

Abstract
Cartilage oligomeric matrix protein (COMP) is a large pentameric glycoprotein that interacts with multiple extracellular matrix proteins in cartilage and other tissues. While, COMP is known to play a role in collagen secretion and fibrillogenesis, chondrocyte proliferation and mechanical strength of tendons, the complete range of COMP functions remains to be defined. COMPopathies describe pseudoachondroplasia (PSACH) and multiple epiphyseal dysplasia (MED), two skeletal dysplasias caused by autosomal dominant COMP mutations. The majority of the mutations are in the calcium binding domains and compromise protein folding. COMPopathies are ER storage disorders in which the retention of COMP in the chondrocyte ER stimulates overwhelming cellular stress. The retention causes oxidative and inflammation processes leading to chondrocyte death and loss of long bone growth. In contrast, dysregulation of wild-type COMP expression is found in numerous diseases including: fibrosis, cardiomyopathy and breast and prostate cancers. The most exciting clinical application is the use of COMP as a biomarker for idiopathic pulmonary fibrosis and cartilage degeneration associated osteoarthritis and rheumatoid and, as a prognostic marker for joint injury. The ever expanding roles of COMP in single gene disorders and multifactorial diseases will lead to a better understanding of its functions in ECM and tissue homeostasis towards the goal of developing new therapeutic avenues.
Keywords: COMP, Pseudoachondroplasia, ER stress, Fibrosis, Adaptor protein and Biomarker
Cartilage oligomeric matrix protein (COMP) – a multifaceted thrombospondin
Cartilage oligomeric matrix protein (COMP) is an extracellular matrix (ECM) glycoprotein, originally isolated from and characterized in cartilage, but later shown to be expressed in a wide variety of tissues: fibroblasts, tendons, ligaments, synovium, vascular smooth cells, myofibroblasts, breast cancer cells, cardiomyocytes and activated platelets [1-10]. COMP, also known as TSP5, is a member of the pentameric subgroup of the thrombospondin gene family, a family of secreted matricellular proteins with diverse functions and common structural organization (Fig. 1) [11-13]. Like other thrombospondins, COMP assembles through its N-terminal domain (NTD). It is a pentameric protein, as are TSP3 and 4 (all belong to subgroup B) and differs from TSP1 and 2, which are trimeric proteins (subgroup A) [14]. As shown in Fig. 1, each monomeric arm contains an N-terminal domain, four type 2 (T2) epidermal growth factorlike repeats, seven type 3 (T3) repeats and a C-terminal globular domain (CTD). The T2 and T3 repeats and CTD bind calcium ions critical to correct protein folding [15, 16]. The T3 repeats contain highly conserved aspartic acid rich motifs, usually containing five aspartic acid residues per motif, with each motif wrapping around two calcium ions. The CTD is a lectin-like-β sandwich domain composed of 15 β-strands and also containing four calcium binding sites [15, 16]. T3 repeats fold together with the CTD to form a tertiary structure called the signature domain that is characteristic of all TSPs and which promotes correct folding and secretion.
Figure 1. Schematic depicting COMP protein domains.

Most COMP mutations are in the T3 repeats with only a few found in the CTD. No mutations have been identified in T2 and NTD domains. NTD = N-terminal domain, T2 = type two repeat, T3 = type 3 repeat, CTD = C-terminal domain.
TSPs are matricellular proteins which mediate cell-matrix and matrix-matrix interactions [17]. Wound healing is enhanced by TSP1 pro-inflammatory activity and inhibited by TSP2 anti-angiogenic effects [17, 18]. Smooth muscle cell (SMC) migration, proliferation and adhesion is supported by TSP1 [19-21]; however, TSP1 inhibits these activities in the presence of elevated nitric oxide (NO) levels [22, 23]. Both TSP1 and 2 inhibit NO-driven proliferation [22], while low levels of NO suppress TSP1 production resulting in a proangiogenic environment [24]. The relationship between TSPs and NO in epithelial cells is complex and dependent on NO concentrations [17]. In muscle cells, TSP1 modulates secretion of type 1 collagen, which affects outgrowth and proliferation of endothelial cells and the migratory capacity of SMCs [25]. In addition to regulating SMCs, TSP1 and TSP4 promote neurite outgrowth [26, 27] and this activity is driven by TSP interaction with multiprotein complexes on neuronal cell surfaces [28].
TSP cell-ECM interactions are numerous in cartilage and bone tissues and regulate mineralization and inflammation; ECM protein interactions are important for protein assembly into matrix [29]. TSP2 promotes preosteoblast mineralization of tissues by facilitating organization of the osteoblast-derived ECM [30], whereas TSP1 inhibits mineralization by osteoblasts in a dose dependent manner in vivo and in vitro [31, 32].
COMP/TSP5, as an extracellular matrix protein secreted by chondrocytes, interacts with other ECM proteins including, but not limited to, collagen types I, II, IX, XII, XIV, matrilin-3, aggrecan and fibronectin [33-37]. The presence of both COMP and type IX collagen are important for incorporation of matrilin-3 into the ECM [38]. COMP's interactions with types II and IX collagens and matrilin-3 play a significant role in the pseudoachondroplasia chondrocyte pathology described below. Interactions with granulin-epithelin precursor (GEP) [39] and metalloproteinases (MMP) MMP-9, MMP-19 and MMP-20 has also been demonstrated [40, 41].
Functional information known about COMP comes from disorders caused either by COMP mutations or, more recently, by the recognition that altered regulation of COMP contributes to disease pathology. While the mechanism(s) causing dysregulation of COMP is not known, the presence of COMP in various disease states suggest important role(s).
Putative COMP functions
Chondrocyte proliferation
COMP has been shown to stimulate chondrocyte proliferation and chondrogenesis [39, 42]. Chondrocyte proliferation occurs through the interaction of the EGF domain of COMP with GEP, since blocking this interaction decreased proliferation [39].
Thrombin inhibition
Thrombin plays an essential role in clot formation by activating platelets and converting fibrinogen in fibrin. Hemostasis is facilitated by gathering activated platelets in blood vessels at the injury site. Thrombosis occurs when fibrin is deposited at the site of injury enmeshing the platelets and forming a blood clot [43]. TSP1 enhances coagulation by promoting platelet adhesion, activation and aggregation through interaction with platelet receptors. TSP1 represents the most abundant protein in the platelets and is released following platelet activation [10]. COMP is stored in platelet α-granules, released and synthesized following platelet activation [44]. COMP binds thrombin slowing clotting by inhibiting thrombin- induced platelet aggregation, activation, and retraction and the thrombin-mediated cleavage of fibrinogen [10]. Consistent with COMP binding to and inhibiting thrombin, injured COMP null mice show accelerated clot formation and shorter bleeding time [10]. The anticoagulant properties of COMP should be assessed as a potential treatment for acute coronary artery diseases or thrombotic disorders that are routinely treated with anticoagulants.
Mechanical stress resistance
Tendon equine studies show that COMP plays an important role in mechanical integrity. COMP is more abundant in weight bearing tendons compared to unloaded tendons, indicating that COMP may enhance the mechanical strength of ECM tissues [36]. Horse studies show that while foal tendons express very little COMP, mechanical loading increases expression with maturity [36, 45]. COMP levels in tendon decline with age and over use and this loss predisposes to equine tendonitis [36, 45]. COMP predominantly associates in tendons with small collagen fibrils specifically at the gap region of the fibril [46] and this structural reinforcement of the gap is likely how COMP increases tendon mechanical strength.
Compopathies
Chondrodysplasias are caused by mutations in COMP
The cartilage-specific functions of COMP have been extensively studied since it was initially thought to be a cartilage-specific protein and because COMP mutations cause two autosomal dominant skeletal dysplasias, pseudoachondroplasia (PSACH,) a severe dwarfing condition and multiple epiphyseal dysplasia (MED), a milder short stature disorder [47-51]. The clinical severity/variability of these disorders ranges from severe to mild and are designated COMPopathies. Whereas all cases of PSACH are cause by mutations in COMP, only 66% of MED cases result from COMP mutations and mutation prevalence doesn't vary with ethnicity or sex [52, 53]. In the remaining MED cases, mutations in COL9A1, COL9A2, COL9A3 and MATN3 have been described, which when compared to COMP-MED, have a later age of onset and affect the major joints without brachydactyly [51, 54-56]. The presence of brachydactyly is a distinguishing finding common only to PSACH and MED-caused by COMP mutations. Importantly, because all PSACH and MED birth parameters are normal, the diagnoses are made between 1-2 years and about 5 years of age, respectively [49, 57].
PSACH is at the severe end of the dwarfism spectrum and is associated with significant limb shortening. The nosologic classification is spondyloepiphyseal dysplasia type pseudoachondroplasia reflecting the involvement of both spine and limb abnormalities [58]. PSACH babies appear normal at birth with the first signs of decelerating linear growth starting by the end of the first year and a waddling gait developing by two years of age. The clinical signs and radiographic findings of long bone shortening, widened and irregular metaphyses, small underossified femoral epiphyses, and platyspondyly assist in the diagnosis [51, 54-56]. Diminished linear growth occurs during the first 8 years resulting in an adult height equivalent to that of an average 6-8 year-old [59]. The face is attractive and has a distinctive and characteristic angular appearance. All joints are affected and are broad and extremely lax. Pain is a significant complication beginning in childhood, likely from the underlying chondrocyte growth plate pathology (described below); whereas, the pain in adulthood reflects osteoarthritic changes that necessitate hip replacement in a majority of adults [57]. MED, the milder skeletal dysplasia, also has characteristic epiphyseal abnormalities and joint pain starting in childhood [51, 53]. The MED facies are also attractive, which aids in the diagnosis; precocious osteoarthritis (OA) especially affecting the hips necessitates hip replacement in early adulthood [49, 57]. While mild to moderate short stature is observed with MED, some individuals have average stature.
