

Abstract
Trimethylamine-N-oxide (TMAO), a microbiota-dependent metabolite derived from trimethylamine (TMA)-containing nutrients that are abundant in a Western diet, enhances both platelet responsiveness and in vivo thrombosis potential in animal models and predicts incident atherothrombotic event risks in clinical studies. Here, utilizing a mechanism-based inhibitor approach targeting a major microbial TMA-generating enzyme (CutC/D), we developed potent, time-dependent and irreversible inhibitors that do not affect commensal viability. In animal models, a single oral dose of a CutC/D inhibitor significantly reduced plasma TMAO levels for up to 3 days and rescued diet-induced enhanced platelet responsiveness and thrombus formation, without observable toxicity or increased bleeding risk. The inhibitor selectively accumulated within intestinal microbes to millimolar levels, a concentration over a million-fold higher than needed for a therapeutic effect. These studies reveal that mechanism-based inhibition of gut microbial TMA/TMAO production reduces thrombosis potential, a critical adverse complication in heart disease. They also offer a generalizable approach for the selective non-lethal targeting of gut microbial enzymes linked to host disease, while limiting systemic exposure of the inhibitor in the host.
Introduction
Recent studies implicate participation of the gut microbiome in numerous facets of human health and disease1–6. For example, less than a decade ago, a link between dietary phosphatidylcholine, a nutrient common in a Western diet, gut microbiota-dependent generation of the metabolite trimethylamine N-oxide (TMAO), and cardiovascular disease (CVD) pathogenesis, was first described7. Since then, multiple human and animal studies supporting both mechanistic and clinical prognostic associations between TMAO formation and cardiometabolic disease risks have been reported8–16. The mechanisms through which TMAO is thought to foster enhanced CVD risks are manifold and include alterations in tissue sterol metabolism7,9,17, enhanced endothelial cell activation and vascular inflammation7,18–20, and stimulation of pro-fibrotic signaling pathways14,15. Historically, gut microbiota are known to impact factors linked to platelet function and hemostasis, including serotonin21, vitamin K22, and von Willebrand factor23. In addition, recent studies reveal TMAO alters calcium signaling in platelets, enhancing responsiveness and in vivo thrombosis potential in animal models15. Parallel clinical studies reveal TMAO levels are associated with thrombotic event risks (heart attack and stroke)15, and clinical interventional studies with choline supplementation in healthy vegan or omnivorous volunteers were shown to both increase circulating TMAO levels and heighten platelet responsiveness to agonists24. Finally, several recent meta-analyses confirm a strong clinical association between increased levels of TMAO and incident adverse cardiovascular event and mortality risks in multiple populations25–27. Thus, there is rapidly growing interest in the therapeutic targeting of gut microbiota-dependent TMAO generation for the potential treatment of CVD28.
TMAO is generated via a meta-organismal pathway that begins with gut microbial conversion of dietary nutrients (e.g. phosphatidylcholine, choline, and carnitine) into trimethylamine (TMA), followed by host liver oxidation to TMAO by flavin monooxygenases (FMOs)29,30. Given the abundance of the choline moiety in both bile31 and common dietary staples (e.g. eggs, meat/fish, and some fruits/vegetables), microbial conversion of choline into TMA likely accounts for a significant portion of TMAO production in subjects, regardless of diet. A pair of microbial proteins encoded by genes of the choline utilization (cut) gene cluster, the catalytic CutC protein and its activating partner, CutD, support choline TMA lyase enzyme activity32–34. We recently reported the use of a natural product, 3,3-dimethyl-1-butanol (DMB), as a tool drug that inhibits microbial choline TMA lyase activity in vitro and in vivo35. When given to atherosclerosis-prone ApoE−/− mice on a choline-supplemented diet, plasma TMAO levels were significantly lowered, and concurrently, macrophage cholesterol accumulation, foam cell formation and atherosclerotic lesion development were attenuated35.
While atherosclerotic plaque development is a defining pathologic feature of coronary artery disease, enhanced platelet reactivity and acute thrombotic occlusion of vessels are the proximate cause of myocardial infarction, stroke and the majority of deaths in patients with CVD36. Use of antiplatelet agents has become a cornerstone for the treatment of CVD because of substantial reduction in CVD events and mortality37,38. However, more widespread use of antiplatelet agents has been limited by the increased risk of bleeding, which also leads to nonadherence39–41. Herein we show that a mechanism-based non-lethal inhibitor of the gut microbial TMAO pathway designed to selectively accumulate within the gut microbial compartment, can serve as a new therapeutic approach for attenuating thrombosis while simultaneously limiting systemic exposure in the host.
Results
DMB, a microbial choline TMA lyase inhibitor, attenuates choline diet-enhanced platelet responsiveness and in vivo rate of thrombus formation
In initial studies, C57BL/6J mice were maintained on a chemically-defined control “Chow” diet versus the same diet supplemented with choline (1% w/w). The choline diet elicited no differences in multiple indices of platelet activation, including surface phosphatidylserine content (p=0.84) in ADP-stimulated washed platelets or levels of von Willebrand factor (p=0.14), alpha granule release (p=0.31), or prothrombotic microvesicle release (p=0.66) in platelet-rich plasma (PRP) in the absence of agonist (Supplementary Figure 1). However, as previously reported15, choline supplementation resulted in 10-fold higher plasma TMAO levels (p<0.0001) and enhanced aggregometry response to submaximal levels of ADP (1 μM) in PRP (p<0.0001; Figure 1a). Moreover, the TMAO enhancing effect on stimulus-dependent platelet aggregation was also observed with washed platelets from mice fed the high choline diet (p=0.0002), and was greatest at submaximal levels of agonist (e.g. ADP, collagen; Supplementary Figure 1).
Figure 1. Proof of concept that microbial choline TMA lyase inhibition can attenuate choline diet-enhanced platelet aggregation and in vivo thrombus formation.

a) Platelet aggregation in PRP of mice fed the indicated diets ± DMB (1.3% v/v) provided in the drinking water for 6 weeks. Platelet aggregation was measured in response to a submaximal concentration of ADP (1 μM). Data points represent aggregation as % of maximum amplitude in PRP recovered from each mouse, and bars represent mean levels for each group. Plasma TMAO levels are also shown and represent mean ± SEM for each group. Significance determined by two-tailed Student’s t-test.
b) Representative vital microscopy images of carotid artery thrombus formation at the indicated time points following FeCl3-induced carotid artery injury in mice fed either a chemically-defined chow or 1% choline diet with or without the addition of DMB (1.3% v/v). The time to complete occlusion is noted in the right-hand panels. Complete study results including replications are shown in Figure 1c.
c) Quantification of in vivo thrombus formation following FeCl3-induced carotid artery injury in mice fed the indicated diets ± DMB (1.3% v/v) provided in the drinking water for 6 weeks. Data points represent the time to cessation of flow for each mouse, and bars represent mean levels for each group. Plasma TMAO levels are also shown and represent mean ± SEM for each group. Significance determined by two-tailed Student’s t-test.
d) Proposed mechanism by which a potential suicide substrate inhibitor of CutC/D, iodomethylcholine (IMC), can form a reactive iodotrimethylamine (I-TMA) product that can promote irreversible CutC/D inhibition via covalent modification of a reactive, nucleophilic active-site residue (Nu).
e) Comparison of the inhibitory potency of IMC (○), DMB (□) and Resveratrol (RESV, △), against (left) wild-type, recombinant P. mirabilis CutC/D lysate, (center) recombinant D. alaskensis CutC/D lysate, and (right) whole-cell (intact live culture) wild-type P. mirabilis. Data points represent the mean ± SEM. Exact numbers used for each data point can be found in the Source Data (n=2–9 technical replicates).
In parallel studies, mice were maintained on the chow versus choline-supplemented diet but also exposed to the tool drug DMB. DMB significantly reduced both TMAO levels (p<0.0001) and stimulus-dependent (ADP) platelet aggregation (p=0.003) in PRP recovered from choline-supplemented mice (Figure 1a). In separate studies, DMB was directly incubated with the PRP preparations, and no effects on the platelet aggregometry responses either alone, or in the presence of agonist (ADP), were observed (Supplementary Figure 2). Importantly, DMB-induced suppression of plasma TMAO levels and ADP-stimulated platelet aggregation responses in PRP recovered from choline-fed mice were completely reversed by direct injection of TMAO (p=0.32; Figure 1a). Representative platelet aggregometry tracings for each group of mice are shown in Supplementary Figure 3.
The carotid artery FeCl3 injury model was next performed to quantify the rate of clot formation and time to vessel occlusion following injury in mice fed either chow or choline-supplemented diets with or without DMB (Figure 1b,c). Choline supplementation resulted in marked TMAO elevation and shortening of vessel occlusion time (p<0.0001) as previously reported15, and provision of DMB attenuated the choline diet-induced rise in TMAO and rate of clot formation (p=0.002; Figure 1b,c). In separate studies, intraperitoneal injection of TMAO completely reversed DMB-dependent inhibition in choline diet-enhanced rate of thrombus formation (Figure 1c). However, despite the high dose of DMB provided, choline diet-associated TMAO elevations, increased platelet aggregation, and shortened in vivo thrombus formation were not fully rescued compared to chow-fed mice (Figure 1a,c). In light of these promising results, we sought to develop second-generation TMA lyase inhibitors with improved therapeutic potential.
Design and development of potent, mechanism-based, non-lethal microbial CutC/D inhibitors
We hypothesized that a suicide substrate mechanism-based inhibitor could possess many desirable advantages, including the selective targeting of a gut microbial pathway in a non-lethal manner, while potentially accumulating within the microbe thereby limiting systemic exposure of the drug in the host (i.e. as choline catabolism within the microbe was inhibited, cytosolic levels would increase, be sensed as an abundant nutrient, and trigger up regulation of the entire choline utilization gene cluster, including choline transporters). We therefore sought to develop an inhibitor that had the following characteristics: (i) it is a high affinity choline analogue inhibitor; (ii) it is non-lethal to the microbe; (iii) it initially is inert (non-reactive); (iv) it can be transported into an intact gut microbe; and (v) it possesses a cryptic reactive moiety that is only revealed upon the unique C-N bond cleavage executed by microbial choline TMA lyase catalysis. This could in theory result in generation of a reactive species within the microbe that could then covalently modify an active site residue, promoting irreversible inhibition. Based on these goals, we designed as a prototypic suicide substrate inhibitor (tool drug) the choline analogue iodomethylcholine (IMC) (Figure 1d). We predicted that after C-N bond cleavage by CutC, the product formed would be highly reactive and could lead to irreversible inactivation of the microbial enzyme since quantum mechanical calculations using the CutC crystal structure derived from D. alaskensis42 (see Methods) predict the rapid (concerted) loss of I− and the potential for covalent attachment to nearby nucleophilic residues (Figure 1d, Supplementary Figure 4).
