

Abstract
Motivation
The ECOD database classifies protein domains based on their evolutionary relationships, considering both remote and close homology. The family group in ECOD provides classification of domains that are closely related to each other based on sequence similarity. Due to different perspectives on domain definition, direct application of existing sequence domain databases, such as Pfam, to ECOD struggles with several shortcomings.
Results
We created multiple sequence alignments and profiles from ECOD domains with the help of structural information in alignment building and boundary delineation. We validated the alignment quality by scoring structure superposition to demonstrate that they are comparable to curated seed alignments in Pfam. Comparison to Pfam and CDD reveals that 27 and 16% of ECOD families are new, but they are also dominated by small families, likely because of the sampling bias from the PDB database. There are 35 and 48% of families whose boundaries are modified comparing to counterparts in Pfam and CDD, respectively.
Availability and implementation
The new families are now integrated in the ECOD website. The aggregate HMMER profile library and alignment are available for download on ECOD website (http://prodata.swmed.edu/ecod).
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
ECOD classifies protein domains based on their evolutionary history and groups remote homologs that share common ancestors in the same Homology group (H-group) while recognizing fine clustering of close homologs by families (F-group) (Cheng et al., 2014). Distant homologs that have diverged significantly in evolution may be beyond the sensitivity of current sequence-based homology detection programs, or they may have evolved with different topologies, a distinction characterized by ECOD Topology group. Sequence families were introduced to represent a group of proteins that are highly similar to each other and usually contain some conserved residues and motifs with implication of function or structural interaction (Sonnhammer et al., 1997). The sequences in a protein family are usually aligned, and a hidden Markov model (HMM) is derived from the multiple sequence alignment to represent the family for search and domain annotation (Letunic and Bork, 2018; Sonnhammer et al., 1997).
Families in ECOD were primarily dependent on the Pfam database (Cheng et al., 2014; Schaeffer et al., 2017). Although Pfam recently expedited their production (Finn et al., 2016), not all structures in the PDB database can be found in Pfam, especially recent depositions. ECOD differs from existing sequence family databases because ECOD domain boundaries take into account structural information. As a domain can be viewed with different perspectives, i.e. functional, structural and homology-based, it naturally leads to inconsistent definitions among protein classification databases. While structural classifications may cut the domain boundary more clearly and coherently, sequence classifications can access larger datasets and delineate the boundary consistently across domains from multiple sequence alignment. We have found that one ECOD domain could be covered by several Pfam domains or vice versa, and the residue coverage of Pfam family on ECOD domains can be improved for many cases (Schaeffer et al., 2016). This complex situation poses an ongoing challenge for consistent classification in ECOD.
Here we aim to build multiple sequence alignments and family profiles from our ECOD structural domains, not only to provide consistent family grouping within ECOD, but also to improve boundary definitions of existing families with structural information.
2 Materials and methods
Domain sequences and structures were taken from ECOD version 178. All sequence clustering to reduce redundancy was performed by CD-hit (Fu et al., 2012) at different identities without length consideration. Pairwise structure alignments with Dali (Holm and Park, 2000), TM-align (Zhang and Skolnick, 2005) and FAST (Zhu and Weng, 2004) were run for representatives in the same family. The structure alignments generated from coordinates were also adjusted to match the domain sequences by adding gaps, as some residues may be incomplete and ignored by the programs. We used these alignments as custom constraints for PROMALS3D (Pei and Grishin, 2014) rather than leave it to search for structure template on its own, since all domains to align have structures.
For large groups with at least five domains, the core was trimmed from both ends of the alignment until the first column that has more than 70% aligned residues. Structurally conserved indexes were predicted for each reside with both structural and sequence information, including secondary structure, carbon beta contacts and PSSM, conservation and gap fraction derived from PSI-BLAST (Altschul, 1997) results as well as secondary structure predicted by PSIPRED (McGuffin et al., 2000). The core of the alignment was conservatively cut at the column where any domain shows an index larger than 0.71, a threshold used on the structurally conserved region prediction server (Huang et al., 2013).
