

Abstract
Purpose
Early treatment is critical for mucopolysaccharidosis type I (MPS I), justifying its incorporation into newborn screening. Enzyme replacement therapy (ERT) treats MPS I, yet presumptions that ERT cannot penetrate the blood-brain barrier (BBB) support recommendations that hematopoietic cell transplantation (HCT) treat the severe, neurodegenerative form (Hurler syndrome). Ethics preclude randomized comparison of ERT to HCT, but insight into this comparison is presented with an international cohort of patients with Hurler syndrome who received long-term ERT from a young age.
Methods
Long-term survival and neurologic outcomes were compared among three groups of patients with Hurler syndrome: 18 treated with ERT monotherapy (ERT group), 54 who underwent HCT (HCT group), and 23 who received no therapy (Untreated). All were followed starting before age 5 years. A sensitivity analysis restricted age below 3 years.
Results
Survival was worse when comparing ERT versus HCT, and Untreated versus ERT. The cumulative incidences of hydrocephalus and cervical spinal cord compression were greater in ERT versus HCT. Findings persisted in the sensitivity analysis.
Conclusion
As newborn screening widens treatment opportunity for Hurler syndrome, this examination of early treatment quantifies some ERT benefit, supports presumptions about BBB impenetrability, and aligns with current guidelines to treat with HCT.
Keywords: mucopolysaccharidosis, enzyme replacement therapy, hematopoietic cell transplantation, neurodegenerative, newborn screening
Introduction
Incorporation of mucopolysaccharidosis type I (MPS I) into newborn screening in the United States and internationally has been driven by an ever-growing body of evidence that early treatment leads to more favorable outcomes1. Early treatment inhibits the generally irreversible, progressive pathology of MPS I, and thus newborn screening affords greater numbers of patients better opportunity for reducing or preventing cognitive or physical disability. MPS I is an autosomal recessive disorder resulting from a deficiency of the lysosomal enzyme α-L-iduronidase, crucial in a step-wise pathway of glycosaminoglycan (GAG) degradation. Accumulating GAG in nearly all organ systems 2-4 causes clinical symptoms beginning in infancy5 that progress to worsening disability due to cardiac, airway, pulmonary, orthopedic, ophthalmologic and auditory problems, hepatosplenomegaly, and neurologic dysfunction including increased risk for hydrocephalus and spinal cord compression. In contrast to the attenuated forms of MPS I, the severe form, Hurler syndrome (MPS IH), involves rapid and dramatic neurologic deterioration during early childhood including intellectual developmental decline, as well as death between 5 to 10 years of age.
Enzyme replacement therapy (ERT) with laronidase was approved for all phenotypes of MPS I in 2003 in Europe and the United States, and in 2005 and 2006 in Brazil and Japan, respectively. However, the presumption that the blood-brain barrier (BBB) is impenetrable to intravenously delivered ERT has guided the continued recommendation that MPS IH be treated with hematopoietic cell transplantation (HCT), a therapy that was first utilized for this disease more than two decades prior6. HCT has long been shown to arrest neurologic deterioration, stabilize cognition, improve metabolic correction and extend survival2-4,6-11. Given these HCT outcomes combined with presumptions about the impermeable BBB, a formal randomized comparison of the efficacy of relatively newer ERT as monotherapy to that of HCT for MPS IH was never ethically possible to pursue11. Short-term safety studies evaluating ERT as well as longer-term case reports and case series have suggested therapeutic benefit that is less effective than HCT12-14, although long-term outcomes of ERT in larger groups, particularly those who were treated from a young age, has not been understood. ERT from birth, or at higher doses, in animal models has demonstrated efficacy in affected organ systems that are typically impervious to ERT, including the central nervous system (CNS)15-17. Given the capacity of newborn screening to shift the window of intervention earlier, clarifying long-term efficacy of systemic therapies when initiated early in life has become more pressing. This study presents a benchmark examination of the long-term outcomes of ERT monotherapy in a unique international cohort of patients with Hurler syndrome who were treated exclusively with ERT from a young age, before significant disease progression was apparent. All but 3 patients in this unusual group were first described a decade ago in a 1-year safety study of ERT14, thus enabling a rare opportunity for longitudinal tracking of survival and cardinal central nervous system manifestations.