The majority of COMP-PSACH mutations are observed in the calcium binding T3 repeats with only a few reported in the CTD (Fig. 1) [50]. Interestingly, deletion of one of five aspartic acid residues between amino acid residues 469 and 473 (referred to as D469del or D469del) in the seventh T3 repeat accounts for 30% of PSACH cases [47, 49, 60, 61]. While there is no strict genotype–phenotype correlation, missense mutations and in-frame insertion/deletion of single residue in T35-7 cause PSACH, while missense mutations in T33-4 more often cause MED [50]. COMP mutations preferentially affect high affinity calcium binding residues, which results in loss of tertiary structure of T3 and CTD domains, thereby causing protein misfolding [62]. Rotary shadowing studies confirm that mutant subunits are longer than wild-type confirming the protein folding abnormality [63].
In 1973, electron microscopy studies of PSACH iliac crest biopsies showed massively dilated endoplasmic reticulum (ER) filled with lamellar appearing material suggesting that PSACH was an ER storage disorder [47, 48, 64]. Following the discovery that COMP mutations cause PSACH, COMP was shown to be retained in the PSACH ER [65]. Surprisingly, other ECM proteins, types II and IX collagens and matrilin-3, were also retained forming an ordered intracellular matrix in vitro and in vivo [66, 67] (Fig. 2). Figure 2 shows the deconvolution microscopy images of the intracellular matrix that is formed in the ER of human PSACH chondrocytes as a result of mutant COMP expression. The retained insoluble matrix was postulated to be resistant to cellular degradation processes, or at the very least, the rate of accumulation exceeds the rate of degradation [68, 69]. This finding was confirmed in our Ind-D469del-Tg mouse model expressing mutant COMP [67].
Figure 2. Mutant COMP-induced matrix in ER of PSACH chondrocytes.

Fluorescence deconvolution microscopy with image reconstruction was used to visualize the ordered matrix involving type II collagen (yellow), type IX collagen (red), COMP (green) and matrilin-3 (MATN3 blue) in the ER of human and mouse mutant growth plate chondrocytes. Matrix assembles in response to stalling of mutant COMP in the ER.
Although the natural history of PSACH has been well–characterized over the last five decades, little information has been gleamed about the chondrocyte and ligament pathology because of the inaccessibility of relevant tissues for study. This was rectified by the use of murine models that show the hallmarks of PSACH: 1) intracellular ER COMP retention, 2) premature chondrocyte death and 3) limb shortening. Different approaches and mutations, summarized in Table 1, have been used to generate four mouse models of PSACH. While PSACH results from a heterozygous mutation, the phenotype in some mice manifests only in the presence of homozygous (biallelic) mutations. For example, mild limb shortening and intracellular retention of mutant COMP occurs only in the presence of homozygous (biallelic) D469del (D469del-KI) or T585M (T585M-KI) knock-in mutations [70, 71] (Table 1) indicating that that expression levels of mutant COMP must be high and exceed the heterozygous endogenous level in order to mimic the PSACH phenotype in mice. In contrast, the transgenic D469del mouse with the BM40 signal peptide that enhances secretion showed no retention (D469del-BM40-Tg) [72], while the transgenic Ind-D469del-Tg mouse with the nascent signal peptide and high mutant COMP expression showed both intracellular retention and limb shortening [67] (Table 1).
Table 1. Pathology in mutant COMP murine models.
| Mutation in T3 repeat-PSACH | Mutation in CTD mild PSACH-MED | |||
|---|---|---|---|---|
| Mutant COMP mouse model | D469del BM40 [70] | D469del KI [72] | Inducible D469del Tg [66] | T585M KI [69] |
| ER retention | yes-/-WT-COMP | yes+/+mut | yes | no |

|
No ↓Peroxiredoxin ↑Oxidative stress |
↑PERK-CHOP ↓Mitochondrial metabolism ↑Oxidative stress ↑Inflammation |
↑ATF6-CHOP | |
| Chondrocyte cell death | yes | yes | yes | yes |
| Limb shortening | 8% only males | 6%* | 12% | 4%* |
= homozygous mutation
While there have been varying degrees of success generating the PSACH phenotype in these mice, Figure 3 lists how each mouse model contribute to our overall understanding of the growth plate chondrocyte pathology and functional changes in the bone, cartilage and joints. Retention of mutant COMP in the ER of growth plate chondrocytes is the triggering event that ultimately causes decreased chondrocyte proliferation and viability [67, 70-72]. Stalling of COMP initiates ER stress and the unfolded protein response (UPR), which is responsible for clearing misfolded protein by refolding or degradation in order to maintain ER homoeostasis [67, 70, 71]. Despite select activation of UPR responses, clearance of the ER does not occur, resulting in oxidative stress, inflammation, increased cell death and decreased chondrocyte proliferation [67, 71, 72]. Under normal conditions, binding immunoglobulin protein (BiP) suppresses the UPR by preventing activation of ATF6, PERK and IRE1 [73-75]. Only the T585M-KI mouse with mild ER stress had early upregulation of BiP in the ER [70] (Fig. 3). In contrast, PERK, ATF6, CHOP and calreticulin were upregulated in the other mouse models [67, 71, 72] (Fig. 3). These cellular stresses together cause cell cycle arrest, chondrocyte death and decreased proliferation, which ultimately leads to growth plate abnormalities [67, 70-72].
Figure 3. Pathology associated with mutant COMP.
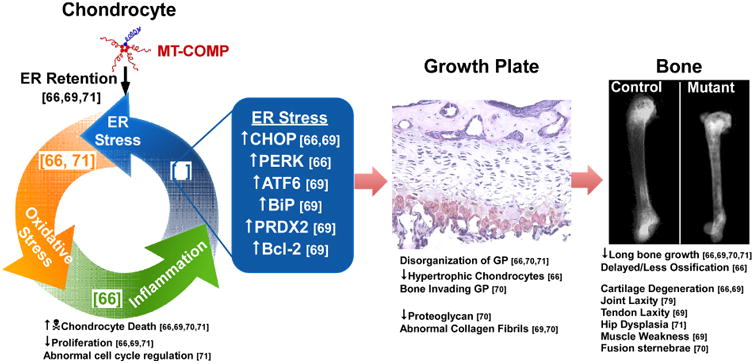
The mechanism in chondrocytes and resulting changes in growth plate, bone, cartilage and joints, are shown.
The growth plate, the structure responsible for linear growth, is compromised by mutant COMP accumulation in growth plate chondrocytes. Mutant COMP in mice disrupts columnar organization of growth plate chondrocytes (Fig. 3) [67, 70, 71] similar to PSACH growth plate chondrocytes that are clustered [69]. Hypertrophic chondrocytes are crucial for growth as they provide the matrix that will eventually be mineralized and reorganized into bone [69]. Fewer hypertrophic chondrocytes were observed in mutant COMP mice [67] and PSACH growth plates [69], which likely compromises the matrix synthesis capacity of the growth plate as evidenced by diminished proteoglycans [72] (Fig. 3). Additionally, abnormal collagen fibrils were found in the ECM surrounding growth plate chondrocytes [70, 72], which may reflect the absence of COMP in the matrix needed to organize matrix collagens [76]. The deleterious effect that mutant COMP has on chondrocytes and the growth plate translates into linear bone growth inhibition.
In addition, to diminished long bone growth [67, 70-72], there are changes in ossification, bone mineral density (BMD), cartilage health and longevity, joint laxity and muscle weakness. Recent work found that mutant COMP negatively impacts BMD by increasing miR223, which disturbs the balance between osteogenesis and adipogenesis delaying and impairing ossification [77] (Fig. 3). Abnormalities in the PSACH femoral head and precocious OA lead to hip replacement in early adulthood. Similarly, the T585M-KI mouse showed hip dysplasia [71], the Ind-D469del-Tg mouse had thin knee articular cartilage at 16 weeks [77] and the D469del-KI mouse had severely eroded articular cartilage with little to no proteoglycans at 16 months [70]. These findings are consistent with early severe OA reported in PSACH [57] (Fig. 3). While there is little information about the causes of PSACH joint laxity, joint [77] and tendon laxity [70] have been demonstrated in mutant mice. The D469del -KI mouse showed thicker fused collagen fibrils in tendons and ligaments and the Achilles tendon had increased laxity (∼17%) by cyclic strain testing (Fig. 3) [70]. Consistent with the joint laxity in PSACH, the Ind-D469del-Tg knee joints showed 2.23- and 1.45-fold increased laxity in varus-valgus and anterior-posterior direction respectively [77]. Interestingly, myopathy has been reported with MED CTD mutations and muscle weakness was shown in the T585M-KI mice with increased number of abnormal muscle fibers with central nuclei [70] (Fig. 3). The muscle pathology in the T585M-KI mouse was restricted to the site of connection between tendon and muscle (perimysium and myotendinous junction). CTD MED mutations are a rare cause of MED [78].