Protein sequences from UniProt reference proteomes were downloaded in May 2017 containing 9123 proteomes. Profile built with alignment of structural domains was searched against 80% redundancy reference proteome dataset using HMMSEARCH with an inclusion threshold of 1e-10 to construct the seed alignment. Then family HMM profile was built from seed alignment using reference columns deducted from alignment of structural domains. A full alignment was created by searching the family profile against the whole reference proteomes database with an inclusion threshold of 1e-3.
Pfam dataset for LGA computation was constructed by taking Pfam alignments with annotated PDB information. The sequence extracted from the mapped PDB and range was checked with sequence in the alignment and inconsistent sequences were disregarded. In some cases, different isoforms are recorded by PDB authors than the default UniProt isoform used by Pfam. Then, for each family, all pairs of aligned sequences in the multiple sequence alignment were extracted, and aligned positions were converted to PDB index ranges for LGA to score (Zemla, 2003). Scores for all pairs were averaged, and average scores for each family were collected for comparison.
HHsearch (Soding, 2005) profiles of Pfam version 31 and CDD domains were built with default parameters from downloaded alignments. E-value of 1e-5 was used as threshold for hit acceptance. But hits up to probability 90% were considered for database crosslinking and used for automatic naming. Non-overlapping hits were accepted if the overlap is less than 10 residues or less than 10% of the length of accepted hits. To be labeled as an identical family, the length of the profile-to-profile alignment needs to be more than 80% or within 10 residues of either the length of query or hit.
To generate domain structures colored by conservation, individual domain sequences were aligned and added to its family seed alignment with MAFFT (Katoh and Standley, 2013). Then, the sequence conservation was calculated and written into the B-factor column of the domain PDB by AL2CO (Pei and Grishin, 2001). For users’ convenience, ECOD website provides the Pymol session file for download that wraps the PDB format file and does basic visualization on startup.
3 Results
3.1 Construction of ECOD family alignments and profiles
The whole process of family determination is summarized as shown in Figure 1. The classification of ECOD sequence families and our temporary solutions for domains that cannot be mapped to existing families were described in an initial ECOD publication and our recent updates (Cheng et al., 2014; Schaeffer et al., 2017). Briefly, we assigned Pfam version 27 families to classified ECOD domain when possible with HMMER 3.1b2 (Eddy, 2011). Unmapped domains were clustered and served as provisional sequence families.
Fig. 1.

Flowchart of the pipeline to build ECOD family alignment and profile. Domains binned into the same Pfam or provisional families are collected and aligned by PROMALS3D with pairwise structure alignments as constraints and then possibly trimmed to the core region by consensus gaps or prediction. Then seed alignments and HMM profiles are obtained by searching against UniProt reference proteomes with profiles built from alignment of structural domains
For each ECOD F-group, which is either a provisional family or can be mapped to one or more non-overlapping Pfam families, we clustered domains to 70% sequence redundancy and ran all-vs-all pairwise structure alignments for representatives using Dali (Holm and Park, 2000), TM-align (Zhang and Skolnick, 2005) and FAST (Zhu and Weng, 2004). The aggregated list of the structure alignments was then used as custom constraints for PROMALS3D (Pei and Grishin, 2014) to build multiple sequence alignment for sets of all and non-redundant domains.
Next, we attempted to define the boundaries of the core alignment before building profiles. For larger groups, the beginning and end could be decided based on consensus gaps of the alignment. For small groups or singletons, we resorted to the software developed in lab to predict structurally conserved regions, which utilizes both sequence and structure information including secondary structure, contacts, sequence conservation, etc. (Huang et al., 2013). This process also assists in removal of non-homologous regions at the ends of domain, such as linkers and expression tags, which could introduce contamination in profile construction. The performance of our core definition was evaluated by distribution of the percentage of the alignment that is cut (Supplementary Fig. S1). For most groups, the trimmed proportion is less than 20%, and if the percentage exceeds 50%, which are mostly derived from prediction results, we simply kept the alignment intact.