Patients and Methods
Patients
Eighteen patients who were treated exclusively and continuously with ERT from a young age until last follow-up or death (ERT group) were identified from three sources: 15 patients from a larger published international study involving sites in the United Kingdom, France, Germany, and the Netherlands14, two patients from Brazil (courtesy of R. Giugliani and C. de Souza), and one patient from the United States who has been previously described13. The clinical diagnosis of MPS IH was confirmed with genotyping which has been previously reported in all patients13,14, except for the two patients from Brazil, one whose genotype was W402X/W402X and the other whose genotype was p.R621x IVS9-12_/-4delCAGGCCCCG. All patients received the standard dose of ERT except for the one previously reported, whose dose was increased13. No other patients were known to meet the three inclusion criteria for this ERT group: 1) a genotypic diagnosis of MPS IH, 2) ERT initiated before age 5 years, and 3) no history of HCT.
The outcomes of the ERT group were compared to those of two additional groups. The HCT group comprised 54 sequential MPS IH patients transplanted at the University of Minnesota since 2002. As transplant is a complex treatment, a single-institution and recent cohort (reflecting modern HCT practice) was chosen. In the HCT group, 42 of the 54 patients received ERT in the peri-transplant period, generally through 8 weeks post transplant18, but not long-term. The remaining 12 did not receive ERT. The second comparison group (Untreated group) comprised 21 historical patients who did not receive any treatment due to lack of available HCT donors in the pre-ERT era. In addition, one patient in each of the treatment groups received treatment after age 5; thus they were censored at date of treatment initiation and added to the Untreated group to reflect their untreated clinical course during their first 5 years of life. One patient in the ERT group eventually underwent HCT at one of the international sites at age 3 and was censored at that time point and maintained in the ERT group. Institutional Review Board and parallel human subjects protection boards at international sites provided approval for review of medical files.
Study Design and Endpoints
This observational study involved retrospective chart review of endpoints that relate to the lethality and neurologic pathology of MPS IH:survival as well as emergence of hydrocephalus and cervical spinal cord compression. These endpoints were reported from the international centers. Long term IQ data were not available, as IQ was not measured in most of the ERT group over time. Age at the emergence of hydrocephalus and cervical cord compression was chosen because these two, commonly-assessed, cardinal CNS manifestations of MPS IH significantly impact functioning and quality of life4,19,20. Both outcomes were defined according to standard clinical guidelines at each institution, including neuro-imaging, and all patients proceeded to surgical intervention, except for 2 (both in the ERT group) for whom a procedure was thought to be ill-advised based on the status of the patient. Specifically, all patients who developed hydrocephalus were treated with shunt placement except for one whose hydrocephalus was discovered by MRI and neuro-ophthalmologic exam at end-of-life; therefore a VP shunt was not placed. All patients who developed cervical cord compression underwent decompression surgery, except for one who was diagnosed via MRI, abnormal somatosensory evoked potentials and pyramidal tract signs, but this child was receiving palliative care and the risk of anesthesia was felt to be higher than expected benefits of the surgery. Records on these CNS outcomes were available on all patients in the HCT group and most patients in the ERT group, but they were not for the untreated historical controls. Some ERT patients did not undergo routine neuro-imaging, and thus due to missing exams, 1 patient was excluded from outcome analysis of hydrocephalus, and 5 patients were excluded from outcome analysis of cervical cord compression. Patients who developed hydrocephalus or cervical cord compression prior to initiation of treatment were also excluded from those analyses (hydrocephalus N = 2; cervical cord compression N = 0).
Statistics
Descriptive statistics were tabulated per treatment group. Continuous variables were summarized with mean and range while categorical variables were summarized with frequency and percentage. Survival curves were evaluated using Kaplan-Meier estimates while comparisons between groups were based on hazard ratios as estimated by unadjusted Cox proportional hazards models and robust variance estimation. Hydrocephalus and cervical cord compression were evaluated separately based on cumulative incidence functions due to the competing risk of death with comparisons based on the subdistribution between groups21.
R v3.2.4 was used for all analyses (R Foundation for Statistical Computing, Vienna, Austria).
Results
Patient characteristics are summarized in Table 1. Mean age at treatment was younger in the HCT group than the ERT group (1.5 years, 2.6 years, respectively), although the ranges of treatment age were similar between the two groups. A sensitivity analysis was conducted in which age at treatment was restricted to younger than 3 years, to make the groups more comparable in that regard: Median age at treatment was identical for the ERT and HCT groups in this analysis (1.3 years).