Together, mutant COMP mouse models suggest that both apoptosis and lower chondrocyte proliferation contribute to depletion of the chondrocyte pool available for cartilage synthesis. This pathological process is stimulated by ER stress from intracellular retention of COMP along with oxidative stress and inflammation overwhelming chondrocyte viability (Table 1; Fig. 3). Some have suggested that extracellular pathology from mutant COMP expression plays a significant role in PSACH [70, 79]. The fact that ER growth plate chondrocyte retention of mutant thyroid protein (thyroglobulin) causes murine dwarfism refutes this claim [80]. Mutant COMP in the extracellular matrix may compromise cartilage and tendon tissue but this not the primary molecular mechanism of PSACH pathology [79, 81]. Collectively, mouse studies demonstrate that intracellular retention of COMP drives limb shortening in both mice and humans. The loss of chondrocytes leads to changes in the growth plate, matrix and ultimately alterations of bone, cartilage and joint and muscle function (Fig. 3).
Conditions involving COMP
Cartilage destruction biomarker
COMP is a well-established marker of cartilage turnover and is associated with joint degeneration [82, 83]. A comprehensive review of OA biomarkers reported that COMP, osteocalcin and carboxy-terminal cross-linked fragment of type II collagen (CTX-II) are currently the most reliable markers of OA [84]. High levels of COMP in serum and synovial fluid are correlated with early stage OA and rheumatoid arthritis (RA), the degree of cartilage destruction and disease progression [82, 83, 85-90]. Serum COMP levels were not elevated in the late stages of OA [87]. In late stage OA, COMP is selectively upregulated in chondrocytes adjacent to damaged cartilage but not in the chondrocytes in neighboring normal tissue [86]. These findings suggest that COMP is upregulated in late OA either to repair damage or replenish the matrix, whereas high levels of serum COMP in the early stages of OA are the result of cartilage degradation.
COMP is elevated in RA, an autoimmune disorder in which systemic chronic inflammation causes joint degradation [85, 91-99]; synovial fluid and serum COMP levels are correlated with the joint destruction. Two RA inflammatory markers, erythrocyte sedimentation rate and the presence of rheumatoid nodule, correlate with serum COMP levels, as well as the disease activity score [85]. Lower serum COMP levels correlated with mild RA disease and decreased level of inflammatory markers. In contrast, higher COMP synovial fluid levels were found in patients requiring extensive surgical intervention [93]. Additionally, high serum COMP levels in scleroderma patients are associated with higher joint tenderness as measured by the Ritchie articular index and suggests possible RA development [100]. COMP is elevated in RA, an autoimmune disorder in which systemic chronic inflammation causes joint degradation [85, 91-99]; synovial fluid and serum COMP levels are correlated with the joint destruction. Two RA inflammatory markers, erythrocyte sedimentation rate and the presence of rheumatoid nodule, correlate with serum COMP levels as well as the disease activity score [85]. Lower serum COMP levels correlated with mild RA disease and decreased level of inflammatory markers. In contrast, higher COMP synovial fluid levels were found in patients requiring extensive surgical intervention [93].
Normal physical activity causes a transient increase in serum COMP, which resolves within 3 hours post exercise [101, 102]. With exercise, COMP peaks twice, by one hour and then at 5 hours. The increase in COMP after physical activity likely reflects articular cartilage surface wear and the later increase may be the result of COMP synthesis needed for joint resurfacing. Consistent with this hypothesis, 4-fold high baseline levels of serum COMP were found in marathon and ultra-marathon runners [90, 103]. COMP levels returned to baseline within 24-48 hours after the marathon; however, inflammatory markers remained elevated 1-2 days after the marathon [90]. Similarly, elevated COMP levels were found with joint injury and post-traumatic arthritis [90]. COMP in the synovial fluid from the injured knee was increased up to 10-fold compared to the uninjured knee [104]. Patients with knee pain but no visible injury with arthroscopy also had abnormally high levels of COMP [87]. Collectively, these studies show that COMP is a sensitive marker of cartilage wear and elevated COMP levels associated with OA and RA likely correlate with continuous joint wear and sustained attempts at resurfacing the articular cartilage.
The most valuable biomarkers are diagnostic and predictive of outcome. Increased COMP synovial fluid levels are predictive of poor outcome in the very early stages of disease when knee and hip radiographs are generally normal [87]. Higher baseline levels of COMP are associated with patients that progress rapidly and have a worse 5 year disease prognosis [91, 92, 97] suggesting that COMP levels reflect joint degeneration and can be used to predict long-term prognosis.
Fibrosis
COMP has been shown to play a role in fibrosis, a process characterized by excess ECM deposition, including type I collagen [105]. The excess matrix, in fibrosis, stiffens the ECM causing cellular dysfunction and organ failure. Activated fibroblasts and myofibroblasts in the fibrotic process are responsible for the overexpression of type I collagen [105]. The activation and conversion of fibroblasts to myofibroblasts takes place following infiltration of inflammatory cells and/or excessive mechanical stretch of these tissues [105].
Importantly, COMP plays a critical role in the structural organization of the ECM in skin modulating type 1 collagen synthesis and fibrillogenesis, particularly in the vicinity of anchoring plaques needed for cohesion between upper dermis and basement membrane [106]. COMP interacts with other ECM proteins types I, XII and I collagens, the most abundant component in the skin ECM [107]. In wound healing, COMP was not detected when fibroblasts were activated and converted into myofibroblasts and collagen deposition was increased. In contrast, COMP expression in fibrotic conditions was increased following activation of skin fibroblasts and lung epithelial cells by transforming growth factor-β (TGFβ) [108, 109] (Fig. 4). COMP is among th most upregulated gene in idiopathic pulmonary fibrosis (IPF) [110], a chronic and progressive inflammatory lung disease [111]. Once the fibrotic process is initiated, a self-perpetuating cycle is created with COMP increasing TGFβ activity and TGFβ increasing COMP expression [108, 112] (Fig. 4). TGFβ converts more skin fibroblasts and lung epithelial cells to myofibroblasts which increases ECM synthesis perpetuating the fibrosis cycle [108, 111, 112]. Constant COMP expression upregulated TGFβ signaling and the modulation of COMP expression by TGFβ constitutes a positive feedback loop in IPF and fibrotic skin [108, 109, 112]. COMP increases type I collagen expression and reorganizes the collagen fibrils stiffening the ECM [109], which corresponded to decreased force vital capacity in the lungs [109]. Additionally, higher levels of fibrosis in systemic sclerosis (scleroderma) is associated with higher levels of serum COMP and higher levels of inflammatory markers (eosinophils and higher chemokine (C-X-C motif) ligand 8) in bronchoalveolar lavage fluid [113].
Figure 4. COMP and fibrosis.
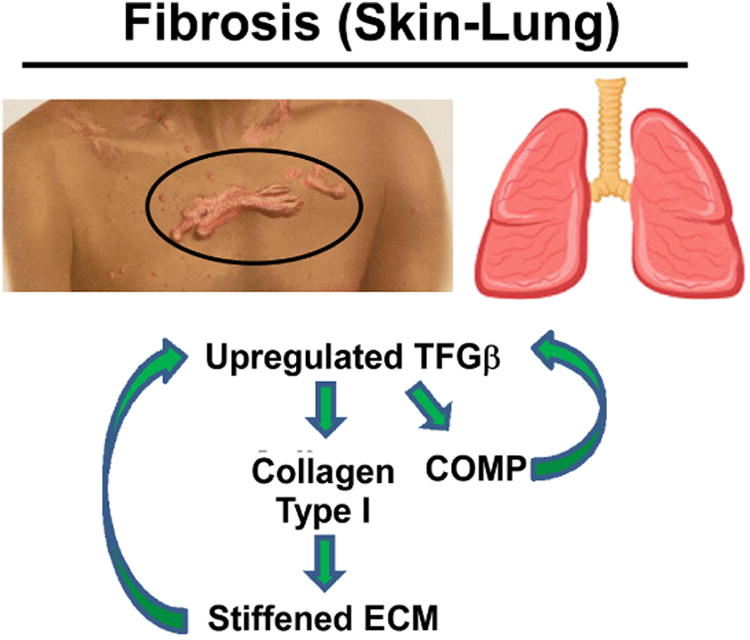
COMP upregulates TGFβ stimulating additional COMP expression. COMP assists in type I collagen secretion which results in stiffening of the ECM, an important component in fibrosis pathology.
In contrast, COMP expression in the fibrotic liver was elevated by reactive oxygen species, chemokines, growth factors, matrix stiffness, matricellular proteins or DAMPs [7, 114] but TGFβ mRNA and proteins levels were not changed [115]. COMP increased type 1 collagen synthesis in hepatic stellate cells through CD36 receptor signaling and activation of the MEK1/2 – pERK1/2 pathway [115]. Similar to lung and skin fibrosis, increased type I collagen synthesis in the liver stiffens the ECM contributing to cellular and organ dysfunction.
The fibrotic response has been extensively studied in COMP null mice. COMP null fibroblasts do not deposit type I and XII collagens in the ECM [116]. Instead, these collagens were retained of in the ER suggesting COMP is necessary for their secretion [116]. Absence of COMP negatively impacts the assembly of collagen and blunts fibrosis [108, 116].
Collectively, these findings indicate that COMP plays a major role in fibrosis by abnormally increasing type I collagen synthesis and matrix reorganization. This suggests that increased COMP levels can be used as a marker for multiple fibrotic conditions [108].
