

Abstract
Summary
DNA methylation is an important mechanism regulating gene transcription, and its role in carcinogenesis has been extensively studied. Hyper and hypomethylation of genes is a major mechanism of gene expression deregulation in a wide range of diseases. At the same time, high-throughput DNA methylation assays have been developed generating vast amounts of genome wide DNA methylation measurements. We developed MethylMix, an algorithm implemented in R to identify disease specific hyper and hypomethylated genes. Here we present a new version of MethylMix that automates the construction of DNA-methylation and gene expression datasets from The Cancer Genome Atlas (TCGA). More precisely, MethylMix 2.0 incorporates two major updates: the automated downloading of DNA methylation and gene expression datasets from TCGA and the automated preprocessing of such datasets: value imputation, batch correction and CpG sites clustering within each gene. The resulting datasets can subsequently be analyzed with MethylMix to identify transcriptionally predictive methylation states. We show that the Differential Methylation Values created by MethylMix can be used for cancer subtyping.
Availability and implementation
MethylMix 2.0 was implemented as an R package and is available in bioconductor. https://www.bioconductor.org/packages/release/bioc/html/MethylMix.html
1 Introduction
DNA methylation is the best studied epigenetic aberration underlying oncogenesis. Besides genetic mutations, hypermethylation and hypomethylation of genes (increased and decreased methylation in a disease relative to a normal state) is an alternative mechanism that is capable of altering the normal transcriptional state and driving a wide range of diseases. Prior studies have focused on the analysis of DNA methylation data, for example in breast cancer or to identify differentially methylated regions for specific DNA methylation platforms (Wang et al., 2012; Warden et al., 2013). A recent pancancer study of DNA-methylation (Gevaert et al., 2015) revealed 10 pancancer clusters reflecting new similarities across malignantly transformed tissues. Furthermore, several computational tools have been developed incorporating state-of-the-art statistical techniques for the analysis of DNA methylation data (Aryee et al., 2014).
In Gevaert (2015), we introduced MethylMix: an algorithm that integrates DNA methylation from normal and disease samples and matched gene expression data to identify likely DNA methylation driven genes in diseases. The main output of MethylMix is a novel metric called ‘Differential Methylation value’ or ‘DM-value’ defined as the difference of an abnormal methylation state (Hypermethylated or hypomethylated) from the normal methylation state. These methylation states are computed using a beta mixture model on the beta-values.
Here, we present MethylMix 2.0, the updated version of MethylMix that features functions for downloading and preprocessing DNA methylation and gene expression datasets from all cancer sites in The Cancer Genome Atlas (TCGA). We also demonstrate an application of MethylMix for cancer subtyping and show how to use the DM-values for clustering using the R package ConsensusClusterPlus (Wilkerson and Hayes, 2010).
2 Algorithm
MethylMix 2.0 identifies DNA methylation driven genes by modeling DNA methylation data in cancer versus normal and looking for homogeneous subpopulations. In addition, matched gene expression data can be used to identify transcriptionally predictive DNA methylation events by requiring a negative correlation between methylation and gene expression of a particular gene. Therefore, MethylMix 2.0 requires DNA methylation from normal and disease samples and matched disease gene expression data. In MethylMix 2.0, we have automated the construction of the methylation and gene expression datasets from TCGA via a three-step algorithm:
Step 1: Automated Downloading from TCGA: DNA methylation datasets and Gene expression Datasets are downloaded automatically from TCGA by supplying the TCGA cancer code. We have provided the functionality to study any of the 33 TCGA cancer sites that are currently available.
Step 2: Automated preprocessing: The preprocessing steps include eliminating samples and genes with too many missing values, imputing remaining missing values and batch correction across technical batches.
Step 3: Clustering of the CpG probes: The methylation data produced by the Illumina 450k methylation array consists of multiple CpG probes for each gene. Since the probes are highly correlated, we clustered them prior to learning a mixture model. We used a complete linkage hierarchical clustering algorithm for all probes of a single gene to cluster the probes into CpG clusters. Then we cut off the hierarchical tree at a Pearson correlation threshold of 0.7.
The outputs of these functions are numeric matrices (METcancer, METnormal and GEcancer) with genes in rows and samples in columns to be used as inputs in MethylMix for further analysis.