Table 1. Descriptive data: values are mean (SD), median (range), or N (%) where indicated.
| Untreated | ERT | HCT | |
|---|---|---|---|
| Full sample N | 23 | 18 | 54 |
| Male | 12 (52%) | 12 (67%) | 30 (56%) |
| Age at treatment (years) | |||
| Mean (SD) | NA | 2.6 (1.3) | 1.5 (0.80) |
| Median (range) | NA | 2.6 (0.5-4.7) | 1.3 (0.4-4.8) |
| Birth years | 1980-1994 | 1998-2010 | 1999-2013 |
| Under 3a years N | NA | 10 | 53 |
| Male | 6 (60%) | 30 (57%) | |
| Age at treatment (years) | |||
| Mean (SD) | NA | 1.6 (0.74) | 1.4 (0.65) |
| Median (range) | NA | 1.3 (0.5-2.7) | 1.3 (0.4-2.9) |
At initiation of ERT or time of HCT
Survival
Survival was significantly worse in the Untreated group compared to the ERT group (Hazard Ratio = 2.3; p = 0.008; Figure 1). Survival was also worse in the ERT group, compared to the HCT group (HR = 2.6; P = 0.033). Survival differences when age at treatment was restricted to <3 (N = 53 and 10 in the HCT and ERT groups, respectively)remained qualitatively similar for the Untreated group compared to the ERT group and for the ERT group compared to the HCT group (HR = 2.4, P=0.046 and 2.50, P=0.089 respectively), wherein point estimates were nearly identical and the P-value was slightly higher as expected due to a smaller sample size (Figure S1). In this study the HCT group had the longest survival, with an estimated survival beyond 10 years of age of 83% (95% CI: 73%, 93%), while survival estimates for the ERT and Untreated groups were 45% (95% CI: 21%, 70%) and 10% (95% CI: 0%, 23%) respectively. The Untreated group had a median age of death of 6.4 years, while that of the ERT group was 9.0 years. This finding does not change significantly if ERT is initiated prior to age 3 (8.9 years).
Figure 1.
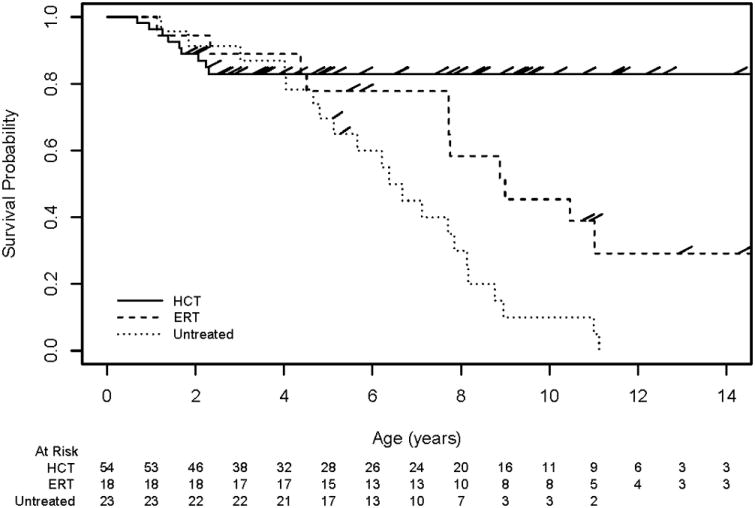
Survival in Hurler syndrome. Survival curves depict differences in clinical course for patients with Hurler syndrome who received HCT (HCT), ERT monotherapy (ERT), or no systemic therapy (Untreated).
CNS Outcomes
At 14 years follow-up, the cumulative incidence of hydrocephalus in the ERT group was 27%, compared to 0% in the HCT group (P=0.036; Figure 2). The cumulative incidence of cervical cord compression in the ERT group was 51%, compared to 16% in the HCT group (P=0.039; Figure 3). Restricting age at treatment to <3 years did not significantly change results: The cumulative incidence of hydrocephalus in the ERT group was 40%, compared to 0% in the HCT group (P=0.010), while the cumulative incidence of cord compression in the ERT group was 67%, compared to 16% in the HCT group (P=0.013).
Figure 2.
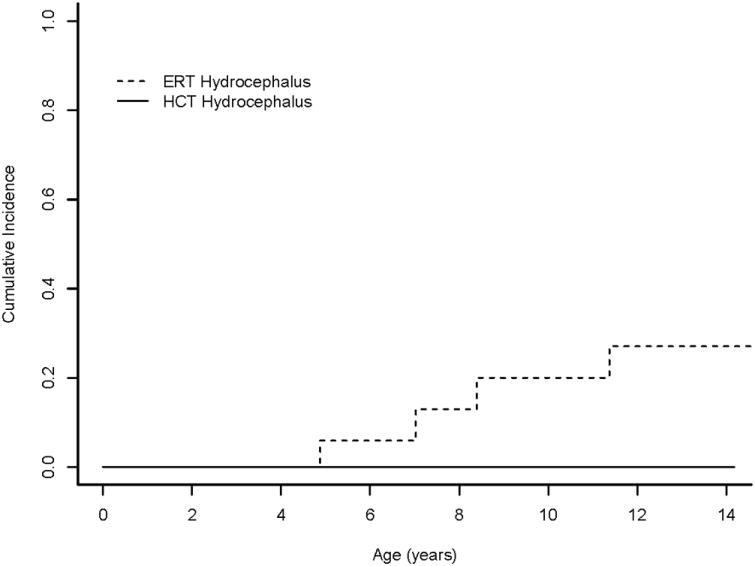
Cumulative incidence of hydrocephalus in Hurler syndrome. Cumulative incidence functions show differences in development of hydrocephalus for patients with Hurler syndrome who received HCT (HCT) or ERT monotherapy (ERT). As no patients who underwent HCT developed this symptom, the function is flat.
