

Abstract
Functional pain syndromes, such as fibromyalgia and temporomandibular disorder, are associated with enhanced catecholamine tone and decreased levels of catechol-O-methyltransferase (COMT; an enzyme that metabolizes catecholamines). Consistent with clinical syndromes, our lab has shown that sustained 14-day delivery of the COMT inhibitor OR486 in rodents results in pain at multiple body sites and pain-related volitional behaviors. The onset of COMT-dependent functional pain is mediated by peripheral β2- and β3-adrenergic receptors (β2- and β3ARs) through the release of the pro-inflammatory cytokines tumor necrosis factor α (TNFα), interleukin-1β (IL-1β), and interleukin-6 (IL-6). Here, we first sought to investigate the role of β2- and β3ARs and downstream mediators in the maintenance of persistent functional pain. We then aimed to characterize the resulting persistent inflammation in neural tissues (neuroinflammation), characterized by activated glial cells and phosphorylation of the mitogen-activated protein kinases (MAPKs) p38 and extracellular signal-regulated kinase (ERK). Separate groups of rats were implanted with subcutaneous osmotic mini-pumps to deliver OR486 (15 mg/kg/day) or vehicle for 14 days. The β2AR antagonist ICI118,551 and β3AR antagonist SR59230A were co-administrated subcutaneously with OR486 or vehicle either on day 0 or day 7. The TNFα inhibitor Etanercept, the p38 inhibitor SB203580, or the ERK inhibitor U0126 were delivered intrathecally following OR486 cessation on day 14. Behavioral responses, pro-inflammatory cytokine levels, glial cell activation, and MAPK phosphorylation were measured over the course of 35 days. Our results demonstrate that systemic delivery of OR486 leads to mechanical hypersensitivity that persists for at least 3 weeks after OR486 cessation. Corresponding increases in spinal TNFα, IL-1β, and IL-6 levels, microglia and astrocyte activation, and neuronal p38 and ERK phosphorylation were observed on days 14-35. Persistent functional pain was alleviated by systemic delivery of ICI118,551 and SR59230A beginning on day 0, but not day 7, and by spinal delivery of Etanercept or SB203580 beginning on day 14. These results suggest that peripheral β2- and β3ARs drive persistent COMT-dependent functional pain via increased activation of immune cells and production of pro-inflammatory cytokines, which promote neuroinflammation and nociceptor activation. Thus, therapies that resolve neuroinflammation may prove useful in the management of functional pain syndromes.
Keywords: beta-adrenergic receptor (βAR); beta 2-adrenergic receptor (β2AR); beta 3-adrenergic receptor (β3AR); catechol-O-methyltransferase (COMT); catecholamines; epinephrine; norepinephrine; allodynia; hyperalgesia; p38; extracellular signal-regulated kinases (ERK); mitogen-activated protein kinase (MAPK); microglia; astrocyte; neuroinflammation; tumor necrosis factor alpha (TNFα); chronic pain, idiopathic pain, functional pain
1. Introduction
Functional pain syndromes (FPS), including fibromyalgia (FM), temporomandibular disorder (TMD), tension-type headache (TTH), and irritable bowel syndrome (IBS), represent a significant healthcare problem that affect nearly one in every three Americans (Clauw, 2014; Dzau and Pizzo, 2014; Rey and Talley, 2009; Slade et al., 2013; Tsang et al., 2008; Wesselmann et al., 2014). These conditions are characterized by persistent pain in the absence of tissue damage and often co-occur, thereby affecting multiple body sites. Accumulating evidence suggests that the origins of FPS are linked to abnormalities in catecholaminergic tone. Patients with FPS have increased levels of the catecholamines epinephrine and norepinephrine (Bote et al., 2014; Evaskus and Laskin, 1972; Perry et al., 1989; Torpy et al., 2000) alongside decreased levels of catechol-O-methyltransferase (COMT) (Marbach and Levitt, 1976; Smith et al., 2014), a ubiuitously expressed enzyme that metabolizes catecholamines (Mannisto and Kaakkola, 1999). Consistent with clinical findings, rodents receiving sustained delivery of the COMT inhibitor OR486 exhibit heightened mechanical sensitivity at multiple body sites, including the hindpaw and the abdomen (Ciszek et al., 2016; Kline et al., 2015). The development of this COMT-dependent functional pain is mediated by stimulation of peripheral β2- and β3-adrenergic receptors (β2- and β3ARs) (Ciszek et al., 2016).
β2- and β3ARs are G protein-coupled receptors (GPCRs) implicated in the transmission of pain. β2- and β3ARs can directly produce pain by increasing the excitability of primary afferent nociceptors on which they reside (Aley et al., 2001; Favaro-Moreira et al., 2012; Kanno et al., 2010; Khasar et al., 2003; Khasar et al., 1999a; Khasar et al., 1999b; Zhang et al., 2014). β2- and β3ARs can also influence pain through activation of immunoregulatory cells. Increased catecholamine levels following stress or pharmacologic manipulation lead to β2AR-mediated activation of T-cells (Laukova et al., 2012; Slota et al., 2015), mast cells (Chi et al., 2004), and macrophages (Chiarella et al., 2014; Kim et al., 2014) and β3AR-mediated activation of adipocytes (Fu et al., 2007; Kralisch et al., 2006; Mohamed-Ali et al., 2001), so as to increase the production of pro-inflammatory cytokines that sensitize nociceptors (Binshtok et al., 2008; Czeschik et al., 2008; Obreja et al., 2005; Obreja et al., 2002). Previously, our group demonstrated that acute delivery of OR486 leads to β2- and β3AR-mediated increases in plasma levels of the pro-inflammatory cytokines tumor necrosis factor α (TNFα), interleukin-1β (IL-1β), and interleukin-6 (IL-6) within 3 hours and that these cytokines were required for the development of functional pain (Hartung et al., 2014).
While acute increases in pro-inflammatory cytokines elicit acute pain and promote tissue repair, sustained elevations elicit persistent pain and maladaptive tissue pathology (Bennett and Schultz, 1993; Dinarello et al., 1990; Gharaee-Kermani and Phan, 2001). Sustained peripheral inflammation leads to increased release of excitatory neurotransmitters from the central terminals of primary afferents in the spinal cord and increased phosphorylation of mitogen-activated protein kinases (MAPKs), producing a state of neuronal hyperactivity in central pain-coding pathways (Woolf, 1983; Woolf and Salter, 2000). This state of ‘central sensitization’ is associated with inflammation in neural tissues (neuroinflammation), characterized by increased activation of glial cells and production of pro-inflammatory cytokines in neural tissues (Ji et al., 2016).
The present study sought to determine the relevance of these neuro-immune interactions to the maintenance of functional pain. We hypothesized that β2- and β3ARs drive persistent COMT-dependent functional pain via increased activation of immune cells and production of pro-inflammatory cytokines, which promote neuroinflammation and nociceptor activation. To test this hypothesis, we measured circulating and spinal levels of pro-inflammatory cytokines; activation of spinal microglia, astrocytes, and neurons; phosphorylation of spinal p38 and ERK MAPKs; and functional pain over the course of 35 days in rats receiving sustained subcutaneous delivery of β2- and β3AR antagonists on day 0 prior to or on day 7 following subcutaneous delivery of OR486 for 14 days. In addition, separate groups received sustained intrathecal delivery of TNFα, p38, or ERK inhibitors on day 14 following OR486 in order to determine the role of these effectors downstream of β2- and β3ARs in the maintenance of functional pain.
2. Materials and Methods
2.1. Subjects
Adult male and female Sprague-Dawley rats (N=210) were purchased (Charles River Laboratories, USA) and bred in-house. Rats weighed between 200 and 400g for all experimental studies. Rats had ad libitum access to standard laboratory chow and water. All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at Duke University and the University of North Carolina at Chapel Hill, and conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
2.2. General experimental design
First, after establishing the time course for acute COMT-dependent pain, we determined the effects of sustained β2- and β3AR activation on pain and spinal cord glial and neuronal activity in rats receiving the COMT inhibitor OR486 or vehicle, together with the β2AR-specific antagonist ICI118,551 + the β3AR-specific antagonist SR59230A or vehicle over the course of 14 days. Pain behavior was measured prior to (day 0), during (days 1, 3, 7, and 14), and 1-3 weeks following (days 21, 28, and 35) drug delivery. Spinal cord glial and MAPK activity were measured in separate groups of rats on day 14 during drug delivery or on day 21 (1 week) following drug delivery.
Next, we evaluated the contribution of β2- and β3ARs to the maintenance of pain and spinal cord glial and neuronal activity in rats receiving ICI118,551 + SR59230A or vehicle on day 7 following sustained delivery of OR486 or vehicle. Pain behavior was measured prior to (day 0), during (days 1, 3, 7, and 14), and 1-3 weeks following (days 21, 28, and 35) OR486 or vehicle delivery. Spinal cord glial and MAPK activity were measured in separate groups of rats on day 21 (1 week) following OR486 or vehicle delivery.
Finally, we evaluated the contribution of the pro-inflammatory cytokine TNFα and the MAPKs p38 and ERK in the maintenance of pain in rats by administering the TNFα inhibitor Etanercept, the p38 inhibitor SB203580, the ERK inhibitor U0126, or vehicle on day 14 following sustained delivery of OR486 or vehicle. Pain behavior was measured prior to (day 0), during (days 1, 3, 7, and 14), and 1-3 weeks following (days 21, 28, and 35) OR486 or vehicle delivery.
2.3. Drug preparation
OR486 (Tocris Bioscience, Bristol, UK) was dissolved in a vehicle consisting of a 5:2:3 ratio of dimethylsulfoxide (DMSO, DMSO Store, Fort Lauderdale, FL, USA), ethanol (Decon Labs Inc, King of Prussia, PA, USA) and 0.9% saline (Hospira, San Jose, CA, USA). ICI118,551 and SR59230A (Tocris Bioscience) were dissolved in a vehicle consisting of a 1:4 ratio of DMSO and 0.9% saline. Etanercept (Amgen Inc., Thousand Oaks, CA, USA), SB203580 (EMD Millipore, Burlington, MA, USA), and U0126 (EMD Millipore) were dissolved in a vehicle consisting of a 1:1 ratio of DMSO and 0.9% saline. For acute delivery, OR486 (30 mg/kg) was injected intraperitoneally (ip) as previously described (Hartung et al., 2014). For sustained delivery, drug or vehicle solutions were injected into Alzet osmotic mini-pumps (model 2002; Durect Corporation, Cupertino, CA, USA), which have a 0.5 μl/h infusion rate and a 200 μl reservoir volume. Mini-pumps were placed in 15 mL conical tubes containing sterile 0.9% saline and primed overnight in a dry heat bath (Lab Armor, Cornelius, OR, USA) at 37°C. Subcutaneous delivery of OR486 (15 mg/kg/day) and ICI118,551 (1.5 mg/kg/day) + SR59230A (1.67 mg/kg/day) was at a constant rate over 14 days. Intrathecal delivery of SB203580 (12 μg/day), U0126 (12 μg/day) and Etanercept (10 μg/day) was at a constant rate over 14 days.
For the maintenance studies, the Lynch method (Lynch et al., 1980) was used to delay delivery of ICI118,551 + SR59230A until day 7. Briefly, coiled PE50 polyethylene tubing (Scientific Commodities Inc., Lake Havasu City, AZ, USA) was placed onto the stainless steel flow moderator, which delayed drug delivery according to the length and diameter of the tubing. The pumps filled with Etanercept, SB203580, and U0126 were implanted on day 14, at which time the OR486 and vehicle pumps were removed.
2.4. Surgical procedures
Rats were anesthetized by isoflurane (Baxter, San Juan, Puerto Rico) inhalation (5% induction, 1.5-2% maintenance). Incision sites were shaved and disinfected with 70% ethanol and betadine (CVS Health, Woonsocket, RI, USA). Eyes were lubricated (Lubrifresh PM Ophthalmic Ointment; Major Pharmaceuticals, Livonia, MI, USA). Sterile technique was employed throughout the duration of all procedures according to IACUC requirements. For systemic delivery (Ciszek et al., 2016) of OR486, βAR antagonists, or vehicle, a small incision was made over the left shoulder blade of the rat. Hemostats were used to create a small subcutaneous pocket, in which each pump was placed. Several 9 mm stainless steel wound clips (Stoelting Co, Wood Dale, IL, USA) were used to close the incisions.