We developed a tiered screening assay system to test for broad efficacy among phylogenetically diverse commensal bacteria that contain CutC/D and robust choline TMA lyase activity. As previously reported35, we used cloned cutC/D genes from P. mirabilis (ID: 6801039) and D. alaskensis (ID: 3926085) that were separately transformed into competent E. coli cells. Choline TMA lyase activity was monitored in the clarified lysate using isotope-labeled choline (d9[trimethyl]-choline) as the substrate and measuring the production of d9-TMA. We also employed similar lysate and whole-cell culture assays using wild-type P. mirabilis, as well as stable polymicrobial human fecal cultures using a continuous flow bioreactor system (see Methods). Remarkably, IMC displayed an in vitro potency (IC50) approximately 10,000 times greater than DMB against the recombinant P. mirabilis CutC/D lysate, recombinant D. alaskensis CutC/D lysate, and wild-type, whole-cell P. mirabilis in culture (Figure 1e). In contrast to a recent report43, resveratrol was largely ineffective in each of the screening assays employed (Figure 1e) and showed no effect on plasma TMAO levels in vivo when administered to mice on a choline-supplemented diet (p=0.91; data not shown)
Having observed the potent inhibition in TMA production in vitro with IMC, we synthesized multiple alternative halomethylcholines, which as a group were remarkably effective in all lysate and whole-cell assays (Table 1, Figure 2a). Interestingly, the fluorinated choline analogue, fluoromethylcholine (FMC), was the most potent halomethylcholine examined (IC50=900 pM against P. mirabilis CutC/D), and in docking calculations, was predicted to have the tightest interaction with the active site of the CutC crystal structure derived from D. alaskensis (see Methods). FMC was also more potent than IMC against our polymicrobial, human fecal cultures (EC50=7.9 nM and 1.6 μM, respectively; Table 1). In drug metabolism studies in mice, we detected low levels of halomethylbetaines (an oxidative metabolite) following oral halomethylcholine administration. We therefore synthesized each of the corresponding halomethylbetaines and screened them for inhibitory activity. In general, the halomethylbetaines were found to act as CutC/D TMA lyase inhibitors but approximately 2–3 orders of magnitude weaker than their choline counterparts (Table 1). Given the promising results observed with the halomethylcholines, both FMC and IMC were advanced for further mechanistic and preclinical studies.
Table 1.
Summary of the inhibitory potency across multiple screens of halomethylcholine and halomethylbetaine analogues
| Name | in vitro clarified lysate IC50 (nM) | Whole-cell EC50 (nM) | ex vivo EC50 (nM) | in vivo EC50 (mg/kg/day) | |||
|---|---|---|---|---|---|---|---|
| rec. CutC/D P. mirabilis |
rec. CutC/D D. alaskensis |
Wild-type P. mirabilis |
Wild-type P. mirabilis |
Fecal polymicrobial | q24h post-gavage | d9-choline challenge | |
Fluoromethylcholine (FMC)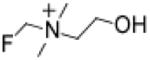
|
0.9 | 1.4 | 2.0 | 56 | 7.9 | 3.4 | 0.01 |
Iodomethylcholine (IMC)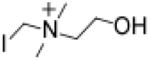
|
1.3 | 2.6 | 1.5 | 48 | 1,600 | 45 | 0.2 |
Chloromethylcholine (CMC)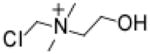
|
3.0 | 2.8 | 8.9 | 45 | 63 | 15.3 | 0.02 |
Bromomethylcholine (BMC)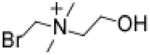
|
7.8 | 5.6 | 4.4 | 40 | 160 | >310 | 0.03 |
Iodomethylbetaine (IMB)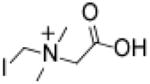
|
400 | 350 | 2,600 | 14,000 | 250,000 | >310 | 19.3 |
Bromomethylbetaine (BMB)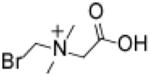
|
2,800 | 3,100 | 1,300 | 9,400 | 130,000 | > 310 | 9.5 |
Fluoromethylbetaine (FMB)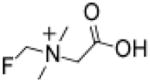
|
9,100 | 14,000 | 16,000 | 320,000 | 79,000 | 300 | 58.9 |
Chloromethylbetaine (CMB)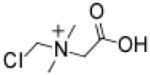
|
9,800 | 11,000 | 5,000 | 81,000 | 50,000 | > 310 | 83.3 |
Numbers shown represent either IC50 or EC50 values for the indicated halomethylcholine or halomethylbetaine compound, as tested in the indicated in vitro enzyme activity screen, in vitro culture of intact individual (P mirabilis) or polymicrobial (human fecal) culture, or two different in vivo screening strategies. For all in vitro or ex vivo studies, calculated IC50 or EC50 values represent results from dose-response curves with between 6–11 different concentrations monitored. Exact numbers used for each data point can be found in the Source Data. For the in vivo studies, calculated EC50 doses use data either from the (i) d9-choline challenge model, which gives an estimate of the oral dose needed to inhibit 50% of microbial d9-choline → d9-TMAO in mice, or (ii) the q24h post-gavage model, which gives an estimate of the oral dose needed to inhibit 50% of plasma level of TMAO at trough time in a chronic, once daily oral gavage dosing regimen in mice maintained on a high choline (1% w/w) diet. In vivo dose response curves employed at least 4–5 animals per dose examined, with 6–10 different inhibitor concentrations examined for each compound. See Source Data for exact sample numbers for each experiment performed and each independent experimental dose-response curve included in the cumulative analysis used to calculate IC50 or EC50 values in each cell in the Table.
Figure 2. IMC and FMC are non-lethal, irreversible and non-competitive CutC/D inhibitors.

a) Dose-response curves for halide-substituted (fluorine, F; iodine, I; chlorine, Cl; bromine, Br) methyl-choline analogues (X-MC) against recombinant P. mirabilis CutC/D lysate. Data points represent the mean ± SEM. Exact numbers used for each data point can be found in the Source Data (n=3–9 technical replicates).
b) The time-dependence of the inhibitory potency as assessed by pre-incubating the indicated concentrations FMC or IMC with recombinant P. mirabilis CutC/D lysate for increasing intervals before the addition of d9-choline substrate and subsequent quantification of choline TMA lyase activity. Relative activity was measured against no inhibitor controls during the initial kinetic phase. Data points represent the mean ± SEM. Exact numbers used for each data point can be found in the Source Data (n=2–9 technical replicates).
c) Characterization of the reversibility of FMC, IMC, and phenylcholine (PhC) inhibition of recombinant P. mirabilis CutC/D lysate. The indicated inhibitors were incubated with P. mirabilis CutC/D at a fully inhibitory concentration (10 μM). After overnight dialysis, recovered CutC/D activity was measured by LC/MS/MS quantification of d9-TMA generated from the d9-choline substrate. Data points represent the mean ± SEM (n=3 technical replicates for each).
d) The impact of FMC (top) and IMC (bottom) on the growth of the indicated human TMA-producing gut commensals was tested by culturing them with or without inhibitor in rich nutrient broth and monitoring growth curves. “No Addition” (NA) used vehicle for comparison. Insets: During the log-phase of growth (OD600=0.5), TMA lyase activity was measured by LC/MS/MS quantification of d9-TMA generated from d9-choline substrate. Growth curves shown are from representative experiments (n=3 technical replicates), and TMA quantification represents the mean ± SEM.
To better understand the mechanism of inhibition, we first characterized the kinetics of IMC- and FMC-dependent inhibition of recombinant CutC/D since time-dependent enhancement in enzyme inactivation is a characteristic feature of suicide substrate inhibitors44,45. As expected, both IMC and FMC demonstrated enhanced inhibitory potency following longer pre-incubation times with the enzyme prior to the addition of d9-choline substrate (Figure 2b). Second, as anticipated for a mechanism-based mode of inhibition, the inactivation of CutC/D by both IMC and FMC were irreversible, since dialysis failed to rescue enzyme activity. In contrast, reversible inhibition of CutC/D activity following dialysis was observed with an alternative, potent inhibitor developed, phenylcholine (IC50=150 nM against P. mirabilis CutC/D) (Figure 2c).
We further examined the behavior of FMC and IMC interaction with recombinant P. mirabilis CutC/D using Michaelis-Menten kinetics. Addition of FMC at increasing concentrations lowered maximal enzyme velocity (Vmax) with no apparent effect on substrate affinity (KM) (Supplementary Figure 5), indicating that FMC acts kinetically as a non-competitive inhibitor. Increasing concentrations of IMC similarly decreased Vmax, but also increased KM (Supplementary Figure 5), indicating IMC acts kinetically as a mixed mechanism inhibitor, with elements of both competitive and non-competitive inhibition. In an effort to demonstrate an enzyme-inhibitor adduct, we performed multiple mass spectrometry studies but were unable to find a covalent adduct between IMC or FMC with either CutC or CutD. Nevertheless, both IMC and FMC display numerous characteristics consistent with a covalent mode of inhibition (time-dependent, irreversible, and non-competitive inhibition), which is also consistent with our initial hypothesized, quantum mechanical-derived suicide substrate-based reaction mechanism (Figure 1d, Supplementary Figure 4).
Importantly, both IMC and FMC were non-lethal, even at high concentrations (1 mM). Moreover, neither affected the growth rate of several known TMA-producing human commensals (P. mirabilis, E. fergusonii, and P. penneri)33,46 when cultured in nutrient-rich media under conditions showing complete inhibition in choline TMA lyase activity (Figure 2d). In studies with prolonged exposures to either IMC or FMC, no reduction in apparent microbial fitness (growth rate or density) were observed (Supplementary Figure 6).