Figure 3.
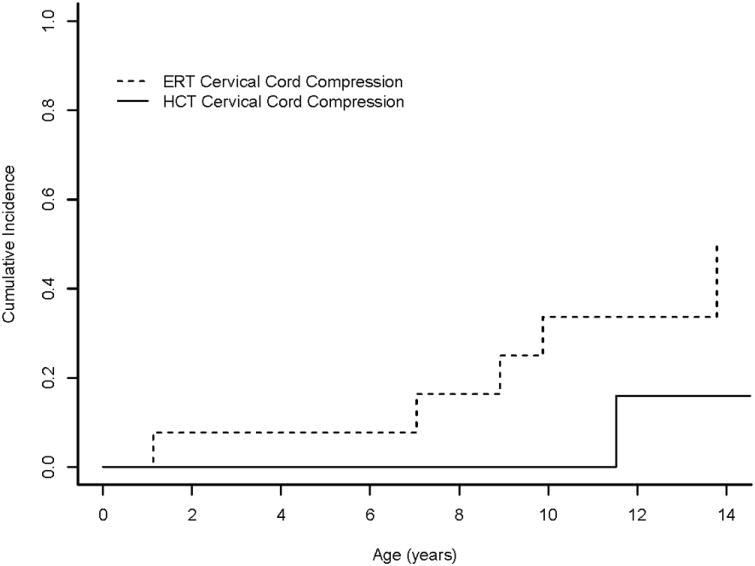
Cumulative incidence of cervical cord compression in Hurler syndrome. Cumulative incidence functions show differences in development of cervical cord compression for patients with Hurler syndrome who received HCT (HCT) or ERT monotherapy (ERT).
Discussion
This is the first clinical study to quantify long-term outcomes for patients with Hurler syndrome comparing ERT, HCT, or no systemic therapy. Until now, comparison of relative clinical efficacy of the two systemic therapies for MPS IH has not been approached, as ethics preclude a prospective trial comparing HCT to an intravenous drug that is thought to be largely excluded from the central nervous system by the blood-brain barrier4,22,23, and few patients with this relatively less common treatment course were followed long-term. With animal model evidence of improved efficacy of ERT when initiated at birth, combined with the shifting window of treatment opportunity owing to newborn screening, there is more pressing need for data-driven comparison of all systemic therapeutic choices for the severe form of MPS I. Therefore, this study evaluated 10-year follow-up data on a group of patients whose 1-year treatment response to early initiated ERT was previously published14, and added data from three additional ERT patients, including one who was previously described13. By comparing this ERT group to those who received HCT, as well as to those who were untreated, this study reveals an alteration in the natural history of Hurler syndrome with use of ERT, provides clinical data further supporting presumptions about the impermeability of the BBB to intravenously delivered enzyme, and also presents evidence of superior HCT outcomes with respect to survival and disease related neurologic pathology, even when ERT is initiated early.
The ERT group had a substantially greater cumulative incidence of hydrocephalus than the HCT group, where no hydrocephalus was observed. The ERT group also had a much greater cumulative incidence of cervical spinal cord compression. Consistent with previous findings9,24, cervical cord compression was not completely absent in the HCT group. Given the importance of early treatment in this disease, a sensitivity analysis was conducted to minimize differences in age at initiation of therapy in the two groups and results remained nearly the same, suggesting these poorer outcomes are not explained by differential delay to treatment. These findings of greater cumulative incidences of negative CNS outcomes in the ERT group, versus the HCT group, support the presumption that intravenous ERT does not penetrate the BBB to benefit the CNS structure to a measurable extent9,22,23.