For intrathecal delivery (Ciszek et al., 2016) of TNFα, p38, and ERK inhibitors or vehicle, a small incision was made on the nape of the neck, and scissors and hemostats were used to lift muscle and expose the atlanto-occipital membrane. The membrane was carefully incised using the tip of a pair of scissors, a 12.3-cm, polyurethane Alzet Short Rat IT Catheter (Durect Corporation) inserted into the intrathecal space, and placed dorsal to the spinal cord. The catheter was sutured to the surrounding tissue using 4.0 silk suture (Oasis Surgical, Scottsdale, AZ, USA) and attached to the osmotic pump, which was subcutaneously implanted over the right shoulder blade. The initial OR486 or vehicle pumps were removed before incision closure. Delivery of the TNFα, p38, and ERK inhibitors or vehicle lasted for 14 days.
2.5. Assessment of pain behaviors
Paw withdrawal threshold was measured using the von Frey up-down method, as described by Chaplan (Chaplan et al., 1994). Nine calibrated von Frey monofilaments (bending forces of 0.69, 1.2, 1.5, 2.1, 3.6, 5.7, 8.7, 11.7, and 15.0 g; Stoelting) with equal logarithmic spacing between filaments were applied to the plantar surface of the hind paw. A series of 6 applications of monofilaments with varying gram forces was applied to the plantar surface of the hindpaw. Testing began with the middle filament in the series (3.6 g). If the response included the withdrawal of the hindpaw, an incrementally lower filament was applied. In the absence of a paw withdrawal, an incrementally higher filament was applied. These data were entered into the Paw Flick module within the National Instruments LabVIEW 2.0 (Austin, TX, USA) software. Mechanical allodynia was defined as a heightened response to a normally innocuous stimulus and was determined as a significant decrease in paw withdrawal threshold from baseline.
After determining paw withdrawal threshold, paw withdrawal frequency to a noxious von Frey monofilament was assessed. The highest gram force filament (15.0 g) was applied to the hind paw 10 times. Stimulus was applied for 1 second followed by a 1-second interval without a stimulus. The number of paw withdrawals was recorded for each hindpaw. Mechanical hyperalgesia was defined as an increase in the number of paw withdrawals to a noxious mechanical stimulus from baseline. In all behavioral experiments, the experimenter was blinded to treatment.
2.6. Assessment of cytokines
Plasma and cerebral spinal fluid (CSF) were collected from separate groups of rats on days 0, 14, 21, and 35 following OR486 delivery. TNFα, IL-1β, and IL-6 were measured using the Rat Cytokine Multiplex Assay from BioRad (Hercules, CA, USA). All plasma samples were diluted at 2x and CSF samples were run neat.
2.7. Assessment of spinal cord glial and neuronal activity
Five minutes prior to tissue collection, mechanical pinch was briefly applied to the hindpaws in order to evoke pERK. Then, rats were deeply anesthetized with Fatal Plus (Vortech Pharmaceuticals, Dearborn, MI, USA) and when no longer responsive to paw- or tail-pinch, they were perfused with 4% paraformaldehyde (PFA; ThermoFisher, Carlsbad, CA, USA) in 0.1 M PBS (Life Technologies, Carlsbad, CA, USA). Spinal cords were collected, placed in 4% PFA post-fixative overnight, and cryoprotected in 30% sucrose (BDH, Kandivali, Mumbai, India) in 0.01M PBS at 4°C. Tissues were then embedded in Optimal Cutting Temperature (OCT) cryomatrix and sectioned by cryostat. Spinal cords were sliced to 30 μm-thick free-floating sections, placed in chilled 0.01M PBS with 0.1% sodium azide, and stored at 4°C until use.
A combination of antibodies was used to determine glial activation and MAPK phosphorylation in spinal cord. For spinal cord staining, anti-p-p38 (1:300, Cell Signaling, Danvers, MA, USA) or anti-pERK (1:600, Cell Signaling) was used in combination with either anti-CD11b (1:500, AbD Serotec, Hercules, CA, USA), anti-GFAP (1:500, Cell Signaling), or anti-NeuN (1:500, Millipore). Secondary antibodies were either anti-rabbit or anti-mouse Alexa Fluor 488 or Alexa Fluor 594 (Jackson Immuno, West Grove, PA, USA). Stained tissues were mounted with Vectashield (Vector Laboratories, Burlingame, CA, USA). Images were collected on a Zeiss 780 confocal microscope. Quantification of immunofluorescence and immunopositive cells for spinal cord was conducted using ImageJ/Fiji software (National Institutes of Health, Bethesda, MD, USA).
For p-p38, CD11b, and GFAP analysis, the spinal dorsal horn area was outlined and fluorescence intensity was quantified using Image J software. Data were normalized to the Veh/Veh group and shown as the percentage compared to Veh/Veh. For pERK analysis, the number of pERK+ cells in dorsal horn were counted manually. Control experiments, including incubation of slices in primary or secondary antibody alone, were conducted for each round of staining and showed only low intensity non-specific binding patterns.
2.8. Assessment of MAPK inhibition
Flash frozen spinal cord tissue was homogenized in a Precellys 24 tissue homogenizer (Bertin Instruments, Montigny-le-Bretonneux, France) with tissue protein extraction reagent (TPER, ThermoFisher). To accommodate for variation in tissue homogenates, protein concentrations were normalized following bicinchoninic acid (BCA; Pierce, Grans Island, NY, USA) measurement of protein content to evenly load protein into wells of 10% SDS-PAGE gels (BioRad). Gels were run using standard SDS-PAGE methods and transferred onto PVDF membrane using the iBlot dry blotting system (Life Technologies). Membranes were then blocked, probed with primary antibodies for GAPDH (1:1000; Cell Signaling), p-ERK1/2 (1:1000; Cell Signaling) or p-p38 (1:100; Cell Signaling), and subsequently probed with a corresponding secondary antibody. Specific bands were visualized with enhanced chemiluminescence (Thermo Scientific, Waltham, MA, USA) and quantified with Image J. The relative levels of p-p38 and pERK were normalized to GAPDH.
2.9. Statistical analysis
Group differences in mechanical allodynia and mechanical hyperalgesia over time were analyzed by two-way ANOVA for repeated measures or by one-way ANOVA for the time-collapsed panel insets. Group differences in cellular activity at a single time point were analyzed by one-way ANOVA. Post-hoc comparisons were performed using the Bonferroni test, which corrected for multiple comparisons. Statistical significance was defined as P < 0.05. All statistical analyses were performed using GraphPad Prism 7 (GraphPad Software, San Diego, CA, USA).
Time-dependent changes in plasma vs. CSF concentrations of the cytokines were analyzed using a general linear model as implemented in SAS PROC GLM (SAS 9.4, SAS Institute Inc, Cary, NC). Before the analysis, plasma concentrations for each cytokine were standardized by subtracting the mean and dividing by the standard deviation. CSF concentrations were standardized in the same way. In the linear model, cytokine concentration was used as a continuous dependent variable. Biomarker source (plasma vs. CSF, coded as a binary variable), batch effect, and time (number of days since the beginning of experiment as a continuous variable), together with source-by-time interaction term, were included as independent variables. The source-by-time interaction term in the model can be interpreted as the difference in time slopes (trends) between standardized plasma and CSF concentrations. A Bonferroni correction was applied to account for testing three cytokines, and the P-value for the interaction term adjusted to a significance threshold of 0.017.
3. Results
3.1. Sustained COMT inhibition results in persistent mechanical allodynia and hyperalgesia mediated by β2- and β3ARs
We first determined the duration of a single ip dose of the COMT inhibitor OR486 (30 mg/kg) on mechanical pain. We found that a single dose of OR486 produced mechanical allodynia (F 1, 80 = 96.27, P < 0.0001) and mechanical hyperalgesia (F 1, 80 = 94.41, P < 0.0001), which peaked at 3h and returned to baseline at 12h. (Supplementary Figure 1). These data suggest that a single dose of COMT inhibitor is not sufficient to drive chronic pain states. As a result, we chose to administer OR486 for continuously over 14 days to assess the effects of sustained reductions in COMT activity. In addition, as the magnitude and duration of mechanical allodynia and hyperalgesia were similar in males and females (P > 0.05), we used male rats for subsequent behavioral experiments.
Our previous work demonstrated that sustained 14-day delivery of the COMT inhibitor OR486 in rats results in mechanical hypersensitivity at multiple body sites, mediated by peripheral β2- and β3ARs (Ciszek et al., 2016; Kline et al., 2015). Here, we show that this β2- and β3AR-mediated pain persists for at least 3 weeks following OR486 cessation. As shown in Figure 1, sustained delivery of OR486 resulted in mechanical allodynia (F1,128=199.7, P < 0.0001) and mechanical hyperalgesia (F1,128=415.7, P < 0.0001) beginning on day 1 and lasting until day 35 (21 days following the cessation of OR486). Co-administration of the β2AR antagonist ICI118,551 and the β3AR antagonist SR59230A together with OR486 on day 0, blocked the development of COMT-dependent mechanical allodynia (F3,208=64.19, P < 0.0001) and mechanical hyperalgesia (F3,208=194.2, P < 0.0001) throughout the 35-day experimental paradigm. The moderate dip in paw withdrawal threshold on days 14 and 28 may be attributed to a lower COMT activity. Co-administration of ICI118,551+SR59230A together with vehicle on day 0 had no effect on pain behavior. Therefore, β2- and β3ARs are required for the onset of functional pain that fails to resolve after removal of the COMT inhibitor.
Figure 1. Sustained stimulation of β2- and β3ARs results in hypersensitivity to mechanical stimuli that persists for 3 weeks following cessation of OR486.
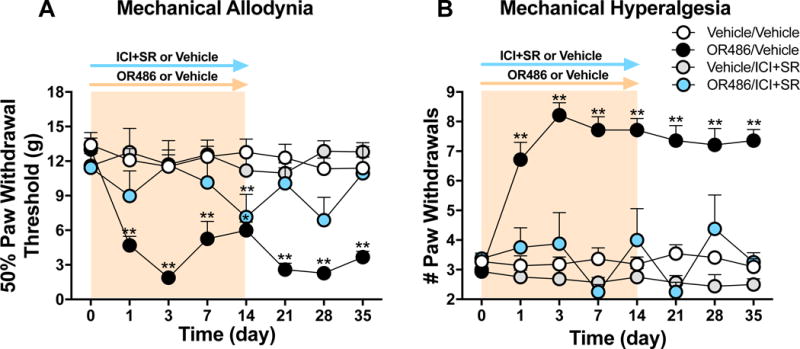
Animals receiving OR486 for 14 days exhibit (A) mechanical allodynia and (B) mechanical hyperalgesia, lasting up to 21 days following the cessation of OR486. The OR486-induced pain behavior is prevented by co-administration of ICI118,551+SR59230A on day 0. Vehicle/Vehicle n=11, OR486/Vehicle n=7; Vehicle/ICI&SR n=8; OR486/ICI&SR n=4 males per group. Data are shown as mean ± SEM. **P < 0.01, *P < 0.05 versus vehicle/vehicle.
3.2. Sustained COMT inhibition results in persistent neuroinflammation
Our previous work demonstrates that peripheral β2- and β3ARs initiate COMT-dependent functional pain through increased circulating levels of the pro-inflammatory cytokines TNFα, IL-1β, and IL-6 (Hartung et al., 2014). Here, we investigated changes in levels of these pro-inflammatory cytokines in CSF as well as circulating blood collected on day 0, on day 14 following OR486 delivery, and on days 21 and 35 (1 and 3 weeks following OR486 cessation). As shown in Figure 2, plasma levels of TNFα decreased while CSF levels increased over the course of 35 days, resulting in a significant source-by-time interaction (P = 0.0078). Similarly, opposite trends in levels of IL-1β and IL-6 were observed in plasma versus CSF, resulting in significant source by time interactions (P = 0.021 and P = 0.0098, respectively). Source-by-time interactions for TNFα and IL-6 remained significant after Bonferroni adjustment for three statistical tests. Figure 2 insets further show the percent change in CSF vs plasma pro-inflammatory cytokine levels on day 35 compared to day 0. By day 35, plasma levels of TNFα, IL-1β, and IL-6 have decreased 30-60%, while CSF levels have increased 50-180%.
Figure 2. Plasma cytokines decrease, while CSF cytokines increase 1-3 weeks following cessation of OR486.