COMP in cancer
Stromal cells create an ECM barrier that tumor cells must breach to become metastatic [8]. COMP did not change adhesion and migration of the breast cancer cells but increased invasiveness of breast cancer cells by upregulating MMP 9 and increasing the ability of the cancer cells to degrade surrounding ECM stroma [8]. Gene expression analyses found higher expression of COMP in cancerous breast and prostate tissues [110, 117] correlating with immunostaining that shows COMP is expressed both in epithelial tumor cells and stroma [8] (Fig. 5). COMP binds cancer cell surface through a region overlapping the T2 and T3 repeats domains. COMP was found to enhance invasion of prostate cancer cells (DU145 cells) through integrin binding. Moreover, COMP transfected breast cancer cells or purified COMP added to media of prostate cancer cells had a lower oxygen consumption rate than those that do not express COMP suggesting the COMP expressing cancer cells switch to aerobic glycolysis to generate energy (known as the Warburg effect) giving a survival advantage to the interior of the tumor which becomes oxygen deprived [117]. Although this process is less efficient at producing ATP, it may enable cancer cells to proliferate by supporting the uptake and incorporation of nutrients into the biomass (e.g., nucleotides, amino acids, and lipids) rather than stimulating apoptosis due to oxygen deprivation [118, 119]. Consistent with these findings, COMP expressing cancer cells produced larger tumors when injected into SCID mice compared to COMP non-expressing cells suggesting that COMP increases survival [8] (Fig. 5). Additionally, COMP is detected in tumors that have metastasized to the lungs and lymph nodes from the original breast site [8]. Higher COMP expression in tumors decreased ER stress, which gave the tumor cells survival advantage despite high level of general protein production (Fig. 5). The reduction in ER stress led to downregulation of apoptosis in cancer cells expressing COMP [8] and this was accomplished by preventing Ca2+ release from the endoplasmic reticulum of prostate cells. Furthermore, COMP may play a similar role as TSP4 which is postulated to have a protective role against ER stress in the heart [120]. TSP4 binds to ATF6 and promotes its activation and shuttling to the nucleus, which upregulates the adaptive ER stress response together with an expansion on the ER [120].
Figure 5. COMP's role in breast cancer metastasis.
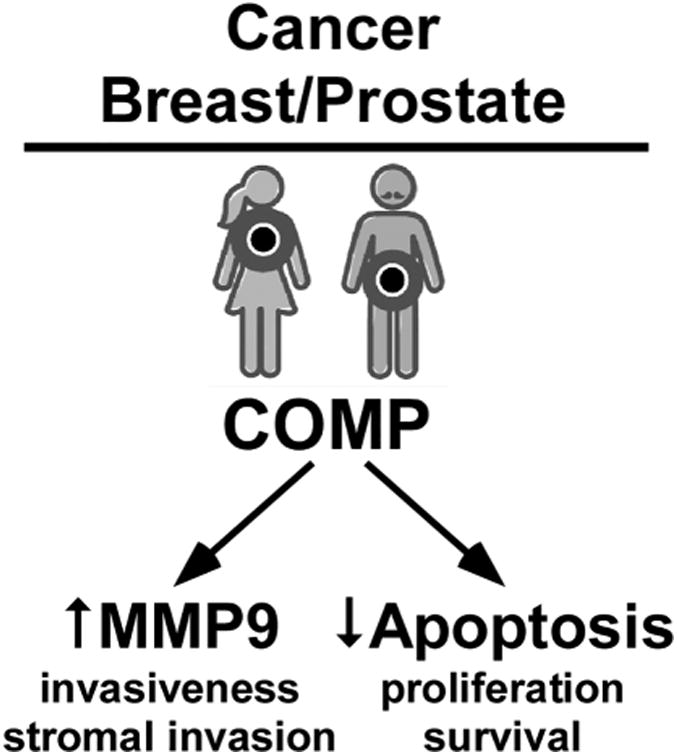
COMP increases MMP9 which breaks down the stromal barrier. COMP reduces apoptosis levels in cancer cells thereby fostering tumor survival.
COMP plays a role in breast cancer by increasing invasiveness by a switch in cell metabolism and decreasing apoptosis potentially through ER stress resistance resulting in larger tumors in vivo and enhanced metastasis (Fig. 5). Finally, high levels of COMP in breast cancer correlate with poor survival and cancer recurrence [8]. These findings suggest that COMP should be investigated as a prognostic marker for breast and prostate cancers [117].
Cardiomyopathy
Dilated cardiomyopathy (DCM) is the third most common cause of congestive heart failure in adults and is the most common form of cardiomyopathy in children [9, 121]. DCM is more frequent in men and half of the individuals affected ultimately require a heart transplant [9, 121]. DCM is characterized by dilatation of ventricular chambers, impaired myocardial contractility and inability to pump blood efficiently [122]. Cardiac cells rely on interactions with the surrounding ECM through integrins to support contractile function(s). Cellular adhesion of the cardiac myocytes to the ECM is required to sustain the mechanical force of heart contraction-relaxation cycles and for the signaling transduction [123]. COMP is expressed in cardiac ECM, and, interestingly, reduced levels of COMP were found in end-stage DCM hearts compared to levels detected in healthy left ventricular tissue [9]. COMP null mice develop DCM between 3 to 5 months, although these mice have few other health issues, normal fertility and longevity [124]. Loss of COMP increased MMP9 but reduced integrin β-1 expression and signaling in cardiomyocytes and connexin 43 protein levels. COMP and integrin β1 co-localize in cardiomyocytes and in cardiac fibroblasts with COMP binding to the proximal β-tail of integrin β1 through its C-terminal domain [9] (Fig. 6). This binding of COMP to β1 integrin on the membrane stabilizes β1 integrin by inhibiting ubiquitination and degradation [9]. Cardiomyocyte death occurs because COMP integrin connection loss and mitochondrial induced apoptosis [9]. Increased expression of TSP1-4 occurs in the heart in response to hypertrophy or injury inducing the adaptive stress response in the cells to protect against cell death [120]. This suggests that all the TSPs contribute to heart function with COMP supporting cardiac function by stabilizing integrin β1.
Figure 6. COMP in cardiac function.

COMP binds to β1 integrin stabilizing the protein and supporting cellular survival.
Conclusions
COMP is an important member of the ECM playing multiple and diverse roles necessary for the integrity of the matricellular network. Current theories postulate that COMP stabilizes the interactions of ECM proteins and increases the mechanical strength of tissues. More matrix research will provide a better understanding of how this multifunctional protein contributes to matrix homeostatsis. Study of genetic disorders revealed that COMP mutations cause stalling of COMP and other ECM proteins with matrix assembly within the chondrocyte ER to the detriment of cellular homeostasis. Interestingly, absence of COMP (COMP-/-) in mice also causes stalling of ECM proteins indicating that COMP plays an important role in the export of these matrix proteins. Thus, two important roles for COMP are export of ECM proteins and correct integration into the ECM. Dysregulation of these processes cause skeletal dysplasias, abnormal wound healing and fibrosis. Taken together, too little or too much COMP is detrimental and likely tissue specific and should be the focus of future studies. Currently, the best characterized clinical application is the use of COMP as a biomarker for cartilage degeneration and fibrosis. The expanding roles of COMP in normal cellular homeostasis and abnormal disease states are exciting and will provide new insights that may open future therapeutic avenues.
Highlights.
COMP is an extracellular matrix protein and the fifth member of the thrombospondin gene family.
COMP facilitates collagen fibrillogenesis and chondrocyte proliferation.
Autosomal dominant mutations in COMP cause two skeletal dysplasias, pseudoachondroplasia and multiple epiphyseal dysplasia.
Dysregulated COMP expression is found in tissue fibrosis, breast and prostate cancer and cardiomyopathy.
COMP is a biomarker idiopathic pulmonary fibrosis and for cartilage degeneration in osteoarthritis and rheumatoid arthritis.