The natural history of Hurler syndrome is defined not just by the progressive involvement of the CNS but also by worsening somatic complications beginning in infancy5and significantly shortened lifespan including death within the first decade of life2. The Untreated group had a median age of death of 6.4 years, while that of the ERT group was 9 years. This finding does not change significantly if ERT is started prior to age 3 (8.9 years). Thus, initiation of ERT early in life appears to extend survival in Hurler syndrome, aligning with evidence that early ERT may delay or reduce disease burden in attenuated forms of MPS I25-27. Of note, the Untreated group was living at a time when medical care of MPS was not as advanced nor were multidisciplinary interventions as refined, and thus the comparison of ERT to Untreated must be interpreted within the context of differential quality of supportive, coordinating therapies. Survival was significantly worse in the ERT group as compared with the HCT group, and this finding was re-demonstrated on the sensitivity analysis, where median age at treatment was identical. Thus poorer survival in the ERT group does not appear confounded by differential delay to therapy.
Beyond questions related to survival and CNS pathology, HCT and ERT are not otherwise equivalent therapies for this complicated, multi-system disease. While ERT has been found to result in reductions in hepatosplenomegaly, urinary GAG excretion and sleep apnea, as well as improvements in growth, physical endurance, and joint range of motion 7, HCT has resulted in better GAG clearance and metabolic correction than with ERT11. Outcome of successful HCT includes improved multi-system functioning 3,9,10,24,28, such that GAG are reduced in soft tissue such as the liver, spleen, lungs, airway, marrow, facies, and the vascular system. Although guidelines recommend HCT for MPS IH, often ERT is used as an adjunctive therapy to improve the health status of patients prior to HCT, and ERT in the peri-transplant period has been used with the intent of decreasing morbidity and mortality associated with HCT 18,29,30. Further, these combined therapies have been associated with less severe cognitive decline following HCT 31.
This study revealed superior outcomes for survival and classic MPS-related CNS pathology with HCT, a treatment for which advances have led to considerable improvements in engraftment and survival in MPS IH the past decade32. There is overwhelming and long-standing evidence that younger age at HCT predicts better outcomes with respect to survival, engraftment, and residual disease burden1,3,18,24,32-34. With more favorable HCT outcomes being made possible by newborn screening, there are still challenges associated with transplant. First, it is recognized that metabolic correction is not uniformly distributed among organ systems, as differential delivery of enzyme to the target organs is difficult to measure, and problems remain, particularly in the skeletal, nervous, and cardiac systems, despite full engraftment of enzyme competent donor hematopoietic cells following transplantation 3,10,24,35,36. Thus many transplanted children show continued cognitive and physical impairments, with severity related to age at HCT3,9,24,34. Second, there is a lack of availability of transplant in many nations worldwide. There may be considerable practical or clinical obstacles for some patients to undergo HCT beyond the USA, such that families may decide not to pursue it 37. Third, while HCT is readily available in the US and EU, it is quite costly 38. On the other hand, the high cost of a single HCT is actually less than the accumulated cost of life-long ERT for these patients.
Limitations
A common problem in rare disease research, small sample size limits this study by restricting the number of factors that can be controlled when predicting outcomes. Differences in genotypes, existing disease burden, failure of first treatment (e.g., need for second transplant or change in ERT dosing), and access to supportive therapies (e.g., occupational therapy, physical therapy, special education, etc.) could have explained some of the findings. Further, the use of ERT as adjunctive therapy in the majority of the HCT sample creates some therapeutic overlap in comparing ERT versus HCT; however the duration of this adjunctive ERT was comparatively quite brief in a study of long-term outcomes. More detailed information on causes of death in the ERT group may have provided additional insights into whether certain organ systems are differentially improved or burdened in the long term. As monitoring anti-drug antibody responses is an increasingly recognized factor in ERT success15,39,40, this study cannot account for the degree to which it affected outcomes of the ERT group. However, a recent meta-analysis demonstrated that antibody titers generally decrease over time during treatment with ERT40, and while this may not be true for every patient, a more comprehensive examination of one of the patients from this group revealed that even though anti-ERT antibodies doubled in a 6-year span, there was no apparent impact on efficacy for her13.
Conclusion
By addressing the lack of long-term outcome comparison between ERT monotherapy and HCT in Hurler syndrome, this study reveals superior outcomes for survival and CNS pathology with HCT. It also provides clinical data to suggest benefit of ERT and to support existing presumptions that the blood-brain barrier is impermeable to standard doses of ERT. Given improved early detection of MPS I thanks to newborn screening, geneticists, pediatricians, and other health practitioners will be faced with new questions, and treatment decisions, for the increasing number of infants with this condition who start appearing in their practices. These findings on suboptimal long-term outcomes of ERT monotherapy in MPS IH, even when begun early in life, are considered in the context of problems of access to HCT worldwide, and highlight the need for better, more accessible, and less expensive somatic and CNS therapy.