3 Functions and examples
MethylMix 2.0 was implemented in the statistical language R and is provided as an R package, and is also available on bioconductor. MethylMix 2.0 was designed to identify transcriptionally predictive DNA methylation events using a beta mixture modeling approach (Gevaert, 2015). MethylMix 2.0 requires three datasets as inputs: cancer DNA methylation data (METcancer), normal DNA methylation data (METnormal) and matched disease gene expression data (GEcancer). These datasets can be downloaded as follows using the appropriate TCGA cancer codes (example OV for Ovarian Cancer):
> library(MethylMix) > cancerSite <- “OV”> targetDirectory <- paste0(getwd(), “/”)> METdirectories <- Download_DNAmethylation(cancerSite, targetDirectory, TRUE)> GEdirectories <- Download_GeneExpression(cancer Site, targetDirectory, TRUE)
For DNA-methylation and Gene Expression, we used the Broad Institute Firehose tool (Firehose, 2016), which includes several preprocessing steps such as removing problematic rows, removing redundant columns, reordering the columns and sorting the data by gene name. MethylMix’s contribution to the preprocessing consists of eliminating samples and genes with too many missing values, imputing remaining missing values and performing batch correction across technical batches within each cancer type.
We used an adjustable missing value threshold for removing samples or genes with too many missing values. The default threshold is a conservative one where genes with more than 20% of missing data are removed and we applied a K-Nearest Neighbors approach with K = 15 to estimate the remaining missing values, as proposed in Troyanskaya et al. (2001). Since TCGA data was generated in sample batches, we implemented batch correction to remove any systematic differences between technical batches. To this end, we used the ComBat algorithm introduced by Johnson et al. (2007) that removes known batch effects by implementing empirical Bayes methods for adjusting for additive, multiplicative and exponential batch effects. These adjustments methods are robust to small sample sizes. Since DNA methylation data generally do not follow a normal distribution, we used the nonparametric version of ComBat to correct the DNA methylation data.
> METProcessedData <- Preprocess_DNAmethylation (cancerSite, METdirectories)> GEProcessedData <- Preprocess_GeneExpression (cancerSite, GEdirectories)
The last step for preprocessing the methylation data is to assign each probe to a gene based on their closest transcription start site. Then for each gene, we cluster all its CpG sites using hierarchical clustering with complete linkage and the Pearson correlation as distance metric. If data for normal samples is provided, some probes might be removed in the normal samples or in the cancer samples due to a high number of missing values. In this case, only overlapping probes between cancer and normal samples are retained because MethylMix provides an analysis of the differential methylation.
> res <- ClusterProbes(METProcessedData[[1]], METProcessedData[[2]])
Probes with SNPs are removed since SNPs within the probe binding sequence prevent methylation of that CpG site. Additionally, MethylMix 2.0 provides a function getData that wraps the functions for downloading and preprocessing DNA methylation and gene expression data, as well as for clustering CpG probes.
> cancerSite <- “OV”> targetDirectory <- paste0(getwd(), “/”)> GetData(cancerSite, targetDirectory)
Next, it should be noted that all these functions are prepared to run in parallel if the user registers a parallel structure, otherwise they run sequentially.
Finally, the user can also use MethylMix with data that is not from TCGA. To use custom data, the user has to provide DNA methylation beta-values of a cancer cohort and optionally normal DNA methylation data and matched gene expression data in the form of a data.matrix object in R with the rows corresponding to the genes and the columns to the sample. In addition, the user can provide batch information in the case where multiple technical batches were used to generate the data. MethylMix can be applied on all Illumina DNA methylation arrays, including the newly released Epic platform and any microarray that outputs beta values. Similarly, sequencing-based methylation data can be modeled, if the data is formatted in proportions, but, as mixture modeling is computationally demanding, MethylMix will require proportionally more time to finish as the number of CpG sites is bigger.
4 Applications
The main output of MethylMix are the ‘DM-values’, which reflect the homogeneous subpopulations of samples with a particular methylation state. An application of the DM-values is to identify DNA methylation subtypes. For instance in lung squamous cell carcinoma (Fig. 1), a DNA hypomethylated subtype featuring genetic inactivation of NSD1 was identified (Brennan et al., 2017b; Campbell et al., 2018). DNA methylation subtypes were discovered by applying consensus clustering (a widely-used algorithm for clustering patients based on molecular data) to the ‘DM-values’ matrix output of MethylMix. Patients are thereby clustered into robust and homogenous groups (putative subtypes) based on their abnormal methylation profiles. Consensus clustering was performed using the ConsensusClusterPlus R package (Version 1.36.0) (Wilkerson and Hayes, 2010), with 1000 rounds of k-means clustering and a maximum of k = 10 clusters. Selection of the best number of clusters was based on the visual inspection of ConsensusClusterPlus output plots.
Fig. 1.
