDS-level CD4 counts, as well as contracted, T-cell deficient lymphoid organs. Missing naïve T-cells are instead found sequestered in large numbers in the bone marrow. This phenomenon characterizes not only GBM but a variety of other cancers, although only when tumors are introduced into the intracranial compartment. T-cell sequestration is accompanied by tumor-imposed loss of S1P1 from the T-cell surface and is reversible upon precluding S1P1 internalization. In murine models of GBM, hindering S1P1 internalization and reversing sequestration licenses T-cell-activating therapies that were previously ineffective. Sequestration of T-cells in bone marrow is therefore a tumor-adaptive mode of T-cell dysfunction, whose reversal may constitute a promising immunotherapeutic adjunct.
INTRODUCTION
Cancer-induced T-cell dysfunction facilitates tumor immune escape1,2 and can be particularly severe in patients with glioblastoma (GBM)3–6. Despite near universal confinement to the intracranial compartment7, GBM frequently depletes systemic T-cells of both number and function. Regarding the former, T-cell lymphopenia is prominent but has remained incompletely explained for four decades8.
Sphingosine-1-phosphate receptor 1 (S1PR1 or S1P1) is one of five G protein-coupled receptors (GPCR) (S1P1 through 5) that bind the lipid second messenger, sphingosine-1-phosphate (S1P)9,10. The S1P-S1P1 axis is increasingly recognized for its role governing lymphocyte trafficking. Naïve T-cell egress from thymus and secondary lymphoid organs cannot occur without functional S1P1 on the cell surface: S1P1 thus serves naive T-cells as a lymphoid organ “exit visa”11,12. Concentrations of S1P are higher in the blood and lymph13, establishing a chemotactic gradient that directs T-cell egress from lymphoid organs into the circulation. Disruptions to this gradient result in T-cell trapping within lymphoid organs and pursuant T-cell lymphopenia14. Such T-cell sequestration is the intended mechanism of action for the drug fingolimod (FTY720), which is FDA-approved for multiple sclerosis (MS). Fingolimod induces rapid S1P1 internalization, confining T-cells to lymphoid organs, where they are prevented from trafficking to the brain and eliciting autoimmunity9.
Classically, surface S1P1 affords T-cell egress from the spleen, lymph node, and thymus11,15–17. A role mediating egress from bone marrow has been shown, however, and this role increases when other lymphoid organs are missing or deficient18. Here, we reveal that T-cell numbers are severely deficient in the blood and contracted lymphoid organs of patients and mice with GBM. “Missing” naïve T-cells are instead found sequestered in large numbers in the bone marrow. This phenomenon characterizes not only GBM, but a variety of cancers, although solely when these tumors are introduced intracranially. Sequestration accompanies tumor-imposed loss of S1P1 from the T-cell surface and is reversible upon precluding receptor internalization. In murine models of GBM, hindering S1P1 internalization and reversing sequestration licenses T-cell-activating therapies that were previously ineffective.
RESULTS
T-cell lymphopenia and splenic contraction in treatment-naïve patients with glioblastoma
We reviewed the records of patients at our institution from the prior 10 years meeting the following criteria: 1) GBM diagnosis; 2) complete blood counts (CBC) at presentation; and 3) CT of the chest/abdomen/pelvis. Lymphocyte counts and splenic volumes were assessed. GBM patient data were compared to all trauma patients evaluated in the emergency department over the same 10-year period fitting the same age range and with a CBC and normal abdominal CT imaging, as determined by a radiologist. Exclusion criteria for both cohorts included history of autoimmune disorder, immune-deficiency, hematologic cancer, splenic injury, active infection, or chemotherapy. Ultimately, 300 patients with GBM and 46 controls satisfied the above inclusion criteria (Supplementary Table 1): Numbers were not determined a priori. Spleen volumes were determinable in 278 patients and 43 controls; dexamethasone exposure/dosing information was available for 284 patients.
Generalized lymphopenia was present in treatment-naïve GBM patients, with treatment-naïve defined as no prior biopsy, resection, chemotherapy, or radiation. As some patients had been diagnosed at outside hospitals prior to presentation, previous dexamethasone exposure varied. Patients were divided into those entirely dexamethasone-naïve versus those receiving at least a single dose of dexamethasone. Lymphopenia was present in 24.7% of all GBM patients (18.2% of dexamethasone-naïve; 37.1% of dexamethasone-experienced) compared to 10.9% of controls, with lymphopenia defined as lymphocyte count < 1000 cells/µL) (Supplementary Fig 1a).
To examine T-cell counts specifically, we prospectively studied a new cohort of treatment-naïve patients with GBM (n=15), as well as controls meeting similar demographics (n=13) (Supplementary Table 2). Patients were dexamethasone-naïve and demonstrated a prevalent, severe reduction in T-cell counts, with a mean CD4 count of 411 cells/µL (control mean 962 cells/µL). CD8 counts were also significantly lower in patients than controls (Fig. 1a). Notably, ~15% of treatment-naïve GBM patients presented with CD4 counts less than 200 cells/µL, the threshold demarcating AIDS in HIV-infected individuals. T-cell loss trended towards being more severe among naïve T-cells (CD27+CD45RA+) than among memory (CD45RO+), with patients exhibiting decreased ratios of naïve to memory T-cells compared to controls (Supplementary Fig. 1b, c).
Fig. 1. T-cell lymphopenia and splenic contraction in treatment-naïve patients with GBM.
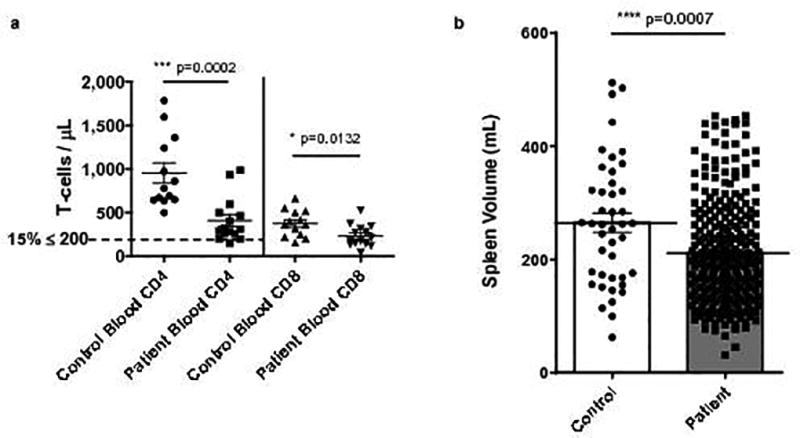
a, Blood CD4+ and CD8+ T-cell counts measured prospectively in n=15 newly diagnosed patients with GBM (prior to therapy) and n=13 age-matched controls. b, Spleen volume on abdominal CT scans performed on n=278 newly diagnosed treatment-naïve patients with GBM and n=43 age-matched controls. All data in a–b are shown as mean ± s.e.m. P values were determined by two-tailed, unpaired Student’s t-test.
We hypothesized that splenic sequestration might explain the T-cell lymphopenia, with resultant splenomegaly. To the contrary, returning to the retrospective dataset, we observed that splenic volume was markedly contracted in GBM patients (32% mean size reduction), with an overall mean of 217.1 milliliters (mL) compared to 317.3 mL in controls (Fig. 1b). Splenic volume in patients was not influenced by dexamethasone exposure (214.4 mL in dexamethasone-naïve; 219.3 mL in dexamethasone-experienced, Supplementary Fig. 1d).
Recapitulated T-cell lymphopenia and lymphoid organ contraction in murine glioma
To assess for similar changes in murine glioma models, SMA-560 or CT2A murine glioma cells were implanted stereotactically into the brains (intracranial = IC) of syngeneic VM/Dk or C57BL/6 mice, respectively. Blood, spleen, cervical lymph nodes (CLN), and thymus were analyzed once tumors had become sizeable (Day 18–20). Mice were exclusively treatment-naïve. Both tumor models demonstrated significant T-cell lymphopenia in the CD4 and CD8 compartments (Fig. 2a, b). As with patients, naïve (CD62LhiCD44lo) T-cell numbers were more prominently diminished. Memory (CD44hi) T-cell counts were not significantly reduced (Supplementary Fig. 2a). The splenic contraction observed in patients with GBM was recapitulated in mice (Fig. 2c), and volume contractions further typified CLN and thymus (thymus depicted in Supplementary Fig. 2c).
Fig. 2. Recapitulated T-cell lymphopenia and lymphoid organ contraction in murine glioma.