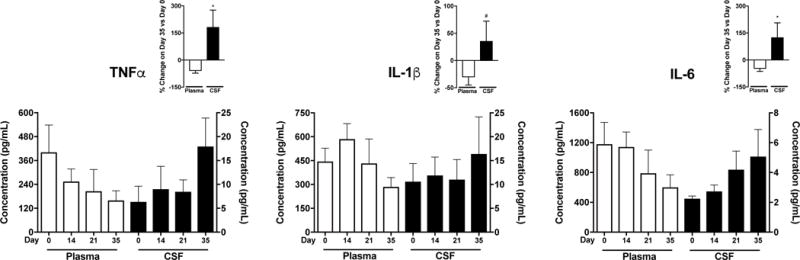
Plasma levels of TNFα, IL-1β, and IL-6 decrease over the course of 35 days, while CSF levels of these pro-inflammatory cytokines increase over the course of 35 days (source-by-time interactions are P = 0.0078, 0.021, and 0.0098, respectively). The insets further show the percent change in CSF vs plasma pro-inflammatory cytokine levels on day 35 compared to day 0. Plasma on days 0-35 n=6, CSF on days 0-35 n=6 males per group. Data are shown as mean ± SEM, *P < 0.05, #P < 0.1 versus plasma.
These and previous findings, together, suggest that COMT inhibition leads to β2- and β3AR-mediated inflammation in peripheral tissues within hours, that resolves and transitions to neuroinflammation over the course of weeks.
3.3. Sustained COMT inhibition results in β2- and β3AR-mediated increases in the activation of spinal cord microglia and astrocytes
Neuroinflammation is characterized by increased activity of microglia and astrocytes, which are glial cells that contribute to chronic pain following nerve injury or inflammation (Gosselin et al., 2010). Here, we sought to investigate alterations in glial cell activity in our model of functional pain.
We first assessed if a single dose of OR486 would lead to glial activation in the dorsal horn. No changes in spinal microglia or astrocyte activation were observed at 3 or 12 hours (P > 0.05, Supplementary Figure 2). However, sustained administration of OR486 for 14 days resulted in increased immunofluorescence of glial markers in the dorsal horn. Compared to vehicle, sustained delivery of OR486 induced microglial activation in the spinal dorsal horn on days 14 (F3,55=26.28, P < 0.0001) and 21 (F3,79=12.44, P < 0.0001) (Figure 3A). COMT-dependent increases in microglial activation were blocked by co-administration of ICI118,551+SR59230A on day 0. Similarly, compared to vehicle, sustained delivery of OR486 induced astrocyte activation in the spinal dorsal horn on days 14 (F3,57=23.41, P < 0.0001) and 21 (F3,74=21.27, P < 0.0001) (Figure 3B). COMT-dependent increases in astrocyte activation were blocked by co-administration of ICI118,551+SR59230A on day 0. Furthermore, we did not observe sex-dependent differences in glial immunoreactivity (Supplementary Figure 3). Finally, co-administration of ICI118,551+SR59230A in vehicle-treated animals had no effect on the activity of microglia or astrocytes.
Figure 3. Sustained stimulation of β2- and β3ARs results in increased microglia and astrocyte activation on days 14 and 21.
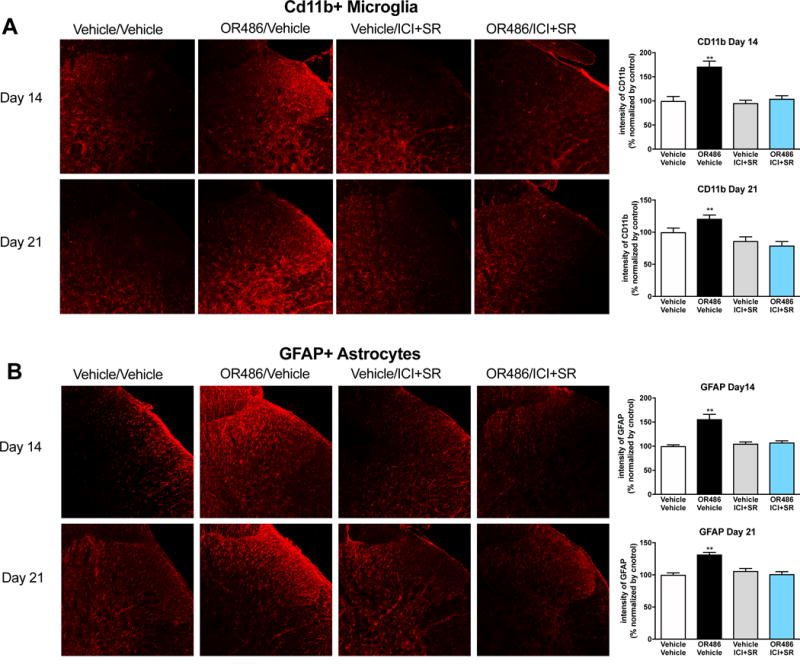
Quantitative analysis of immunofluorescence intensity demonstrates that rats receiving sustained delivery of OR486 for 14 days, exhibit increased activity of (A) CD11b+ microglia and (B) GFAP+ astrocytes in the spinal dorsal horn on days 14 and 21. OR486-induced activation of microglia and astrocytes is blocked by co-administration of ICI-118,551 + SR59230A on day 0. Day 14 groups: Vehicle/Vehicle n=5 (3 males + 2 females); OR486/Vehicle n=6 (3 males + 3 females); OR486/ICI&SR n=6 (3 males + 3 females); Vehicle/ICI&SR n=5 (3 males + 2 females). Day 21 groups: Vehicle/Vehicle n=6 (3 males + 3 females); OR486/Vehicle n=7 (4 males + 3 females); OR486/ICI&SR n=7 (4 males + 3 females); Vehicle/ICI&SR n=6 (3 males + 3 females). Data are expressed as mean ± SEM. **P < 0.01 versus vehicle/vehicle.
3.4. Sustained COMT inhibition results in β2- and β3AR-mediated increases in the phosphorylation of spinal cord p38 and ERK MAPK
As the p38 and ERK MAPKs are important mediators of pain and inflammation (Ji and Suter, 2007), we sought to investigate their alteration in our model of functional pain. Compared to vehicle, sustained delivery of OR486 induced p38 phosphorylation in the spinal cord on days 14 (F3,58=9.617, P < 0.0001) and 21 (F3,77=43.93, P < 0.0001) (Figure 4A). COMT-dependent increases in p38 phosphorylation were blocked by co-administration of ICI118,551+SR59230A on day 0. Similarly, compared to vehicle, sustained delivery of OR486 resulted in pressure-evoked increases in ERK phosphorylation in the spinal dorsal horn on days 14 (F3,28=945.34, P < 0.0001) and 21 (F3,28=1547, P < 0.0001) (Figure 4B). COMT-dependent pressure-evoked increases in ERK phosphorylation were blocked by co-administration of ICI118,551+SR59230A on day 0. Of note, in the absence of mechanical pinch, spinal ERK phosphorylation was not observed in rats receiving vehicle or OR486 (data not shown). Co-administration of ICI118,551+SR59230A in vehicle-treated animals had no effect on the phosphorylation of p38 or ERK.
Figure 4. Sustained stimulation of β2- and β3ARs results in increased phosphorylation of spinal cord p38 and ERK on days 14 and 21.
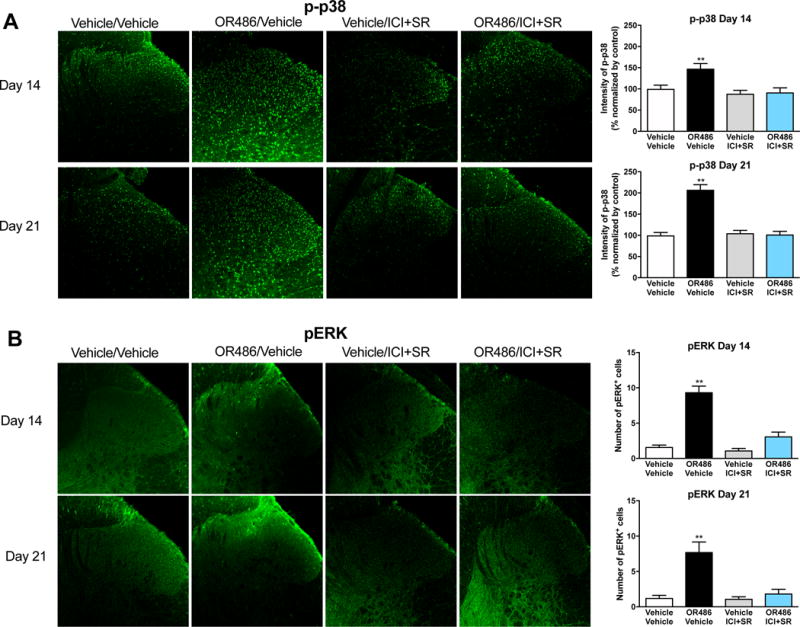
Quantitative analysis of immunofluorescence intensity demonstrates that rats receiving sustained delivery of OR486 for 14 days, exhibit increased expression of (A) p-p38 and (B) pERK in the spinal dorsal horn on days 14 and 21. OR486-induced phosphorylation of p38 and ERK is blocked by co-administration of ICI-118,551 + SR59230A on day 0. Day 14 groups: Vehicle/Vehicle n=5 (3 males + 2 females); OR486/Vehicle n=6 (3 males + 3 females); OR486/ICI&SR n=6 (3 males + 3 females); Vehicle/ICI&SR n=5 (3 males + 2 females). Day 21 groups: Vehicle/Vehicle n=6 (3 males + 3 females); OR486/Vehicle n=7 (4 males + 3 females); OR486/ICI&SR n=7 (4 males + 3 females); Vehicle/ICI&SR n=6 (3 males + 3 females). Data are expressed as mean ± SEM. **P < 0.01 versus vehicle/vehicle.
To determine the cell types that express p-p38 and pERK in our model of functional pain, we performed subsequent double-labeling immunohistochemical experiments using cell-specific markers for microglia (CD11b), astrocytes (GFAP), and neurons (NeuN). OR486-induced increases in p-p38 and pERK were predominantly found in neurons in the spinal dorsal horn. Specifically, 81% of p-p38 was expressed in neurons, 4% in microglia, and 3% in astrocytes (Figure 5A). Similarly, 87% of pERK was expressed in neurons, 9% in microglia, and 17% in astrocytes (Figure 5B). Note that percentages do not add up to exactly 100%, as double-staining was performed in alternate spinal cord sections.
Figure 5. The p-p38 and pERK MAPKs are predominantly co-expressed with spinal neurons.
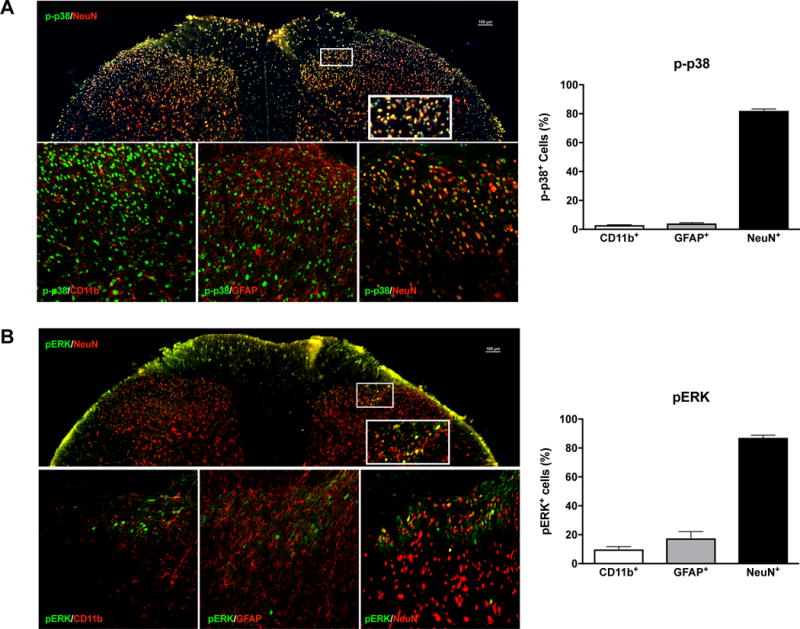
Immunohistochemical staining showing the expression of (A) p-p38 (green) and (B) pERK (green) with cell markers (red) for microglia (CD11b), astrocytes (GFAP), and neurons (NeuN). Quantitative analysis reveals that the majority (>80%) of p-p38 or pERK positive cells were co-expressed with NeuN+ neurons. N=6 (3 males + 3 females) per group. Data are expressed as mean ± SEM.