The trimmed core alignment was then converted into an HMM profile which was subsequently searched against 80% redundancy UniProt reference proteomes (The UniProt, 2017) with HMMER to include sequences without structures. The resulting sequences were used to build the seed HMM profile. A full alignment was then produced by searching the seed profile against the reference proteome database. This approach and the underlying sequence database are similar to the method used by Pfam since version 28 (Finn et al., 2016).
We used HHalign in HHsuite (Soding, 2005) to compare and score all pairwise family profiles in the same H-group and merged redundant families. The scores were also converted to distances and then used to build phylogenic trees to display the relationship of homologous families on the website with the help of pHMM-Tree software (Huo et al., 2017).
3.2 Validation of alignment quality
Traditionally, evaluation of multiple sequence alignment quality uses established benchmarks of manual alignments or ad hoc structural alignments as a gold standard (Pei et al., 2008; Thompson et al., 2005; Van Walle et al., 2005). We sought to evaluate our PROMALS3D alignments with the Local-Global Alignment (LGA) method (Zemla, 2003). LGA is a program frequently used in model evaluation in CASP competition for the global distance test (GDT) and the total score (GDT_TS), which ranges from 0 to 100 and describes the average percentage of residues that can match under different distance thresholds. LGA can also run in sequence-independent analysis mode if equivalent residues are defined.
We utilized LGA to superimpose and score pairs of ECOD domains with the sequence equivalence defined in the alignment and calculated the average GDT_TS score for each family. The distribution of the average GDT_TS score per family is compared between ECOD alignments and Pfam alignments (Fig. 2a). In general, the two distributions are similar with peaks around GDT_TS score of 80. It suggests that the average quality of automatically built ECOD alignments with structural constraints is comparable to that of manually curated Pfam alignments. Similar results were obtained from comparison of only those ECOD families containing PDBs in the Pfam dataset (Supplementary Fig. S2a).
Fig. 2.

Validation of ECOD family alignment. (a) The distribution of the average GDT_TS score per family of all ECOD and Pfam families. (b) An example of Pfam alignment with registry shift mapped on the structures. Aligned residues are colored the same in rainbow from N-terminus. (c) ECOD family alignment of the same protein pair mapped on the structures. Proper gaps are made to handle loops and corresponding elements of different lengths
On the other hand, the distribution of Pfam alignments has a longer tail on the left side with lower scores. In some hard cases, divergent family members had differing insertions and elaborations at different locations, making alignment solely by sequence difficult. Such an example is illustrated in Figure 2b and c, where corresponding residues in the alignment are mapped on the structures with the same color. The Pfam alignments are mostly continuous with few gaps in the middle and contains a registration shift in the alignment, which results in a poor GDT_TS score of 28.8 (Fig. 2b, alignment shown in Supplementary Fig. S2b). The alignment built with Promols3D makes more gaps to take care of corresponding secondary structure elements and loops of differing lengths (Fig. 2c, Supplementary Fig. S2c), which results in a much better GDT_TS score of 71.3.
ECOD family alignments have high average quality based on the structural evaluation criteria. In most cases, close homologs in a family tend to have a similar overall topology, except for those that have large flexible regions or can undergo significant conformational changes, such as the N-terminal domain of chaperone SurA (PDB: 3RFW and 3NRK).
3.3 Statistics and new families
In total, we have determined 12 316 families, each of which is represented by an alignment of sequences with structures, alignments of sequences from UniProt reference proteome, and a HMMER profile. Firstly, we looked at the distribution of the number of sequences in ECOD families. The histogram of the natural logarithm of seed alignment size is plotted in Figure 3a, which shows a striking peak of small families. More specifically, the proportion of singleton families is 8.4%, and families with no more than 10 members constitute 25.2%. We calculated the same statistics for Pfam (Fig. 3b); the distribution also exhibits a single peak, and the percentages of singleton family and families with no more than 10 members are 1.9 and 14.5%, respectively.