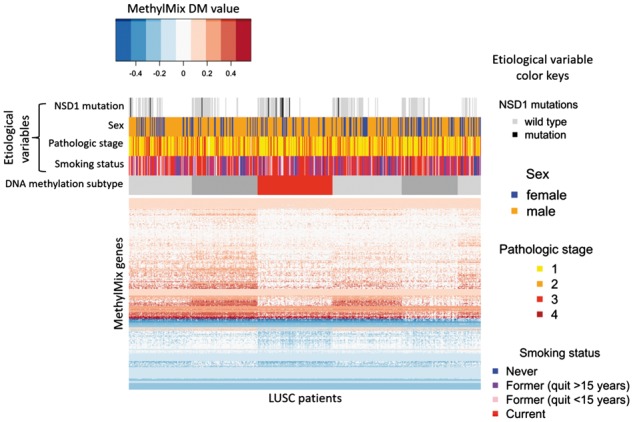
Subtyping of lung squamous cell carcinoma patients based on MethylMix2.0 analysis. This heatmap illustrates ‘DM values’ for 638 MethylMix genes (rows) in 503 LUSC patient primary tumors (columns). Patients are ordered by DNA methylation subtype, indicated in the horizontal sidebar. DNA methylation subtypes represent patient groups with distinct DNA methylation profiles that are homogenous within subtypes. The NSD1-inactivated subtype is highlighted in red, with other (NSD1 proficient) subtypes indicated in grey. Horizontal sidebars indicate the category of each patient with regard to key etiological variables including NSD1 mutations (white space reflects patients without NSD1 mutation data), smoking status, sex, pathological stage and smoking status. MethylMix genes (rows) are ordered by hierarchical clustering
Exploration of somatic mutation and copy number alteration data revealed that one patient cluster (indicated by the red sidebar) represents a DNA hypomethylated subtype that is enriched for inactivating genetic mutations and deletions in the NSD1, encoding a histone lysine methyltransferase. Indeed, six of ten patients with NSD1 mutations in LUSC were within the NSD1 subtype (Chi-squared P = 0.005). This mirrors the phenotype of a similar hypomethylated, NSD1-inactivated subtype that was recently described in head and neck squamous cell carcinoma (Brennan et al., 2017a).
> source(“https://bioconductor.org/biocLite.R”) > biocLite(“ConsensusClusterPlus”)> library(“ConsensusClusterPlus”)> MethylMixResults <- MethylMix(METcancer, GEcancer, METnormal)> DMvalues <- MethylMixResults$MethylationStates> cons_cluster <- ConsensusClusterPlus(d = DMvalues, maxK = 10, reps = 1000, pItem = 0.8, distance=’euclidean’, clusterAlg=“km”)
5 Conclusion
MethylMix 2.0 is an R package that provides automated functionalities that improve upon the original MethylMix algorithm. In MethylMix 2.0 we have implemented an automated process to download and preprocess these datasets directly from TCGA in a few lines of code. In addition, we have demonstrated a key application of MethylMix, identifying robust subgroups. In summary, MethylMix 2.0 offers a tool that facilitates the systematic analysis of methylation-driven genes in pan-cancer studies from TCGA.
Funding
Research was supported by NIBIB, NIDCR and NCI grants: R01EB020527, U01CA199241, U01DE025188 and U01CA217851. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest: none declared.
References
- Aryee M.J. et al. (2014) Minfi: a flexible and comprehensive bioconductor package for the analysis of infinium dna methylation microarrays. Bioinformatics, 30, 1363.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan K. et al. (2017a) Identification of an atypical etiological head and neck squamous carcinoma subtype featuring the cpg island methylator phenotype. EBioMedicine, 17, 223–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan K. et al. (2017b) Nsd1 inactivation defines an immune cold, dna hypomethylated subtype in squamous cell carcinoma. Sci. Rep., 7, 17064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell J.D. et al. (2018) Genomic, pathway network, and immunologic features distinguishing squamous carcinomas. Cell Rep., 23, 194–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firehose (2016) Broad institute tcga genome data analysis center: firehose stddata__2016_01_28 run. Broad Inst. MIT Harvard, doi: 10.7908/C11G0KM9. [Google Scholar]
- Gevaert O. (2015) Methylmix: an r package for identifying dna methylation-driven genes. Bioinformatics, 31, 1839–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gevaert O. et al. (2015) Pancancer analysis of dna methylation-driven genes using methylmix. Genome Biol., 16, 17.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson W.E. et al. (2007) Adjusting batch effects in microarray expression data using empirical bayes methods. Biostatistics, 8, 118.. [DOI] [PubMed] [Google Scholar]
- Troyanskaya O. et al. (2001) Missing value estimation methods for dna microarrays. Bioinformatics, 17, 520.. [DOI] [PubMed] [Google Scholar]
- Wang D. et al. (2012) Ima: an r package for high-throughput analysis of illumina’s 450k infinium methylation data. Bioinformatics, 28, 729–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warden C.D. et al. (2013) Cohcap: an integrative genomic pipeline for single-nucleotide resolution dna methylation analysis. Nucleic Acids Res., 41, e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkerson M.D., Hayes D.N. (2010) Consensusclusterplus: a class discovery tool with confidence assessments and item tracking. Bioinformatics, 26, 1572–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]