3.5. Inhibition of TNFα or p38 reverses COMT-dependent functional pain
The above findings demonstrate that sustained stimulation of β2- and β3ARs results in functional pain and neuroinflammation, characterized by increased pro-inflammatory cytokine production, glial activation, and MAPK phosphorylation in spinal tissues. Next, we targeted β2- and β3ARs and their downstream effectors (TNFα, p38, and ERK) with selective inhibitors to determine the role of these molecules in the maintenance of COMT-dependent functional pain.
To determine the role of β2- and β3ARs in the maintenance of pain, ICI118,551+SR59230A or vehicle were delivered systemically for 14 days, beginning on day 7 following delivery of OR486 or vehicle. Delivery of these β2- and β3AR antagonists on day 7 did not alleviate OR486-induced mechanical allodynia (Figure 6A) or mechanical hyperalgesia (Figure 6B). In addition, β2- and β3AR antagonist delivery beginning on day 7 did not reduce OR486-induced glial activation or MAPK phosphorylation (Supplementary Figure 4).
Figure 6. COMT-dependent functional pain is reversed by intrathecal TNFα or p38 inhibitors, but not by systemic β2- and β3AR antagonists.

Animals receiving OR486 for 14 days exhibit mechanical allodynia and hyperalgesia over the course of 35 days. Systemic delivery of ICI118,551+SR59230A for 14 days beginning on day 7 does not reverse OR486-induced (A) mechanical allodynia or (B) mechanical hyperalgesia. Vehicle/Vehicle n=8; OR486/Vehicle n=7; OR486/ICI+SR n=6 males per group. Intrathecal administration of the TNFα inhibitor Etanercept (C,D) or the p38 inhibitor SB203580 (E,F) for 14 days beginning on day 14 reverses OR486-induced mechanical pain. Insets represent average pain behavior following inhibitor delivery on days 21-35. N=6 males per group. Data are mean ± SEM. **P < 0.01, *P < 0.05 vs. vehicle/vehicle.
To determine the role of spinal TNFα in the maintenance of pain, the TNFα inhibitor Etanercept was delivered intrathecally for 14 days, beginning on day 14 (a time point when mechanical sensitivity and neuroinflammation are more established) following delivery of OR486 or vehicle. Results show that Etanercept completely reversed OR486-induced mechanical allodynia (F3,151=17.62, P < 0.0001; Figure 6C) and hyperalgesia (F3,147=30.44, P < 0.0001; Figure 6D).
Similarly, to determine the role of spinal p38 and ERK in the maintenance of pain, the p38 inhibitor SB203580 or the ERK inhibitor U0126 was delivered intrathecally for 14 days, beginning on day 14 following delivery of OR486 or vehicle. Results show that SB203580 reversed OR486-induced mechanical allodynia (F3,63=16.63, P < 0.0001; Figure 6E) and hyperalgesia (F3,63=16.89, P < 0.0001; Figure 6F). In contrast, U0126 had no effect on OR486-induced pain (Supplementary Figure 5). The OR486/U0126 group exhibited an increase in paw withdrawal threshold prior to delivery of U0126, making interpretation of the effects of U0126 on mechanical allodynia difficult. Nonetheless, we did find that U0126 had no effect on mechanical hyperalgesia (which was robust and consistent throughout the observation period; Supplementary Figure 5), suggesting that transient pERK expression is not sufficient to maintain functional pain. Thus, spinal TNFα and p38, which are key mediators of neuroinflammation, are required for the maintenance of functional pain.
To confirm if the doses for SB203580 and U0126 effectively inhibited the phosphorylation of p38 and ERK, respectively, we performed a Western blot to measure p-p38 and pERK. Sustained delivery of OR486 resulted in significant increases in p-p38 expression in the spinal cord on day 35, which was blocked by SB203580. Though OR486 treatment did not increase the pERK expression in spinal cord (in the absence of mechanical stimuli), the ERK inhibitor U0126 decreased pERK expression (Supplementary Figure 6).
4. Discussion
The present study sought to investigate the role of β2- and β3ARs and downstream mediators in the maintenance of persistent functional pain linked to abnormalities in catecholamine signaling. Our results are the first to show that sustained stimulation of β2- and β3ARs produces functional pain and neuroinflammation that persist for weeks after removal of the causal stimulus (systemic COMT inhibitor). Further, we provide evidence that peripherally-initiated functional pain is centrally-maintained by TNFα and p38 MAPK.
4.1. Sustained stimulation of β2- and β3ARs leads to persistent mechanical allodynia and hyperalgesia
Pain amplification is a hallmark feature of FPS, with patients experiencing hypersensitivity to mechanical stimuli (Maixner et al., 2016). Mechanical hypersensitivity may occur prior to (predicting onset) or following (signifying chronification) the development of FPS, and often occurs at remote body sites. Consistent with clinical studies, our lab has shown that administration of the COMT inhibitor OR486 in rodents produces increased mechanical hypersensitivity at multiple body sites and alters pain-related volitional behaviors (eg, avoidance of painful heat and bright light) (Ciszek et al., 2016; Hartung et al., 2014; Kline et al., 2015; Nackley et al., 2007). In subsequent pharmacologic studies, we found that the development of acute (measured over 3 hours) and persistent (measured over 14 days) OR486-induced mechanical hypersensitivity is mediated by peripheral, but not spinal or supraspinal, β2- and β3ARs (Ciszek et al., 2016; Hartung et al., 2014; Kline et al., 2015; Nackley et al., 2007; Zhang et al., 2018). This finding is in line with that of Kambur and colleagues, who demonstrated that the peripherally-restricted COMT inhibitor nitecapone elicits mechanical allodynia when administered systemically, but not intrathecally (Kambur et al., 2010). Here, we further demonstrated that systemic delivery of OR486 by osmotic mini-pump for 14 days leads to β2- and β3AR-mediated mechanical allodynia and mechanical hyperalgesia that persists for at least 3 weeks after OR486 cessation. These findings suggest that sustained increases in catecholamine signaling at peripheral β2- and β3ARs, strengthen pain-coding pathways such that functional pain continues in the absence of the precipitating cause.
4.2. Sustained stimulation of β2- and β3ARs leads to neuroinflammation
In previous studies, we demonstrated that acute COMT-dependent pain is initiated by peripheral β2- and β3ARs through release of the pro-inflammatory cytokines TNFα, IL-1β, and IL-6 in plasma within several hours (Ciszek et al., 2016; Hartung et al., 2014). Here, we found that plasma levels of TNFα, IL-1β, and IL-6 decreased, while CSF levels of these cytokines increased 1-3 weeks following cessation of OR486. This transition from acute inflammation in peripheral tissues to neuroinflammation has been well-characterized within the context of neuropathic, inflammatory, and cancer-related pain (Ji et al., 2016), and may also play a critical role in the chronification of functional pain.
Cytokines are small intracellular regulatory proteins secreted by immune cells in the periphery and neurons and glia in the central nervous system (Kress and Sommer, 2004; Miller et al., 2009). Pro-inflammatory cytokine levels are elevated in patients with FM (Bazzichi et al., 2007; Gur et al., 2002; Sommer et al., 2008; Wallace et al., 2001; Zhang et al., 2008), TMD (Kaneyama et al., 2002; Matsumoto et al., 2006; Ogura et al., 2010; Slade et al., 2011; Takahashi et al., 1998), TTH (Bo et al., 2009; Della Vedova et al., 2013; Kocer et al., 2010), IBS (Dinan et al., 2008; Liebregts et al., 2007; Rana et al., 2012), and pelvic pain (Liebregts et al., 2007; Lindenlaub and Sommer, 2003; Poole et al., 1999), such that higher levels are associated with greater pain (Gur et al., 2002; Kopp, 1998; Ogura et al., 2010; Shafer et al., 1994; Takahashi et al., 1998). While pro-inflammatory cytokines confer survival advantage in an acute setting by promoting immune responses that limit tissue damage and initiate remodeling (Bennett and Schultz, 1993; Dinarello et al., 1990; Gharaee-Kermani and Phan, 2001), persistent elevations result in tissue pathology and altered nociceptor function. TNFα, IL-1β, and IL-6 increase the activity of nociceptors by direct receptor-mediated actions as well as by inducing the transcription of pain-relevant genes that promote long-term synaptic plasticity (Dansereau et al., 2008; Gao et al., 2009; Jung et al., 2009; Kawasaki et al., 2008; Morales and Gereau, 2007; Oh et al., 2001; Sommer and Kress, 2004). Hyperactive nociceptors, in turn, secrete chemokines and other glial modulators from their central terminals in the spinal dorsal horn, leading to the activation of microglia and astrocytes (Huh et al., 2017; Tsuda et al., 2017).
In the present study, we found that systemic delivery of OR486 for 14 days led to β2- and β3AR-mediated increases in the expression of CD11b, a marker of activated microglia (Ponomarev et al., 2005), and GFAP, a marker of activated astrocytes (Venkatesh et al., 2013), in the spinal dorsal horn on day 14 and on day 21, 1 week following OR486 cessation. Microglia and astrocytes are glial cells known to regulate neuroinflammation and pain. Microglia are erythromyeloid-derived glial cells that serve as the resident macrophages of the spinal cord and brain (Kierdorf and Prinz, 2013). They detect adenosine triphosphate, chemokines, and proteases released from sensory afferents and respond by removing debris and resolving damage. Activated microglia promote inflammation and pain through increases in pro-inflammatory cytokines and growth factors that sensitize nociceptive neurons (Ji et al., 2016). Astrocytes are neuroectoderm-derived glial cells that regulate glutamate release and neurotransmission by adjusting concentrations of K+ and Ca++ (De Leo et al., 2006). Similar to microglia, astrocytes release cytokines and chemokines that increase the activity of nociceptive neurons. Injection of TNFα-activated astrocytes in the spinal cord of naïve mice results in mechanical allodynia that persists for days, thus demonstrating a direct causal role for astrocytes in persistent pain (Gao et al., 2010).
While the exact mechanisms through which microglia and astrocytes contribute to functional pain remain unknown, our data demonstrate that activation of spinal glia precedes increases in CSF pro-inflammatory cytokines. It is possible that the expression of TNFα and other pro-inflammatory cytokines was increased in spinal neurons and glial cells on days 14 and 21, with detection in CSF delayed until day 35. It is also possible that spinal microglia and astrocytes were activated through the release of pro-nociceptive molecules, such as ATP and reactive oxygen species (Basbaum et al., 2009; Sofroniew and Vinters, 2010). Following their stimulation, glia may promote pain transmission via release of inflammatory molecules (eg, cytokines and chemokines) as well as through release of gliotransmitters (eg, glutamate and adenosine triphosphate) that strengthen the responses of first and second order nociceptive neurons (Halassa et al., 2007).
4.3. Sustained stimulation of β2- and β3ARs leads to activation of MAPKs in nociceptors
MAPKs, including p38 and ERK, are intracellular signaling mediators expressed by nociceptors and glial cells that contribute to persistent pain and neuroinflammation following nerve injury or inflammation (Borges et al., 2015; Crown, 2012; Gao and Ji, 2008; Ji et al., 2009), and may also play a role in the chronification of functional pain.
We found that systemic delivery of OR486 for 14 days led to β2- and β3AR-mediated increases in neuronal p-p38 expression that remained high on day 21, 1 week following the cessation of OR486. In addition, systemic delivery of OR486 for 14 days led to β2- and β3AR-mediated increases in evoked pERK expression, predominantly in the superficial lamina of the spinal dorsal horn, on days 14 and 21.