Acknowledgments
Research performed in the authors' laboratory and reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Number RO1-AR057117-05 and the Leah Lewis Family Foundation. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations used
- ATF6
activating transcription factor 6
- ATP
adenosine triphosphate
- BiP
binding immunoglobulin protein
- BMD
bone mineral density
- Chop
Mouse c/ebp homologous protein genes
- CHOP
c/ebp homologous protein
- COL9A1
human collagen type 9 Alpha 1 chain genes
- COL9A2
human collagen type 9 Alpha 2 chain genes
- COL9A3
human collagen type 9 Alpha 3 chain genes
- COMP
Cartilage oligomeric matrix protein
- CTD
C-terminal globular domain
- DCM
dilated cardiomyopathy
- DAMPS
Damage-associated molecular patterns
- ECM
Extracellular matrix
- EGF
epidermal growth factor
- ER
endoplasmic reticulum
- GEP
granulin-epithelin precursor
- IPF
idiopathic pulmonary fibrosis
- IRE1
inositol-requiring enzyme 1
- KI
knock-in
- MATN3
matrilin 3
- MED
multiple epiphyseal dysplasia
- MMP
matrix metalloproteinase
- NTD
N-terminal domain
- OA
osteoarthritis
- PERK
pancreatic ER kinase (PKR)-like ER kinase
- PSACH
pseudoachondroplasia
- RA
rheumatoid arthritis
- SMC
smooth muscle cell
- TGFβ
transforming growth factor-β
- TSP5
thrombospondin 5
- UPR
unfolded protein response
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fife R, Brandt K. Identification of a high-molecular-weight (> 400 000) protein in hyaline cartilage. Biochimica et Biophysica Acta (BBA) - General Subjects. 1984;802(3):506–514. doi: 10.1016/0304-4165(84)90370-2. [DOI] [PubMed] [Google Scholar]
- 2.Hedbom E, Antonsson P, Hjerpe A, Aeschlimann D, Paulsson M, Rosa-Pimentel E, Sommarin Y, Wendel M, Oldberg A, Heinegard D. Cartilage matrix proteins. An acidic oligomeric protein (COMP) detected only in cartilage. J Biol Chem. 1992;267(9):6132–6. [PubMed] [Google Scholar]
- 3.DiCesare P, Hauser N, Lehman D, Pasumarti S, Paulsson M. Cartilage oligomeric matrix protein (COMP) is an abundant component of tendon. FEBS Lett. 1994;354(2):237–40. doi: 10.1016/0014-5793(94)01134-6. [DOI] [PubMed] [Google Scholar]
- 4.Di Cesare PE, Carlson CS, Stollerman ES, Chen FS, Leslie M, Perris R. Expression of cartilage oligomeric matrix protein by human synovium. FEBS Lett. 1997;412(1):249–52. doi: 10.1016/s0014-5793(97)00789-8. [DOI] [PubMed] [Google Scholar]
- 5.Müller G, Michel A, Altenburg E. COMP (Cartilage Oligomeric Matrix Protein) is Synthesized in Ligament, Tendon, Meniscus, and Articular Cartilage. Connective tissue research. 1998;39(4):233–244. doi: 10.3109/03008209809021499. [DOI] [PubMed] [Google Scholar]
- 6.Riessen R, Fenchel M, Chen H, Axel DI, Karsch KR, Lawler J. Cartilage oligomeric matrix protein (thrombospondin-5) is expressed by human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2001;21(1):47–54. doi: 10.1161/01.atv.21.1.47. [DOI] [PubMed] [Google Scholar]
- 7.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214(2):199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Englund E, Bartoschek M, Reitsma B, Jacobsson L, Escudero-Esparza A, Orimo A, Leandersson K, Hagerling C, Aspberg A, Storm P, Okroj M, Mulder H, Jirstrom K, Pietras K, Blom AM. Cartilage oligomeric matrix protein contributes to the development and metastasis of breast cancer. Oncogene. 2016;35(43):5585–5596. doi: 10.1038/onc.2016.98. [DOI] [PubMed] [Google Scholar]
- 9.Huang Y, Xia J, Zheng J, Geng B, Liu P, Yu F, Liu B, Zhang H, Xu M, Ye P, Zhu Y, Xu Q, Wang X, Kong W. Deficiency of cartilage oligomeric matrix protein causes dilated cardiomyopathy. Basic research in cardiology. 2013;108(5):374. doi: 10.1007/s00395-013-0374-9. [DOI] [PubMed] [Google Scholar]
- 10.Liang Y, Fu Y, Qi R, Wang M, Yang N, He L, Yu F, Zhang J, Yun CH, Wang X, Liu J, Kong W. Cartilage oligomeric matrix protein is a natural inhibitor of thrombin. Blood. 2015;126(7):905–14. doi: 10.1182/blood-2015-01-621292. [DOI] [PubMed] [Google Scholar]
- 11.Bornstein P, Sage EH. Matricellular proteins: extracellular modulators of cell function. Curr Opin Cell Biol. 2002;14(5):608–16. doi: 10.1016/s0955-0674(02)00361-7. [DOI] [PubMed] [Google Scholar]
- 12.Adams JC. Thrombospondins: multifunctional regulators of cell interactions. Annu Rev Cell Dev Biol. 2001;17:25–51. doi: 10.1146/annurev.cellbio.17.1.25. [DOI] [PubMed] [Google Scholar]
- 13.Lawler J. The functions of thrombospondin-1 and-2. Curr Opin Cell Biol. 2000;12(5):634–40. doi: 10.1016/s0955-0674(00)00143-5. [DOI] [PubMed] [Google Scholar]
- 14.Bornstein P. Thrombospondins: structure and regulation of expression. Faseb J. 1992;6(14):3290–9. doi: 10.1096/fasebj.6.14.1426766. [DOI] [PubMed] [Google Scholar]
- 15.Carlson CB, Lawler J, Mosher DF. Structures of thrombospondins. Cellular and molecular life sciences : CMLS. 2008;65(5):672–86. doi: 10.1007/s00018-007-7484-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tan K, Duquette M, Joachimiak A, Lawler J. The crystal structure of the signature domain of cartilage oligomeric matrix protein: implications for collagen, glycosaminoglycan and integrin binding. FASEB J. 2009;23(8):2490–501. doi: 10.1096/fj.08-128090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adams JC, Lawler J. The thrombospondins. Cold Spring Harbor perspectives in biology. 2011;3(10):a009712. doi: 10.1101/cshperspect.a009712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Agah A, Kyriakides TR, Lawler J, Bornstein P. The lack of thrombospondin-1 (TSP1) dictates the course of wound healing in double-TSP1/TSP2-null mice. The American journal of pathology. 2002;161(3):831–9. doi: 10.1016/S0002-9440(10)64243-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Majack RA, Cook SC, Bornstein P. Platelet-derived growth factor and heparin-like glycosaminoglycans regulate thrombospondin synthesis and deposition in the matrix by smooth muscle cells. J Cell Biol. 1985;101(3):1059–70. doi: 10.1083/jcb.101.3.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yabkowitz R, Mansfield PJ, Ryan US, Suchard SJ. Thrombospondin mediates migration and potentiates platelet-derived growth factor-dependent migration of calf pulmonary artery smooth muscle cells. J Cell Physiol. 1993;157(1):24–32. doi: 10.1002/jcp.1041570104. [DOI] [PubMed] [Google Scholar]
- 21.Patel MK, Lymn JS, Clunn GF, Hughes AD. Thrombospondin-1 is a potent mitogen and chemoattractant for human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1997;17(10):2107–14. doi: 10.1161/01.atv.17.10.2107. [DOI] [PubMed] [Google Scholar]
- 22.Isenberg JS, Ridnour LA, Perruccio EM, Espey MG, Wink DA, Roberts DD. Thrombospondin-1 inhibits endothelial cell responses to nitric oxide in a cGMP-dependent manner. Proc Natl Acad Sci U S A. 2005;102(37):13141–6. doi: 10.1073/pnas.0502977102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Isenberg JS, Wink DA, Roberts DD. Thrombospondin-1 antagonizes nitric oxide-stimulated vascular smooth muscle cell responses. Cardiovasc Res. 2006;71(4):785–93. doi: 10.1016/j.cardiores.2006.05.024. [DOI] [PubMed] [Google Scholar]
- 24.Ridnour LA, Isenberg JS, Espey MG, Thomas DD, Roberts DD, Wink DA. Nitric oxide regulates angiogenesis through a functional switch involving thrombospondin-1. Proc Natl Acad Sci U S A. 2005;102(37):13147–52. doi: 10.1073/pnas.0502979102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou Y, Poczatek MH, Berecek KH, Murphy-Ullrich JE. Thrombospondin 1 mediates angiotensin II induction of TGF-beta activation by cardiac and renal cells under both high and low glucose conditions. Biochem Biophys Res Commun. 2006;339(2):633–41. doi: 10.1016/j.bbrc.2005.11.060. [DOI] [PubMed] [Google Scholar]
- 26.O'Shea KS, Liu LH, Dixit VM. Thrombospondin and a 140 kd fragment promote adhesion and neurite outgrowth from embryonic central and peripheral neurons and from PC12 cells. Neuron. 1991;7(2):231–7. doi: 10.1016/0896-6273(91)90261-w. [DOI] [PubMed] [Google Scholar]
- 27.Arber S, Caroni P. Thrombospondin-4, an extracellular matrix protein expressed in the developing and adult nervous system promotes neurite outgrowth. J Cell Biol. 1995;131(4):1083–94. doi: 10.1083/jcb.131.4.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barros CS, Franco SJ, Muller U. Extracellular matrix: functions in the nervous system. Cold Spring Harb Perspect Biol. 2011;3(1):a005108. doi: 10.1101/cshperspect.a005108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hankenson KD, Sweetwyne MT, Shitaye H, Posey KL. Thrombospondins and novel TSR-containing proteins, R-spondins, regulate bone formation and remodeling. Curr Osteoporos Rep. 2010;8(2):68–76. doi: 10.1007/s11914-010-0017-0. [DOI] [PubMed] [Google Scholar]