Supplementary Material
Acknowledgments
We are indebted to the patients who enabled this research.
Supported in part by the Center for Neurobehavioral Development, University of Minnesota.
Funding sources: The Lysosomal Disease Network (U54NS065768) is a part of the Rare Diseases Clinical Research Network (RDCRN), an initiative of the Office of Rare Diseases Research (ORDR), and NCATS. This consortium is funded through a collaboration between NCATS, NINDS, and NIDDK. Research reported in this publication was also supported in part by the National Center for Advancing Translational Sciences of the National Institutes of Health Award Number UL1TR000114. The University of Minnesota's Center for Neurobehavioral Development, the Center for Magnetic Resonance Research, and the Minnesota Supercomputer Center provided infrastructure for this research. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supplementary information is available at the Genetics in Medicine website.
Conflicts of Interest: Julie Eisengart has received honoraria, consulting fees, and/or research support from ArmaGen, Regenexbio, Sangamo, and Sanofi Genzyme, and has done contract work for Shapiro Neuropsychology Consulting, LLC; Yong Xue is an employee of Sanofi Genzyme; Paul Orchard has received research support and honoraria from Sanofi Genzyme; Weston Miller will be employed by Sangamo Therapeutics in January 2018; Troy Lund has received research support from Sanofi Genzyme; Simon Jones has received research support and consulting fees from Sanofi Genzyme; SeyfullahGökce has received travel support from Alexion, Sanofi Genzyme, and Shire. Nathalie Guffon has received research support from Sanofi Genzyme and Biomarin, and consulting fees from Sanofi Genzyme; Roberto Giugliani has received speaker honoraria, travel grants and investigator fees from ArmaGen, BioMarin and Sanofi Genzyme; Carolina F. M. Souza has received speaker honoraria, travel grants and investigator fees from BioMarin and Sanofi Genzyme; Elsa Shapiro is a Partner in Shapiro Neuropsychology Consulting, LLC; and Chester B. Whitley has received consulting fees and research support from ArmaGen, Sangamo, BioMarin and Sanofi Genzyme. The other authors have no conflicts of interest to disclose.
References
- 1.Grosse SD, Lam WK, Wiggins LD, Kemper AR. Cognitive outcomes and age of detection of severe mucopolysaccharidosis type 1. Genetics in Medicine. 2017 doi: 10.1038/gim.2016.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neufeld E, Muenzer J. The mucopolysaccharidoses. The metabolic and molecular bases of inherited disease. 2001;8:3421–3452. [Google Scholar]
- 3.Aldenhoven M, Wynn RF, Orchard PJ, et al. Long-term outcome of Hurler syndrome patients after hematopoietic cell transplantation: an international multicenter study. Blood. 2015;125(13):2164–2172. doi: 10.1182/blood-2014-11-608075. [DOI] [PubMed] [Google Scholar]
- 4.Muenzer J, Wraith JE, Clarke LA. Mucopolysaccharidosis I: Management and Treatment Guidelines. Pediatrics. 2009;123(1):19–29. doi: 10.1542/peds.2008-0416. [DOI] [PubMed] [Google Scholar]
- 5.Kiely BT, Kohler JL, Coletti HY, Poe MD, Escolar ML. Early disease progression of Hurler syndrome. Orphanet journal of rare diseases. 2017;12(1):32. doi: 10.1186/s13023-017-0583-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hobbs J, Barrett A, Chambers D, et al. Reversal of clinical features of Hurler's disease and biochemical improvement after treatment by bone-marrow transplantation. The Lancet. 1981;318(8249):709–712. doi: 10.1016/s0140-6736(81)91046-1. [DOI] [PubMed] [Google Scholar]
- 7.Kakkis ED, Muenzer J, Tiller GE, et al. Enzyme-Replacement Therapy in Mucopolysaccharidosis I — NEJM. The New England Journal of Medicine. 2001;344:182–188. doi: 10.1056/NEJM200101183440304. [DOI] [PubMed] [Google Scholar]
- 8.Whitley CB, Ramsay NK, Kersey JH, Krivit W. Bone marrow transplantation for Hurler syndrome: assessment of metabolic correction. Birth Defects Orig Artic Ser. 1986;22(1):7–24. [PubMed] [Google Scholar]