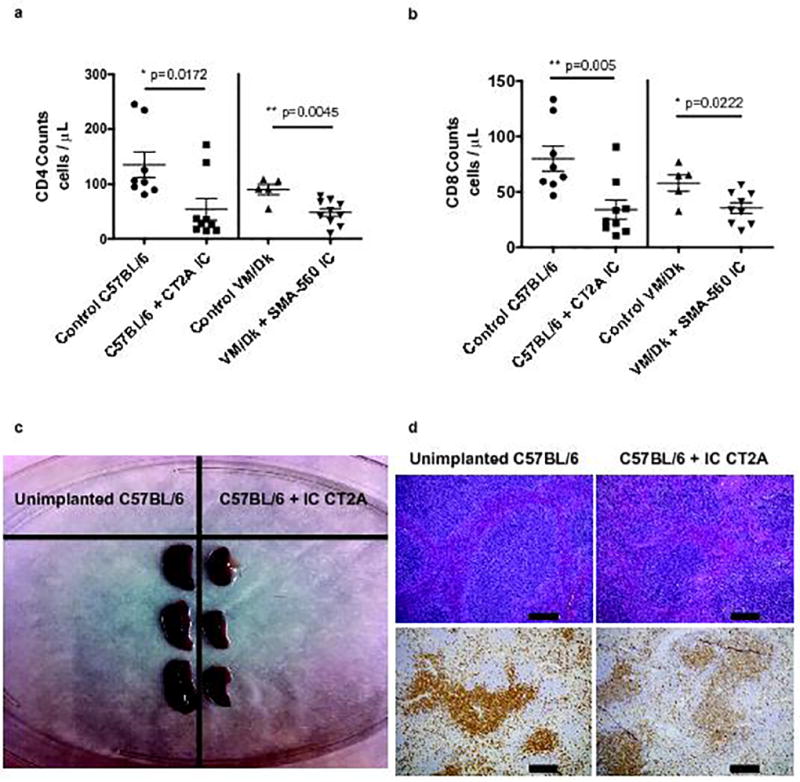
a, Blood CD4 T-cell counts in n=8 control C57BL/6 and n=5 control VM/Dk mice, or n=9 IC CT2A glioma-bearing C57BL/6 mice and n=10 SMA-560 glioma-bearing VM/Dk mice. b, Blood CD8 T-cell counts in n=8 control C57BL/6 and n=5 control VM/Dk mice, or n=9 intracranial (IC) CT2A glioma-bearing C57BL/6 mice and n=9 SMA-560 glioma-bearing VM/Dk mice. Data in a–b are shown as mean ± s.e.m. P values were determined by two-tailed, unpaired Student’s t-test. c, Gross image depicting spleens taken from unimplanted or IC CT2A glioma-bearing C57BL/6 mice. d, Hematoxylin & eosin (H&E) staining (upper panel) or immuno-histochemistry (IHC) for CD3 (lower panel) of formalin-fixed paraffin-embedded (FFPE) spleen taken from unimplanted or IC CT2A glioma-bearing C57BL/6 mice. Histopathologic examination of spleens from IC CT2A mice showed diminution in T-dependent lymphoid areas. These findings accompanied marked organ lymphopenia and lymphoid necrosis. IHC confirmed spleens of IC CT2A mice had marked T-cell lymphopenia, scale bar = 200 µm. All data in a–d are representative findings from one of at least three independently repeated experiments with similar results. Blood (a–b) was drawn and spleens (c–d) were harvested at 18 days following tumor implantation.
Accompanying the volume reductions in lymphoid organs were significant decreases to organ T-cell counts (spleen counts depicted in Supplementary Fig. 2b). Histologic examination and immunohistochemical staining of spleen, thymus, and cervical lymph node revealed marked lymphodepletion, primarily in T-cell-dependent areas. Lymphoid necrosis was also present (Fig. 2d; Supplementary Fig. 2d). Severe T-cell disappearance thus appeared systemic, characterizing both blood and lymphoid organs.
Naïve T-cells accumulate in the bone marrow of mice and patients with GBM
Diminished naïve T-cell counts suggested deficient production, leading us to investigate the bone marrow of glioma-bearing mice for T-cell progenitor frequencies. This analysis instead revealed that naive T-cell disappearance from blood and lymphoid organs was met conversely with 3- to 5-fold expansions of mature, single-positive T-cell numbers within the bone marrow of mice bearing either SMA-560 or CT2A IC (Fig. 3a; sample flow cytometry in Supplementary Fig. 3a). Immune cell accumulation in the bone marrow was T-cell-specific, with no increases observed for NK-cells, B-cells, or granulocytes/monocytes (Supplementary Fig. 3b). Both CD4+ and CD8+ T-cells accumulated (Fig. 3b), albeit disproportionately those with a naïve phenotype (CD44loCD62Lhi) (Fig. 3c). A time course for T-cell accumulation is provided in Supplementary Fig. 3c.
Fig. 3. Naïve T-cells accumulate in the bone marrow of mice and patients with GBM.
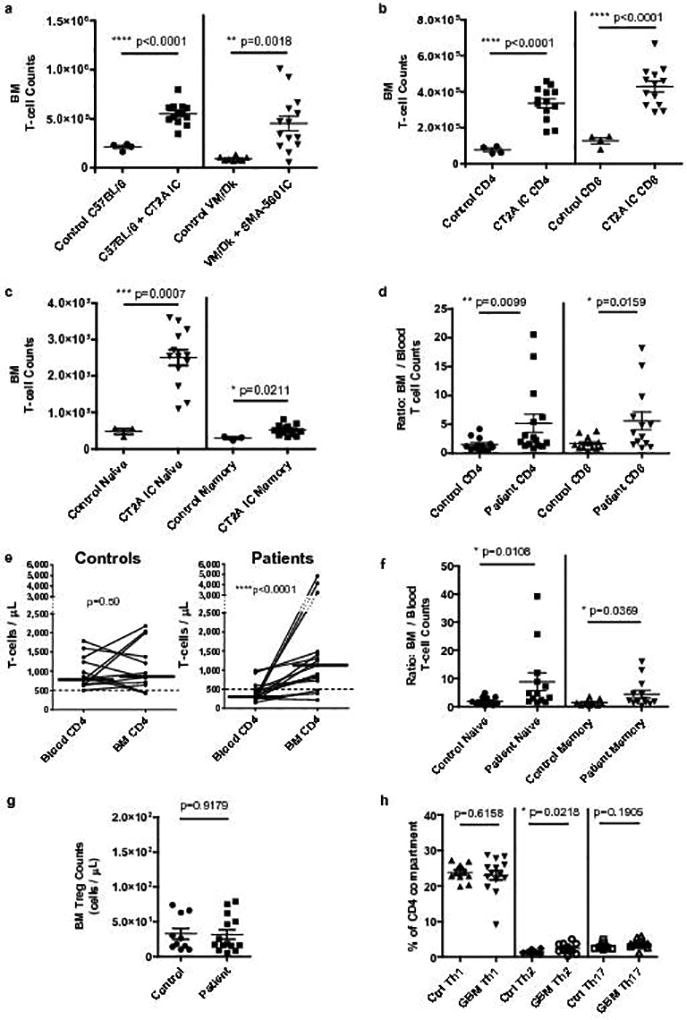
a, Bone marrow T-cell counts from a single hind leg femur and tibia in n=4 control C57BL/6 and n=8 control VM/Dk mice, or n=13 IC CT2A glioma-bearing C57BL/6 mice and n=14 SMA-560 glioma-bearing VM/Dk mice. b, Bone marrow CD4+ and CD8+ T-cell counts in n=4 control C57BL/6 or n=13 IC CT2A mice. c, Bone marrow naïve and memory CD4+ T-cell counts in n=3 control C57BL/6 or n=13 IC CT2A mice. Cumulative data from three experiments are depicted in a–c. d, The ratio of bone marrow to blood CD4 and CD8 counts were calculated for n=15 treatment-naïve GBM patients and n=13 spinal fusion controls. e, For the same n=13 controls and n=15 GBM patients, paired absolute CD4+ T-cell counts in blood and bone marrow are depicted. Median counts in each compartment are identified by horizontal lines. Dashed line demarcates low cut-off of normal CD4 range. Similar results were obtained for CD8+ T-cells. f, For the same n=13 controls and n=15 GBM patients, the ratio of bone marrow to blood naïve and memory T-cells was calculated. g, Regulatory T-cells (Treg) counts in the bone marrow of n=11 controls compared to n=15 GBM patients. h, Relative frequencies of CD4+ T-helper cell subsets: Th1 (CXCR3+CCR6−), Th2 (CXCR3−CCR6−), Th17 (CXCR3−CCR6+) in bone marrow of n=13 controls and n=15 GBM patients. Data in a–d, f–h are shown as mean ± s.e.m. P values were determined by two-tailed, unpaired Student’s t-test (a–c,g–h) and two-tailed, Mann Whitney test with Gaussian approximation (d, f). Blood and bone marrow CD4+ T-cell counts in e were compared using Wilcoxon matched-pairs signed rank tests, P values are depicted.
This finding unexpected, we investigated whether it was mirrored in patients with GBM. Blood and bone marrow aspirates were collected from 15 treatment-naïve GBM patients and 15 healthy controls undergoing spinal fusion (from whom bone marrow aspirates are often collected intra-operatively for employment in fusion constructs). All bone marrow was harvested from patients and controls following the induction of general anesthesia for their respective surgeries (resection or fusion). Aspirates were collected from the iliac crest prior to incision or to administration of any indicated intra-operative steroids. Samples were analyzed by flow cytometry.
In patients with GBM, we uncovered a significant re-allocation of T-cells to bone marrow, compared to blood. While bone marrow T-cell counts varied widely among all individuals, controls typically had matching T-cell counts across bone marrow and blood (median marrow to blood ratio for CD4+ T-cells 1.06:1; for CD8+ T-cells 1.42:1). This homeostasis was disrupted in GBM patients, who nearly universally had higher T-cell counts in their bone marrow, with marrow to blood ratios ranging as high as 20:1 (Fig. 3d). In GBM patients, there was a consistent increase in both CD4 and CD8 counts as one moved from blood to bone marrow (p<0.0001 and p=0.0007, respectively, Wilcoxon matched-pairs signed rank test; CD4+ T-cells depicted in Fig. 3e). Indeed, 14 of 15 GBM patients had higher T-cell counts in bone marrow than in blood, while for controls this was true in only 8 of 15, (p=0.01, Chi Square analysis). As with mice, naïve (CD27+CD45RA+) T-cells were over-represented in the bone marrow (CD4+ T-cells depicted in Fig. 3f, sample flow cytometry depicted in Supplementary Fig. 3d). Exploring CD4 subsets, we found no difference in the counts of bone marrow Tregs across patients and controls (Fig. 3g). Although most T-cells detected in marrow were naïve, we analyzed differentiated CD4+ helper T-cell (Th1, 2, or 17) subsets, finding no substantial differences in the relative representation of each across the bone marrow of patients and controls (Fig. 3h).