FMC and IMC suppress host TMA and TMAO levels for sustained periods of time with limited systemic exposure and without observed toxicity
In initial experiments, mice were placed on a choline-supplemented diet, and subsequent provision of either IMC or FMC as a single oral dose via gastric gavage resulted in marked inhibition of plasma TMAO levels (>95% inhibition, p<0.0001) (Figure 3a). We next progressed to a chronic daily exposure study, treating mice on a choline-supplemented diet with inhibitor via oral gavage once a day for 2 weeks, monitoring plasma TMAO at time of trough (24 hours post-gavage), resulting in virtually complete inhibition in circulating TMAO levels (Figure 3a center, Supplementary Figure 7). In dose-response studies, both FMC and IMC showed dose-dependent suppression in both TMA production (EC50=4.5 and 31 mg/kg, respectively; Supplementary Figure 8) and systemic TMAO levels, with FMC demonstrating potency an order of magnitude greater than IMC (EC50=3.4 and 45 mg/kg, respectively; Table 1, Figure 3a right). We also examined the in vivo dose response curves for microbial choline TMA lyase inhibition by quantifying d9-TMA and d9-TMAO production from d9-choline provided concomitantly with the inhibitor as a single oral gavage. Under these conditions, both FMC and IMC demonstrated remarkable capacity to suppress production of d9-TMA (EC50=0.008 and 0.5 mg/kg, respectively; Supplementary Figure 8) and d9-TMAO (EC50=0.01 mg/kg and 0.2 mg/kg, respectively; Table 1, Figure 3a right).
Figure 3. The in vivo pharmacokinetic and pharmacodynamic properties of FMC and IMC.

a) (Left) Plasma TMAO levels 24 hours post-gavage of vehicle, 100 mg/kg FMC, or 100 mg/kg IMC in mice maintained on a choline-supplemented (1% w/w) diet. Significance determined by two-tailed Student’s t-test. (Center) Plasma TMAO levels (determined at 24 hours post-gavage) on the indicated days over a course of daily FMC or IMC treatment for 14 days. (Right) Relative activity (compared to vehicle controls) in mice treated by oral gavage with either vehicle (“No Addition”, NA) or the indicated range of doses (0.0001 – 310 mg/kg) of either FMC or IMC. For each inhibitor, two groups of mice were tested. In the “d9-choline challenge” group (⋄), mice were maintained on a chemically-defined chow diet (0.08% w/w total choline) and simultaneously gavaged with 10 mg/mL d9-choline plus the indicated dose of FMC or IMC as described in Methods; the relative activity was measured in blood collected 3 hours post-gavage as the amount of d9-TMAO produced relative to the vehicle control (100%). In the “q24h post-gavage” groups (O), mice were maintained on a choline-supplemented diet (1% w/w) and treated with once-daily oral gavages of the indicated inhibitor and dose for 4 days; the relative activity represents plasma TMAO levels relative to the vehicle control in blood collected 24 hours after the last gavage (on day 5). Data points represent the mean ± SEM. Exact numbers of mice used for each data point can be found in the Source Data (n=4–14).
b) Plasma levels of (left) FMC and FMB and (right) choline, betaine, TMA, and TMAO at the indicated time points after a single oral gavage of FMC (100 mg/kg) in mice maintained on a choline-supplemented diet (1% w/w) for 3 weeks. Center, levels of FMC and FMB in fresh fecal samples, normalized to the dry weight of the samples. Data points represent the mean ± SEM for the indicated number of mice.
c) Concentrations of FMC, IMC, choline and TMA within the indicated intestinal luminal compartment 4 hours after a single gavage of either vehicle, 100 mg/kg FMC, or 100 mg/kg IMC in mice maintained on a choline-supplemented diet (1% w/w) for 3 weeks. Bars represent the mean ± SEM for the indicated number of mice.
We next examined both the pharmacokinetics and functional metabolic effects of oral FMC and IMC provision by measuring plasma, fecal, and urinary levels of the drugs, their metabolites, and both choline and its microbial- and host-derived metabolites over time in mice fed a high-choline diet. Results with FMC (Figure 3, Supplementary Figure 9) and IMC (Supplementary Figure 10) were similar, with FMC showing enhanced potency and sustained duration of inhibition. FMC and IMC were detectable at low (μM) levels only for the first few hours in plasma, but their halomethylbetaine oxidation products, fluoromethylbetaine (FMB) and iodomethylbetaine (IMB), were ~10-fold more abundant, reaching their peak level 2 hours post-gavage (Figure 3b left, Supplementary Figure 10). Despite intensive screening at multiple time points, other potential metabolites, including the halide-substituted versions of TMA, and the predicted intermediate trimethylhalide-imines (see Supplementary Figure 4), were not detected in plasma, urine or fecal samples. Importantly, analyses of feces revealed the majority of FMC and IMC remain in the gut luminal compartment, with only nominal levels of FMB or IMB present in fecal samples (Figure 3b center, Supplementary Figure 10). Modest but relatively equal amounts of FMC and FMB (Supplementary Figure 9) and IMC and IMB (Supplementary Figure 10) were observed in urine.
Both FMC and IMC induced an almost complete reduction in plasma TMA and TMAO levels following a single oral dose (via gavage) for a sustained period, with FMC demonstrating a striking trough that persisted for over 3 days (Figure 3b right), and IMC for over 2 days post-gavage (Supplementary Figure 10). These same trends for TMA and TMAO were observed in the urine and feces following administration of either FMC or IMC (Supplementary Figures 9,10). Plasma betaine levels were modestly increased, before returning to baseline, following gavage of either FMC (Figure 3b right) or IMC (Supplementary Figure 10). Choline (via betaine) can enter into one-carbon methyl donor-related metabolic pathways; however, following single or chronic dosing with FMC or IMC, no changes were noted in plasma levels for homocysteine, dimethylglycine, or folate pathway-related metabolites (all p>0.05, data not shown). Analyses of fecal samples revealed significant increases in choline levels following FMC or IMC administration, which before addition of the inhibitor were extremely low (Supplementary Figures 9,10). Importantly, no significant effects on plasma choline levels were observed (Figure 3b right, Supplementary Figure 10). These results are interesting in light of recent reports by Rey and colleagues33,47 suggesting gut microbiota, via the CutC/D pathway, may play a significant role in choline bioavailability in the host.
No signs of toxicity were observed in mice following chronic exposure to wholly effective doses of FMC (10 mg/kg) or IMC (100 mg/kg) (Supplementary Figure 11). This included no indications of body weight loss, grooming or other behavioral problems, or adverse effects on renal functional measures (serum creatinine, BUN), liver function tests (ALT, AST, BUN), and hematology related measures (hemoglobin, hematocrit, and complete blood cell count with differential that remained within the normal range)48. In addition, plasma levels of choline and betaine were not significantly different (from vehicle) with chronic exposure to either FMC or IMC, and neither FMB nor IMB showed evidence of accumulation in plasma (Supplementary Figure 12). In alternative studies in both male and female mice with more prolonged exposures (15–20 weeks) to IMC, no adverse effects were observed, including normal indices of renal function, liver function, and CBC with differential (data not shown). Screening of IMC and FMC in a battery of pharmacological safety tests failed to show: (i) inhibition in hERG channel function; (ii) toxicity to mitochondria in HepG2 cells cultured in glucose- or galactose-supplemented media; (iii) adverse effects on the viability of HK-2 cells in culture; or (iv) adverse signals during AMES testing at levels up to 1000 μg/well (Supplementary Table 1).
IMC and FMC are preferentially sequestered within gut microbes
As part of our drug development design, we hypothesized that sustained inhibitory activity could be achieved following single dose exposures if the inhibitors were non-lethal, and selectively accumulated within the microbe by induction of the cut gene cluster, which contains an active choline transporter. In other words, with active transport of the inhibitor into the microbe and accumulation to high levels, it would take multiple bacterial divisions to slowly and progressively deplete microbe intracellular inhibitor concentration until it was below the therapeutic threshold before TMA production from choline would occur. To test for this, we performed experiments in which gut microbes were recovered from different segments of the intestines 4 hours after a single oral gavage with either FMC, IMC, or vehicle in mice fed a high-choline diet. Strikingly, within the large intestine (cecum and colon), the anatomic location where the majority of choline TMA lyase harboring commensals reside49, we observed the selective accumulation of each inhibitor to the millimolar level (2–6 mM), virtually complete elimination in detectable luminal TMA (product), and a similar marked increase in intestinal microbial choline (substrate) levels (Figure 3c). Levels of halomethylbetaines in all intestinal segments were significantly lower than their halomethylcholine counterparts, and there was no significant effect on gut luminal betaine or TMAO levels (Supplementary Figure 13).
Inhibition of gut microbial choline TMA lyase activity suppresses choline diet-enhanced platelet aggregation and in vivo rate of thrombus formation without increased bleeding time
In further studies mice were placed on either chow or choline-supplemented diets in the absence or presence of inhibitors to determine effects on platelet aggregation. Results confirmed complete suppression of choline diet-enhanced TMAO generation and accompanying enhancement in ADP-dependent platelet aggregation response with provision of either FMC (p=0.0001) or IMC (p=0.001; Figure 4a). Representative platelet aggregometry tracings from each group are shown in Supplementary Figure 3. Furthermore, provision of IMC to the mice was shown to reverse choline diet-induced effects on not only multiple indices of platelet responsiveness using distinct agonists (e.g. ADP, collagen) in PRP, but also in ADP-stimulated isolated platelets (Supplementary Figure 14). In additional control studies, FMC and IMC were directly incubated with PRP from both mice and humans (with or without agonist), and no effects on platelet aggregometry responses were observed. Their primary in vivo metabolites, FMB and IMB, also did not affect platelet aggregometry responses in human PRP (Supplementary Figure 2). We next examined the effect of IMC on choline diet-dependent enhancement in platelet adhesion within whole blood using a microfluidic device under conditions of physiological levels of shear stress. Provision of IMC reversed choline diet-dependent increases in both TMAO levels and platelet adherence to collagen matrix to levels observed in chow-fed mice (Figure 4b).