Fig. 3.

Characterization of small families and new families in ECOD. (a) The logarithmic distribution of the number of sequences in ECOD families, showing a peak at very small size. (b) The logarithmic distribution of the number of sequences in Pfam families for comparison. (c) The size distribution of only those families that cannot find a significant hit (>90% HHSearch probability) to Pfam by HHsearch. (d) The pie graph illustrates the proportion of four kinds of families when compared with Pfam. Identical family hits a Pfam family with comparable length. Modified family has a Pfam counterpart, but lengths differ substantially. Merged family has multiple non-overlapping Pfam hits. New family means no good Pfam hits. (e) An HHsearch alignment of an omega toxin family against Pfam as an example to show the difficulty to detect sequence similarity for small family, especially those domains with few secondary structure elements. Small family has a thin profile and does not exhibit too much conservation pattern. (f) An unrooted tree of all families in ECOD omega toxin-related topology group with identical families to Pfam colored in blue. New families are scattered and distributed with Pfam families, and the distances between families are comparable with distances between Pfam families
In order to explore what constitutes the small families, we used HHsearch (Soding, 2005) to compare ECOD family profiles with the latest Pfam family profiles (Finn et al., 2016) and as well as the CDD database (Marchler-Bauer et al., 2015), which incorporates many domain databases including Pfam and some curated families at NCBI. Depending on whether there are hits passing the score threshold and the coverage of the hits (see ‘Materials and methods’ section), ECOD families can be divided into four categories: families with no hits are referred to as ‘new families’; families with one hit of comparable length are referred to as ‘identical families’; families with one hit of significantly different length are referred to as ‘modified families’; families with multiple non-overlapping hits are referred to as ‘merged families’. When we compared the sizes of new families to Pfam, it shows a strong bias towards small families (Fig. 3c). Among these new families, 21.8% of them are singletons and 52.4% have no more than 10 members. Similar results were obtained from comparison with the CDD database (Supplementary Fig. S3a), where fewer families are new, but the percentage of small families is higher, with 32.5% for singletons and 70.8% for families no larger than 10. This suggests that the overrepresentation of small families is due to the sampling bias of structures deposited in the PDB database, and these new small families are initiated by structures that cannot be found in existing families.
The summary statistics of the Pfam comparisons are summarized in Figure 3d. 36% of families are essentially identical to existing Pfam families. An equivalent proportion of families have some similarity to counterpart families in Pfam, but their domain boundary is fundamentally different. This boundary difference could represent a domain boundary extension, domain split, or domain merge, most likely reflecting how much structural information from ECOD domains help to improve domain boundary definition.
The most interesting category is the new families, which are as plentiful as 3321 when compared with Pfam (Fig. 3d) and 1977 when compared with CDD (Supplementary Fig. S3b). We have shown that most of them are small due to PDB bias, but is it an artifact of the sequence selection in structure determination experiments? If many proteins are from isolated phylogenetic branches, it would be difficult to detect their homologs, resulting in a bias of small and new families. We checked the phylogenetic distribution of sequences in the alignment and assigned each family a category if the sequences are mainly (>90%) from one kingdom or superkingdom. It turned out that the taxonomy distributions between all ECOD families and new families do not show much difference (Supplementary Fig. S4).
We further examined the most populated ECOD homology groups containing new families. Compared with the most populated H-groups (Cheng et al., 2014), the rank of H-groups with new families overlaps greatly, with Helix-turn-helix domains and Immunoglobulin-like domains containing the most new families. However, small domains, i.e. domains with few residues, such as ‘omega toxin-related’ and ‘beta-beta-alpha zinc fingers’ are overrepresented. This could indicate specialized functions, but more likely implies that such domains required specialized methods for similarity comparisons. Additionally, many of the families in these groups are also very small in size, which suggests the alignment and scoring are highly biased by cysteines and their relative position, because there is little conservation in thin profiles or even singletons (Fig. 3e). The unrooted tree of families in omega toxin-related topology group (the major topology group in this homology group) is shown in Figure 3f with ‘identical families’ of Pfam families colored in blue. New ECOD families are often grouped with a known family in Pfam, and the distance between families in the same clade are similar for both Pfam and ECOD families. This suggests that new families in ECOD are close relatives of known Pfam families or they could even be from the same family given the limitation of current methodology. At a minimum, they are consistent with Pfam family definition in this homologous group.