- 30.Alford AI, Terkhorn SP, Reddy AB, Hankenson KD. Thrombospondin-2 regulates matrix mineralization in MC3T3-E1 pre-osteoblasts. Bone. 2010;46(2):464–71. doi: 10.1016/j.bone.2009.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Canfield AE, Sutton AB, Hoyland JA, Schor AM. Association of thrombospondin-1 with osteogenic differentiation of retinal pericytes in vitro. J Cell Sci. 1996;109(Pt 2):343–53. doi: 10.1242/jcs.109.2.343. [DOI] [PubMed] [Google Scholar]
- 32.Ueno A, Miwa Y, Miyoshi K, Horiguchi T, Inoue H, Ruspita I, Abe K, Yamashita K, Hayashi E, Noma T. Constitutive expression of thrombospondin 1 in MC3T3-E1 osteoblastic cells inhibits mineralization. J Cell Physiol. 2006;209(2):322–32. doi: 10.1002/jcp.20735. [DOI] [PubMed] [Google Scholar]
- 33.Holden P, Meadows RS, Chapman KL, Grant ME, Kadler KE, Briggs MD. Cartilage oligomeric matrix protein interacts with type IX collagen, and disruptions to these interactions identify a pathogenetic mechanism in a bone dysplasia family. J Biol Chem. 2001;276(8):6046–55. doi: 10.1074/jbc.M009507200. [DOI] [PubMed] [Google Scholar]
- 34.Thur J, Rosenberg K, Nitsche DP, Pihlajamaa T, Ala-Kokko L, Heinegard D, Paulsson M, Maurer P. Mutations in cartilage oligomeric matrix protein causing pseudoachondroplasia and multiple epiphyseal dysplasia affect binding of calcium and collagen I, II, and IX. J Biol Chem. 2001;276(9):6083–92. doi: 10.1074/jbc.M009512200. [DOI] [PubMed] [Google Scholar]
- 35.Chen FH, Herndon ME, Patel N, Hecht JT, Tuan RS, Lawler J. Interaction of cartilage oligomeric matrix protein/thrombospondin 5 with aggrecan. J Biol Chem. 2007;282(34):24591–8. doi: 10.1074/jbc.M611390200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mann HH, Ozbek S, Engel J, Paulsson M, Wagener R. Interactions between the cartilage oligomeric matrix protein and matrilins. Implications for matrix assembly and the pathogenesis of chondrodysplasias. J Biol Chem. 2004;279(24):25294–8. doi: 10.1074/jbc.M403778200. [DOI] [PubMed] [Google Scholar]
- 37.Di Cesare PE, Chen FS, Moergelin M, Carlson CS, Leslie MP, Perris R, Fang C. Matrix-matrix interaction of cartilage oligomeric matrix protein and fibronectin. Matrix biology : journal of the International Society for Matrix Biology. 2002;21(5):461–70. doi: 10.1016/s0945-053x(02)00015-x. [DOI] [PubMed] [Google Scholar]
- 38.Blumbach K, Bastiaansen-Jenniskens YM, DeGroot J, Paulsson M, van Osch GJ, Zaucke F. Combined role of type IX collagen and cartilage oligomeric matrix protein in cartilage matrix assembly: cartilage oligomeric matrix protein counteracts type IX collagen-induced limitation of cartilage collagen fibril growth in mouse chondrocyte cultures. Arthritis Rheum. 2009;60(12):3676–85. doi: 10.1002/art.24979. [DOI] [PubMed] [Google Scholar]
- 39.Xu K, Zhang Y, Ilalov K, Carlson CS, Feng JQ, Di Cesare PE, Liu CJ. Cartilage oligomeric matrix protein associates with granulin-epithelin precursor (GEP) and potentiates GEP-stimulated chondrocyte proliferation. J Biol Chem. 2007;282(15):11347–55. doi: 10.1074/jbc.M608744200. [DOI] [PubMed] [Google Scholar]
- 40.Stracke JO, Fosang AJ, Last K, Mercuri FA, Pendas AM, Llano E, Perris R, Di Cesare PE, Murphy G, Knauper V. Matrix metalloproteinases 19 and 20 cleave aggrecan and cartilage oligomeric matrix protein (COMP) FEBS Lett. 2000;478(1-2):52–6. doi: 10.1016/s0014-5793(00)01819-6. [DOI] [PubMed] [Google Scholar]
- 41.Dickinson SC, Vankemmelbeke MN, Buttle DJ, Rosenberg K, Heinegard D, Hollander AP. Cleavage of cartilage oligomeric matrix protein (thrombospondin-5) by matrix metalloproteinases and a disintegrin and metalloproteinase with thrombospondin motifs. Matrix Biol. 2003;22(3):267–78. doi: 10.1016/s0945-053x(03)00034-9. [DOI] [PubMed] [Google Scholar]
- 42.Kipnes J, Carlberg AL, Loredo GA, Lawler J, Tuan RS, Hall DJ. Effect of cartilage oligomeric matrix protein on mesenchymal chondrogenesis in vitro, Osteoarthritis and cartilage / OARS. Osteoarthritis Research Society. 2003;11(6):442–54. doi: 10.1016/s1063-4584(03)00055-4. [DOI] [PubMed] [Google Scholar]
- 43.Mackman N, Tilley RE, Key NS. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler Thromb Vasc Biol. 2007;27(8):1687–93. doi: 10.1161/ATVBAHA.107.141911. [DOI] [PubMed] [Google Scholar]
- 44.Metharom P, Berndt MC. COMP: an endogenous thrombin inhibitor. Blood. 2015;126(7):831–2. doi: 10.1182/blood-2015-06-650846. [DOI] [PubMed] [Google Scholar]
- 45.Smith RK, Gerard M, Dowling B, Dart AJ, Birch HL, Goodship AE. Correlation of cartilage oligomeric matrix protein (COMP) levels in equine tendon with mechanical properties: a proposed role for COMP in determining function-specific mechanical characteristics of locomotor tendons. Equine Vet J Suppl. 2002;(34):241–4. doi: 10.1111/j.2042-3306.2002.tb05426.x. [DOI] [PubMed] [Google Scholar]
- 46.Sodersten F, Ekman S, Eloranta ML, Heinegard D, Dudhia J, Hultenby K. Ultrastructural immunolocalization of cartilage oligomeric matrix protein (COMP) in relation to collagen fibrils in the equine tendon. Matrix biology : journal of the International Society for Matrix Biology. 2005;24(5):376–85. doi: 10.1016/j.matbio.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 47.Hecht JT, Nelson LD, Crowder E, Wang Y, Elder FFB, Harrison WR, Francomano CA, Prange CK, Lennon GG, Deere M, Lawler J. Mutations in exon 17B of cartilage oligomeric matrix protein (COMP) cause pseudoachondroplasia. Nature Genetics. 1995;10(3):325–329. doi: 10.1038/ng0795-325. [DOI] [PubMed] [Google Scholar]
- 48.Briggs MD, Hoffman SMG, King LM, Olsen AS, Mohrenweiser H, Leroy JG, Mortier GR, Rimoin DL, Lachman RS, Gaines ES, Cekleniak JA, Knowlton RG, Cohn DH. Pseudoachondroplasia and multiple epiphyseal dysplasia due to mutations in the cartilage oligomeric matrix protein gene. Nature Genetics. 1995;10(3):330–336. doi: 10.1038/ng0795-330. [DOI] [PubMed] [Google Scholar]
- 49.Unger S, Hecht JT. Pseudoachondroplasia and multiple epiphyseal dysplasia: New etiologic developments. American Journal of Medical Genetics. 2001;106(4):244. [PubMed] [Google Scholar]
- 50.Briggs MD, Brock J, Ramsden SC, Bell PA. Genotype to phenotype correlations in cartilage oligomeric matrix protein associated chondrodysplasias. Eur J Hum Genet. 2014;22(11):1278–82. doi: 10.1038/ejhg.2014.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Briggs MD, Wright MJ. Pseudoachondroplasia. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Ledbetter N, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews(R) Seattle (WA): 1993. [Google Scholar]
- 52.Jackson GC, Mittaz-Crettol L, Taylor JA, Mortier GR, Spranger J, Zabel B, Le Merrer M, Cormier-Daire V, Hall CM, Offiah A, Wright MJ, Savarirayan R, Nishimura G, Ramsden SC, Elles R, Bonafe L, Superti-Furga A, Unger S, Zankl A, Briggs MD. Pseudoachondroplasia and multiple epiphyseal dysplasia: a 7-year comprehensive analysis of the known disease genes identify novel and recurrent mutations and provides an accurate assessment of their relative contribution. Hum Mutat. 2012;33(1):144–57. doi: 10.1002/humu.21611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Briggs MD, Wright MJ, Mortier GR. Multiple Epiphyseal Dysplasia, Autosomal Dominant. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mefford HC, Stephens K, Amemiya A, Ledbetter N, editors. GeneReviews(R) Seattle (WA): 1993. [PubMed] [Google Scholar]
- 54.Posey KL, Alcorn JL, Hecht JT. Pseudoachondroplasia/COMP - translating from the bench to the bedside. Matrix biology : journal of the International Society for Matrix Biology. 2014;37:167–73. doi: 10.1016/j.matbio.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Posey KL, Hecht JT. Novel therapeutic interventions for pseudoachondroplasia. Bone. 2017;102:60–68. doi: 10.1016/j.bone.2017.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gamble C, Nguyen J, Hashmi SS, Hecht JT. Pseudoachondroplasia and painful sequelae. American journal of medical genetics Part A. 2015;167(11):2618–22. doi: 10.1002/ajmg.a.37253. [DOI] [PubMed] [Google Scholar]
- 57.McKeand J, Rotta J, Hecht JT. Natural history study of pseudoachondroplasia. American Journal of Medical Genetics. 1996;63(2):406–410. doi: 10.1002/(SICI)1096-8628(19960517)63:2<406::AID-AJMG16>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 58.Bonafe L, Cormier-Daire V, Hall C, Lachman R, Mortier G, Mundlos S, Nishimura G, Sangiorgi L, Savarirayan R, Sillence D, Spranger J, Superti-Furga A, Warman M, Unger S. Nosology and classification of genetic skeletal disorders: 2015 revision. American journal of medical genetics Part A. 2015;167A(12):2869–92. doi: 10.1002/ajmg.a.37365. [DOI] [PubMed] [Google Scholar]