Figure 4. The mechanism based CutC/D inhibitors IMC and FMC reverse choline diet-enhanced platelet responsiveness and thrombus formation.

a) Platelet aggregation in PRP of mice fed the indicated diets ± FMC (0.006% w/w) or IMC (0.06% w/w) for 2 weeks. Platelet aggregation was measured in response to a submaximal concentration of ADP (1 μM). Data points represent aggregation as % of maximum amplitude in PRP recovered from each mouse, and bars represent mean levels for each group. Plasma TMAO levels are also shown and represent mean ± SEM for the indicated number of mice for each group. Significance determined by two-tailed Student’s t-test.
b) Platelet (fluorescently-labeled) adherence in whole blood samples to a collagen-coated microfluidic biochip under physiological shear stress from mice fed the indicated diets (0.06% w/w IMC) for 2 weeks. Data points represent the mean ± SEM for the indicated numbers of mice. Plasma TMAO levels are also shown and represent mean ± SEM for each group. Significance between “Choline” and “Choline + IMC” groups determined by two-tailed Student’s t-test.
c) Representative vital microscopy images of carotid artery thrombus formation at the indicated time points following FeCl3-induced carotid artery injury in mice fed the indicated diets for 2 weeks. The time to complete vessel occlusion is noted in the right-hand panels. Cumulative study results for the indicated number of mice in each group are shown in Figure 4d.
d) Quantification of in vivo thrombus formation following FeCl3-induced carotid artery injury in mice fed the indicated diets ± FMC (0.006% w/w) or IMC (0.06% w/w) for 2 weeks. Data points represent the time to cessation of flow for each mouse, and bars represent mean levels for each group. Plasma TMAO levels are also shown and represent mean ± SEM for each group. Significance determined by two-tailed Student’s t-test.
(e) The cecal contents were harvested from the mice used in Figure 4d, and qPCR was used to quantify the relative expression of microbial cutC as described under Methods. Data points represent the relative cutC expression for each mouse, and bars represent mean levels for each group. Significance determined by two-tailed Student’s t-test.
f) Bleeding time following tail-tip amputation in the indicated number of mice fed the indicated diets (0.006% w/w FMC; 0.06% w/w IMC) for 1 week. Data points represent the cumulative bleeding time over 10 minutes for each mouse, and bars represent mean levels for each group. Plasma TMAO levels are also shown and represent mean ± SEM for each group. Significance was determined using two-way ANOVA.
The impact of FMC and IMC on in vivo thrombus formation was next examined using the carotid artery FeCl3-induced injury model. When mice were placed on either chow or choline-supplemented diets, in the absence or presence of inhibitor (0.06% IMC or 0.006% FMC), provision of either FMC (p<0.0001) or IMC (p<0.0001) completely blocked choline-supplemented diet-induced increases in both plasma TMAO and in vivo rate of clot formation (Figure 4c,d). Importantly, neither FMC (p=0.85) nor IMC (p=0.73) led to a prolongation in occlusion time beyond that observed with normal chow. In these animals, we also investigated potential effects of FMC and IMC on the regulation of the microbial cut gene cluster within the cecal polymicrobial community. As a prototypic gene within the cluster, we quantified cutC expression via recovered cecal microbial RNA by quantitative PCR. Notably, global cecal microbial cutC expression was significantly upregulated by the addition of either FMC or IMC in chow diet in the absence of added choline (Figure 4e). Similar enhanced cutC expression was also seen with choline supplementation relative to chow. Addition of FMC or IMC to the choline-supplemented diet groups failed to further increase microbial expression of cutC (Figure 4e).
In further studies, we examined the impact of FMC and IMC on in vivo bleeding time. Mice were again placed on either chow or choline-supplemented diets with or without FMC or IMC, and then bleeding time was determined using a tail tip amputation model50. While TMAO levels were markedly suppressed, neither FMC nor IMC induced a significant change in bleeding potential, as monitored by multiple different measures including cumulative bleeding time (pANOVA=0.90) (Figure 4f), hemoglobin loss (pANOVA=0.14), and weight loss from bleeding (pANOVA=0.51) (Supplementary Figure 15).
Inhibition of microbial choline TMA lyase activity shifts intestinal microbial communities
While the halomethylcholines developed are non-lethal to cultured microbes when alternative nutrients are available (Figure 2d), we sought to determine whether they induce a shift in gut microbial composition in vivo. Cecal microbial DNA encoding 16S ribosomal RNA was sequenced from mice examining the impact of IMC on in vivo thrombus formation (a subset of mice in Figure 4d). Principal Coordinates Analysis of microbial taxa revealed distinct clusters, indicating both dietary choline supplementation and IMC exposure each induced significant (p=0.001) rearrangements in microbial composition (Figure 5a). Performance of Linear Discriminant Analysis coupled with effect size measurements permitted further identification of microbial taxa whose proportions accounted for significant characteristic differences observed in the chow versus choline-supplemented diets, as well as IMC-treated versus non-IMC-treated groups (Figure 5b, Supplementary Figure 16).
Figure 5. Microbial choline TMA lyase inhibitor reverses diet-induced changes in cecal microbial community composition associated with plasma TMAO levels, platelet responsiveness, and in vivo thrombosis potential.

a–b) Groups of mice were maintained on the indicated diets ± IMC (0.06% w/w) for 2 weeks. Cecal contents were harvested and intestinal microbial community composition was assessed by (a) Principal Coordinates Analysis or (b) Linear Discriminant Analysis (LDA) effect size (LEfSe).
c) (Top) Scheme illustrating the relationship between human gut commensal choline TMA lyase activity, TMA and TMAO generation, and enhanced platelet responsiveness and thrombosis risk in the host. (Bottom) Illustration of the impact of halomethylcholine mechanism-based microbial choline TMA lyase inhibitors (XMC) on human commensal TMA generation, host TMAO generation, platelet responsiveness, and thrombosis potential. Upon irreversible enzymatic inhibition of CutC, microbial cytosolic choline increases. Choline is sensed as an abundant nutrient, leading to upregulation of the cut gene cluster, including cutC and a choline active-transporter. Choline and the halomethylcholine inhibitor are actively pumped into the microbe and accumulate. Sequestration of choline in the microbe also depletes the levels of choline available to neighboring microbes, further preventing production of TMA from the gut microbial community and contributing to a reduction in systemic TMA and TMAO levels in the host. The net effect is reduction in platelet aggregation responsiveness to multiple agonists and reduced thrombosis potential in the host.
We also examined whether the proportions of any of the detected cecal genera within all groups of mice were significantly correlated with both plasma TMAO levels and in vivo thrombosis potential as quantified by time to carotid artery vessel occlusion during the FeCl3-induced injury model. Provision of a high-choline diet was associated with both significant increases (e.g. Dehalobacterium and Adlercreutzia) and reductions (e.g. Bifidobacterium) in the proportions of several genera, which were each also significantly associated with both TMAO levels and time to vessel occlusion; moreover, these shifts in proportions were reversed with the addition of IMC (p<0.0001; Supplementary Figure 17). In parallel studies, the impact of FMC treatment on choline diet-induced changes in cecal microbial composition showed analogous results (Supplementary Figure 18). Interestingly, both IMC and FMC treatment in choline-supplemented mice induced a significant increase in the proportions of Akkermansia (Supplementary Figure 17,18), a genus of considerable interest because of its reported links to improvement in obesity and metabolic health51.
Discussion
The growing recognition of a contributory role of gut microbiota to both health and disease susceptibility promises that efforts to “drug the microbiome”, which are still in their infancy, may someday become commonplace in medicine. Herein we describe the development and characterization of potent inhibitors of gut microbiota-dependent TMA/TMAO generation that might serve as therapeutic agents capable of reducing thrombosis risk (Figure 5c). In contrast to current antiplatelet drugs, which target mammalian enzymes (e.g. COX1) or receptors (e.g. P2Y12, PAR1)52 and are limited in use by their potential for untoward bleeding, inhibition in microbial TMAO generation would not hypothetically suppress platelet function “below normal” and thus heighten bleeding risks. Further, given the unorthodox characteristics of a gut microbial pharmacological target, one can envision developing drugs that optimally target a gut microbial pathway mechanistically linked to disease susceptibility while limiting systemic exposure in the host.
One potential theoretical difficulty in “drugging the microbiome” is that if every related microbial enzyme with similar function is not inhibited, there exists potential for compensation from alternative species, possibly limiting overall change in total microbial community activity. To overcome this issue, our drug development program incorporated several features. First, recognizing that microbial catabolic enzyme systems, such as the choline utilization (cut) cluster, typically contain coordinately regulated genes, including a microbial transporter that actively transports the substrate into the microbe53,54 to enable rapid exploitation of increased availability of an abundant nutrient55, we speculated that development of a substrate analogue inhibitor that irreversibly inactivated its target, while retaining ability to be used by the microbial transporters, could produce a rise in microbial cytosolic substrate concentration. This might produce a “feed forward” cycle whereby both the substrate and the drug (substrate analogue) are actively transported and sequestered within the microbe in proportion to the extent of inhibition. Indeed, we saw that by blocking choline catabolism with the potent CutC/D inhibitors, cutC expression is significantly increased in cecal microbes without dietary choline supplementation. And both choline and the inhibitors reached remarkably high gut microbial intracellular levels in the cecum and colon. Thus, our system of inhibition appears to create a positive feedback loop whereby the microbes in which the halomethylcholines potently blocks choline catabolism are transformed into virtual “vacuum sweepers” for both choline and inhibitor accumulation. Microbes in the immediate environment that are not as potently inhibited directly might still thus have reduced TMA generation as they become starved of extracellular choline substrate.