Lastly, we examined and annotated the large new families (when compared against CDD) which have more than 100 sequences in the seed alignment (Supplementary Table S1). Top 10 families are also shown in Table 1. Most of these new families represent a separate branch of domains in a specific protein family which shares no significant sequence similarity to other families in the homologous group; only few families have distant sequence similarity to known families, for example the MucBP_like family, or are the singlet family in its ECOD H-group, for example the DPY family. There are several recently discovered enzyme families that are recorded in CAZy (Lombard et al., 2014), but not yet in domain databases, such as glycoside hydrolase 95, 109, 120, glucuronoyl esterase, polysaccharide lyase 14.
Table 1.
Top 10 new ECOD families comparing with the CDD database
| Family accession | Number of sequences | Family ID | Description | Representative ECOD domain | Representative UniProt sequence |
|---|---|---|---|---|---|
| EF20768 | 884 | Cucumisin_fn3 | Fibronectin III-like domain of cucumisin | e3vtaA3[A: 628-730] | SBT11_ARATH/675-772 |
| EF18709 | 845 | Glyco_hydro_95_C | Glycoside hydrolase family 95 C-terminal domain | e2eabA3[A: 780-896] | Q9KEL0_BACHD/694-789 |
| EF18980 | 665 | DPY | DPY domain of the Dumpy protein | e1oigA1[A: 1-24] | M9PB30_DROME/9941-9964 |
| EF24417 | 462 | AFP_R2 | Marinomonas primoryensis antifreeze protein highly repetitive Region II | e4p99A1[A: 2-104] | Q6D230_PECAS/273-363 |
| EF09551 | 460 | MucBP_like | Mucin-binding protein domain | e3lyyA1[A: 1-102] | Q5FJ43_LACAC/76-164 |
| EF19702 | 431 | MSMEG_5817 | A bacteria family homologous to sterol carrier proteins | e4nssB1[B: 8-128] | A0R4F7_MYCS2/10-126 |
| EF17528 | 411 | CE15 | Carbohydrate esterase family 15; Glucuronoyl esterase | e4g4gA1[A: 31-397] | GCE_HYPJQ/165-431 |
| EF20492 | 388 | Polysacc_lyase_14 | Polysaccharide lyase family 14 | e3a0nA1[A: 2-243] | F4PN81_DICFS/107-341 |
| EF20386 | 382 | Trp_halogenase_C_1 | Tryptophan halogenase C-terminal domain | e3i3lA2[A: 181-278] | Q4KCZ0_PSEF5/193-303 |
| EF21944 | 378 | MmeI_S | DNA methyltransferase MmeI specificity domain | e5hr4J3[J: 621-906] | W6LYZ1_9GAMM/639-891 |
4 Discussion
In this work, we described our procedure to build multiple sequence alignments and profiles based on ECOD domains. ECOD families take advantage of the manual domain boundaries in ECOD, use structural information to build alignments, and follow similar processes to create profiles and add sequences as Pfam (Finn et al., 2016). We also demonstrated that the quality of our alignments is comparable to Pfam alignments. Profile-to-profile comparison results suggest that the domain boundaries of a large proportion of existing families were adjusted and presumably improved with the ECOD domain definitions. The Pfam comparison also discovered an unexpected number of new families. Investigation of these new families revealed that most of them have few sequences, likely resulting from bias from the PDB database. However, it is worth noting another factor which confounds the analysis and sequence search in general, simply that the shorter the domain length, the more difficult to get statistically significant scores. As in many cases of the domain boundary conflicts between ECOD and Pfam, ECOD usually further splits domains into individual evolutionary units (Cheng et al., 2015). Especially when a short domain is split out of a much longer domain, the score of the model for the long region, sometimes a whole protein, tends to be lower. For disulfide bond-rich domains and zinc fingers, it is intrinsically difficult since the sequence similarity signal is usually dominated by cysteines and their spacing, which calls for specialized methods to study the relationship in the future.