- 59.Horton WA, Hall JG, Scott CI, Pyeritz RE, Rimoin DL. Growth curves for height for diastrophic dysplasia, spondyloepiphyseal dysplasia congenita, and pseudoachondroplasia. Am J Dis Child. 1982;136(4):316–9. doi: 10.1001/archpedi.1982.03970400034010. [DOI] [PubMed] [Google Scholar]
- 60.Briggs MD, Chapman KL. Pseudoachondroplasia and multiple epiphyseal dysplasia: mutation review, molecular interactions, and genotype to phenotype correlations. Hum Mutat. 2002;19(5):465–78. doi: 10.1002/humu.10066. [DOI] [PubMed] [Google Scholar]
- 61.Mabuchi A, Manabe N, Haga N, Kitoh H, Ikeda T, Kawaji H, Tamai K, Hamada J, Nakamura S, Brunetti-Pierri N, Kimizuka M, Takatori Y, Nakamura K, Nishimura G, Ohashi H, Ikegawa S. Novel types of COMP mutations and genotype-phenotype association in pseudoachondroplasia and multiple epiphyseal dysplasia. Hum Genet. 2003;112(1):84–90. doi: 10.1007/s00439-002-0845-9. [DOI] [PubMed] [Google Scholar]
- 62.Kvansakul M, Adams JC, Hohenester E. Structure of a thrombospondin C-terminal fragment reveals a novel calcium core in the type 3 repeats. Embo J. 2004;23(6):1223–33. doi: 10.1038/sj.emboj.7600166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen H, Deere M, Hecht JT, Lawler J. Cartilage oligomeric matrix protein is a calcium-binding protein, and a mutation in its type 3 repeats causes conformational changes. J Biol Chem. 2000;275(34):26538–44. doi: 10.1074/jbc.M909780199. [DOI] [PubMed] [Google Scholar]
- 64.Cooper RR, Ponseti IV, Maynard JA. Pseudoachondroplastic dwarfism. A rough-surfaced endoplasmic reticulum storage disorder. Journal of Bone & Joint Surgery - American. 1973;55(3):475–84. [PubMed] [Google Scholar]
- 65.Hecht JT, Montufar-Solis D, Decker G, Lawler J, Daniels K, Duke PJ. Retention of cartilage oligomeric matrix protein (COMP) and cell death in redifferentiated pseudoachondroplasia chondrocytes. Matrix Biology. 1998;17(8-9):625–633. doi: 10.1016/s0945-053x(98)90113-5. [DOI] [PubMed] [Google Scholar]
- 66.Merritt TM, Bick R, Poindexter BJ, Alcorn JL, Hecht JT. Unique Matrix Structure in the Rough Endoplasmic Reticulum Cisternae of Pseudoachondroplasia Chondrocytes. The American journal of pathology. 2007;170(1):293–300. doi: 10.2353/ajpath.2007.060530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Posey KL, Veerisetty AC, Liu P, Wang HR, Poindexter BJ, Bick R, Alcorn JL, Hecht JT. An Inducible Cartilage Oligomeric Matrix Protein Mouse Model Recapitulates Human Pseudoachondroplasia Phenotype. The American journal of pathology. 2009;175(4):1555–1563. doi: 10.2353/ajpath.2009.090184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hecht JT, Hayes E, Haynes R, Cole WG. COMP mutations, chondrocyte function and cartilage matrix. Matrix biology : journal of the International Society for Matrix Biology. 2005;23(8):525–33. doi: 10.1016/j.matbio.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 69.Hecht JT, Makitie O, Hayes E, Haynes R, Susic M, Montufar-Solis D, Duke PJ, Cole WG. Chondrocyte cell death and intracellular distribution of COMP and type IX collagen in the pseudoachondroplasia growth plate. J Orthop Res. 2004;22(4):759–67. doi: 10.1016/j.orthres.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 70.Pirog-Garcia KA, Meadows RS, Knowles L, Heinegard D, Thornton DJ, Kadler KE, Boot-Handford RP, Briggs MD. Reduced cell proliferation and increased apoptosis are significant pathological mechanisms in a murine model of mild pseudoachondroplasia resulting from a mutation in the C-terminal domain of COMP. Human molecular genetics. 2007;16(17):2072–2088. doi: 10.1093/hmg/ddm155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Suleman F, Gualeni B, Gregson HJ, Leighton MP, Pirog KA, Edwards S, Holden P, Boot-Handford RP, Briggs MD. A novel form of chondrocyte stress is triggered by a COMP mutation causing pseudoachondroplasia. Human mutation. 2012;33(1):218–31. doi: 10.1002/humu.21631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schmitz M, Niehoff A, Miosge N, Smyth N, Paulsson M, Zaucke F. Transgenic mice expressing D469Delta mutated cartilage oligomeric matrix protein (COMP) show growth plate abnormalities and sternal malformations. Matrix biology : journal of the International Society for Matrix Biology. 2008;27(2):67–85. doi: 10.1016/j.matbio.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 73.Malhotra JD, Kaufman RJ. The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol. 2007;18(6):716–31. doi: 10.1016/j.semcdb.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8(7):519–29. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 75.Schroder M. Endoplasmic reticulum stress responses. Cellular and molecular life sciences : CMLS. 2008;65(6):862–94. doi: 10.1007/s00018-007-7383-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Halasz K, Kassner A, Morgelin M, Heinegard D. COMP acts as a catalyst in collagen fibrillogenesis. J Biol Chem. 2007;282(43):31166–73. doi: 10.1074/jbc.M705735200. [DOI] [PubMed] [Google Scholar]
- 77.Coustry F, Posey KL, Maerz T, Baker K, Abraham AM, Ambrose CG, Nobakhti S, Shefelbine SJ, Bi X, Newton M, Gawronski K, Remer L, Veerisetty AC, Hossain MG, Chiu F, Hecht JT. Mutant cartilage oligomeric matrix cartilage (COMP) compromises bone integrity, joint function and the balance between adipogenesis and osteogenesis. Matrix biology : journal of the International Society for Matrix Biology. 2018 doi: 10.1016/j.matbio.2017.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pirog KA, Katakura Y, Mironov A, Briggs MD. Mild myopathy is associated with COMP but not MATN3 mutations in mouse models of genetic skeletal diseases. PloS one. 2013;8(11):e82412. doi: 10.1371/journal.pone.0082412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dinser R, Zaucke F, Kreppel F, Hultenby K, Kochanek S, Paulsson M, Maurer P. Pseudoachondroplasia is caused through both intra- and extracellular pathogenic pathways. J Clin Invest. 2002;110(4):505–13. doi: 10.1172/JCI14386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gualeni B, Rajpar MH, Kellogg A, Bell PA, Arvan P, Boot-Handford RP, Briggs MD. A novel transgenic mouse model of growth plate dysplasia reveals that decreased chondrocyte proliferation due to chronic ER stress is a key factor in reduced bone growth. Disease models & mechanisms. 2013;6(6):1414–25. doi: 10.1242/dmm.013342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schmitz M, Becker A, Schmitz A, Weirich C, Paulsson M, Zaucke F, Dinser R. Disruption of extracellular matrix structure may cause pseudoachondroplasia phenotypes in the absence of impaired cartilage oligomeric matrix protein secretion. J Biol Chem. 2006;281(43):32587–95. doi: 10.1074/jbc.M601976200. [DOI] [PubMed] [Google Scholar]
- 82.Saxne T, Heinegard D. Cartilage oligomeric matrix protein: a novel marker of cartilage turnover detectable in synovial fluid and blood. Br J Rheumatol. 1992;31(9):583–91. doi: 10.1093/rheumatology/31.9.583. [DOI] [PubMed] [Google Scholar]
- 83.Urakami T, Manki A, Inoue T, Oda M, Tanaka H, Morishima T. Clinical significance of decreased serum concentration of cartilage oligomeric matrix protein in systemic juvenile idiopathic arthritis. J Rheumatol. 2006;33(5):996–1000. [PubMed] [Google Scholar]
- 84.Bay-Jensen AC, Reker D, Kjelgaard-Petersen CF, Mobasheri A, Karsdal MA, Ladel C, Henrotin Y, Thudium CS. Osteoarthritis year in review 2015: soluble biomarkers and the BIPED criteria. Osteoarthritis Cartilage. 2016;24(1):9–20. doi: 10.1016/j.joca.2015.10.014. [DOI] [PubMed] [Google Scholar]
- 85.Wislowska M, Jablonska B. Serum cartilage oligomeric matrix protein (COMP) in rheumatoid arthritis and knee osteoarthritis. Clin Rheumatol. 2005;24(3):278–84. doi: 10.1007/s10067-004-1000-x. [DOI] [PubMed] [Google Scholar]
- 86.Koelling S, Clauditz TS, Kaste M, Miosge N. Cartilage oligomeric matrix protein is involved in human limb development and in the pathogenesis of osteoarthritis. Arthritis Res Ther. 2006;8(3):R56. doi: 10.1186/ar1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lohmander LS, Saxne T, Heinegard DK. Release of cartilage oligomeric matrix protein (COMP) into joint fluid after knee injury and in osteoarthritis. Ann Rheum Dis. 1994;53(1):8–13. doi: 10.1136/ard.53.1.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Neidhart M, Hauser N, Paulsson M, DiCesare PE, Michel BA, Hauselmann HJ. Small fragments of cartilage oligomeric matrix protein in synovial fluid and serum as markers for cartilage degradation. Br J Rheumatol. 1997;36(11):1151–60. doi: 10.1093/rheumatology/36.11.1151. [DOI] [PubMed] [Google Scholar]