T-cell accumulation in bone marrow reflects intracranial tumor location rather than tumor histologic type
We examined whether accumulation of T-cells in bone marrow characterized cancer more generally or, rather, was specific to either glioma or the intracranial tumor environment. To test this, E0771 breast carcinoma, B16F10 melanoma, Lewis lung carcinoma (LLC), or CT2A gliomas were each implanted either IC or subcutaneously (SC) into syngeneic C57BL/6 mice and bone marrow T-cell frequency assessed. Notably, each IC tumor provoked significant accumulation of T-cells in bone marrow, regardless of the primary tumor type. Conversely, none of the SC-situated tumors, including glioma, evoked the same phenomenon (Fig. 4a). Control IC injections with saline and methycellulose produced no increase in bone marrow T-cell numbers (Supplementary Fig. 4a). CD4+ and CD8+ T-cells accumulated in the bone marrow in approximately equal proportions across all tumor types (Fig 4b), but for all models, accumulating T-cells were disproportionatley naïve (CD44loCD62Lhi) (Fig. 4c).
Fig. 4. T-cell accumulation in bone marrow reflects intracranial tumor location rather than tumor histologic type.
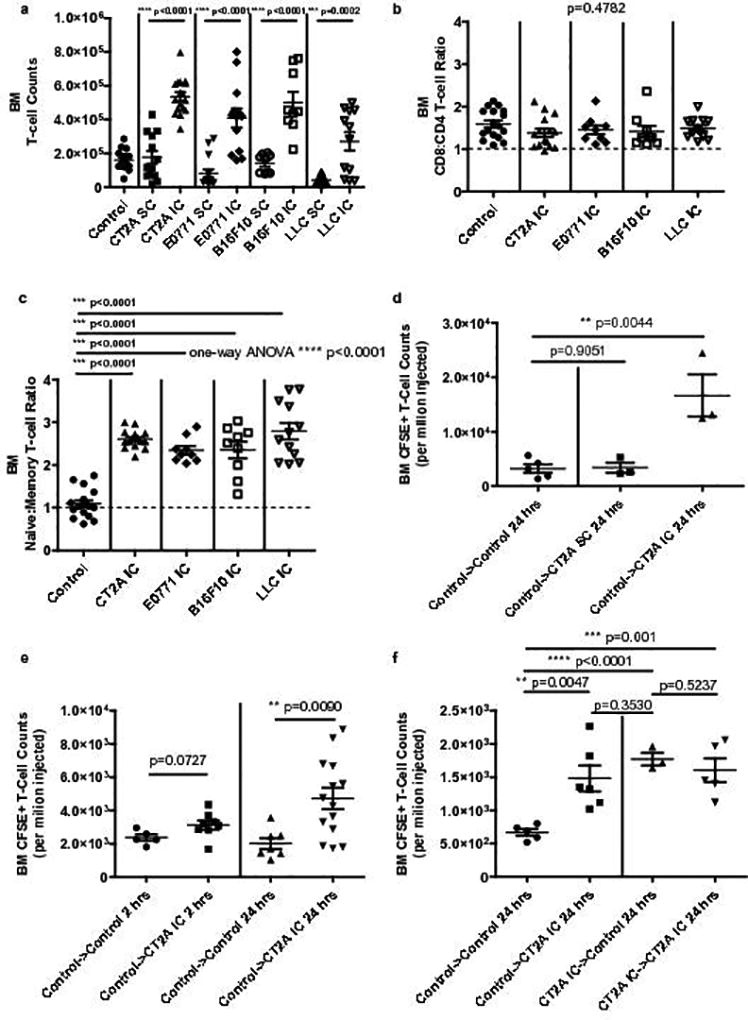
a, Bone marrow T-cell counts in n=13 SC and n=17 IC CT2A glioma-bearing C57BL/6 mice, or n=15 SC and n=13 IC E0771 breast carcinoma-bearing mice, or n=9 SC and n=9 IC B16F10 melanoma-bearing mice, or n=13 SC and n=12 IC Lewis Lung carcinomas (LLC)-bearing mice. b, CD8:CD4 ratios in the bone marrow of n=15 IC CT2A, n=9 IC E0771, n=9 IC B16F10, or n=12 IC LLC-bearing mice. c, Naïve:Memory T-cells ratios in the bone marrow of the same tumor-bearing mice as in b. Counts and ratios in a–c were compared to those in the bone marrow of n=17 control C57BL/6 mice. Data in a–c are cumulative results from a minimum of two experiments with each tumor type. d, Accumulation of adoptively transferred CFSE-labeled T-cells in the bone marrow of n=5 recipient control C57BL/6 or CT2A glioma-bearing C57BL/6 mice. Glioma bearing mice harbored tumor in either the IC or SC compartment (n=3 tumor-bearing mice per group). T-cell counts were assessed 24 hours following adoptive transfer. Transferred cells were splenocytes from naïve C57BL/6 (control) donors. e. Accumulation of adoptively transferred CFSE-labeled T-cells in the bone marrow of n=5 control recipient mice and n= 8 IC CT2A-bearing (CT2A IC) recipient mice at 2 hours (left) post-transfer, or n=7 control recipient mice and n=14 IC CT2A-bearing (CT2A IC) recipient mice at 24 hours (right) post-transfer. Transferred cells were splenocytes from naïve C57BL/6 (control) donors. f. Accumulation of adoptively transferred CFSE-labeled T-cells in the bone marrow of n=5 control recipient mice and n=6 CT2A IC recipient mice 24 hours after transfer (left). Transferred cells were splenocytes from naïve C57BL/6 (control) donors. Accumulation of adoptively transferred CFSE-labeled T-cells in the bone marrow of n=3 control recipient mice and n=5 CT2A IC recipient mice 24 hours after transfer (right). Transferred cells were bone marrow cells from CT2A IC mice. Data in d–f are representative findings from one of a minimum of two independently repeated experiments with similar results. All data in a–f are shown as mean ± s.e.m. P values in a, d–f were determined by two-tailed, unpaired Student’s t-test. Ratios in b, c were compared using one-way ANOVA, with post hoc Tukey’s test when applicable. P values are depicted.
The accumulation of largely naïve T-cells in the bone marrow suggested homing or sequestration, rather than in situ antigen-driven expansion. We therefore investigated whether adoptively transferred naïve T-cells would likewise preferentially collect in the bone marrow of glioma-bearing mice. Naïve C57BL/6 spleens were harvested as a source of donor leukocytes. Cells (1×107) were CFSE-labeled and injected via tail vein into naïve control mice or mice bearing CT2A glioma IC or SC. At 24-hours, analysis revealed increased numbers of labeled T-cells in the bone marrow uniquely in hosts bearing CT2A IC, and not in hosts bearing CT2A SC (Fig. 4d). This experiment was repeated with IC CT2A recipient mice, assessing at time-points 2- and 24-hours post-transfer. Although present again in marrow at 24-hours, labeled T-cells failed to accumulate in the bone marrow at the 2-hour time point, suggesting T-cell trapping or sequestration rather than direct bone marrow homing (Fig. 4e).
As a crossover, T-cells that had accumulated within the bone marrow of glioma-bearing mice were harvested, enriched, labeled with CFSE, and injected into tail veins of naïve control mice. T-cells that had accumulated in the bone marrow of glioma-bearing mice re-accumulated within the marrow of naïve mice with equivalent efficiency. Transferring the same cells into tumor-bearing hosts yielded no further increase in marrow accumulation (Fig. 4f). These experiments suggested the acquisition of T-cell phenotypic changes precipitating their sequestration rather than changes to the bone marrow itself (schematic in Supplementary Fig. 4b).
Loss of surface S1P1 on T-cells directs their sequestration in bone marrow in the setting of intracranial tumor
As the experiments in Figs. 4d–f suggested that T-cells acquire alterations facilitating their sequestration in the glioma-bearing state, we hypothesized the relevant alteration might be diminished levels of surface S1P1. (We had previously investigated the CXCR4-CXCL12 axis, failing to find a relationship) (Supplementary Fig. 5a). A role for the S1P-S1P1 axis mediating bone marrow T-cell egress has been observed in alymphoplastic mice lacking thymus and SLO18, a phenotype paralleling the lymphoid organ contraction we observed with GBM. We conjectured that a similar shift in control of T-cell bone marrow egress to the S1P-S1P1 axis might occur with GBM, with disruption to that axis instead fostering T-cell sequestration within the marrow.
Surface S1P1 levels were assessed on T-cells in the bone marrow of control mice and mice bearing IC CT2A glioma. For the detection of otherwise fleeting S1P1 on the cell surface by flow cytometry, we employed a reliable technique in which all harvested tissues were immediately placed into a fixative solution to cross link surface molecules, and in which no solutions contained fetal calf serum in order to avoid ligand-induced internalization19. Mice with IC CT2A demonstrated markedly reduced T-cell S1P1 levels in bone marrow (Fig. 5a, b) and moderately reduced T-cell S1P1 levels in contracted spleens and CLN (Supplementary Fig. 5b).
Fig. 5. Loss of surface S1P1 on T-cells directs their sequestration in bone marrow in the setting of intracranial tumor.