Our efforts to develop inhibitors were also helped by the fact that there are no known mammalian enzymes that can cleave the C-N bond of choline54. We therefore sought to develop selective, non-reactive choline analogues that contain a cryptic reactive group that might become liberated upon enzymatic cleavage by the unique microbial enzymes that produce TMA, the precursor for TMAO generation (i.e. suicide substrate mechanism-based inhibitors). We also utilized a comprehensive screening strategy that included phylogenetically diverse recombinant microbial choline TMA lyase enzymes and both mono- and poly-microbial (human fecal) assays to ensure the broad efficacy needed to inhibit a range of microbial choline TMA lyase enzymes from diverse taxa, while also increasing the chances for development of highly potent inhibitors in vivo. Although we designed IMC to be the prototypical mechanism-based inhibitor, FMC proved to be more potent. Indeed, fluorine substitution can confer properties beneficial to the chemistry of suicide substrate inhibitors and their potential use as pharmaceuticals56.
Kinetic analyses demonstrate that IMC and FMC are non-competitive, time-dependent and irreversible, hallmarks of mechanism-based inhibitors. However, despite extensive efforts, we were not able to find a covalent inhibitor-enzyme adduct via mass spectrometry. Upon observing that IMC shows kinetic evidence of both competitive and non-competitive forms of inhibition, we hypothesize that there exist several potential avenues for halomethylcholines to serve as suicide substrate inhibitors and promote irreversible, covalent attachment to multiple nucleophilic amino acid targets (Supplemental Figure 4). This potential, along with their high inhibitory potency make detection more elusive. Nonetheless, both FMC and IMC proved non-lethal to human commensals when cultured in nutrient-rich broth under conditions demonstrating full inhibition of microbial choline TMA lyase activity. In mice, FMC and IMC lowered plasma TMAO levels over a prolonged period with limited systemic exposure. Moreover, by inhibiting gut microbial choline consumption, fecal choline content significantly increased without impacting plasma choline levels. These results suggest that inhibition in microbial choline utilization will not only suppress TMA/TMAO generation, but also may help enhance choline bioavailability for the host.
It is also notable that both IMC and FMC induced a shift in mouse cecal microbiota composition, particularly on a choline-supplemented diet, reversing choline-induced changes in the proportions of certain taxa. Thus, eliminating selective pressures may prove elusive when targeting the microbiome, even if the inhibitors are non-lethal to cultured bacteria in nutrient-rich broths and fail to demonstrate reduction in microbial fitness with prolonged exposures. Since the proportions of several taxa reduced by inhibitor exposure were also associated with both TMAO levels and shortened time to arterial occlusion, with chronic inhibitor use, it appears likely that some of the potential efficacy of this family of inhibitors in suppressing TMAO levels also occurs by shifting of microbial composition to one less prone to produce TMA. This effect of the inhibitors appears to be secondary, and it is worth noting that significant reductions in TMA/TMAO production can be detected rapidly at 1 hour post oral gavage of inhibitor concurrently with isotope-labeled choline precursor (Supplementary Figure 19). It is also noteworthy that many drugs induce shifts in microbial community composition57,58. In a recent examination of over 1,000 clinically used drugs and their impact on a broad selection of human gut commensals, nearly a quarter demonstrated antibiotic-like side effects59. And in a recent elegant study with metformin, a shift in microbial community structure was suggested to contribute in part to its observed beneficial metabolic effects60.
The present studies suggest development of poorly absorbed suicide substrate inhibitors of gut microbial catabolic pathways mechanistically linked with disease pathogenesis deserves further investigation as a potentially generalizable approach to achieve therapeutic outcomes in the host. In the present case, by inhibiting gut microbial production of TMA, we suppressed plasma TMAO levels for prolonged periods with limited systemic exposure of the drugs within the host, reversing choline diet-induced increases in platelet responses to agonists and thrombus formation in vivo. We observed an almost complete reduction in TMA at the site of action in the large intestine with a parallel buildup of inhibitor within the microbe to concentrations a million-fold higher than necessary to achieve inhibition within the microbe. A shift in cecal microbial community induced by the inhibitor was noted that chronically may contribute to reduction in systemic TMA/TMAO levels in the host. Importantly, no evidence of systemic toxicity was observed and our treatment did not adversely affect bleeding potential, a common side effect of traditional antiplatelet therapies. These studies suggest a new potential target for the treatment of subjects at risk for thrombotic complications and cardiovascular disease.
Methods
Ethical considerations
All animal model studies were approved by the Institutional Animal Care and Use Committee at the Cleveland Clinic. All study protocols and informed consent for human subjects were approved by the Cleveland Clinic Institutional Review Board. Informed consents were obtained for all subject samples. All studies complied with all relevant animal and human use guidelines and ethical regulations.
In vivo carotid artery thrombosis models
C57BL/6J female mice (6–10 weeks of age) were fed a chemically-defined diet comparable to normal chow (0.08% total choline) versus the same chemically-defined diet supplemented with 0.5% or 1% w/w choline (Envigo TD.140294 and TD.09041, respectively). Half of the mice were treated with TMA lyase inhibitors (DMB, IMC or FMC) in their diet for more than 1 week. Mice (>12 weeks of age) were anaesthetized (100 mg/kg ketamine and 10 mg/kg xylazine) and subjected to common carotid artery injury by application of 10% FeCl3 for 1 minute as previously described15.
Mouse ex vivo platelet aggregometry studies
C57BL/6J female mice (6–10 weeks of age) were fed a chemically-defined diet comparable to normal chow (0.08% total choline) versus the same chemically-defined diet supplemented with either 0.5% or 1% w/w choline (Envigo TD.140294 and TD.09041, respectively as indicated). Half of the mice were treated with TMA lyase inhibitors (DMB, IMC or FMC) in their diet for more than 1 week. Mice (>12 weeks of age) were anesthetized with ketamine (100 mg/kg) and xylazine (10 mg/kg), and blood samples were collected and analyzed for ex vivo aggregation response as previously described15.
Recombinant TMA lyase inhibition assays
Enzymatic assays were conducted to measure the conversion of choline to TMA using an isotope-labeled version of choline (d9[trimethyl]-choline). Clarified lysate was diluted to 5 mg/mL in lysis buffer and the necessary co-factors were added (1 mM SAM, 10 mM NaDT, and 2 mM NADH). Individual 400 μL aliquots were added to 13 × 100 mm threaded glass tubes with gas-tight mininert caps. Potential inhibitor compounds were serially diluted (10-fold) and added to the reaction mixtures. Each inhibitor concentration was done in triplicate. The mixture was then allowed to incubate for 15 minutes before the addition of substrate (300 μM final) to initiate the reaction, which was carried out for 2 hours at room temperature in the dark. Whole-cell assays were conducted similarly at 37 °C. The relevant cell cultures were diluted 1:100 in LB medium and shaken at 37 °C until an OD600=0.5 was reached. At this point, the cells were aliquoted (400 μL) in 13 × 100 mm threaded glass tubes with gas-tight mininert caps. Without the need for co-factors, the reaction was initiated with the addition of d9-labeled substrate after a period of incubation with potential inhibitor compounds. Whole-cell reactions were carried out at 37 °C for 2 hours. For each experiment, “no addition” (vehicle) controls were used to calculate 100% relative activity. Reactions were quenched 15 minutes after substrate addition (during the initial, linear kinetic phase) for the analysis of time-dependence and Michaelis-Menten kinetics.
The reversibility of inhibitors was tested by overnight dialysis. Crude lysate was prepared, as previously described (with or without addition of inhibitor), prior to addition to 3,500 MWCO Slide-A-Lyzer Mini Dialysis Units (Pierce, Rockford, IL) in triplicate. Each unit was floated on ~500 mL of lysis buffer for 2 hours with gentle stirring at 4 °C. The buffer was then changed before continuing stirring overnight. Reaction samples were also prepared without dialysis and left overnight at 4 °C as a control. Reactions were initiated the next day with the addition of substrate and carried out as previously described above.
The reactions were quenched with the addition of 200 μL of 1 M NaOH, and 8 μL of 25 μM [13C315N1]TMA (Sigma Chemicals, St. Louis, MO) was added as the internal standard before placing on ice. After 15 minutes, 1.6 mL lysis buffer, 2 mL hexane, and 1 mL butanol were added to each reaction mixture. The tubes were then vortexed for 1 minute and centrifuged at 2500 × g for 10 minutes. The top, organic layer was then transferred to 12 × 75 mm PTFE capped threaded glass tube, acidified with the addition of 200 μL of 0.2 N formic acid, vortexed, and the organic and aqueous layers were separated by centrifugation. After centrifugation, an aliquot of formic acid containing the d9-TMA product was transferred to a mass spectrometry vial containing a plastic insert for quantification by stable isotope dilution LC/MS/MS analysis.
All data analyses were performed in GraphPad Prism. For IC50 calculations, the data were fit to a nonlinear regression equation (normalized response, variable slope). For time-dependence, the data were fit to the dissociation (one phase exponential decay) equation. For Michaelis-Menten, the curves were fit (along with calculations of KM and Vmax) by nonlinear regression (“Michaelis-Menten”).
Quantification of d9-TMA
Samples were injected onto a reverse phase C18 HPLC column and resolved with a linear gradient between 0.2% formic acid in water and 0.2% formic acid in an acetonitrile/methanol mixture (95:5 v/v). The effluent was analyzed by an AB Sciex 5500 QTRAP mass spectrometer using electrospray ionization in the positive-ion mode. TMA was quantified by measuring the peak area ratio of the precursor to product ion transition of d9-TMA (m/z 69 → 49) to the internal standard [13C315N1]TMA (m/z 64 → 47).
Human fecal polymicrobial assay
Human fecal samples were collected from healthy volunteers with no known chronic illnesses, blood borne diseases or active infections. The volunteers had not received antibiotics within two months of donation and provided written informed consent. Samples were diluted to make a 20% (w/v) fecal slurry by resuspension of the feces in a media containing 3% (w/v) tryptic soy broth, 1% (w/v) trehalose, pH 7.3. The fecal slurry was homogenized and filtered by hand using a stomacher bag with an integrated 170 μm membrane. DMSO (5% (w/v) was added to the filtered slurry and aliquots were stored in cryogenic vials at −80°C until use. Frozen fecal slurries were diluted to 0.2% (w/v) with M9 media (Na2HPO4 (6 g/L), KH2PO4 (3 g/L), NaCl (0.5 g/L) with addition of 0.1 mM CaCl2 and 1 mM MgSO4) and dispensed (1 mL) into deep well 96-well plates. Diluted fecal slurries containing 50 μM d9-choline chloride and halomethylcholine and halomethylbetaine compounds in doses ranging from 3.81 nM to 250 μM were sealed and incubated at 37 °C with shaking. After 20 hours, an aliquot of the fecal polymicrobial community was analyzed for viability using Prestoblue cell viability reagent following the manufacturer’s instructions (Thermo Fisher Scientific). The reaction plates were subsequently centrifuged (4000 × g at 4 °C for 12 minutes) to pellet fecal material and 150 μL aliquots were transferred and quenched with addition of formic acid to 1% (v/v). All fecal processing and polymicrobial assay steps were performed in an anaerobic environment. The products were determined by LC/MS/MS and IC50 values were calculated as described previously for detection and analysis of TMA and d9-TMA in wild-type P. mirabilis cell lysates.