Compared with sequence domain databases such as Pfam, ECOD families generally should have more consistent domain boundaries with support from structural information. As an extension of the ECOD classification, ECOD families can be used to map protein domains to ECOD and study their evolutionary relationship. Since they are derived from structural domains, the scope of the families is biased towards structures in the PDB database. It uniquely covers new and important proteins that are the focus of recent research but also misses families that do not yet have a member with known structure or are intrinsically disordered.
ECOD families are now used in the ECOD update pipeline for family assignment. The aggregate HMM library and alignment file are available for download on the ECOD website together with distributable files of ECOD versions. Each family has a dedicated webpage showing various alignments interactively using MSAViewer (Yachdav et al., 2016), taxonomy distribution of sequences and relationship to other ECOD families, Pfam and CDD families. The family information page is also linked to the family level on the tree view page which displays the classification hierarchically.
ECOD domain pages add pre-calculated Pymol session files with domain structures colored by conservation for download, which is deduced from family sequence alignment by AL2CO (Pei and Grishin, 2001). These pages aid users in understanding their proteins of interest by readily combining sequence information and structural information. If the conserved residues tend to cluster in space, it may suggest a catalytic site or an interaction surface. Such an example is illustrated in Figure 4. TagF is an associated protein in the hemolysin co-regulated secretion island I-encoded type VI secretion system (H1-T6SS) of Pseudomonas aeruginosa. It posttranslationally represses the activity of H1-T6SS in a novel fashion that is independent of the previously known threonine phosphorylation-dependent pathway (Silverman et al., 2011). The detailed molecular mechanism remains unknown, although the structure of TagF was solved by structural genomics in 2007 (PDB: 2QNU). When the sequence conservation is colored on the TagF structure, it clearly shows a cluster of several most conserved residues, i.e. Gly8, Asp15, Phe16, Asp81 and Arg85 (Fig. 4a), and they seem to form a potential catalytic pocket (Fig. 4b), proposing a sound hypothesis for experimental testing.
Fig. 4.
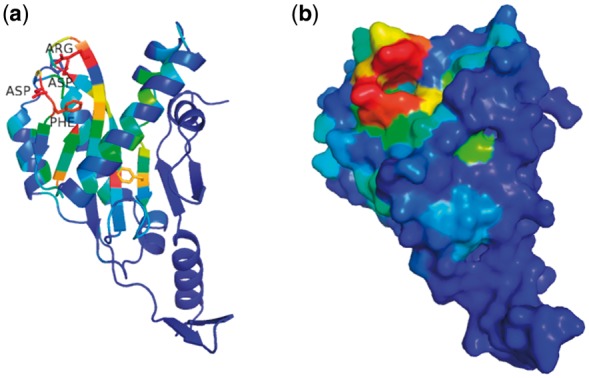
Structure of TagF colored by conservation shows a potential function site. (a) The structure of TagF (PDB: 2QNU) is rendered in cartoon and colored in rainbow by sequence conservation derived from family alignment. The conservation index is normalized, and red means the most conserved region. Side chains of the conserved residues are shown, and the names of several residues forming a pocket are labeled. (b) The surface of the same colored TagF structure is shown, highlighting a conserved pocket, which could be function-related
Our pipeline can be used to continually create new families from unmapped domains in ECOD. Through periodic updates, it will not only help ECOD to classify domains consistently and will also facilitate dedicated studies about specific families and protein annotations as a complementary resource to existing domain databases.