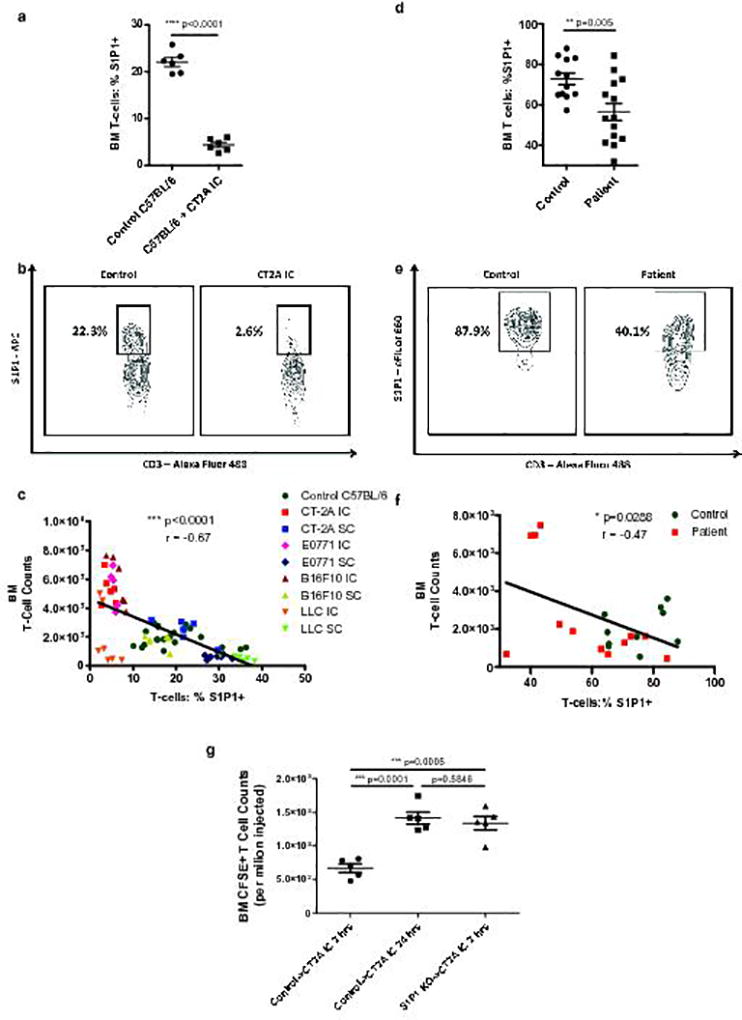
a, The percentage of nascent T-cells expressing surface S1P1 was assessed by flow cytometry in the bone marrow of n=6 control C57BL/6 mice or n=6 mice bearing IC CT2A on Day 18 following tumor implantation. b, Representative flow cytometry plot of data depicted in a. c, Negative correlation between bone marrow T-cell counts and S1P1 levels on bone marrow T-cells across IC and SC murine tumor models. Data in c were obtained from n=6 IC CT2A, n=5 IC E0771, n=6 IC B16F10, n=7 IC LLC, n=6 SC CT2A, n=7 SC E0771, n=6 SC B16F10, and n=7 SC LLC tumor-bearing mice. N=21 control C57BL/6 were also included. Data in c are cumulative results from a minimum of two experiments with each tumor type. d, The percentage of nascent T-cells expressing surface S1P1 was assessed by flow cytometry in the bone marrow of n=14 GBM patients or n=12 age-matched controls. e, Representative flow cytometry plot of data depicted in d. f, Negative correlation between bone marrow T-cell counts and surface S1P1 levels on bone marrow T-cells in n=12 GBM patients and n=10 age-matched controls. g, Relative sequestration of adoptively transferred CFSE-labeled T-cells within the bone marrow of IC CT2A recipient mice either 2 or 24 hours after transfer (n=5 mice per group). As indicated, transferred cells were splenocytes from either control C57BL/6 donors (control) or from S1P1 conditional knockout (S1P1 KO) donors. Data in g are representative findings from one of a minimum of two independently repeated experiments with similar results. All data in a, d, and g are shown as mean ± s.e.m. P values in a, d, and g were determined by two-tailed, unpaired Student’s t-test. Two-tailed, p values and Pearson coefficients for c, f are depicted.
Loss of S1P1 might result from changes to gene expression or from alterations at the protein level (increased receptor internalization or decreased recycling). To assay for altered S1pr1 expression (the gene encoding S1P1), we performed qRT-PCR of T-cells sorted from the spleens of control and glioma-bearing mice. No differences in S1pr1 transcript numbers were detected (Supplementary Fig. 5c). Likewise, RNA flow cytometry of T-cells revealed no differences in levels of the upstream S1pr1 modulators CD69, KLF2, or STAT3 (Supplementary Fig. 5d).
As S1P1 receptor loss or internalization might accompany increased levels of S1P ligand, we assessed S1P concentrations in the plasma and tumors of control and glioma-bearing mice by liquid chromatography – tandem mass spectrometry (LC-MS/MS). No differences were seen in the plasma, and IC CT2A gliomas instead showed slightly decreased levels of S1P compared to normal brain (Supplementary Figs. 5e,f).
We next looked for an association between T-cell S1P1 levels and their sequestration in bone marrow across our various IC and SC tumor models. A strong inverse relationship was uncovered between T-cell S1P1 levels and T-cell numbers in bone marrow (Fig. 5c). Furthermore, bone marrow T-cell sequestration was associated with the presence of contracted spleens and thymuses (Supplementary Fig. 5f,g), suggesting such lymphoid organ contraction as contextually important.
We explored whether similar alterations in S1P1 were present in the bone marrow of patients with GBM, using flow cytometry. The results paralleled our findings in the murine models, with GBM patients exhibiting decreased levels of S1P1 on the T-cell surface compared to healthy, age-matched controls (Fig. 5d,e). Likewise, an inverse relationship emerged between bone marrow T-cell counts and surface S1P1 levels across GBM patients and controls (Fig. 5f).
Given these associations, we investigated whether forced loss of surface S1P1 on T-cells might be sufficient to facilitate their sequestration. In Figs. 4d–f, we demonstrated that transferred T-cells accumulated in the bone marrow of IC glioma-bearing mice after 24-hours. This accumulation had not yet occurred at 2-hours following transfer, which would have otherwise suggested active T-cell homing to marrow. We theorized that the 24-hour delay was a function of the time needed for T-cells to lose surface S1P1 when transferred into glioma-bearing recipients. Conversely, we hypothesized that T-cells with prior loss of surface S1P1 would be subject to more immediate sequestration in mice bearing glioma.
To test this, we employed an S1P1 conditional knockout (KO) mouse. Mice with loxP sites flanking exon 2 of S1pr1 were crossed with mice possessing inducible Cre recombinase20. When treated with tamoxifen, these mice demonstrate a decrease in S1P1 protein levels. Donor splenocytes were harvested from tamoxifen-treated S1P1-KO mice and labeled with CSFE. These were injected via tail vein into IC CT2A-bearing recipients, and accumulation in bone marrow assessed at 2- and 24-hours post-injection. T-cells from S1P1-KO mice accumulated in the bone marrow within 2-hours, while cells from WT C57BL/6 (control) donors did not (Fig 5g). Similar results were obtained when S1P1 loss was instead imposed pharmacologically by treating recipient mice with the S1P1 functional antagonist FTY720 at the time of adoptive transfer (Supplementary Fig. 5i).
Hindering S1P1 internalization abrogates T-cell sequestration in bone marrow
We next examined whether increased/stabilized surface S1P1 might abrogate bone marrow T-cell sequestration in glioma-bearing mice. There currently exist no means for pharmacologically fixing S1P1 on the T-cell surface. We thus employed an S1P1 “knock-in” (S1P1-KI) mouse strain in which lymphocyte S1P1 internalization is hindered (B6.129P2-S1pr1tm1.1Cys/J), resulting in stabilized cell surface receptor levels21. The S1P1 receptor in these mice has disrupted serine residues on the intracellular domain, precluding GRK2 phosphorylation, β-arrestin recruitment, and clathirin-mediated endocytosis.
We tested whether T-cells possessing stabilized, internalization-deficient S1P1 would resist sequestration when adoptively transferred into glioma-bearing mice. Recipient mice were C57BL/6 mice bearing IC CT2A. Donor T-cells were harvested from WT or S1P1-KI mice, CSFE-labeled, and injected IV. Bone marrow of recipient mice was analyzed at 2- and 24-hours post-transfer. At both time-points, T-cells from S1P1-KI donors failed to become appreciably sequestered within bone marrow when compared to T-cells from WT donors (Fig 6a). Likewise, S1P1-KI mice themselves directly implanted with IC CT2A proved similarly resistant to bone marrow T-cell sequestration (Fig. 6b).
Fig. 6. Hindering S1P1 internalization abrogates T-cell sequestration in bone marrow.