- 9.Aldenhoven M, Boelens J, de Koning TJ. The clinical outcome of Hurler syndrome after stem cell transplantation. Biology of Blood and Marrow Transplantation. 2008;14(5):485–498. doi: 10.1016/j.bbmt.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 10.Escolar ML, Aldenhoven M. Leukodystrophies and Lysosomal Storage Disorders. In: Boelens JJ, Wynn R, editors. Stem Cell Biology and Regenerative Medicine. New York: Springer; 2013. pp. 63–125. [Google Scholar]
- 11.Wynn RF, Wraith JE, Mercer J, et al. Improved metabolic correction in patients with lysosomal storage disease treated with hematopoietic stem cell transplant compared with enzyme replacement therapy. The Journal of pediatrics. 2009;154(4):609–611. doi: 10.1016/j.jpeds.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 12.da Silva Franco JF, El Dib R, Agarwal A, et al. Mucopolysaccharidosis type I, II and VI and response to enzyme replacement therapy: Results from a single-center case series study. Intractable & rare diseases research. 2017;6(3):183–190. doi: 10.5582/irdr.2017.01036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eisengart JB, Jarnes J, Ahmed A, et al. Long-term cognitive and somatic outcomes of enzyme replacement therapy in untransplanted Hurler syndrome. Molecular Genetics and Metabolism Reports. 2017;13:64–68. doi: 10.1016/j.ymgmr.2017.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wraith J, Beck M, Lane R, et al. Enzyme replacement therapy in patients who have mucopolysaccharidosis I and are younger than 5 years: results of a multinational study of recombinant human {alpha}-L-iduronidase (laronidase) Pediatrics. 2007;120(1):e37. doi: 10.1542/peds.2006-2156. [DOI] [PubMed] [Google Scholar]
- 15.Ou L, Herzog T, Koniar BL, Gunther R, Whitley CB. High-dose enzyme replacement therapy in murine Hurler syndrome. Molecular genetics and metabolism. 2014;111(2):116–122. doi: 10.1016/j.ymgme.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pievani A, Azario I, Antolini L, et al. Neonatal bone marrow transplantation prevents bone pathology in a mouse model of mucopolysaccharidosis type I. Blood. 2015;125(10):1662–1671. doi: 10.1182/blood-2014-06-581207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baldo G, Mayer FQ, Martinelli BZ, et al. Enzyme replacement therapy started at birth improves outcome in difficult-to-treat organs in mucopolysaccharidosis I mice - 1-s2.0-S1096719213000942-main.pdf. Molecular Genetics and Metabolism. 2013;109:33–40. doi: 10.1016/j.ymgme.2013.03.005. [DOI] [PubMed] [Google Scholar]
- 18.Ghosh A, Miller W, Orchard PJ, et al. Enzyme replacement therapy prior to haematopoietic stem cell transplantation in Mucopolysaccharidosis Type I: 10year combined experience of 2 centres. Molecular genetics and metabolism. 2016;117(3):373–377. doi: 10.1016/j.ymgme.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Horovitz DDG, Magalhães TDSPC, e Costa AP, et al. Spinal cord compression in young children with type VI mucopolysaccharidosis. Molecular genetics and metabolism. 2011;104:295–300. doi: 10.1016/j.ymgme.2011.07.019. [DOI] [PubMed] [Google Scholar]
- 20.Kulkami A, Shams I. Quality of life in children with hydrocephalus: Results from the Hospital for Sick Children, Toronto. Journal of Neurosurgery: Pediatrics. 2007;107:76–85. doi: 10.3171/PED-07/11/358. [DOI] [PubMed] [Google Scholar]
- 21.Gray R. A class of K-sample tests for comparing the cumulative incidence of a competing risk. Annals of Statistics. 1988;16:1141–1154. [Google Scholar]
- 22.Thomas JA, Jacobs S, Kierstein J, Van Hove J. Outcome after three years of laronidase enzyme replacement therapy in a patient with Hurler syndrome. J Inherit Metab Dis. 2006;29(6):762. doi: 10.1007/s10545-006-0457-y. [DOI] [PubMed] [Google Scholar]
- 23.Tokic V, Barisic I, Huzjak N, Petkovic G, Fumic K, Paschke E. Enzyme replacement therapy in two patients with an advanced severe (Hurler) phenotype of mucopolysaccharidosis I. Eur J Pediatr. 2007;166(7):727–732. doi: 10.1007/s00431-006-0316-8. [DOI] [PubMed] [Google Scholar]