The p38 and ERK MAPKs are activated by β2- and β3ARs, through Gi-dependent (Daaka et al., 1997; Schmitt and Stork, 2000; Soeder et al., 1999) and G protein-independent (Aley et al., 2001; Azzi et al., 2003; Cao et al., 2000) mechanisms, as well as by pro-inflammatory cytokines and other nociceptive mediators released from cells on which β2- and β3ARs reside (Ji et al., 2009; Takahashi et al., 2006; Willemen et al., 2010). Upon activation, p38 and ERK stimulate the transcription of pain-relevant genes and modify target proteins to drive peripheral and central sensitization (Chi et al., 2004; Grace et al., 2014; Ji et al., 2009; Koj, 1996; McMahon et al., 2005; Obata and Noguchi, 2004; Seger and Krebs, 1995). For example, inflammatory mediators, injury, and cellular stress activate p38 in DRG neurons, spinal neurons, and spinal microglia (Crown et al., 2008; Jin et al., 2003; Kumar et al., 2003; Moon et al., 2014; Takahashi et al., 2006; Willemen et al., 2010). Activated p38 then translocates to the nucleus and phosphorylates transcription factors (eg, cAMP response element-binding protein; CREB) to stimulate transcription of pro-inflammatory cytokines such as TNFα (Kumar et al., 2003). Activated p38 in spinal interneurons has also been shown to mediate TNFα –induced impairments in the inhibition of pain signals. Similar to p38, activation of ERK following inflammation or injury (Cheng et al., 2008; Gao and Ji, 2010; Ji et al., 2002; Ji et al., 2009; Song et al., 2005; Takahashi et al., 2006; Wei et al., 2006) results in upregulation of CREB-mediated transcription (Song et al., 2005) and glial production of pro-inflammatory molecules (Ji et al., 2009). In contrast to p38, ERK activation is more dynamic, requiring external stimulation (eg, mechanical pinch), and more specific, so that it is restricted to neurons in the superficial dorsal horn (Gao and Ji, 2009). pERK is also considered a marker of central sensitization, as expression levels are correlated with pain progression and chronicity (Ji et al., 2009; McMahon et al., 2005). Inhibition of either p38 or ERK is able to reduce cytokine production, glial activation, and pain (Ji et al., 2009; McMahon et al., 2005).
In line with these investigations, our data show that sustained administration of OR486 over a 14 day period leads to β2- and β3AR-mediated increases in p-p38 and pERK in spinal cord neurons, coincident with the activation of microglia and astrocytes in spinal cord and increased levels of pro-inflammatory cytokines in CSF. Thus, p-p38 and pERK represent downstream effectors of βARs that may contribute to the maintenance of persistent functional pain.
4.4. Contribution of βARs, TNFα, and MAPKs to the maintenance of functional pain
This study demonstrates that sustained stimulation of β2- and β3ARs produces functional pain that persists after removal of the causal stimulus, as well as corresponding increases in spinal pro-inflammatory cytokine production, glial activation, and MAPK phosphorylation. To determine the role of β2- and β3ARs and their downstream effectors (eg, TNFα and MAPKs) in the maintenance of functional pain, we evaluated the ability of specific inhibitors to alleviate pain after it had been established.
While co-administration of the β2AR antagonist ICI118,551 and the β3AR antagonist SR59230A blocked COMT-dependent pain and neuroinflammation when delivered on day 0, they did not reverse the pain when delivered on day 7. This finding suggests that peripheral β2- and β3ARs are required for the development, but not the maintenance of functional pain. Consistent with the behavioral results, co-administration of β2- and β3AR antagonists failed to alter COMT-dependent glial activation and MAPKs phosphorylation. Thus, downstream effectors that regulate central sensitization may be more critical for the maintenance of functional pain.
Indeed, we found that intrathecal administration of either the TNFα inhibitor Etanercept or the p38 inhibitor SB203580 on day 14 reversed COMT-dependent mechanical allodynia and hyperalgesia. Thus, TNFα, a key regulator of immune function, and p-p38, a stably expressed MAPK, are essential for the maintenance of functional pain. Etanercept-mediated analgesia on days 21-35 was likely due to inhibition of TNFα in spinal neurons and/or glial cells, as TNFα in CSF was not elevated until day 35. In contrast to TNFα and p38 inhibitors, intrathecal administration of the ERK inhibitor U0126 had no effect on mechanical hypersensitivity. The minimal contribution of pERK to functional pain may be due to the transient nature of its activation only in response to external stimuli.
While Etanercept and other TNFα antibodies have not been evaluated for the treatment of FPS, these anti-TNFα agents are used clinically for the treatment of inflammatory pain conditions, such as arthritis and inflammatory bowel disease (Li et al., 2017), and neuropathic pain conditions, such as lumbar disk hernia and sciatica (Freeman et al., 2013; Tobinick, 2009). However, 40% of patients do not respond to TNFα antibodies (Li et al., 2017). Among responders, long-term administration is associated with increased risk of infection and other adverse events related to off-target activity and antibody-induced immune sensitization (Curtis et al., 2014; Dixon et al., 2006; Feyen et al., 2008; Kroesen et al., 2003; Li et al., 2017). Thus, pharmacologic agents that selectively target intracellular elements within the TNFα signaling network may produce superior therapeutic outcomes for patients with chronic pain in the absence of side-effects associated with long-term antibody regimens.
In line with findings from studies of inflammatory and neuropathic pain (Latremoliere and Woolf, 2009), our results suggest that functional pain is maintained in the central nervous system long after the precipitating cause is removed. Sustained peripheral inflammation can produce neuroplastic changes in the spinal cord and brain, so as to increase central responses to peripheral stimuli. This phenomenon may explain why patients with FPS experience sensory abnormalities at regions remote from the original painful site.
5. Conclusions
Collectively, these findings illustrate the importance of β2- and β3ARs in driving neuroimmune signaling events that may underlie functional pain. The onset of COMT-dependent functional pain requires coincident activation of peripheral β2- and β3ARs, while the maintenance requires spinal TNFα and p-p38. Treatments that reduce neuroinflammation linked to abnormalities in catecholaminergic tone (eg, anti-TNFα agents) may prove useful in the management of FPS.
Supplementary Material
Highlights.
Sustained stimulation of β2- and β3ARs leads to persistent functional pain
The pain is accompanied by activation of glia and neuronal MAPKs in the spinal cord
Inhibition of spinal TNFα or p38 resolves functional pain
Acknowledgments
This work was funded by the NIH/NINDS R01 NS072205 and NIH/NINDS R03 NS106166 to A.N. Biomarker profiling was performed under the management of Dr. David Barrow at the University of North Carolina Cytokine Analysis Facility and under the direction of Dr. Andrew N. Macintyre and direction of Dr. Gregory D. Sempowski in the Immunology Unit of the Duke Regional Biocontainment Laboratory (RBL). The RBL received partial support for construction from the NIH/NIAID UC6-AI058607.
Funding Statement: This work was funded by the NIH/NINDS R01 NS072205, NIH/NINDS P01 NS045685, and NIH/NIDCR R56 DE025296 to A.N.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest: The authors declare no competing interests.
Data Statement
The authors of this manuscript will provide behavioral and immunohistochemical data upon request.
References
- Borges G, Berrocoso E, Mico JA, Neto F. ERK1/2: Function, signaling and implication in pain and pain-related anxio-depressive disorders. Progress in neuro-psychopharmacology & biological psychiatry. 2015;60:77–92. doi: 10.1016/j.pnpbp.2015.02.010. [DOI] [PubMed] [Google Scholar]
- Ciszek BP, O’Buckley SC, Nackley AG. Persistent Catechol-O-methyltransferase-dependent Pain Is Initiated by Peripheral beta-Adrenergic Receptors. Anesthesiology. 2016;124:1122–1135. doi: 10.1097/ALN.0000000000001070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crown ED. The role of mitogen activated protein kinase signaling in microglia and neurons in the initiation and maintenance of chronic pain. Experimental neurology. 2012;234:330–339. doi: 10.1016/j.expneurol.2011.10.019. [DOI] [PubMed] [Google Scholar]
- Crown ED, Gwak YS, Ye Z, Johnson KM, Hulsebosch CE. Activation of p38 MAP kinase is involved in central neuropathic pain following spinal cord injury. Experimental neurology. 2008;213:257–267. doi: 10.1016/j.expneurol.2008.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ, Ji RR. Activation of JNK pathway in persistent pain. Neuroscience letters. 2008;437:180–183. doi: 10.1016/j.neulet.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ, Ji RR. c-Fos and pERK, which is a better marker for neuronal activation and central sensitization after noxious stimulation and tissue injury? The open pain journal. 2009;2:11–17. doi: 10.2174/1876386300902010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ, Ji RR. Light touch induces ERK activation in superficial dorsal horn neurons after inflammation: involvement of spinal astrocytes and JNK signaling in touch-evoked central sensitization and mechanical allodynia. Journal of neurochemistry. 2010;115:505–514. doi: 10.1111/j.1471-4159.2010.06946.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosselin RD, Suter MR, Ji RR, Decosterd I. Glial cells and chronic pain. The Neuroscientist: a review journal bringing neurobiology, neurology and psychiatry. 2010;16:519–531. doi: 10.1177/1073858409360822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartung JE, Ciszek BP, Nackley AG. beta2- and beta3-adrenergic receptors drive COMT-dependent pain by increasing production of nitric oxide and cytokines. Pain. 2014;155:1346–1355. doi: 10.1016/j.pain.2014.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh Y, Ji RR, Chen G. Neuroinflammation, Bone Marrow Stem Cells, and Chronic Pain. Front Immunol. 2017;8:1014. doi: 10.3389/fimmu.2017.01014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Befort K, Brenner GJ, Woolf CJ. ERK MAP kinase activation in superficial spinal cord neurons induces prodynorphin and NK-1 upregulation and contributes to persistent inflammatory pain hypersensitivity. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2002;22:478–485. doi: 10.1523/JNEUROSCI.22-02-00478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Chamessian A, Zhang YQ. Pain regulation by non-neuronal cells and inflammation. Science. 2016;354:572–577. doi: 10.1126/science.aaf8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Gereau RWt, Malcangio M, Strichartz GR. MAP kinase and pain. Brain research reviews. 2009;60:135–148. doi: 10.1016/j.brainresrev.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Suter MR. p38 MAPK, microglial signaling, and neuropathic pain. Molecular pain. 2007;3:33. doi: 10.1186/1744-8069-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Xu ZZ, Gao YJ. Emerging targets in neuroinflammation-driven chronic pain. Nature reviews Drug discovery. 2014;13:533–548. doi: 10.1038/nrd4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SX, Zhuang ZY, Woolf CJ, Ji RR. p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2003;23:4017–4022. doi: 10.1523/JNEUROSCI.23-10-04017.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki Y, Zhang L, Cheng JK, Ji RR. Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2008;28:5189–5194. doi: 10.1523/JNEUROSCI.3338-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. The journal of pain: official journal of the American Pain Society. 2009;10:895–926. doi: 10.1016/j.jpain.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar NL, Murrell GA, McInnes IB. Inflammatory mechanisms in tendinopathy -towards translation. Nat Rev Rheumatol. 2017;13:110–122. doi: 10.1038/nrrheum.2016.213. [DOI] [PubMed] [Google Scholar]
- Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nature reviews Neuroscience. 2009;10:23–36. doi: 10.1038/nrn2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon JY, Roh DH, Yoon SY, Choi SR, Kwon SG, Choi HS, Kang SY, Han HJ, Beitz AJ, Oh SB, Lee JH. sigma1 receptors activate astrocytes via p38 MAPK phosphorylation leading to the development of mechanical allodynia in a mouse model of neuropathic pain. British journal of pharmacology. 2014;171:5881–5897. doi: 10.1111/bph.12893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nackley AG, Tan KS, Fecho K, Flood P, Diatchenko L, Maixner W. Catechol-O-methyltransferase inhibition increases pain sensitivity through activation of both beta2- and beta3-adrenergic receptors. Pain. 2007;128:199–208. doi: 10.1016/j.pain.2006.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanzello CR. Chemokines and inflammation in osteoarthritis: Insights from patients and animal models. J Orthop Res. 2017;35:735–739. doi: 10.1002/jor.23471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song XS, Cao JL, Xu YB, He JH, Zhang LC, Zeng YM. Activation of ERK/CREB pathway in spinal cord contributes to chronic constrictive injury-induced neuropathic pain in rats. Acta pharmacologica Sinica. 2005;26:789–798. doi: 10.1111/j.1745-7254.2005.00123.x. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Koga K, Chen T, Zhuo M. Neuronal and microglial mechanisms for neuropathic pain in the spinal dorsal horn and anterior cingulate cortex. Journal of neurochemistry. 2017;141:486–498. doi: 10.1111/jnc.14001. [DOI] [PubMed] [Google Scholar]
- Wei F, Vadakkan KI, Toyoda H, Wu LJ, Zhao MG, Xu H, Shum FW, Jia YH, Zhuo M. Calcium calmodulin-stimulated adenylyl cyclases contribute to activation of extracellular signal-regulated kinase in spinal dorsal horn neurons in adult rats and mice. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2006;26:851–861. doi: 10.1523/JNEUROSCI.3292-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xanthos DN, Sandkuhler J. Neurogenic neuroinflammation: inflammatory CNS reactions in response to neuronal activity. Nature reviews Neuroscience. 2014;15:43–53. doi: 10.1038/nrn3617. [DOI] [PubMed] [Google Scholar]
- Yang Y, Li H, Li TT, Luo H, Gu XY, Lu N, Ji RR, Zhang YQ. Delayed activation of spinal microglia contributes to the maintenance of bone cancer pain in female Wistar rats via P2X7 receptor and IL-18. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2015;35:7950–7963. doi: 10.1523/JNEUROSCI.5250-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Dougherty PM. Acute inhibition of signalling phenotype of spinal GABAergic neurons by tumour necrosis factor-alpha. The Journal of physiology. 2011;589:4511–4526. doi: 10.1113/jphysiol.2011.215301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang ZY, Gerner P, Woolf CJ, Ji RR. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain. 2005;114:149–159. doi: 10.1016/j.pain.2004.12.022. [DOI] [PubMed] [Google Scholar]
- Ciszek BP, O’Buckley SC, Nackley AG. Persistent Catechol-O-methyltransferase-dependent Pain Is Initiated by Peripheral beta-Adrenergic Receptors. Anesthesiology. 2016;124:1122–1135. doi: 10.1097/ALN.0000000000001070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosselin RD, Suter MR, Ji RR, Decosterd I. Glial cells and chronic pain. The Neuroscientist: a review journal bringing neurobiology, neurology and psychiatry. 2010;16:519–531. doi: 10.1177/1073858409360822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartung JE, Ciszek BP, Nackley AG. beta2- and beta3-adrenergic receptors drive COMT-dependent pain by increasing production of nitric oxide and cytokines. Pain. 2014;155:1346–1355. doi: 10.1016/j.pain.2014.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Suter MR. p38 MAPK, microglial signaling, and neuropathic pain. Molecular pain. 2007;3:33. doi: 10.1186/1744-8069-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline RHt, Exposto FG, O’Buckley SC, Westlund KN, Nackley AG. Catechol-O-methyltransferase inhibition alters pain and anxiety-related volitional behaviors through activation of beta-adrenergic receptors in the rat. Neuroscience. 2015;290:561–569. doi: 10.1016/j.neuroscience.2015.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. Journal of neuroscience methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Lynch HJ, Rivest RW, Wurtman RJ. Artificial induction of melatonin rhythms by programmed microinfusion. Neuroendocrinology. 1980;31:106–111. doi: 10.1159/000123059. [DOI] [PubMed] [Google Scholar]
- Aley KO, Martin A, McMahon T, Mok J, Levine JD, Messing RO. Nociceptor sensitization by extracellular signal-regulated kinases. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2001;21:6933–6939. doi: 10.1523/JNEUROSCI.21-17-06933.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzi M, Charest PG, Angers S, Rousseau G, Kohout T, Bouvier M, Pineyro G. Beta-arrestin-mediated activation of MAPK by inverse agonists reveals distinct active conformations for G protein-coupled receptors. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:11406–11411. doi: 10.1073/pnas.1936664100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284. doi: 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzichi L, Rossi A, Massimetti G, Giannaccini G, Giuliano T, De Feo F, Ciapparelli A, Dell’Osso L, Bombardieri S. Cytokine patterns in fibromyalgia and their correlation with clinical manifestations. Clin Exp Rheumatol. 2007;25:225–230. [PubMed] [Google Scholar]
- Bennett NT, Schultz GS. Growth factors and wound healing: biochemical properties of growth factors and their receptors. Am J Surg. 1993;165:728–737. doi: 10.1016/s0002-9610(05)80797-4. [DOI] [PubMed] [Google Scholar]
- Binshtok AM, Wang H, Zimmermann K, Amaya F, Vardeh D, Shi L, Brenner GJ, Ji RR, Bean BP, Woolf CJ, Samad TA. Nociceptors are interleukin-1β sensors. J Neurosci. 2008;28:14062–14073. doi: 10.1523/JNEUROSCI.3795-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bo SH, Davidsen EM, Gulbrandsen P, Dietrichs E, Bovim G, Stovner LJ, White LR. Cerebrospinal fluid cytokine levels in migraine, tension-type headache and cervicogenic headache. Cephalalgia: an international journal of headache. 2009;29:365–372. doi: 10.1111/j.1468-2982.2008.01727.x. [DOI] [PubMed] [Google Scholar]
- Borges G, Berrocoso E, Mico JA, Neto F. ERK1/2: Function, signaling and implication in pain and pain-related anxio-depressive disorders. Progress in neuro-psychopharmacology & biological psychiatry. 2015;60:77–92. doi: 10.1016/j.pnpbp.2015.02.010. [DOI] [PubMed] [Google Scholar]
- Bote ME, Garcia JJ, Hinchado MD, Ortega E. An exploratory study of the effect of regular aquatic exercise on the function of neutrophils from women with fibromyalgia: role of IL-8 and noradrenaline. Brain Behav Immun. 2014;39:107–112. doi: 10.1016/j.bbi.2013.11.009. [DOI] [PubMed] [Google Scholar]
- Cao W, Luttrell LM, Medvedev AV, Pierce KL, Daniel KW, Dixon TM, Lefkowitz RJ, Collins S. Direct binding of activated c-Src to the beta 3-adrenergic receptor is required for MAP kinase activation. The Journal of biological chemistry. 2000;275:38131–38134. doi: 10.1074/jbc.C000592200. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. Journal of neuroscience methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Cheng HT, Suzuki M, Hegarty DM, Xu Q, Weyerbacher AR, South SM, Ohata M, Inturrisi CE. Inflammatory pain-induced signaling events following a conditional deletion of the N-methyl-D-aspartate receptor in spinal cord dorsal horn. Neuroscience. 2008;155:948–958. doi: 10.1016/j.neuroscience.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi DS, Fitzgerald SM, Pitts S, Cantor K, King E, Lee SA, Huang SK, Krishnaswamy G. MAPK-dependent regulation of IL-1- and beta-adrenoreceptor-induced inflammatory cytokine production from mast cells: implications for the stress response. BMC immunology. 2004;5:22. doi: 10.1186/1471-2172-5-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarella SE, Soberanes S, Urich D, Morales-Nebreda L, Nigdelioglu R, Green D, Young JB, Gonzalez A, Rosario C, Misharin AV, Ghio AJ, Wunderink RG, Donnelly HK, Radigan KA, Perlman H, Chandel NS, Budinger GR, Mutlu GM. beta(2)-Adrenergic agonists augment air pollution-induced IL-6 release and thrombosis. J Clin Invest. 2014;124:2935–2946. doi: 10.1172/JCI75157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciszek BP, O’Buckley SC, Nackley AG. Persistent Catechol-O-methyltransferase-dependent Pain Is Initiated by Peripheral beta-Adrenergic Receptors. Anesthesiology. 2016;124:1122–1135. doi: 10.1097/ALN.0000000000001070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clauw DJ. Fibromyalgia: a clinical review. JAMA: the journal of the American Medical Association. 2014;311:1547–1555. doi: 10.1001/jama.2014.3266. [DOI] [PubMed] [Google Scholar]
- Crown ED. The role of mitogen activated protein kinase signaling in microglia and neurons in the initiation and maintenance of chronic pain. Experimental neurology. 2012;234:330–339. doi: 10.1016/j.expneurol.2011.10.019. [DOI] [PubMed] [Google Scholar]
- Crown ED, Gwak YS, Ye Z, Johnson KM, Hulsebosch CE. Activation of p38 MAP kinase is involved in central neuropathic pain following spinal cord injury. Exp Neurol. 2008;213:257–267. doi: 10.1016/j.expneurol.2008.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis JR, Yang S, Patkar NM, Chen L, Singh JA, Cannon GW, Mikuls TR, Delzell E, Saag KG, Safford MM, DuVall S, Alexander K, Napalkov P, Winthrop KL, Burton MJ, Kamauu A, Baddley JW. Risk of hospitalized bacterial infections associated with biologic treatment among US veterans with rheumatoid arthritis. Arthritis Care Res (Hoboken) 2014;66:990–997. doi: 10.1002/acr.22281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czeschik JC, Hagenacker T, Schafers M, Busselberg D. TNF-alpha differentially modulates ion channels of nociceptive neurons. Neuroscience letters. 2008;434:293–298. doi: 10.1016/j.neulet.2008.01.070. [DOI] [PubMed] [Google Scholar]
- Daaka Y, Luttrell LM, Lefkowitz RJ. Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase A. Nature. 1997;390:88–91. doi: 10.1038/36362. [DOI] [PubMed] [Google Scholar]
- Dansereau MA, Gosselin RD, Pohl M, Pommier B, Mechighel P, Mauborgne A, Rostene W, Kitabgi P, Beaudet N, Sarret P, Melik-Parsadaniantz S. Spinal CCL2 pronociceptive action is no longer effective in CCR2 receptor antagonist-treated rats. J Neurochem. 2008;106:757–769. doi: 10.1111/j.1471-4159.2008.05429.x. [DOI] [PubMed] [Google Scholar]
- De Leo JA, Tawfik VL, LaCroix-Fralish ML. The tetrapartite synapse: path to CNS sensitization and chronic pain. Pain. 2006;122:17–21. doi: 10.1016/j.pain.2006.02.034. [DOI] [PubMed] [Google Scholar]
- Della Vedova C, Cathcart S, Dohnalek A, Lee V, Hutchinson MR, Immink MA, Hayball J. Peripheral interleukin-1ss levels are elevated in chronic tension-type headache patients. Pain research & management: the journal of the Canadian Pain Society = journal de la societe canadienne pour le traitement de la douleur. 2013;18:301–306. doi: 10.1155/2013/796161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinan TG, Clarke G, Quigley EM, Scott LV, Shanahan F, Cryan J, Cooney J, Keeling PW. Enhanced cholinergic-mediated increase in the pro-inflammatory cytokine IL-6 in irritable bowel syndrome: role of muscarinic receptors. The American journal of gastroenterology. 2008;103:2570–2576. doi: 10.1111/j.1572-0241.2008.01871.x. [DOI] [PubMed] [Google Scholar]
- Dinarello CA, Kluger MJ, Oppenheim JJ, Powanda MC. The Physiological and Pathological Effects of Cytokines. John Wiley & Sons Inc; Liss, New York: 1990. [Google Scholar]
- Dixon WG, Watson K, Lunt M, Hyrich KL, Silman AJ, Symmons DP, British Society for Rheumatology Biologics, R Rates of serious infection, including site-specific and bacterial intracellular infection, in rheumatoid arthritis patients receiving anti-tumor necrosis factor therapy: results from the British Society for Rheumatology Biologics Register. Arthritis Rheum. 2006;54:2368–2376. doi: 10.1002/art.21978. [DOI] [PubMed] [Google Scholar]