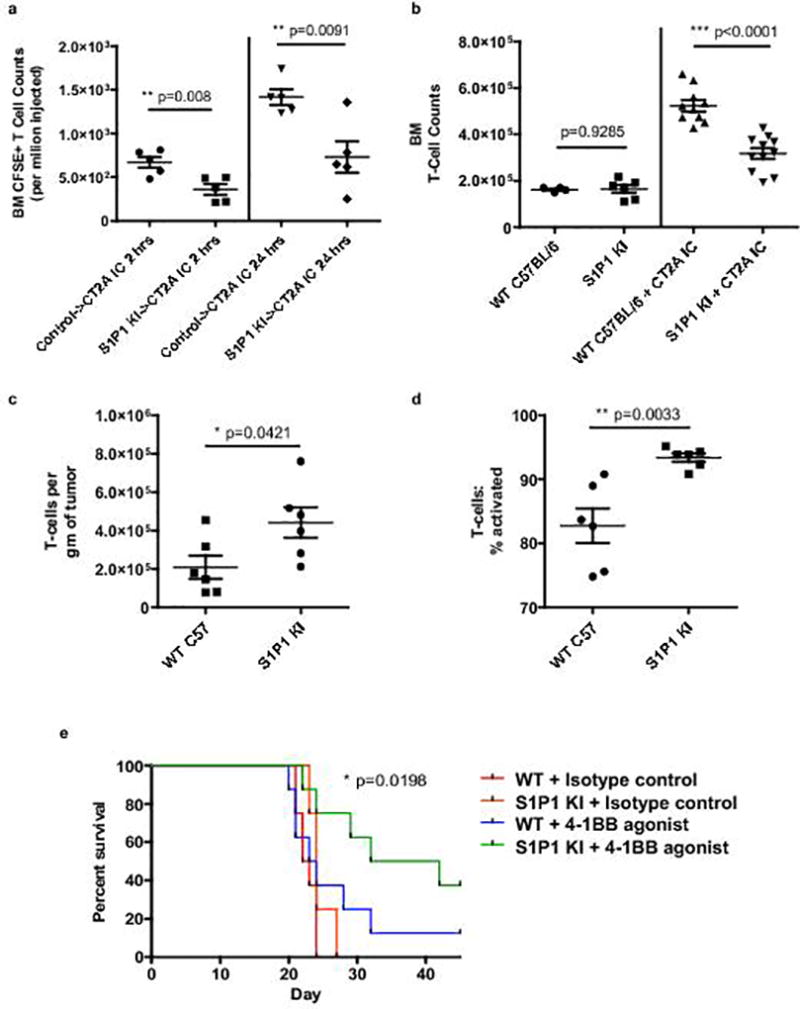
a, Relative sequestration of adoptively transferred CFSE-labeled T-cells within the bone marrow of CT2A IC recipient mice at 2 hours (left) or 24 hours (right) after transfer. As indicated, transferred cells were splenocytes from either control C57BL/6 donors (control) or from S1P1 stabilized “knock-in” (S1P1 KI) donors (n=5 recipient mice per group). Data in a are representative findings from one of a minimum of two independently repeated experiments with similar results. b, T-cell counts in the bone marrow of n=10 IC CT2A-bearing wild type (WT) C57BL/6 or n=11 S1P1 KI mice. Counts were assessed 18 days following tumor implantation and are shown relative to baseline counts in n=6 tumor-naïve control WT or n=6 tumor-naïve S1P1 KI mice. Cumulative data from three experiments are depicted in b. c, IC CT2A tumors were harvested from n=6 WT C57BL/6 (WT) or n=6 S1P1 KI mice 18 days following tumor implantation. TIL were assessed by flow cytometry and the number of total T-cells per gram of tumor quantified. d, The frequency of activated effector (CD44hiCD62Llo) T-cells in IC CT2A tumors from the same n=6 WT C57BL/6 (WT) or n=6 S1P1 KI mice in c was also quantified. Data in c, d are representative findings from one of a minimum of three independently repeated experiments with similar results. e, C57BL/6 (WT) or S1P1 KI mice were implanted with IC CT2A tumors and treated with a 4-1BB agonist antibody or isotype control (n=8 per group). All data in a–d are shown as mean ± s.e.m. P values in a–d were determined by two-tailed, unpaired Student’s t-test. Survival in e was assessed by two-tailed generalized Wilcoxon test. P value for overall comparison is depicted.
We therefore explored whether T-cells “liberated” from sequestration by S1P1 stabilization would travel to the intracranial compartment and effect an anti-tumor response. We examined IC CT2A tumors from both WT and S1P1-KI glioma-bearing mice. TIL were analyzed by flow cytometry and their number and phenotype characterized. Tumors from S1P1-KI mice contained higher numbers of CD3+ TIL than those from WT mice (Fig 6c). Likewise, CT2A-bearing S1P1-KI mice demonstrated increased proportions of CD3+ TIL possessing an activated, effector CD44hiCD62Llo phenotype (Fig. 6d).
Despite displaying higher numbers of activated TIL, tumor-bearing S1P1-KI mice that underwent no further intervention failed to consistently show improved survival. We hypothesized, however, that coupling S1P1 stabilization to T-cell activating therapies, such as 4-1BB agonism and/or checkpoint blockade, would have a synergistic effect, licensing the anti-tumor capacities of the newly freed T-cells. Indeed, S1P1-stabilized (KI) mice treated with a 4-1BB agonist demonstrated improved survival compared to the effects seen with either stabilized S1P1 or with 4-1BB agonism in WT mice alone (Fig. 6e). Furthermore, in S1P1-KI mice themselves, whereas PD-1 blockade was ineffectual as monotherapy, the effects of 4-1BB agonism and checkpoint blockade proved additive, with the combination prolonging median survival and producing a 50% long-term survival rate (Fig. 6f).
Again, as no pharmacologic means exist for stabilizing S1P1 on the T cell surface, we explored alternative translatable means for freeing sequestered T-cells. We uncovered that treating CT2A glioma-bearing mice with G-CSF decreased bone marrow T-cell counts and reversed T-cell lymphopenia (Supplementary Fig 6a, b). As with S1P1 stabilization alone, G-CSF monotherapy failed to consistenty impact survival. When combined with 4-1BB agonism, however, a similar additive effect was achieved, yielding approximately 40% long-term survival. (Supplementary Fig 6c).
DISCUSSION
The data reported here introduce sequestration as a novel mode of T-cell dysfunction in cancer, specifically intracranial tumors. The S1P-S1P1 axis is proposed as the contributing mediator, with S1P1 loss on naïve T-cells fostering their sequestration in bone marrow. Disturbances to T-cell S1P1 are not previously reported in cancer, and T-cell sequestration remains a mostly unaddressed mode of T-cell dysfunction. While studies have revealed that cancer vaccines can elicit counterproductive T-cell sequestration at sites of vaccine delivery22, there is little known about T-cell sequestration that is tumor-imposed. Sequestration of T-cells may instigate their resultant antigenic ignorance, limiting their anti-tumor capacities23. Our data suggest the need for a new treatment scope within immunotherapy: strategies to reverse T-cell ignorance. Given the results reported here, these strategies are anticipated to include S1P1 stabilization or overexpression, modalities not previously tested in cancer.
The role of the S1P-S1P1 axis in immunology continues to emerge. S1P1 and S1P4 are highly expressed by T-cells, with S1P1 regulating T-cell chemotactic responses11,24, but also impacting resident memory commitment25, Treg-induction26, and IL-6-dependent pathways27,28. S1P1’s chemotactic function has made it a newer treatment target for the MS drug fingolimod27,29–31, whose aim is forced internalization of the S1P1 receptor on T-cells to effect their sequestration in SLO and decrease their transit to brain. Ironically, our data suggest that tumors of the intracranial compartment may usurp a previously unrecognized CNS capacity for eliciting this same phenomenon. Such a capacity may play a physiologic role limiting T-cell access to the CNS and contribute to immune privilege.
Subsets of patients with relapsing remitting MS (RRMS) experience a paradoxical exacerbation of MS and increase in brain-infiltrating Th17 CD4+ T-cells when treated with fingolimod. Such patients frequently harbor a phosphorylation-defective S1P1, similar to our S1P1-KI mice, which likewise demonstrate an increase in TIL numbers27. We do not observe these to be Th17-polarized, however, which in the RRMS patients may instead be a function of strong fingolimod agonism and resultant S1P1-signaling through the JAK-STAT3-IL17 pathway27,28. This agonism is not present in our therapy model (we did not administer fingolimod to S1P1-KI mice), suggesting that interventions targeting S1P1 internalization more specifically (and not signaling) may be effective at guiding increased numbers of functional T-cells into intracranial tumors. As S1P1 internalization is arbitrated by recruitment of β-arrestin to the receptor’s intracytoplasmic components following GRK2-mediated phosphorylation32, GRK2 and β-arrestin may represent new and promising therapeutic targets.
A possible detriment to S1P1 stabilization would be reductions to numbers of tissue resident memory T-cells (TRM), whose ability to establish themselves in non-lymphoid tissues (NLT) depends on transcriptional downregulation of S1P125. While such loss of TRM might prove restrictive for immunity at barrier tissue sites (skin, gastrointestinal or respiratory tract), the role of TRM in mediating immune responses in the brain is less clear33. Although TRM may accumulate in the brain during an acute viral infection34, no data are available regarding their prevalence or role in brain tumors. Our survival data suggest that loss of any present TRM within the tumor does not obviate a benefit to retaining S1P1 on the T-cell surface. Likewise, the efficacy of fingolimod for MS suggests further that S1P1+ T-cells are quite capable of trafficking to brain and eliciting local immune responses.
The mechanism of observed systemic T-cell S1P1 loss remains an arena for future study. Both the lack of observed differences in S1P1 transcript levels in T-cells from tumor-bearing mice, and the improved S1P1 levels seen with hindered receptor internalization, suggest the defect is at the protein level, with the disturbance being either increased receptor internalization or delayed/failed receptor recycling. One study has reported increased levels of S1P ligand in GBM (a possible mechanism)35, although this study used normal gray matter rather than white matter as a control, and we saw no such increase in ligand levels in either tumors or plasma by both ELISA and LC-MS/MS. Likewise, blockade of known transcriptional down-regulators of S1P1 that are prevalent in GBM, such as TGF-β, produced no effect on sequestration in our hands (data not shown).