Quantitation of in vivo TMAO, TMA, choline, betaine, IMC, IMB, FMC, and FMB
Stable isotope dilution liquid chromatography with on-line tandem mass spectrometry (LC/MS/MS) was used for quantification of levels of TMAO, TMA, choline, and betaine in plasma, urine, fecal and tissue as previously described7,35,61. Their isotope-labeled (d9-) analogues were used as internal standards. For IMC quantification, d2-IMC was used as an internal standard, and for FMC, d9-choline was used as an internal standard. For IMB and FMB quantification, d9-betaine was used as an internal standard. LC/MS/MS analyses were performed on a Shimadzu 8050 triple quadrupole mass spectrometer. Although not observed in vivo, levels of iodo-trimethylamine (I-TMA), iodo-trimethylamine-N-oxide (I-TMAO), fluoro-trimethylamine (F-TMA), and fluoro-trimethylamine-N-oxide (F-TMAO) were monitored at predicted transitions using multiple reaction monitoring of precursor and characteristic from CID spectra of synthetic standards product ions: m/z 186.1 → 44.2 for I-TMA; m/z 202 → 184 for I-TMAO; m/z 78.1 → 44.2 for F-TMA; m/z 94 → 76 for F-TMAO. Levels of TMAO, d9-TMAO, TMA, d9-TMA, choline, d9-choline, betaine, d9-betaine, IMC d2-IMC, IMB, FMC, and FMB were monitored using multiple reaction monitoring of precursor and characteristic product ions: m/z 76.0 → 58.1 for TMAO; m/z 85.0 → 66.25 for d9-TMAO; m/z 60.2 → 44.2 for TMA; m/z 69.0 → 49.1 for d9-TMA; m/z 104.0 → 60.15 for choline; m/z 113.1 → 69.2 for d9-choline; m/z 118.0 → 58.1 for betaine; m/z 127.0 → 66.2 for d9-betaine; m/z 230.0 → 58.05 for IMC; m/z 232.0 → 60.1 for d2-IMC; m/z 244.0 → 58.1 for IMB; m/z 122.1 → 58.05 for FMC; m/z 136 → 58.1 for FMB.
Effects on plasma TMAO levels
For our “q24h post-gavage” model, C57BL/6J female mice (12 weeks of age; obtained from Jackson Laboratory #0664) were placed on 1% w/w choline diet (Teklad #TD.09041) and given either vehicle, IMC or FMC at concentrations ranging from 1.8 to 310 mg/kg once daily via gavage. Blood was collected 24 hours post-gavage at the indicated days. For our “d9-choline challenge” model, C57BL/6J female mice (18–20 g average body weight) fed normal chow were fasted for 1 hour. At which point, 2 mg d9-choline in 0.2 mL of sterile water was given via oral gavage in combination with either vehicle, IMC or FMC at concentrations ranging from 0.0001 to 100 mg/kg. The dietary fast was continued for 1 hour post-gavage. Blood was collected 3 hours post-gavage. For blood collection, mice were restrained in a rodent restraint tube. A 26G needle was used to lance the saphenous vein. Blood was collected into a heparin treated capillary tube and was spun in a capillary centrifuge (12,000 rpm, 5 minutes, 4 °C) to separate plasma. Samples were acidified (60 mM HCl final) for analysis of TMA levels prior to flash freezing and storage in gas-tight vials at −80 °C.
In vivo toxicity studies
C57BL/6J female mice (8–12 weeks of age, obtained from Jackson Laboratory #664) were placed on 1% w/w choline diet (Teklad #TD.09041) and given inhibitor or vehicle (water) once daily via gavage. Blood and urine were collected at the indicated days (4, 11, and 14 days after diet start). On day 14, mice were humanely euthanized by anesthesia overdose (>300 mg/kg ketamine + 30 mg/kg xylazine) and blood was collected for analysis of physiologic markers of toxicity. EDTA treated whole-blood from mice treated for 14 days was run on a Siemens Advia 120 Hematology Analyzer for complete blood count (CBC) with differential. Plasma was analyzed for liver (ALT, AST) and kidney function (BUN) using a Roche Cobas C 311 analyzer, and CBC with differential was determined using a Bayer Advia hematology analyzer.
Pharmacokinetics
C57BL/6J female mice (13 weeks of age, obtained from Jackson Laboratory #664) were placed on 1% w/w choline-supplemented diet (Teklad TD.09041) for at least 1 week prior to the start of the experiment. Mice were given a single oral dose of inhibitor via gastric gavage at 100 mg/kg. Whole-blood samples were collected at the indicated time-points post-gavage and treated with EDTA or heparin. Samples were either centrifuged at 4 °C and 4,000 rpm for 20 minutes or collected into a heparin treated capillary tube and was spun in a capillary centrifuge (12,000 rpm, 5 minutes, 4 °C) to separate and collect plasma before 1:4 dilution in cold methanol (containing internal standards). Urine samples were collected in 1.5 mL Eppendorf tubes. Samples were diluted 1:32 in sterile water and then diluted 1:4 in cold methanol (containing internal standards). Fecal samples were collected and vacuum-dried and weighed. Samples were homogenized in 2 mL cryovials with the addition of 1 mL sterile water and ~200 μL acid-washed glass beads (< 106 μm). Samples were then lysed using a TissueLyser II (Qiagen) at 30 m/s for 12 minutes before centrifugation (12,000 rpm, 4 °C, 30 minutes). Aliquots of the supernatants were diluted 1:4 in cold methanol (containing internal standards). LC/MS/MS was used to measure levels of plasma, urine, and fecal metabolites.
Intestinal luminal content analyses
C57BL/6J female mice (13 weeks of age; obtained from Jackson Laboratory #664) were placed on 1% w/w choline diet (Teklad # TD. 09041) for 3 weeks prior to the start of the experiment. Mice were given a single dose of vehicle or 100 mg/kg of compound via gavage. Four hours post-gavage, animals were humanely euthanized by anesthesia overdose (>300 mg/kg ketamine + 30 mg/kg xylazine) and the small intestine, cecum and colon were collected. Starting at the pyloric sphincter, the small intestine was segmented into 3 equal segments (duodenum, jejunum, ileum) 1 mL of buffer (200 mM HEPES, 50 mM NaCl, 10 mM MgCl2, pH 8.0) was used to flush lumen and contents were expelled by force. Cecum and colon were separated, flushed and content expelled by force. Precise concentrations of metabolites was measured by calculating a dilution factor based on the mass of buffer (assuming density = 1 mg/mL) and the change in mass before and after flushing of luminal contents and removal of intestinal tissue. Intestinal contents were transferred to a 2 mL cryovial with the addition of ~200 μL acid-washed glass beads (< 106 μm). Samples were then lysed using a TissueLyser II (Qiagen) at 30 m/s for 12 minutes before centrifugation (12,000 rpm, 4 °C, 30 minutes). Aliquots of the supernatants were diluted 1:4 in cold methanol (containing internal standards). Levels of metabolites were quantified by LC/MS/MS as described above.
Quantification of cutC RNA via qPCR
Quantitative real-time PCR (qPCR) was used to amplify cutC using degenerate primers using a method adapted from genomic analysis of cutC abundance. C57BL/6J female mice (10–13 weeks of age) were started on diets of either minimal choline or choline-supplemented (1% w/w) with or without the addition of FMC (0.006%) or IMC (0.06%) for 2 weeks. Mice were humanely euthanized (300 mg/kg ketamine and 30 mg/kg xylazine). Whole ceca were harvested and flash frozen in liquid nitrogen and stored at −80 °C. The cecal contents were excised on dry ice to minimize thawing of the sample. RNA isolation was performed using the PowerMicrobiome™ RNA Isolation Kit (Qiagen). Frozen cecal contents (~10 mg) were added to the provided tubes. 1.5 mL of chilled QIAzol (Qiagen) was added to each sample. Samples were immediately homogenized on the TissueLyzer LT (Qiagen) at 50 m/s. Three rounds of two minute lysing periods were each separated by one minute on ice to minimize RNA degradation. Chloroform (0.3 mL) was added to each sample and vortexed, and then samples were centrifuged at 4 °C for 10 minutes. The aqueous phase was isolated and used to carry out the remainder of the RNA isolation as described in the PowerMicrobiome™ RNA Isolation Kit instruction manual. RNA concentration was quantified using a Nanodrop 1000 spectrophotometer (Thermo Fisher). RNA (400 ng) was converted to cDNA using High Capacity cDNA Reverse Transcriptase Kit with random primers (Applied Biosystems). Transcript abundance of cutC was normalized to universal 16S ribosomal RNA abundance. Degenerate primer sequences used were62: cutC (forward) 5′-AGRGTTHGATYMTGGCTCAG-3′ and cutC (reverse) 5′-TGCTGCCTCCCGTAGGAGT-3′, and 16S (forward) 5′-TTYGCIGGITAYCARCCNTT and 16S (reverse) 5′-TGNGGYTCIACRCAICCCAT-3′. Reactions were run in triplicate using KAPA SYBR® FAST qPCR Master Mix (Roche) with 8 ng of cDNA per reaction and 1.5 μM primer concentration. Quantification was performed on the LightCycler 480 (Roche) with an initial 95°C step for 5 minutes was followed by 45 cycles of denaturation at 95 °C for 45 seconds, annealing at 57 °C for 45 seconds, and an extension at 72 °C for 45 seconds. Melting curves were performed by 95 °C for 5 seconds, followed by 65 °C for 60 seconds, and a continuous reading step of seven acquisitions per second between 65 °C and 97 °C. Results are expressed as averages of three independent reactions.