- Dzau VJ, Pizzo PA. Relieving pain in America: insights from an Institute of Medicine committee. JAMA: the journal of the American Medical Association. 2014;312:1507–1508. doi: 10.1001/jama.2014.12986. [DOI] [PubMed] [Google Scholar]
- Evaskus DS, Laskin DM. A biochemical measure of stress in patients with myofascial pain-dysfunction syndrome. J Dent Res. 1972;51:1464–1466. doi: 10.1177/00220345720510053501. [DOI] [PubMed] [Google Scholar]
- Favaro-Moreira NC, Parada CA, Tambeli CH. Blockade of beta(1)-, beta(2)- and beta(3)-adrenoceptors in the temporomandibular joint induces antinociception especially in female rats. European journal of pain. 2012;16:1302–1310. doi: 10.1002/j.1532-2149.2012.00132.x. [DOI] [PubMed] [Google Scholar]
- Feyen O, Lueking A, Kowald A, Stephan C, Meyer HE, Gobel U, Niehues T. Off-target activity of TNF-alpha inhibitors characterized by protein biochips. Anal Bioanal Chem. 2008;391:1713–1720. doi: 10.1007/s00216-008-1938-7. [DOI] [PubMed] [Google Scholar]
- Freeman BJ, Ludbrook GL, Hall S, Cousins M, Mitchell B, Jaros M, Wyand M, Gorman JR. Randomized, double-blind, placebo-controlled, trial of transforaminal epidural etanercept for the treatment of symptomatic lumbar disc herniation. Spine (Phila Pa 1976) 2013;38:1986–1994. doi: 10.1097/01.brs.0000435140.61593.4c. [DOI] [PubMed] [Google Scholar]
- Fu L, Isobe K, Zeng Q, Suzukawa K, Takekoshi K, Kawakami Y. beta-adrenoceptor agonists downregulate adiponectin, but upregulate adiponectin receptor 2 and tumor necrosis factor-alpha expression in adipocytes. European journal of pharmacology. 2007;569:155–162. doi: 10.1016/j.ejphar.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Gao YJ, Ji RR. Activation of JNK pathway in persistent pain. Neuroscience letters. 2008;437:180–183. doi: 10.1016/j.neulet.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ, Ji RR. c-Fos and pERK, which is a better marker for neuronal activation and central sensitization after noxious stimulation and tissue injury? The open pain journal. 2009;2:11–17. doi: 10.2174/1876386300902010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ, Ji RR. Light touch induces ERK activation in superficial dorsal horn neurons after inflammation: involvement of spinal astrocytes and JNK signaling in touch-evoked central sensitization and mechanical allodynia. Journal of neurochemistry. 2010;115:505–514. doi: 10.1111/j.1471-4159.2010.06946.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ, Zhang L, Ji RR. Spinal injection of TNF-alpha-activated astrocytes produces persistent pain symptom mechanical allodynia by releasing monocyte chemoattractant protein-1. Glia. 2010;58:1871–1880. doi: 10.1002/glia.21056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ, Zhang L, Samad OA, Suter MR, Yasuhiko K, Xu ZZ, Park JY, Lind AL, Ma Q, Ji RR. JNK-induced MCP-1 production in spinal cord astrocytes contributes to central sensitization and neuropathic pain. J Neurosci. 2009;29:4096–4108. doi: 10.1523/JNEUROSCI.3623-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gharaee-Kermani M, Phan SH. Role of cytokines and cytokine therapy in wound healing and fibrotic diseases. Curr Pharm Des. 2001;7:1083–1103. doi: 10.2174/1381612013397573. [DOI] [PubMed] [Google Scholar]
- Gosselin RD, Suter MR, Ji RR, Decosterd I. Glial cells and chronic pain. The Neuroscientist: a review journal bringing neurobiology, neurology and psychiatry. 2010;16:519–531. doi: 10.1177/1073858409360822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace PM, Hutchinson MR, Maier SF, Watkins LR. Pathological pain and the neuroimmune interface. Nat Rev Immunol. 2014;14:217–231. doi: 10.1038/nri3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gur A, Karakoc M, Nas K, Remzi Cevik, Denli A, Sarac J. Cytokines and depression in cases with fibromyalgia. J Rheumatol. 2002;29:358–361. [PubMed] [Google Scholar]
- Halassa MM, Fellin T, Haydon PG. The tripartite synapse: roles for gliotransmission in health and disease. Trends in Molecular Medicine. 2007;13:54–63. doi: 10.1016/j.molmed.2006.12.005. [DOI] [PubMed] [Google Scholar]
- Hartung JE, Ciszek BP, Nackley AG. beta2- and beta3-adrenergic receptors drive COMT-dependent pain by increasing production of nitric oxide and cytokines. Pain. 2014;155:1346–1355. doi: 10.1016/j.pain.2014.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh Y, Ji RR, Chen G. Neuroinflammation, Bone Marrow Stem Cells, and Chronic Pain. Front Immunol. 2017;8:1014. doi: 10.3389/fimmu.2017.01014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Befort K, Brenner GJ, Woolf CJ. ERK MAP kinase activation in superficial spinal cord neurons induces prodynorphin and NK-1 upregulation and contributes to persistent inflammatory pain hypersensitivity. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2002;22:478–485. doi: 10.1523/JNEUROSCI.22-02-00478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Chamessian A, Zhang YQ. Pain regulation by non-neuronal cells and inflammation. Science. 2016;354:572–577. doi: 10.1126/science.aaf8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Gereau RWt, Malcangio M, Strichartz GR. MAP kinase and pain. Brain research reviews. 2009;60:135–148. doi: 10.1016/j.brainresrev.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Suter MR. p38 MAPK, microglial signaling, and neuropathic pain. Molecular pain. 2007;3:33. doi: 10.1186/1744-8069-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SX, Zhuang ZY, Woolf CJ, Ji RR. p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2003;23:4017–4022. doi: 10.1523/JNEUROSCI.23-10-04017.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H, Bhangoo S, Banisadr G, Freitag C, Ren D, White FA, Miller RJ. Visualization of chemokine receptor activation in transgenic mice reveals peripheral activation of CCR2 receptors in states of neuropathic pain. J Neurosci. 2009;29:8051–8062. doi: 10.1523/JNEUROSCI.0485-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kambur O, Talka R, Ansah OB, Kontinen VK, Pertovaara A, Kalso E, Mannisto PT. Inhibitors of catechol-O-methyltransferase sensitize mice to pain. Br J Pharmacol. 2010;161:1553–1565. doi: 10.1111/j.1476-5381.2010.00999.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneyama K, Segami N, Nishimura M, Suzuki T, Sato J. Importance of proinflammatory cytokines in synovial fluid from 121 joints with temporomandibular disorders. Br J Oral Maxillofac Surg. 2002;40:418–423. [PubMed] [Google Scholar]
- Kanno T, Yaguchi T, Nishizaki T. Noradrenaline stimulates ATP release from DRG neurons by targeting beta(3) adrenoceptors as a factor of neuropathic pain. Journal of cellular physiology. 2010;224:345–351. doi: 10.1002/jcp.22114. [DOI] [PubMed] [Google Scholar]
- Kawasaki Y, Zhang L, Cheng JK, Ji RR. Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2008;28:5189–5194. doi: 10.1523/JNEUROSCI.3338-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasar SG, Green PG, Miao FJ, Levine JD. Vagal modulation of nociception is mediated by adrenomedullary epinephrine in the rat. Eur J Neurosci. 2003;17:909–915. doi: 10.1046/j.1460-9568.2003.02503.x. [DOI] [PubMed] [Google Scholar]
- Khasar SG, Lin YH, Martin A, Dadgar J, McMahon T, Wang D, Hundle B, Aley KO, Isenberg W, McCarter G, Green PG, Hodge CW, Levine JD, Messing RO. A novel nociceptor signaling pathway revealed in protein kinase C epsilon mutant mice. Neuron. 1999a;24:253–260. doi: 10.1016/s0896-6273(00)80837-5. [DOI] [PubMed] [Google Scholar]
- Khasar SG, McCarter G, Levine JD. Epinephrine produces a beta-adrenergic receptor-mediated mechanical hyperalgesia and in vitro sensitization of rat nociceptors. Journal of neurophysiology. 1999b;81:1104–1112. doi: 10.1152/jn.1999.81.3.1104. [DOI] [PubMed] [Google Scholar]
- Kierdorf K, Prinz M. Factors regulating microglia activation. Frontiers in cellular neuroscience. 2013;7:44. doi: 10.3389/fncel.2013.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MH, Gorouhi F, Ramirez S, Granick JL, Byrne BA, Soulika AM, Simon SI, Isseroff RR. Catecholamine stress alters neutrophil trafficking and impairs wound healing by beta2-adrenergic receptor-mediated upregulation of IL-6. J Invest Dermatol. 2014;134:809–817. doi: 10.1038/jid.2013.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline RHt, Exposto FG, O’Buckley SC, Westlund KN, Nackley AG. Catechol-O-methyltransferase inhibition alters pain and anxiety-related volitional behaviors through activation of beta-adrenergic receptors in the rat. Neuroscience. 2015;290:561–569. doi: 10.1016/j.neuroscience.2015.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocer A, Kocer E, Memisogullari R, Domac FM, Yuksel H. Interleukin-6 levels in tension headache patients. Clin J Pain. 2010;26:690–693. doi: 10.1097/AJP.0b013e3181e8d9b6. [DOI] [PubMed] [Google Scholar]
- Koj A. Initiation of acute phase response and synthesis of cytokines. Biochimica et biophysica acta. 1996;1317:84–94. doi: 10.1016/s0925-4439(96)00048-8. [DOI] [PubMed] [Google Scholar]
- Kopp S. The influence of neuropeptides, serotonin, and interleukin 1beta on temporomandibular joint pain and inflammation. J Oral Maxillofac Surg. 1998;56:189–191. doi: 10.1016/s0278-2391(98)90867-9. [DOI] [PubMed] [Google Scholar]
- Kralisch S, Klein J, Lossner U, Bluher M, Paschke R, Stumvoll M, Fasshauer M. Isoproterenol stimulates monocyte chemoattractant protein-1 expression and secretion in 3T3-L1 adipocytes. Regulatory peptides. 2006;135:12–16. doi: 10.1016/j.regpep.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Kress M, Sommer C. Neuroimmunology and Pain: Peripheral Effects of Proinflammatory Cytokines. In: Brune K, Handwerker HO, editors. Hyperalgesia: Molecular Mechanisms and Clinical Implications. IASP Press; 2004. pp. 57–65. [Google Scholar]
- Kroesen S, Widmer AF, Tyndall A, Hasler P. Serious bacterial infections in patients with rheumatoid arthritis under anti-TNF-alpha therapy. Rheumatology (Oxford) 2003;42:617–621. doi: 10.1093/rheumatology/keg263. [DOI] [PubMed] [Google Scholar]
- Kumar S, Boehm J, Lee JC. p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nature reviews Drug discovery. 2003;2:717–726. doi: 10.1038/nrd1177. [DOI] [PubMed] [Google Scholar]
- Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. The journal of pain: official journal of the American Pain Society. 2009;10:895–926. doi: 10.1016/j.jpain.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laukova M, Vargovic P, Csaderova L, Chovanova L, Vlcek M, Imrich R, Krizanova O, Kvetnansky R. Acute stress differently modulates beta1, beta2 and beta3 adrenoceptors in T cells, but not in B cells, from the rat spleen. Neuroimmunomodulation. 2012;19:69–78. doi: 10.1159/000329002. [DOI] [PubMed] [Google Scholar]
- Li P, Zheng Y, Chen X. Drugs for Autoimmune Inflammatory Diseases: From Small Molecule Compounds to Anti-TNF Biologics. Frontiers in pharmacology. 2017;8:460. doi: 10.3389/fphar.2017.00460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebregts T, Adam B, Bredack C, Roth A, Heinzel S, Lester S, Downie-Doyle S, Smith E, Drew P, Talley NJ, Holtmann G. Immune activation in patients with irritable bowel syndrome. Gastroenterology. 2007;132:913–920. doi: 10.1053/j.gastro.2007.01.046. [DOI] [PubMed] [Google Scholar]
- Lindenlaub T, Sommer C. Cytokines in sural nerve biopsies from inflammatory and non-inflammatory neuropathies. Acta Neuropathol. 2003;105:593–602. doi: 10.1007/s00401-003-0689-y. [DOI] [PubMed] [Google Scholar]
- Lynch HJ, Rivest RW, Wurtman RJ. Artificial induction of melatonin rhythms by programmed microinfusion. Neuroendocrinology. 1980;31:106–111. doi: 10.1159/000123059. [DOI] [PubMed] [Google Scholar]
- Maixner W, Fillingim RB, Williams DA, Smith SB, Slade GD. Overlapping Chronic Pain Conditions: Implications for Diagnosis and Classification. J Pain. 2016;17:T93–t107. doi: 10.1016/j.jpain.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannisto PT, Kaakkola S. Catechol-O-methyltransferase (COMT): biochemistry, molecular biology, pharmacology, and clinical efficacy of the new selective COMT inhibitors. Pharmacol Rev. 1999;51:593–628. [PubMed] [Google Scholar]
- Marbach JJ, Levitt M. Erythrocyte catechol-O-methyltransferase activity in facial pain patients. Journal of dental research. 1976;55:711. doi: 10.1177/00220345760550043801. [DOI] [PubMed] [Google Scholar]