S1P1 loss and sequestration characterized predominantly naïve T-cells in our studies. As such, it is perhaps not surprising that stabilizing S1P1 alone to free naive T-cells from the marrow had limited impact on survival as a mono-intervention. S1P1 stabilization, however, did license 41BB agonism and PD-1 blockade, the latter of which has already failed in clinical trials for recurrent GBM as a monotherapy36. The synergy we observed suggests that reduced T-cell numbers are indeed a limiting factor for immunotherapeutic efficacy against intracranial tumors, and that reversal of T-cell sequestration may serve as a useful therapeutic adjunct. The persistent benefits seen when genetic S1P1 stabilization was replaced with G-CSF imply there may be available pharmacologic strategies for averting T-cell sequestration.
Our findings also suggest that T-cell sequestration may be a contributing factor to T-cell lymphopenia in patients with GBM. Numerous studies have highlighted T-cell lymphopenia in this population37–40, but many attribute the observation to the effects of treatment41. Our finding of pre-treatment T-cell lymphopenia contrasts with the work of others41,42, but aligns with historical studies43. While radiation, temozolomide, and dexamethasone are certainly exacerbating, we reveal that T-cell disappearances occur earlier and more severely than previously thought, extending to thymus and SLO.
Lastly, our data indicate that a variety of tumors placed intracranially elicit bone marrow T-cell sequestration, while none of the same tumors placed peripherally exhibit the same proclivity (Fig. 4a). Others have reported differing capacities for the intracranial compartment to influence systemic immune function44. Given our findings, though, we anticipate that studies to understand and reverse sequestration will impact immunotherapeutic design not only for GBM patients, but for patients with intracranial metastases as well, a far larger population45. Other future goals will be to understand the observed lymphoid organ contraction, elucidate tumor-imposed S1P1 loss, and develop pharmacologic means for preventing S1P1 internalization.
ONLINE METHODS
Clinical Studies and Specimen Processing
All studies were conducted with approval from the Massachusetts General Hospital Cancer Center Institutional Review Board. We certify that all applicable institutional regulations concerning the ethical use of information from human patients were followed during this research. For retrospective studies, records for all patients with a diagnosis of GBM over a ten-year period at Massachusetts General Hospital were reviewed. Criteria for inclusion and exclusion are as outlined in the results section and patient characteristics are found in Supplementary Table 1. For prospective studies, 15 treatment-naïve GBM patients and 15 healthy age-matched controls undergoing spinal fusion were included in the prospective collection of whole blood and bone marrow aspirates. Informed consent was obtained from all subjects. Bone marrow aspirates were collected under general anesthesia from the iliac crest. Using a 14-gauge needle, a total volume of 5 mL was collected. Both blood and bone marrow specimens were collected into purple top, EDTA-containing tubes. All blood and bone marrow were stored at room temperature and processed within 12-hours. All samples were labeled directly with antibodies for use in flow cytometry, and red blood cells subsequently lysed using eBioscience RBC lysis buffer (eBioscience, San Diego, CA). Cells were washed, fixed, and analyzed on an LSRII FORTESSA flow cytometer (BD Biosciences).
Reagents
For human studies, fluorochrome-conjugated antibodies to CD3 (Cat#557705, Clone: SP34-2, Lot#5352959, 1:20; Cat#558117, Clone: UCHT1, Lot#3186876, 1:100; Cat#557851, Clone: SK7, Lot#3193549), CD4 (Cat#558116, Clone: RPA-T4, Lot#6224744, 1:100; Cat#557695, Clone: RPA-T4, 1:20), CD8 (Cat#565310, Clone: SK1, Lot#7003689, 1:20; Cat#557746, Clone: RPA-T8, Lot#79151, 1:20; Cat#558207, Clone: RPA-T8, 1:100), CD45RO (Cat#563722, Clone: UCHL1, Lot#7096923, 1:20), CD25 (Cat#562403, Clone: M-A251, Lot#7088762, 1:20), CD27 (Cat#558664, Clone: M-T271, Lot#7136657, 1:5), CD127 (Cat#563225, Clone: HIL-7R-M21, Lot#7012862, 1:20), CCR6 (Cat#559562, Clone: 11A9, Lot#7019800, 1:100), CCR7 (Cat#557648, Clone: 3D12, Lot#3186974, 1:20), and CXCR4 (Cat#560669, Clone: 12G5, 1:20) were obtained from BD Biosciences (San Diego, CA). Antibodies to human CD45RA (Cat#304128, Clone: HI100, 1:20) and CXCR3 (Cat#353738, Clone: G025H7, Lot#B228065, 1:100) were obtained from BioLegend (San Diego, CA). Antibodies to human S1P1 (Cat#50-3639-42, Clone: SW4GYPP, Lot#4299074, 1:20) were obtained from eBioscience (San Diego, CA). For murine studies, fluorochrome-conjugated antibodies to CD3 (Cat#557666, Clone: 145-2C11, Lot#7096805, 1:100; Cat#553066, Clone: 145-2C11, Lot#7150784, 1:100), CD4 (Cat#553049, Clone: RM4-5, Lot#4189673, 1:100; Cat#558107, Clone: RM4-5, 1:100), CD8 (Cat#551162, Clone: 53-6.7, Lot#4275549, 1:100; Cat#563234, Clone: 53-6.7, Lot#7047617, 1:100), CD44 (Cat#562464, Clone: IM7, Lot#6205542, 1:100; Cat#559250, Clone: IM7, Lot#25892, 1:100), CD62L (Cat#553152, Clone: MEL-14, Lot#40865, 1:100), NK1.1 (Cat#553164, Clone: PK136, Lot#80219, 1:100), B220 (Cat#558108, Clone: RA3-6B2, Lot#6175996, 1:100), and GR-1 (Cat#553128, Clone: RB6-8C5, Lot#09439, 1:100) were obtained from BD Biosciences (San Diego, CA). Antibodies to murine S1P1 (Cat#FAB7089A, Clone: 713412, Lot#ACNG0216051, 1:10) were obtained from R&D systems (Minneapolis, MN). Probes for RNA PrimeFlow for mouse CD69, KLF2, and STAT3 were obtained from Life Technologies (Carlsbad, CA). For qRT-PCR, total RNA was isolated by RNeasy Mini Kit (Cat#74104) from Qiagen (Germantown, MD). The assays were perfomed with Mouse S1P1 TaqMan (Cat#Mm02619656_s1) and Mouse GAPDH TaqMan (Cat#Mm03302249_g1) from ThermoFisher (Waltham, MA). In vivo therapeutic antibodies (anti-mouse PD-1 (Cat#BE0146, clone: RMP 1-14, Lot#640517M2) and 4-1BB agonist antibody (Cat#BE0169, clone: LOB12.3, Lot#647417M1)) were obtained from Bio-X-cell (West Lebanon, NH).
Mice
Female C57BL/6, VM/Dk, and B6.129P2-S1pr1tm1.2Cys/J S1P1-KI mice were used at 6–12 weeks of age. The generation of B6.129P2-S1pr1tm1.2Cys/J (S1P1-KI) mice has been described46. S1P1-KI mice carry a Thr-Ser-Ser (TSS) to Ala-Ala-Ala (AAA) mutation in the C-terminus (the last 12 amino acids) of the sphingosine-1-phosphate receptor 1 (S1P1). This mutation leads to a loss in sensitivity for ligand-mediated receptor down-modulation, leading to the partial block in the desensitization process, resulting in resistance to S1P-mediated S1P1 internalization in naive T-cells. Parental transgenic mice are acquired from the Jackson Laboratory (Bar Harbor, ME) with in-house breeding colony expansion. C57BL/6 mice purchased from Charles River Laboratories (Wilmington, MA) were used as wild-type controls. S1P1 conditional knockout mice were created by crossing B6.129S6(FVB)-S1pr1tm2.1Rlp/J, which contains loxP sites flanking exon 2 of S1pr1 gene (JAX Stock #019141), with B6.Cg-Tg(UBC-cre/ERT2)1Ejb/1J (JAX Stock #007001), which contains tamoxifen-inducible Cre. These two mice were obtained from the Jackson Laboratory (Bar Harbor, ME) and crossed and then back-crossed to obtain mice with the genotype flox/flox Cre (+/−). The mice were then treated with tamoxifen to induce recombination. VM/Dk mice are bred and maintained as a colony at Duke University. Animals were maintained under specific pathogen-free conditions at Cancer Center Isolation Facility (CCIF) of Duke University Medical Center. All experimental procedures were approved by the Institutional Animal Care and Use Committee.
Cell Lines
Cell lines studied included murine SMA-560 malignant glioma, CT-2A malignant glioma, E0771 breast medullary adenocarcinoma, B16F10 melanoma, and Lewis Lung Carcinoma (LLC). SMA-560 cells are syngeneic on the VM/Dk mouse background, while all others are syngeneic in C57BL/6 mice. SMA-560, CT-2A, B16F10, and LLC cells were grown in vitro in Dulbecco's Modified Eagle Medium (DMEM) with 2 mM 1-glutamine and 4.5 mg/mL glucose (Gibco) containing 10% fetal bovine serum (Gemini Bio-Products). E0771 cells were grown in vitro in RPMI 1640 (Gibco) containing 10% fetal bovine serum plus 1% HEPES (Gibco). Cells were harvested in the logarithmic growth phase. For intracranial implantation, tumor cells in PBS were then mixed 1:1 with 3% methylcellulose and loaded into a 250 µL syringe (Hamilton, Reno, NV). The needle was positioned 2 mm to the right of the bregma and 4 mm below the surface of the skull at the coronal suture using a stereotactic frame. 1 × 104 SMA-560, CT-2A, E0771, and LLC cells or 1 × 103 B16F10 cells were delivered in a total volume of 5 µL per mouse. For subcutaneous implantation, 5 × 105 SMA-560, CT-2A, E0771, and LLC cells or 2.5 × 105 B16F10 cells were delivered in a total volume of 200 µL per mouse into the subcutaneous tissues of the left flank. All cell lines have been authenticated by using NIST published 9 species-specific STR markers to establish genetic profiles. Interspecies contamination check for human, mouse, rat, African green monkey and Chinese hamster was also performed for each cell line. All cell lines have been tested negative for Mycoplasma spp. and karyotyped, and none are among the ICLAC database of commonly misidentified cell lines. The CellCheck Mouse Plus™ cell line authentication and Mycoplasma spp. testing services were provided by IDEXX Laboratories (Westbrook, ME).