Supplementary Material
Acknowledgements
The authors thank Dr. Yun Qing for critically reading of the manuscript.
Funding
This work was supported in part by the National Institute of General Medical Sciences of the National Institutes of Health [grant number GM094575 to NVG]; and the Welch Foundation [grant number I-1505 to NVG].
Conflict of Interest: none declared.
References
- Altschul S.F. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H. et al. (2014) ECOD: an evolutionary classification of protein domains. PLoS Comput. Biol., 10, e1003926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H. et al. (2015) Manual classification strategies in the ECOD database. Proteins, 83, 1238–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy S.R. (2011) Accelerated profile HMM searches. PLoS Comput. Biol., 7, e1002195.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn R.D. et al. (2016) The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res., 44, D279–D285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L. et al. (2012) CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics, 28, 3150–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L., Park J. (2000) DaliLite workbench for protein structure comparison. Bioinformatics, 16, 566–567. [DOI] [PubMed] [Google Scholar]
- Huang I.K. et al. (2013) Defining and predicting structurally conserved regions in protein superfamilies. Bioinformatics, 29, 175–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo L. et al. (2017) pHMM-tree: phylogeny of profile hidden Markov models. Bioinformatics, 33, 1093–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., Standley D.M. (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol., 30, 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I., Bork P. (2018) 20 years of the SMART protein domain annotation resource. Nucleic Acids Res., 46, D493–D496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard V. et al. (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res., 42, D490–D495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A. et al. (2015) CDD: nCBI's conserved domain database. Nucleic Acids Res., 43, D222–D226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuffin L.J. et al. (2000) The PSIPRED protein structure prediction server. Bioinformatics, 16, 404–405. [DOI] [PubMed] [Google Scholar]
- Pei J., Grishin N.V. (2001) AL2CO: calculation of positional conservation in a protein sequence alignment. Bioinformatics, 17, 700–712. [DOI] [PubMed] [Google Scholar]
- Pei J., Grishin N.V. (2014) PROMALS3D: multiple protein sequence alignment enhanced with evolutionary and three-dimensional structural information. Methods Mol. Biol., 1079, 263–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei J. et al. (2008) PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res., 36, 2295–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer R.D. et al. (2016) Classification of proteins with shared motifs and internal repeats in the ECOD database. Protein Sci. Publ. Protein Soc., 25, 1188–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer R.D. et al. (2017) ECOD: new developments in the evolutionary classification of domains. Nucleic Acids Res., 45, D296–D302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman J.M. et al. (2011) Separate inputs modulate phosphorylation-dependent and -independent type VI secretion activation. Mol. Microbiol., 82, 1277–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soding J. (2005) Protein homology detection by HMM-HMM comparison. Bioinformatics, 21, 951–960. [DOI] [PubMed] [Google Scholar]
- Sonnhammer E.L. et al. (1997) Pfam: a comprehensive database of protein domain families based on seed alignments. Proteins, 28, 405–420. [DOI] [PubMed] [Google Scholar]
- The UniProt C. (2017) UniProt: the universal protein knowledgebase. Nucleic Acids Res., 45, D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J.D. et al. (2005) BAliBASE 3.0: latest developments of the multiple sequence alignment benchmark. Proteins, 61, 127–136. [DOI] [PubMed] [Google Scholar]
- Van Walle I. et al. (2005) SABmark–a benchmark for sequence alignment that covers the entire known fold space. Bioinformatics, 21, 1267–1268. [DOI] [PubMed] [Google Scholar]
- Yachdav G. et al. (2016) MSAViewer: interactive JavaScript visualization of multiple sequence alignments. Bioinformatics, 32, 3501–3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemla A. (2003) LGA: a method for finding 3D similarities in protein structures. Nucleic Acids Res., 31, 3370–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Skolnick J. (2005) TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res., 33, 2302–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Weng Z. (2004) FAST: a novel protein structure alignment algorithm. Proteins, 58, 618–627. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.