Bleeding time analyses
C57BL/6J female mice (Jackson #664) 8–12 weeks of age were placed on diets +/− 1% w/w choline (Envigo, TD. 09041; TD 130104) and +/− 0.06% IMC or 0.006% FMC for 1 week. Mice were anesthetized (100 mg/kg ketamine + 10 mg/kg xylazine) and maintained on a warming pad to maintain body temperature. A consistent injury was made 3 mm from the tip of the tail. The tail was immersed in 15 mL of 37 °C saline and the cumulative bleeding time was recorded during a 10 minute period. Body weights were recorded before and after bleeding (including the excised tail) to quantify blood loss. Hemoglobin content was measured by spectrophotometry. Red blood cells were isolated from the tail blood-containing saline samples by centrifuging (4,000 rpm, 5 minutes) and removing the supernatant. Pelleted cells were then lysed by hypotonic lysis. Cells were initially re-suspended in 900 μL H2O. After 17 seconds, 100 μL of 10x PBS was added and inverted to mix. The solution was then centrifuged at 10,000 rpm for 5 minutes. The supernatant was saved, and the optical density at 550 nm (OD550) was measured.
Statistical analyses
Unless otherwise indicated, one- or two-tailed Student’s t-tests or a Wilcoxon non parametric test were used to compare group means as deemed appropriate. The analysis of variance (ANOVA, if normally distributed) or Kruskal-Wallis test (if not normally distributed) was used for multiple group comparisons of continuous variables and a Chi-square test was used for categorical variables. A robust Hotelling T2 test was used to examine the difference in the proportion of specific bacterial genera along with TMAO levels or occlusion times between the different dietary and treatment groups. The indicated number of biological replicates were used in all experiments. Results from all animals in a given experiment were included in the analyses. Investigators performing quantitative analyses of endpoints (e.g., plasma, urine, or fecal metabolite levels) were blinded to group allocation with samples labeled by code only. Investigators were not blinded to mouse group allocation during the performance of animal husbandry requirements for experiments. Unless otherwise noted, most data were analyzed using R software version 2.15 and Prism (GraphPad Software).
Reporting Summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Data are available from the corresponding author upon reasonable request. The 16s sequencing datasets generated and analyzed during the current study are available in the NCBI Sequence Read Archive, https://www.ncbi.nlm.nih.gov/bioproject/PRJNA471699.
Supplementary Material
Acknowledgments
This research was supported by grants from the National Institutes of Health and the Office of Dietary Supplements (HL103866, HL126827 and DK106000 (to S.L.H.), HL122283 and AA024333 (to J.M.B.), and HL28481 and HL30568 (to A.J.L.)). S.L.H. reports being supported in part by a grant from the Leducq Foundation. V.G. acknowledges a Faculty Research Development Award from Cleveland State University. A.B.R. was supported in part by a grant from the American Heart Association (15POST25750053). W.Z. was supported in part by grants from the American Heart Association, and the American Stroke Association. S.M.S. was supported in part by training grant T32DK007470 from the NIH – NIDDK. Some of the toxicology/safety studies were performed by Pharmaron. Mass spectrometry studies were performed on instrumentation housed in a facility supported in part through a Shimadzu Center of Excellence award. Computational resources were provided by the Extreme Science and Engineering Discovery Environment (National Science Foundation). We appreciate the aid of Judith A. Drazba and Gauravi Deshpande of the Lerner Research Institute Imaging Core in studies using the Cellix microfluidic system.
Footnotes
Author Contributions
A.B.R, J.A.B. and X.G designed, performed, and analyzed data from most of the studies. They also helped to write the manuscript with input from all authors. A.G.H., A.D., V.G., A.J.M. and B.S.L aided in chemical synthesis and characterization of all compounds, computational drug design efforts, docking analyses and quantum mechanical calculations. Z.W., and X.F. helped with design and performance of mass spectrometry analyses. W.Z., N.G. and M.W.R. helped in the design and performance of platelet functional studies, in vivo thrombosis and other mouse experiments. S.M.S., J.M.L., L.L, W.T.B., and A.J.L. participated in microbe composition analyses and cut gene cluster transcription quantification studies. D.B.C., J.M.R. and J.C.G.-G. helped design and perform some of the human commensal and polymicrobial bioreactor studies characterizing inhibitor efficacy. J.A.D. provided overall advice, as well as performing plasmid cloning and construction use in multiple bacterial inhibitor studies. S.R., J.M.B., and J.C.G.-G. provided critical scientific input and discussions. S.L.H conceived, designed, and supervised all studies, and participated in the drafting and editing of the manuscript. All authors contributed to the critical review of the manuscript.
Competing Financial Interests
The authors declare the following competing interests: Drs. Hazen, Gu, Wang and Levison are named as co-inventors on pending and issued patents held by the Cleveland Clinic relating to cardiovascular diagnostics or therapeutics. Drs. Hazen, Wang and Levison report having the right to receive royalty payment for inventions or discoveries related to cardiovascular diagnostics from Cleveland Heart Lab, Inc. and Quest Diagnostics. Dr. Hazen also reports having been paid as a consultant for P&G, and receiving research funds from Astra Zeneca, P&G, Pfizer Inc., and Roche Diagnostics.
References
- 1.Kau AL, Ahern PP, Griffin NW, Goodman AL, Gordon JI. Human nutrition, the gut microbiome and the immune system. Nature. 2011;474:327–336. doi: 10.1038/nature10213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blaser MJ. The microbiome revolution. J Clin Invest. 2014;124:4162–4165. doi: 10.1172/JCI78366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fischbach MA, Segre JA. Signaling in Host-Associated Microbial Communities. Cell. 2016;164:1288–1300. doi: 10.1016/j.cell.2016.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aron-Wisnewsky J, Clément K. The gut microbiome, diet, and links to cardiometabolic and chronic disorders. Nat Rev Nephrol. 2016;12:169–181. doi: 10.1038/nrneph.2015.191. [DOI] [PubMed] [Google Scholar]
- 5.Schroeder BO, Bäckhed F. Signals from the gut microbiota to distant organs in physiology and disease. Nat Med. 2016;22:1079–1089. doi: 10.1038/nm.4185. [DOI] [PubMed] [Google Scholar]
- 6.Koopen AM, Groen AK, Nieuwdorp M. Human microbiome as therapeutic intervention target to reduce cardiovascular disease risk. Curr Opin Lipidol. 2016;27:615–622. doi: 10.1097/MOL.0000000000000357. [DOI] [PubMed] [Google Scholar]
- 7.Wang Z, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tang WHW, et al. Intestinal Microbial Metabolism of Phosphatidylcholine and Cardiovascular Risk. N Engl J Med. 2013;368:1575–1584. doi: 10.1056/NEJMoa1109400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koeth RA, et al. Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19:576–585. doi: 10.1038/nm.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Z, et al. Prognostic value of choline and betaine depends on intestinal microbiota-generated metabolite trimethylamine-N-oxide. Eur Heart J. 2014;35:904–910. doi: 10.1093/eurheartj/ehu002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tang WHW, et al. Prognostic Value of Elevated Levels of Intestinal Microbe-Generated Metabolite Trimethylamine-N-Oxide in Patients With Heart Failure: Refining the Gut Hypothesis. J Am Coll Cardiol. 2014;64:1908–1914. doi: 10.1016/j.jacc.2014.02.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang WHW, et al. Gut Microbiota-Dependent Trimethylamine N-Oxide (TMAO) Pathway Contributes to Both Development of Renal Insufficiency and Mortality Risk in Chronic Kidney DiseaseNovelty and Significance. Circ Res. 2015;116:448–455. doi: 10.1161/CIRCRESAHA.116.305360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang WHW, et al. Intestinal Microbiota-Dependent Phosphatidylcholine Metabolites, Diastolic Dysfunction, and Adverse Clinical Outcomes in Chronic Systolic Heart Failure. J Card Fail. 2015;21:91–96. doi: 10.1016/j.cardfail.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Organ CL, et al. Choline Diet and Its Gut Microbe–Derived Metabolite, Trimethylamine N-Oxide, Exacerbate Pressure Overload–Induced Heart FailureCLINICAL PERSPECTIVE. Circ Heart Fail. 2016;9:e002314. doi: 10.1161/CIRCHEARTFAILURE.115.002314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu W, et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell. 2016;165:111–124. doi: 10.1016/j.cell.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Senthong V, et al. Intestinal Microbiota-Generated Metabolite Trimethylamine-N-Oxide and 5-Year Mortality Risk in Stable Coronary Artery Disease: The Contributory Role of Intestinal Microbiota in a COURAGE-Like Patient Cohort. J Am Heart Assoc Cardiovasc Cerebrovasc Dis. 2016;5:e002816. doi: 10.1161/JAHA.115.002816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Warrier M, et al. The TMAO-Generating Enzyme Flavin Monooxygenase 3 Is a Central Regulator of Cholesterol Balance. Cell Rep. 2015;10:326–338. doi: 10.1016/j.celrep.2014.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seldin MM, et al. Trimethylamine N-Oxide Promotes Vascular Inflammation Through Signaling of Mitogen-Activated Protein Kinase and Nuclear Factor-κB. J Am Heart Assoc. 2016;5:e002767. doi: 10.1161/JAHA.115.002767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li T, Chen Y, Gua C, Li X. Elevated Circulating Trimethylamine N-Oxide Levels Contribute to Endothelial Dysfunction in Aged Rats through Vascular Inflammation and Oxidative Stress. Front Physiol. 2017;8 doi: 10.3389/fphys.2017.00350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yue C, et al. Trimethylamine N-oxide prime NLRP3 inflammasome via inhibiting ATG16L1-induced autophagy in colonic epithelial cells. Biochem Biophys Res Commun. 2017;490:541–551. doi: 10.1016/j.bbrc.2017.06.075. [DOI] [PubMed] [Google Scholar]