Murine Tissue Harvest
Spleen, thymus, cervical lymph nodes, and long bones of the legs (femur and tibia) were collected at defined and/or humane endpoints, in accordance with protocol. For intracranial tumor-bearing animals, humane endpoints include inability to ambulate two steps forward with prompting. For subcutaneous tumor-bearing animals, humane endpoints include tumor size greater than 20 mm in one dimension, 2000 mm3 in total volume, or tumor ulceration or necrosis. Spleens and thymuses were weighed prior to processing. Briefly, tissues were processed in RPMI, minced into single cell suspensions, cell-strained, counted, stained with antibodies, and analyzed via flow cytometry. Bone marrow cells were flushed out from one femur and one tibia. Blood samples were directly labeled with antibodies and red blood cells subsequently lysed using eBioscience RBC lysis buffer (eBioscience, San Diego, CA) or BD Pharm Lyse (BD Biosciences). Spleen and bone marrow were subjected to RBC lysis prior to antibody-labeling, while lymph nodes and thymus were labeled once single cell suspensions were created.
S1P1 Flow Cytometry
For S1P1 staining, all cell suspensions were prepared in staining buffer with the fixative agent (0.1% Buffered Neutral Formalin (BNF) (Sigma-Aldrich), 0.5% Bovine Serum Albumin (Sigma-Aldrich), and 2mM EDTA (Gibco) in PBS). Cells were passed through 40 µm nylon mesh cell strainers. After removing RBCs by BD Pharm Lyse lysing solution (BD Biosciences), cells were re-suspended at a density of 5 × 106 to 2×107 cells per mL in the same staining buffer as described above and were aliquoted in a volume of 100 µL. Cells were then incubated with either rat anti-mouse S1P1 APC-conjugated antibody (R&D systems) or mouse anti-human S1P1 eFlour 660-conjugated antibody (eBioscience) for one hour at 4°c and were washed once. Next, samples were incubated for 30 minutes at 4°c with relevant antibody cocktails consisting of antibodies to additional markers (see Reagents). Cells were analyzed with an LSRFortessa (BD Biosciences) and data were analyzed with FlowJo software (Ashland, OR).
Adoptive Cell Transfer
For tracking cells in vivo, the spleens from naïve C57BL/6 mice were processed into single-cell suspensions in RPMI 1640 (Gibco) containing 10% fetal bovine serum (Gemini Bio-Products). Bone marrow single-cell suspensions from intracranial CT-2A tumor-bearing C57BL/6 mice were acquired from two femurs, two tibias, two humeri, and sternum to achieve maximum yield. Bone marrow cells were then enriched for T-cells via the AutoMACS Pro Separator using the Pan T-Cell Isolation Kit II, mouse with DEPLETE program (Miltenyi Biotec, Auburn, CA). Cells from spleens and bone marrow were labeled with CellTrace CFSE (Life Technologies). The labeled cells were transferred IV via tail veins (1 × 107 cells in 200 µL of PBS per mouse) into tumor-free or intracranial CT-2A tumor bearing C57BL/6 day 18 after tumor implantation. The numbers of CFSE-positive T-cells in the bone marrow were assessed by flow cytometry at specified time points following transfer.
ELISA
Relevant mice underwent retro-orbital bleed at pre-determined time-points using heparin-coated capillary tubes (VWR). Heparinized blood was then centrifuged and aliquots of plasma were stored at −80°c. S1P levels in murine plasma were analyzed using a S1P competitive ELISA kit (Echelon Biosciences, Salt Lake City, UT) according to the manufacturer’s instructions.
Liquid Chromatography- Tandem Mass Spectometry (LC-MS/MS)
Bone marrow was harvested by removing mouse tibia and femurs, removing the ends of the long bones to expose the marrow cavity, placing the long bones inside a centrifuge tube with a hole in the tip and then nesting it inside another centrifuge tube, and spinning for 10,000 g for 15 seconds to produce a pellet. Sample was then frozen at −80°c. Brains were harvested, frozen with liquid nitrogen, and homogenized using mortar and pestle. Plasma was also collected in EDTA-coated tubes. All samples were delivered to Duke Proteomics and Metabolomics Shared Resource and were analyzed by LC-MS/MS according to previously published protocol47.
Statistical Analysis
For human studies, the sample size of 15 patients and 15 controls was chosen so that a two-tailed t-test comparing groups has 80% power to detect a difference that is 1.1 times the standard deviation of the outcome variable in each group. For animal studies sample sizes were chosen based on historical experience and were variable based on numbers of surviving mice available at experimental time-points or technical limitations. Female mice aged 6–12 weeks were included in studies, without additional exclusion criteria employed. Mice were pooled and then sequentially assigned to each pertinent group. Animal studies were not blinded. For statistical comparisons, two-tailed paired and unpaired t-tests were generally used to compare groups. When underlying assumptions for these statistical tests were violated, nonparametric alternatives, such as the Wilcoxon signed rank or Wilcoxon rank sum test, were used. Analysis of variance with interaction, χ2 tests, and correlational analyses were also conducted. Bar graphs and dot plots are used to graphically display data, with dot plots used preferentially when group sizes are smaller or data demonstrate non-Gaussian distributions. Bar graphs and dot plots display the mean +/− the standard error of the mean. Survival comparisons were made by Gehan-Breslow-Wilcoxon test. The specific statistical method employed for each data presentation is denoted in the respective figure legends.
Ethics statement
All studies performed in research animals and human research participants are in compliance with all relevant ethical regulations.
Reporting Summary
Further information on experimental design is available in the Nature Research Life Sciences Reporting Summary linked to this article.
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Supplementary Material
Acknowledgments
We would like to thank G.E. Archer, K.A. Batich, T.A. Chewning, K.L. Congdon, K.A. Keith, R.J. Lefkowitz, P.K. Norberg, E.A. Reap, L.A.M. Rein, K.E. Rhodin, A. Seas, S.H. Shen, R.J. Schmittling, D.J. Snyder, C.M. Suryadevara, A.M. Swartz, W.H. Tomaszewski, D.S. Wilkinson, W. Xie, H. Yang and members of Duke Brain Tumor Immunotherapy Program for their helpful insights throughout the study. We would like to thank M. Foster and J.W. Thompson from Proteomics and Metabolomics Shared Resource, Duke Center for Genomic and Computational Biology for invaluable help with liquid chromatography-tandem mass spectrometry analyses. We would like to thank M.R. Llewellyn for the excellent preparation of medical illustrations. This work was supported by institutional start-up funds from Duke University Medical Center, The Sontag Foundation Distinguished Scientist Award, and National Institute of Health (NIH) R01NS099096 to P.E.F.