- 24.Shapiro EG, Nestrasil I, Rudser K, et al. Neurocognition across the spectrum of mucopolysaccharidosis type I: age, severity, and treatment. Molecular genetics and metabolism. 2015;116(1):61–68. doi: 10.1016/j.ymgme.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Al-Sannaa NA, Bay L, Barbouth DS, et al. Early treatment with laronidase improves clinical outcomes in patients with attenuated MPS I: a retrospective case series analysis of nine sibships. Orphanet journal of rare diseases. 2015;10(1):131. doi: 10.1186/s13023-015-0344-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laraway S, Breen C, Mercer J, Jones S, Wraith, James E. Does early use of enzyme replacement therapy alter the natural history of mucopolysaccharidosis I? Experience in three siblings. Molecular Genetics and Metabolism. 2013;109(3):315–316. doi: 10.1016/j.ymgme.2013.04.023. [DOI] [PubMed] [Google Scholar]
- 27.Gabrielli O, Clarke LA, Bruni S, Coppa GV. Enzyme-Replacement Therapy in a 5-Month-Old Boy With Attenuated Presymptomatic MPS I: 5-Year Follow-up. Pediatrics. 2010;125(1):e183–e187. doi: 10.1542/peds.2009-1728. [DOI] [PubMed] [Google Scholar]
- 28.Shapiro E, Lockman L, Balthazor M, Krivit W. Neuropsychological outcomes of several storage diseases with and without bone marrow transplantation. Journal of inherited metabolic disease. 1995;18(4):413–429. doi: 10.1007/BF00710053. [DOI] [PubMed] [Google Scholar]
- 29.Tolar J, Grewal S, Bjoraker K, et al. Combination of enzyme replacement and hematopoietic stem cell transplantation as therapy for Hurler syndrome. Bone marrow transplantation. 2007;41(6):531–535. doi: 10.1038/sj.bmt.1705934. [DOI] [PubMed] [Google Scholar]
- 30.Wynn R, Mercer J, Page J, Carr T, Jones S, Wraith J. Use of enzyme replacement therapy (Laronidase) before hematopoietic stem cell transplantation for mucopolysaccharidosis I: experience in 18 patients. J Pediatr. 2009;154:135–139. doi: 10.1016/j.jpeds.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 31.Eisengart JB, Rudser KD, Tolar J, et al. Enzyme replacement is associated with better cognitive outcomes after transplant in Hurler syndrome. The Journal of pediatrics. 2013;162(2):375–380. e371. doi: 10.1016/j.jpeds.2012.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lum SH, Miller WP, Jones S, et al. The Changing Patterns of Graft Failure in MPS1H, Hurler Syndrome: A Review of 30-Years Experience. Blood. 2016;128(22):4700–4700. [Google Scholar]
- 33.Orchard P, Milla C, Braunlin E, et al. Pre-transplant risk factors affecting outcome in Hurler syndrome. Bone marrow transplantation. 2010;45(7):1239–1246. doi: 10.1038/bmt.2009.319. [DOI] [PubMed] [Google Scholar]
- 34.Poe MD, Chagnon SL, Escolar ML. Early treatment is associated with improved cognition in Hurler syndrome. Annals of Neurology. 2014;76:747–753. doi: 10.1002/ana.24246. [DOI] [PubMed] [Google Scholar]
- 35.Polgreen L, Tolar J, Plog M, et al. Growth and endocrine function in patients with Hurler syndrome after hematopoietic stem cell transplantation. Bone marrow transplantation. 2008;41(12):1005–1011. doi: 10.1038/bmt.2008.20. [DOI] [PubMed] [Google Scholar]
- 36.Braunlin EA, Harmatz PR, Scarpa M, et al. Cardiac disease in patients with mucopolysaccharidosis: presentation, diagnosis and management. Journal of inherited metabolic disease. 2011;34(6):1183–1197. doi: 10.1007/s10545-011-9359-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dornelles AD, Pinto LLdC, Paula ACd, et al. Enzyme replacement therapy for Mucopolysaccharidosis Type I among patients followed within the MPS Brazil Network. Genetics and molecular biology. 2014;37(1):23–29. doi: 10.1590/s1415-47572014000100006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Majhail NS, Mothukuri JM, MacMillan ML, et al. Costs of pediatric allogeneic hematopoietic-cell transplantation. Pediatric blood & cancer. 2010;54(1):138–143. doi: 10.1002/pbc.22250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Giugliani R, Vieira TA, Carvalho CG, et al. Immune tolerance induction for laronidase treatment in mucopolysaccharidosis I. Molecular Genetics and Metabolism Reports. 2017;10:61–66. doi: 10.1016/j.ymgmr.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xue Y, Richards SM, Mahmood A, Cox GF. Effect of anti-laronidase antibodies on efficacy and safety of laronidase enzyme replacement therapy for MPS I: a comprehensive meta-analysis of pooled data from multiple studies. Molecular genetics and metabolism. 2016;117(4):419–426. doi: 10.1016/j.ymgme.2016.02.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.