- 21.Yano JM, et al. Indigenous Bacteria from the Gut Microbiota Regulate Host Serotonin Biosynthesis. Cell. 2015;161:264–276. doi: 10.1016/j.cell.2015.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fusaro M, et al. Vitamin K plasma levels determination in human health. Clin Chem Lab Med CCLM. 2016;55:789–799. doi: 10.1515/cclm-2016-0783. [DOI] [PubMed] [Google Scholar]
- 23.Jäckel S, et al. Gut microbiota regulate hepatic von Willebrand factor synthesis and arterial thrombus formation via Toll-like receptor-2. Blood. 2017;130:542–553. doi: 10.1182/blood-2016-11-754416. [DOI] [PubMed] [Google Scholar]
- 24.Zhu W, Wang Z, Tang WHW, Hazen SL. Gut Microbe-Generated Trimethylamine N-Oxide From Dietary Choline Is Prothrombotic in Subjects. Circulation. 2017;135:1671–1673. doi: 10.1161/CIRCULATIONAHA.116.025338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heianza Y, Ma W, Manson JE, Rexrode KM, Qi L. Gut Microbiota Metabolites and Risk of Major Adverse Cardiovascular Disease Events and Death: A Systematic Review and Meta-Analysis of Prospective Studies. J Am Heart Assoc. 2017;6:e004947. doi: 10.1161/JAHA.116.004947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schiattarella GG, et al. Gut microbe-generated metabolite trimethylamine-N-oxide as cardiovascular risk biomarker: a systematic review and dose-response meta-analysis. Eur Heart J. 2017;38:2948–2956. doi: 10.1093/eurheartj/ehx342. [DOI] [PubMed] [Google Scholar]
- 27.Qi J, et al. Circulating trimethylamine N-oxide and the risk of cardiovascular diseases: a systematic review and meta-analysis of 11 prospective cohort studies. J Cell Mol Med. 2017;22:185–194. doi: 10.1111/jcmm.13307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brown JM, Hazen SL. Targeting of microbe-derived metabolites to improve human health: The next frontier for drug discovery. J Biol Chem. 2017;292:8560–8568. doi: 10.1074/jbc.R116.765388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dolphin CT, Janmohamed A, Smith RL, Shephard EA, Phillips lan R. Missense mutation in flavin-containing mono-oxygenase 3 gene, FMO3, underlies fish-odour syndrome. Nat Genet. 1997;17:491–494. doi: 10.1038/ng1297-491. [DOI] [PubMed] [Google Scholar]
- 30.Bennett BJ, et al. Trimethylamine-N-Oxide, a Metabolite Associated with Atherosclerosis, Exhibits Complex Genetic and Dietary Regulation. Cell Metab. 2013;17:49–60. doi: 10.1016/j.cmet.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Phillips GB. The lipid composition of human bile. Biochim Biophys Acta. 1960;41:361–363. doi: 10.1016/0006-3002(60)90026-3. [DOI] [PubMed] [Google Scholar]
- 32.Craciun S, Marks JA, Balskus EP. Characterization of choline trimethylamine-lyase expands the chemistry of glycyl radical enzymes. ACS Chem Biol. 2014;9:1408–1413. doi: 10.1021/cb500113p. [DOI] [PubMed] [Google Scholar]
- 33.Romano KA, Vivas EI, Amador-Noguez D, Rey FE. Intestinal Microbiota Composition Modulates Choline Bioavailability from Diet and Accumulation of the Proatherogenic Metabolite Trimethylamine-N-Oxide. mBio. 2015;6 doi: 10.1128/mBio.02481-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martínez-del Campo A, et al. Characterization and Detection of a Widely Distributed Gene Cluster That Predicts Anaerobic Choline Utilization by Human Gut Bacteria. mBio. 2015;6 doi: 10.1128/mBio.00042-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Z, et al. Non-lethal Inhibition of Gut Microbial Trimethylamine Production for the Treatment of Atherosclerosis. Cell. 2015;163:1585–1595. doi: 10.1016/j.cell.2015.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Furie B, Furie BC. Mechanisms of Thrombus Formation. N Engl J Med. 2008;359:938–949. doi: 10.1056/NEJMra0801082. [DOI] [PubMed] [Google Scholar]
- 37.Bobadilla RV. Acute Coronary Syndrome: Focus on Antiplatelet Therapy. Crit Care Nurse. 2016;36:15–27. doi: 10.4037/ccn2016497. [DOI] [PubMed] [Google Scholar]
- 38.Levine GN, et al. 2016 ACC/AHA guideline focused update on duration of dual antiplatelet therapy in patients with coronary artery disease. J Thorac Cardiovasc Surg. 2016;152:1243–1275. doi: 10.1016/j.jtcvs.2016.07.044. [DOI] [PubMed] [Google Scholar]
- 39.Jennings LK. Mechanisms of platelet activation: Need for new strategies to protect against platelet-mediated atherothrombosis. Thromb Haemost. 2009;102:248–257. doi: 10.1160/TH09-03-0192. [DOI] [PubMed] [Google Scholar]
- 40.Manchikanti L, et al. Assessment of bleeding risk of interventional techniques: a best evidence synthesis of practice patterns and perioperative management of anticoagulant and antithrombotic therapy. Pain Physician. 2013;16:SE261–318. [PubMed] [Google Scholar]
- 41.Cohen M. Expanding the Recognition and Assessment of Bleeding Events Associated With Antiplatelet Therapy in Primary Care. Mayo Clin Proc. 2009;84:149–160. doi: 10.4065/84.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bodea S, Funk MA, Balskus EP, Drennan CL. Molecular Basis of C-N Bond Cleavage by the Glycyl Radical Enzyme Choline Trimethylamine-Lyase. Cell Chem Biol. 2016;23:1206–1216. doi: 10.1016/j.chembiol.2016.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen M, et al. Resveratrol Attenuates Trimethylamine-N-Oxide (TMAO)-Induced Atherosclerosis by Regulating TMAO Synthesis and Bile Acid Metabolism via Remodeling of the Gut Microbiota. mBio. 2016;7:e02210–15. doi: 10.1128/mBio.02210-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Walsh CT. Suicide Substrates, Mechanism-Based Enzyme Inactivators: Recent Developments. Annu Rev Biochem. 1984;53:493–535. doi: 10.1146/annurev.bi.53.070184.002425. [DOI] [PubMed] [Google Scholar]
- 45.Hazen SL, Zupan LA, Weiss RH, Getman DP, Gross RW. Suicide inhibition of canine myocardial cytosolic calcium-independent phospholipase A2. Mechanism-based discrimination between calcium-dependent and -independent phospholipases A2. J Biol Chem. 1991;266:7227–7232. [PubMed] [Google Scholar]
- 46.Sandhu SS, Chase T. Aerobic degradation of choline by Proteus mirabilis: enzymatic requirements and pathway. Can J Microbiol. 1986;32:743–750. doi: 10.1139/m86-135. [DOI] [PubMed] [Google Scholar]
- 47.Romano KA, et al. Metabolic, Epigenetic, and Transgenerational Effects of Gut Bacterial Choline Consumption. Cell Host Microbe. 2017;22:279–290.e7. doi: 10.1016/j.chom.2017.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nemzek JA, Bolgos GL, Williams BA, Remick DG. Differences in normal values for murine white blood cell counts and other hematological parameters based on sampling site. Inflamm Res. 2001;50:523–527. doi: 10.1007/PL00000229. [DOI] [PubMed] [Google Scholar]
- 49.Koeth RA, et al. γ-Butyrobetaine Is a Proatherogenic Intermediate in Gut Microbial Metabolism of L-Carnitine to TMAO. Cell Metab. 2014;20:799–812. doi: 10.1016/j.cmet.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu Y, Jennings NL, Dart AM, Du XJ. Standardizing a simpler, more sensitive and accurate tail bleeding assay in mice. World J Exp Med. 2012;2:30–36. doi: 10.5493/wjem.v2.i2.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Derrien M, Belzer C, de Vos WM. Akkermansia muciniphila and its role in regulating host functions. Microb Pathog. 2017;106:171–181. doi: 10.1016/j.micpath.2016.02.005. [DOI] [PubMed] [Google Scholar]
- 52.Gachet C. Antiplatelet drugs: which targets for which treatments? J Thromb Haemost. 2015;13:S313–S322. doi: 10.1111/jth.12947. [DOI] [PubMed] [Google Scholar]
- 53.Osbourn AE, Field B. Operons. Cell Mol Life Sci. 2009;66:3755–3775. doi: 10.1007/s00018-009-0114-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Craciun S, Balskus EP. Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proc Natl Acad Sci. 2012;109:21307–21312. doi: 10.1073/pnas.1215689109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Berthoumieux S, et al. Shared control of gene expression in bacteria by transcription factors and global physiology of the cell. Mol Syst Biol. 2013;9:634. doi: 10.1038/msb.2012.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Clarke DD. Fluoroacetate and fluorocitrate: mechanism of action. Neurochem Res. 1991;16:1055–1058. doi: 10.1007/BF00965850. [DOI] [PubMed] [Google Scholar]
- 57.Imhann F, et al. Proton pump inhibitors affect the gut microbiome. Gut. 2016;65:740–748. doi: 10.1136/gutjnl-2015-310376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rogers MAM, Aronoff DM. The influence of non-steroidal anti-inflammatory drugs on the gut microbiome. Clin Microbiol Infect. 2016;22:178.e1–178.e9. doi: 10.1016/j.cmi.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maier L, et al. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature. 2018;555:623–628. doi: 10.1038/nature25979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu H, et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat Med. 2017;23:850–858. doi: 10.1038/nm.4345. [DOI] [PubMed] [Google Scholar]
- 61.Wang Z, et al. Measurement of trimethylamine-N-oxide by stable isotope dilution liquid chromatography tandem mass spectrometry. Anal Biochem. 2014;455:35–40. doi: 10.1016/j.ab.2014.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rath S, Heidrich B, Pieper DH, Vital M. Uncovering the trimethylamine-producing bacteria of the human gut microbiota. Microbiome. 2017;5:54. doi: 10.1186/s40168-017-0271-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available from the corresponding author upon reasonable request. The 16s sequencing datasets generated and analyzed during the current study are available in the NCBI Sequence Read Archive, https://www.ncbi.nlm.nih.gov/bioproject/PRJNA471699.