- Matsumoto K, Honda K, Ohshima M, Yamaguchi Y, Nakajima I, Micke P, Otsuka K. Cytokine profile in synovial fluid from patients with internal derangement of the temporomandibular joint: a preliminary study. Dentomaxillofac Radiol. 2006;35:432–441. doi: 10.1259/dmfr/77288976. [DOI] [PubMed] [Google Scholar]
- McMahon SB, Cafferty WB, Marchand F. Immune and glial cell factors as pain mediators and modulators. Experimental neurology. 2005;192:444–462. doi: 10.1016/j.expneurol.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Miller RJ, Jung H, Bhangoo SK, White FA. Cytokine and chemokine regulation of sensory neuron function. Handb Exp Pharmacol. 2009:417–449. doi: 10.1007/978-3-540-79090-7_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed-Ali V, Flower L, Sethi J, Hotamisligil G, Gray R, Humphries SE, York DA, Pinkney J. beta-Adrenergic regulation of IL-6 release from adipose tissue: in vivo and in vitro studies. J Clin Endocrinol Metab. 2001;86:5864–5869. doi: 10.1210/jcem.86.12.8104. [DOI] [PubMed] [Google Scholar]
- Moon JY, Roh DH, Yoon SY, Choi SR, Kwon SG, Choi HS, Kang SY, Han HJ, Beitz AJ, Oh SB, Lee JH. sigma1 receptors activate astrocytes via p38 MAPK phosphorylation leading to the development of mechanical allodynia in a mouse model of neuropathic pain. Br J Pharmacol. 2014;171:5881–5897. doi: 10.1111/bph.12893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales MEP, Gereau RWI. Cytokine regulation of ion channels in the pain pathway. In: deLeo JA, Sorkin LS, Watkins LR, editors. Immune and Glial Regulation of Pain. IASP Press; Seattle, WA: 2007. pp. 67–83. [Google Scholar]
- Nackley AG, Tan KS, Fecho K, Flood P, Diatchenko L, Maixner W. Catechol-O-methyltransferase inhibition increases pain sensitivity through activation of both beta2- and beta3-adrenergic receptors. Pain. 2007;128:199–208. doi: 10.1016/j.pain.2006.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata K, Noguchi K. MAPK activation in nociceptive neurons and pain hypersensitivity. Life sciences. 2004;74:2643–2653. doi: 10.1016/j.lfs.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Obreja O, Biasio W, Andratsch M, Lips KS, Rathee PK, Ludwig A, Rose-John S, Kress M. Fast modulation of heat-activated ionic current by proinflammatory interleukin 6 in rat sensory neurons. Brain. 2005;128:1634–1641. doi: 10.1093/brain/awh490. [DOI] [PubMed] [Google Scholar]
- Obreja O, Rathee PK, Lips KS, Distler C, Kress M. IL-1B potentiates heat-activated currents in rat sensory neurons: involvement of IL-1RI, tyrosine kinase, and protein kinase C. FASEB. 2002;16:1497–1503. doi: 10.1096/fj.02-0101com. [DOI] [PubMed] [Google Scholar]
- Ogura N, Satoh K, Akutsu M, Tobe M, Kuyama K, Kuboyama N, Sakamaki H, Kujiraoka H, Kondoh T. MCP-1 production in temporomandibular joint inflammation. J Dent Res. 2010;89:1117–1122. doi: 10.1177/0022034510376041. [DOI] [PubMed] [Google Scholar]
- Oh SB, Tran PB, Gillard SE, Hurley RW, Hammond DL, Miller RJ. Chemokines and glycoprotein120 produce pain hypersensitivity by directly exciting primary nociceptive neurons. J Neurosci. 2001;21:5027–5035. doi: 10.1523/JNEUROSCI.21-14-05027.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry F, Heller PH, Kamiya J, Levine JD. Altered autonomic function in patients with arthritis or with chronic myofascial pain. Pain. 1989;39:77–84. doi: 10.1016/0304-3959(89)90177-2. [DOI] [PubMed] [Google Scholar]
- Ponomarev ED, Shriver LP, Maresz K, Dittel BN. Microglial cell activation and proliferation precedes the onset of CNS autoimmunity. J Neurosci Res. 2005;81:374–389. doi: 10.1002/jnr.20488. [DOI] [PubMed] [Google Scholar]
- Poole S, Lorenzetti BB, Cunha JM, Cunha FQ, Ferreira SH. Bradykinin B1 and B2 receptors, tumour necrosis factor alpha and inflammatory hyperalgesia. Br J Pharmacol. 1999;126:649–656. doi: 10.1038/sj.bjp.0702347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana SV, Sharma S, Sinha SK, Parsad KK, Malik A, Singh K. Pro-inflammatory and anti-inflammatory cytokine response in diarrhoea-predominant irritable bowel syndrome patients. Tropical gastroenterology: official journal of the Digestive Diseases Foundation. 2012;33:251–256. doi: 10.7869/tg.2012.66. [DOI] [PubMed] [Google Scholar]
- Rey E, Talley NJ. Irritable bowel syndrome: novel views on the epidemiology and potential risk factors. Dig Liver Dis. 2009;41:772–780. doi: 10.1016/j.dld.2009.07.005. [DOI] [PubMed] [Google Scholar]
- Schmitt JM, Stork PJ. beta 2-adrenergic receptor activates extracellular signal-regulated kinases (ERKs) via the small G protein rap1 and the serine/threonine kinase B-Raf. The Journal of biological chemistry. 2000;275:25342–25350. doi: 10.1074/jbc.M003213200. [DOI] [PubMed] [Google Scholar]
- Seger R, Krebs EG. The MAPK signaling cascade. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 1995;9:726–735. [PubMed] [Google Scholar]
- Shafer DM, Assael L, White LB, Rossomando EF. Tumor necrosis factor-alpha as a biochemical marker of pain and outcome in temporomandibular joints with internal derangements. J Oral Maxillofac Surg. 1994;52:786–791. doi: 10.1016/0278-2391(94)90217-8. discussion 791-782. [DOI] [PubMed] [Google Scholar]
- Slade GD, Bair E, Greenspan JD, Dubner R, Fillingim RB, Diatchenko L, Maixner W, Knott C, Ohrbach R. Signs and symptoms of first-onset TMD and sociodemographic predictors of its development: the OPPERA prospective cohort study. J Pain. 2013;14:T20–32. e21–23. doi: 10.1016/j.jpain.2013.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slade GD, Conrad MS, Diatchenko L, Rashid NU, Zhong S, Smith S, Rhodes J, Medvedev A, Makarov S, Maixner W, Nackley AG. Cytokine biomarkers and chronic pain: association of genes, transcription, and circulating proteins with temporomandibular disorders and widespread palpation tenderness. Pain. 2011;152:2802–2812. doi: 10.1016/j.pain.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slota C, Shi A, Chen G, Bevans M, Weng NP. Norepinephrine preferentially modulates memory CD8 T cell function inducing inflammatory cytokine production and reducing proliferation in response to activation. Brain, behavior, and immunity. 2015;46:168–179. doi: 10.1016/j.bbi.2015.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SB, Reenila I, Mannisto PT, Slade GD, Maixner W, Diatchenko L, Nackley AG. Epistasis Between Polymorphisms in COMT, ESR1, and GCH1 Influences COMT Enzyme Activity and Pain. Pain. 2014 doi: 10.1016/j.pain.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soeder KJ, Snedden SK, Cao W, Della Rocca GJ, Daniel KW, Luttrell LM, Collins S. The beta3-adrenergic receptor activates mitogen-activated protein kinase in adipocytes through a Gi-dependent mechanism. The Journal of biological chemistry. 1999;274:12017–12022. doi: 10.1074/jbc.274.17.12017. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer C, Hauser W, Gerhold K, Joraschky P, Petzke F, Tolle T, Uceyler N, Winkelmann A, Thieme K. Etiology and pathophysiology of fibromyalgia syndrome and chronic widespread pain. Schmerz. 2008;22:267–282. doi: 10.1007/s00482-008-0672-6. [DOI] [PubMed] [Google Scholar]
- Sommer C, Kress M. Recent findings on how proinflammatory cytokines cause pain: peripheral mechanisms in inflammatory and neuropathic hyperalgesia. Neurosci Lett. 2004;361:184–187. doi: 10.1016/j.neulet.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Song XS, Cao JL, Xu YB, He JH, Zhang LC, Zeng YM. Activation of ERK/CREB pathway in spinal cord contributes to chronic constrictive injury-induced neuropathic pain in rats. Acta pharmacologica Sinica. 2005;26:789–798. doi: 10.1111/j.1745-7254.2005.00123.x. [DOI] [PubMed] [Google Scholar]
- Takahashi N, Kikuchi S, Shubayev VI, Campana WM, Myers RR. TNF-alpha and phosphorylation of ERK in DRG and spinal cord: insights into mechanisms of sciatica. Spine. 2006;31:523–529. doi: 10.1097/01.brs.0000201305.01522.17. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Kondoh T, Fukuda M, Yamazaki Y, Toyosaki T, Suzuki R. Proinflammatory cytokines detectable in synovial fluids from patients with temporomandibular disorders. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1998;85:135–141. doi: 10.1016/s1079-2104(98)90415-2. [DOI] [PubMed] [Google Scholar]
- Tobinick E. Perispinal etanercept for neuroinflammatory disorders. Drug Discov Today. 2009;14:168–177. doi: 10.1016/j.drudis.2008.10.005. [DOI] [PubMed] [Google Scholar]
- Torpy DJ, Papanicolaou DA, Lotsikas AJ, Wilder RL, Chrousos GP, Pillemer SR. Responses of the sympathetic nervous system and the hypothalamic-pituitary-adrenal axis to interleukin-6: a pilot study in fibromyalgia. Arthritis and rheumatism. 2000;43:872–880. doi: 10.1002/1529-0131(200004)43:4<872::AID-ANR19>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Tsang A, Von Korff M, Lee S, Alonso J, Karam E, Angermeyer MC, Borges GL, Bromet EJ, Demytteneare K, de Girolamo G, de Graaf R, Gureje O, Lepine JP, Haro JM, Levinson D, Oakley Browne MA, Posada-Villa J, Seedat S, Watanabe M. Common chronic pain conditions in developed and developing countries: gender and age differences and comorbidity with depression-anxiety disorders. The journal of pain: official journal of the American Pain Society. 2008;9:883–891. doi: 10.1016/j.jpain.2008.05.005. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Koga K, Chen T, Zhuo M. Neuronal and microglial mechanisms for neuropathic pain in the spinal dorsal horn and anterior cingulate cortex. Journal of neurochemistry. 2017;141:486–498. doi: 10.1111/jnc.14001. [DOI] [PubMed] [Google Scholar]
- Venkatesh K, Srikanth L, Vengamma B, Chandrasekhar C, Sanjeevkumar A, Mouleshwara Prasad BC, Sarma PV. In vitro differentiation of cultured human CD34+ cells into astrocytes. Neurology India. 2013;61:383–388. doi: 10.4103/0028-3886.117615. [DOI] [PubMed] [Google Scholar]
- Wallace DJ, Linker-Israeli M, Hallegua D, Silverman S, Silver D, Weisman MH. Cytokines play an aetiopathogenetic role in fibromyalgia: a hypothesis and pilot study. Rheumatology (Oxford) 2001;40:743–749. doi: 10.1093/rheumatology/40.7.743. [DOI] [PubMed] [Google Scholar]
- Wei F, Vadakkan KI, Toyoda H, Wu LJ, Zhao MG, Xu H, Shum FW, Jia YH, Zhuo M. Calcium calmodulin-stimulated adenylyl cyclases contribute to activation of extracellular signal-regulated kinase in spinal dorsal horn neurons in adult rats and mice. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2006;26:851–861. doi: 10.1523/JNEUROSCI.3292-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesselmann U, Bonham A, Foster D. Vulvodynia: Current state of the biological science. Pain. 2014;155:1696–1701. doi: 10.1016/j.pain.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willemen HL, Eijkelkamp N, Wang H, Dantzer R, Dorn GW, 2nd, Kelley KW, Heijnen CJ, Kavelaars A. Microglial/macrophage GRK2 determines duration of peripheral IL-1beta-induced hyperalgesia: contribution of spinal cord CX3CR1, p38 and IL-1 signaling. Pain. 2010;150:550–560. doi: 10.1016/j.pain.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ. Evidence for a central component of post-injury pain hypersensitivity. Nature. 1983;306:686–688. doi: 10.1038/306686a0. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- Zhang C, Rui YY, Zhou YY, Ju Z, Zhang HH, Hu CY, Xiao Y, Xu GY. Adrenergic beta2-receptors mediates visceral hypersensitivity induced by heterotypic intermittent stress in rats. PLoS One. 2014;9:e94726. doi: 10.1371/journal.pone.0094726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Lu Y, Zhang G, Gao S, Sun D, Zhong Y. Bupivacaine-loaded biodegradable poly(lactic-co-glycolic) acid microspheres I. Optimization of the drug incorporation into the polymer matrix and modelling of drug release. International journal of pharmaceutics. 2008;351:244–249. doi: 10.1016/j.ijpharm.2007.10.004. [DOI] [PubMed] [Google Scholar]
- Zhang X, Kim S, O’Buckley S, Ballard H, Nackley AG. 2018 APS Scientific Summit. American Pain Society; Anaheim, CA: 2018. Activation of peripheral β2- and β3ARs leads to increased nociceptor activity. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.