Footnotes
AUTHOR CONTRIBUTIONS
P.C., C.J., S.K., F.L., N.C.S., J.-V.C., B.V.N., and P.E.F obtained and/or analyzed human data. P.C., C.J., S.K., X.C., S.H.F., K.W., A.A.E., C.A.D., H.R.K., L.S.-P., T.A.C., and P.E.F. designed, carried out, and/or analyzed in vitro and in vivo animal experiments. J.I.E. provided pathological characterization and immunohistochemistry analyses. J.E.H. provided biostatistics consultation. J.H.S., M.D.G., R.L.M., G.D., W.T.C., and P.E.F. provided feedback and supervised all research. P.C., C.A.D., and P.E.F. wrote the manuscript. All authors read, revised, and approved the final manuscipt.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat. Immunol. 2002;3:991–998. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 2.Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–360. doi: 10.1146/annurev.immunol.22.012703.104803. [DOI] [PubMed] [Google Scholar]
- 3.Dix AR, Brooks WH, Roszman TL, Morford LA. Immune defects observed in patients with primary malignant brain tumors. J. Neuroimmunol. 1999;100:216–232. doi: 10.1016/s0165-5728(99)00203-9. [DOI] [PubMed] [Google Scholar]
- 4.Dunn GP, Fecci PE, Curry WT. Cancer immunoediting in malignant glioma. Neurosurgery. 2012;71:201–222. doi: 10.1227/NEU.0b013e31824f840d. discussion 222-203. [DOI] [PubMed] [Google Scholar]
- 5.Fecci PE, Heimberger AB, Sampson JH. Immunotherapy for primary brain tumors: no longer a matter of privilege. Clin Cancer Res. 2014;20:5620–5629. doi: 10.1158/1078-0432.CCR-14-0832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woroniecka KI, Rhodin KE, Chongsathidkiet P, Keith KA, Fecci PE. T-Cell Dysfunction in Glioblastoma: Applying a New Framework. Clin Cancer Res. 2018 doi: 10.1158/1078-0432.CCR-18-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ostrom QT, et al. CBTRUS Statistical Report: Primary brain and other central nervous system tumors diagnosed in the United States in 2010–2014. Neuro Oncol. 2017;19:v1–v88. doi: 10.1093/neuonc/nox158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brooks WH, Roszman TL, Mahaley MS, Woosley RE. Immunobiology of primary intracranial tumours. II. Analysis of lymphocyte subpopulations in patients with primary brain tumours. Clin. Exp. Immunol. 1977;29:61–66. [PMC free article] [PubMed] [Google Scholar]
- 9.Garris CS, Blaho VA, Hla T, Han MH. Sphingosine-1-phosphate receptor 1 signalling in T cells: trafficking and beyond. Immunology. 2014;142:347–353. doi: 10.1111/imm.12272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosen H, Gonzalez-Cabrera PJ, Sanna MG, Brown S. Sphingosine 1-phosphate receptor signaling. Annu. Rev. Biochem. 2009;78:743–768. doi: 10.1146/annurev.biochem.78.072407.103733. [DOI] [PubMed] [Google Scholar]
- 11.Matloubian M, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427:355–360. doi: 10.1038/nature02284. [DOI] [PubMed] [Google Scholar]
- 12.Drouillard A, et al. Human Naive and Memory T Cells Display Opposite Migratory Responses to Sphingosine-1 Phosphate. J Immunol. 2018;200:551–557. doi: 10.4049/jimmunol.1701278. [DOI] [PubMed] [Google Scholar]
- 13.Spiegel S, Milstien S. Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat. Rev. Mol. Cell Biol. 2003;4:397–407. doi: 10.1038/nrm1103. [DOI] [PubMed] [Google Scholar]
- 14.Schwab SR, et al. Lymphocyte sequestration through S1P lyase inhibition and disruption of S1P gradients. Science. 2005;309:1735–1739. doi: 10.1126/science.1113640. [DOI] [PubMed] [Google Scholar]
- 15.Cyster JG. Chemokines, sphingosine-1-phosphate, and cell migration in secondary lymphoid organs. Annu Rev Immunol. 2005;23:127–159. doi: 10.1146/annurev.immunol.23.021704.115628. [DOI] [PubMed] [Google Scholar]
- 16.Lo CG, Xu Y, Proia RL, Cyster JG. Cyclical modulation of sphingosine-1-phosphate receptor 1 surface expression during lymphocyte recirculation and relationship to lymphoid organ transit. J Exp Med. 2005;201:291–301. doi: 10.1084/jem.20041509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zajac AJ, Harrington LE. Tissue-resident T cells lose their S1P1 exit visas. Cellular & molecular immunology. 2014;11:221–223. doi: 10.1038/cmi.2014.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maeda Y, Seki N, Sato N, Sugahara K, Chiba K. Sphingosine 1-phosphate receptor type 1 regulates egress of mature T cells from mouse bone marrow. Int Immunol. 2010;22:515–525. doi: 10.1093/intimm/dxq036. [DOI] [PubMed] [Google Scholar]
- 19.Blaho VA, et al. HDL-bound sphingosine-1-phosphate restrains lymphopoiesis and neuroinflammation. Nature. 2015;523:342–346. doi: 10.1038/nature14462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Allende ML, Yamashita T, Proia RL. G-protein-coupled receptor S1P1 acts within endothelial cells to regulate vascular maturation. Blood. 2003;102:3665–3667. doi: 10.1182/blood-2003-02-0460. [DOI] [PubMed] [Google Scholar]
- 21.Arnon TI, et al. GRK2-dependent S1PR1 desensitization is required for lymphocytes to overcome their attraction to blood. Science. 2011;333:1898–1903. doi: 10.1126/science.1208248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hailemichael Y, et al. Persistent antigen at vaccination sites induces tumor-specific CD8(+) T cell sequestration, dysfunction and deletion. Nat Med. 2013;19:465–472. doi: 10.1038/nm.3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schietinger A, Greenberg PD. Tolerance and exhaustion: defining mechanisms of T cell dysfunction. Trends Immunol. 2014;35:51–60. doi: 10.1016/j.it.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chi H, Flavell RA. Cutting edge: regulation of T cell trafficking and primary immune responses by sphingosine 1-phosphate receptor 1. J Immunol. 2005;174:2485–2488. doi: 10.4049/jimmunol.174.5.2485. [DOI] [PubMed] [Google Scholar]
- 25.Skon CN, et al. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat Immunol. 2013;14:1285–1293. doi: 10.1038/ni.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu G, Yang K, Burns S, Shrestha S, Chi H. The S1P(1)-mTOR axis directs the reciprocal differentiation of T(H)1 and T(reg) cells. Nat Immunol. 2010;11:1047–1056. doi: 10.1038/ni.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garris CS, et al. Defective sphingosine 1-phosphate receptor 1 (S1P1) phosphorylation exacerbates TH17-mediated autoimmune neuroinflammation. Nat Immunol. 2013;14:1166–1172. doi: 10.1038/ni.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee H, et al. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat Med. 2010;16:1421–1428. doi: 10.1038/nm.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deng H, et al. Discovery of Clinical Candidate GSK1842799 As a Selective S1P1 Receptor Agonist (Prodrug) for Multiple Sclerosis. ACS medicinal chemistry letters. 2013;4:942–947. doi: 10.1021/ml400194r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jin J, et al. Development of a selective S1P1 receptor agonist, Syl930, as a potential therapeutic agent for autoimmune encephalitis. Biochemical pharmacology. 2014;90:50–61. doi: 10.1016/j.bcp.2014.04.010. [DOI] [PubMed] [Google Scholar]
- 31.Yamamoto R, et al. ASP4058, a novel agonist for sphingosine 1-phosphate receptors 1 and 5, ameliorates rodent experimental autoimmune encephalomyelitis with a favorable safety profile. PloS one. 2014;9:e110819. doi: 10.1371/journal.pone.0110819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reeves PM, Kang YL, Kirchhausen T. Endocytosis of Ligand-Activated Sphingosine 1-Phosphate Receptor 1 Mediated by the Clathrin-Pathway. Traffic (Copenhagen, Denmark) 2016;17:40–52. doi: 10.1111/tra.12343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park CO, Kupper TS. The emerging role of resident memory T cells in protective immunity and inflammatory disease. Nat Med. 2015;21:688–697. doi: 10.1038/nm.3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wakim LM, et al. The molecular signature of tissue resident memory CD8 T cells isolated from the brain. J Immunol. 2012;189:3462–3471. doi: 10.4049/jimmunol.1201305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abuhusain HJ, et al. A metabolic shift favoring sphingosine 1-phosphate at the expense of ceramide controls glioblastoma angiogenesis. J Biol Chem. 2013;288:37355–37364. doi: 10.1074/jbc.M113.494740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Omuro A, et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: results from exploratory phase 1 cohorts of CheckMate 143. Neuro Oncol. 2017 doi: 10.1093/neuonc/nox208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brooks WH, et al. Immunobiology of primary intracranial tumors. Part 5: Correlation of a lymphocyte index and clinical status. Journal of Neurosurgery. 1981;54:331–337. doi: 10.3171/jns.1981.54.3.0331. [DOI] [PubMed] [Google Scholar]
- 38.Brooks WH, Roszman TL, Mahaley MS, Woosley RE. Immunobiology of primary intracranial tumours. II. Analysis of lymphocyte subpopulations in patients with primary brain tumours. Clin Exp Immunol. 1977;29:61–66. [PMC free article] [PubMed] [Google Scholar]
- 39.Fecci PE, et al. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 2006;66:3294–3302. doi: 10.1158/0008-5472.CAN-05-3773. [DOI] [PubMed] [Google Scholar]
- 40.Morford LA, Dix AR, Brooks WH, Roszman TL. Apoptotic elimination of peripheral T lymphocytes in patients with primary intracranial tumors. Journal of Neurosurgery. 1999;91:935–946. doi: 10.3171/jns.1999.91.6.0935. [DOI] [PubMed] [Google Scholar]
- 41.Hughes MA, Parisi M, Grossman S, Kleinberg L. Primary brain tumors treated with steroids and radiotherapy: low CD4 counts and risk of infection. Int J Radiat Oncol Biol Phys. 2005;62:1423–1426. doi: 10.1016/j.ijrobp.2004.12.085. [DOI] [PubMed] [Google Scholar]
- 42.Grossman SA, et al. Immunosuppression in patients with high-grade gliomas treated with radiation and temozolomide. Clin Cancer Res. 2011;17:5473–5480. doi: 10.1158/1078-0432.CCR-11-0774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mahaley MS, Jr, et al. Immunobiology of primary intracranial tumors. Part 1: studies of the cellular and humoral general immune competence of brain-tumor patients. Journal of Neurosurgery. 1977;46:467–476. doi: 10.3171/jns.1977.46.4.0467. [DOI] [PubMed] [Google Scholar]
- 44.Jackson CM, et al. Systemic Tolerance Mediated by Melanoma Brain Tumors Is Reversible by Radiotherapy and Vaccination. Clin Cancer Res. 2016;22:1161–1172. doi: 10.1158/1078-0432.CCR-15-1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ostrom QT, et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro Oncol. 2014;16(Suppl 4):iv1–63. doi: 10.1093/neuonc/nou223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arnon TI, et al. GRK2-dependent S1PR1 desensitization is required for lymphocytes to overcome their attraction to blood. Science. 2011;333:1898–1903. doi: 10.1126/science.1208248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frej C, et al. Quantification of sphingosine 1-phosphate by validated LC-MS/MS method revealing strong correlation with apolipoprotein M in plasma but not in serum due to platelet activation during blood coagulation. Anal Bioanal Chem. 2015;407:8533–8542. doi: 10.1007/s00216-015-9008-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.