- 89.Sharif M, Kirwan JR, Elson CJ, Granell R, Clarke S. Suggestion of nonlinear or phasic progression of knee osteoarthritis based on measurements of serum cartilage oligomeric matrix protein levels over five years. Arthritis Rheum. 2004;50(8):2479–88. doi: 10.1002/art.20365. [DOI] [PubMed] [Google Scholar]
- 90.Neidhart M, Muller-Ladner U, Frey W, Bosserhoff AK, Colombani PC, Frey-Rindova P, Hummel KM, Gay RE, Hauselmann H, Gay S. Increased serum levels of non-collagenous matrix proteins (cartilage oligomeric matrix protein and melanoma inhibitory activity) in marathon runners, Osteoarthritis and cartilage / OARS. Osteoarthritis Research Society. 2000;8(3):222–9. doi: 10.1053/joca.1999.0293. [DOI] [PubMed] [Google Scholar]
- 91.Forsblad d'Elia H, Christgau S, Mattsson LA, Saxne T, Ohlsson C, Nordborg E, Carlsten H. Hormone replacement therapy, calcium and vitamin D3 versus calcium and vitamin D3 alone decreases markers of cartilage and bone metabolism in rheumatoid arthritis: a randomized controlled trial [ISRCTN46523456] Arthritis Res Ther. 2004;6(5):R457–68. doi: 10.1186/ar1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lindqvist E, Eberhardt K, Bendtzen K, Heinegard D, Saxne T. Prognostic laboratory markers of joint damage in rheumatoid arthritis. Ann Rheum Dis. 2005;64(2):196–201. doi: 10.1136/ard.2003.019992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Momohara S, Yamanaka H, Holledge MM, Mizumura T, Ikari K, Okada N, Kamatani N, Tomatsu T. Cartilage oligomeric matrix protein in serum and synovial fluid of rheumatoid arthritis: potential use as a marker for joint cartilage damage. Mod Rheumatol. 2004;14(5):356–60. doi: 10.1007/s10165-004-0323-4. [DOI] [PubMed] [Google Scholar]
- 94.Skoumal M, Kolarz G, Klingler A. Serum levels of cartilage oligomeric matrix protein. A predicting factor and a valuable parameter for disease management in rheumatoid arthritis. Scand J Rheumatol. 2003;32(3):156–61. doi: 10.1080/03009740310002498. [DOI] [PubMed] [Google Scholar]
- 95.Weitoft T, Larsson A, Saxne T, Ronnblom L. Changes of cartilage and bone markers after intra-articular glucocorticoid treatment with and without postinjection rest in patients with rheumatoid arthritis. Ann Rheum Dis. 2005;64(12):1750–3. doi: 10.1136/ard.2004.035022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wollheim FA, Eberhardt KB, Johnson U, Saxne T. HLA DRB1* typing and cartilage oligomeric matrix protein (COMP) as predictors of joint destruction in recent-onset rheumatoid arthritis. Br J Rheumatol. 1997;36(8):847–9. doi: 10.1093/rheumatology/36.8.847. [DOI] [PubMed] [Google Scholar]
- 97.Mansson B, Gulfe A, Geborek P, Heinegard D, Saxne T. Release of cartilage and bone macromolecules into synovial fluid: differences between psoriatic arthritis and rheumatoid arthritis. Ann Rheum Dis. 2001;60(1):27–31. doi: 10.1136/ard.60.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wagner E, Ammer K, Kolarz G, Krajnc I, Palkonyai E, Scherak O, Schodl C, Singer F, Temesvari P, Wottawa A. Predicting factors for severity of rheumatoid arthritis: a prospective multicenter cohort study of 172 patients over 3 years. Rheumatol Int. 2007;27(11):1041–8. doi: 10.1007/s00296-007-0343-4. [DOI] [PubMed] [Google Scholar]
- 99.H ED. Clinical features of rheumatoid arthritis. Philadelphia. 2005 [Google Scholar]
- 100.Gheita TA, Hussein H. Cartilage Oligomeric Matrix Protein (COMP) in systemic sclerosis (SSc): role in disease severity and subclinical rheumatoid arthritis overlap. Joint, bone, spine : revue du rhumatisme. 2012;79(1):51–6. doi: 10.1016/j.jbspin.2011.02.022. [DOI] [PubMed] [Google Scholar]
- 101.Andersson ML, Thorstensson CA, Roos EM, Petersson IF, Heinegard D, Saxne T. Serum levels of cartilage oligomeric matrix protein (COMP) increase temporarily after physical exercise in patients with knee osteoarthritis. BMC Musculoskelet Disord. 2006;7:98. doi: 10.1186/1471-2474-7-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mundermann A, Dyrby CO, Andriacchi TP, King KB. Serum concentration of cartilage oligomeric matrix protein (COMP) is sensitive to physiological cyclic loading in healthy adults, Osteoarthritis and cartilage / OARS. Osteoarthritis Research Society. 2005;13(1):34–8. doi: 10.1016/j.joca.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 103.Kim HJ, Lee YH, Kim CK. Biomarkers of muscle and cartilage damage and inflammation during a 200 km run. Eur J Appl Physiol. 2007;99(4):443–7. doi: 10.1007/s00421-006-0362-y. [DOI] [PubMed] [Google Scholar]
- 104.Fang C, Johnson D, Leslie MP, Carlson CS, Robbins M, Di Cesare PE. Tissue distribution and measurement of cartilage oligomeric matrix protein in patients with magnetic resonance imaging-detected bone bruises after acute anterior cruciate ligament tears. J Orthop Res. 2001;19(4):634–41. doi: 10.1016/S0736-0266(00)00039-5. [DOI] [PubMed] [Google Scholar]
- 105.Rockey DC, Bell PD, Hill JA. Fibrosis--a common pathway to organ injury and failure. N Engl J Med. 2015;372(12):1138–49. doi: 10.1056/NEJMra1300575. [DOI] [PubMed] [Google Scholar]
- 106.Agarwal P, Zwolanek D, Keene DR, Schulz JN, Blumbach K, Heinegard D, Zaucke F, Paulsson M, Krieg T, Koch M, Eckes B. Collagen XII and XIV, new partners of Cartilage oligomeric matrix protein in the skin extracellular matrix suprastructure. J Biol Chem. 2012;287(27):22549–59. doi: 10.1074/jbc.M111.335935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Agarwal P, Zwolanek D, Keene DR, Schulz JN, Blumbach K, Heinegard D, Zaucke F, Paulsson M, Krieg T, Koch M, Eckes B. Collagen XII and XIV, new partners of cartilage oligomeric matrix protein in the skin extracellular matrix suprastructure. The Journal of biological chemistry. 2012;287(27):22549–59. doi: 10.1074/jbc.M111.335935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Agarwal P, Schulz JN, Blumbach K, Andreasson K, Heinegard D, Paulsson M, Mauch C, Eming SA, Eckes B, Krieg T. Enhanced deposition of cartilage oligomeric matrix protein is a common feature in fibrotic skin pathologies. Matrix biology : journal of the International Society for Matrix Biology. 2013;32(6):325–31. doi: 10.1016/j.matbio.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 109.Vuga LJ, Milosevic J, Pandit K, Ben-Yehudah A, Chu Y, Richards T, Sciurba J, Myerburg M, Zhang Y, Parwani AV, Gibson KF, Kaminski N. Cartilage oligomeric matrix protein in idiopathic pulmonary fibrosis. PloS one. 2013;8(12):e83120. doi: 10.1371/journal.pone.0083120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Barrett T, Suzek TO, Troup DB, Wilhite SE, Ngau WC, Ledoux P, Rudnev D, Lash AE, Fujibuchi W, Edgar R. NCBI GEO: mining millions of expression profiles--database and tools. Nucleic Acids Res. 2005;33(Database issue):D562–6. doi: 10.1093/nar/gki022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Martinez FJ, Collard HR, Pardo A, Raghu G, Richeldi L, Selman M, Swigris JJ, Taniguchi H, Wells AU. Idiopathic pulmonary fibrosis. Nat Rev Dis Primers. 2017;3:17074. doi: 10.1038/nrdp.2017.74. [DOI] [PubMed] [Google Scholar]
- 112.Farina G, Lemaire R, Korn JH, Widom RL. Cartilage oligomeric matrix protein is overexpressed by scleroderma dermal fibroblasts. Matrix biology : journal of the International Society for Matrix Biology. 2006;25(4):213–22. doi: 10.1016/j.matbio.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 113.Hesselstrand R, Wildt M, Bozovic G, Andersson-Sjoland A, Andreasson K, Scheja A, Westergren-Thorsson G, Bjermer L, Wuttge DM. Biomarkers from bronchoalveolar lavage fluid in systemic sclerosis patients with interstitial lung disease relate to severity of lung fibrosis. Respir Med. 2013;107(7):1079–86. doi: 10.1016/j.rmed.2013.03.015. [DOI] [PubMed] [Google Scholar]
- 114.Novo E, Cannito S, Paternostro C, Bocca C, Miglietta A, Parola M. Cellular and molecular mechanisms in liver fibrogenesis. Arch Biochem Biophys. 2014;548:20–37. doi: 10.1016/j.abb.2014.02.015. [DOI] [PubMed] [Google Scholar]
- 115.Magdaleno F, Arriazu E, Ruiz de Galarreta M, Chen Y, Ge X, Conde de la Rosa L, Nieto N. Cartilage oligomeric matrix protein participates in the pathogenesis of liver fibrosis. J Hepatol. 2016;65(5):963–971. doi: 10.1016/j.jhep.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schulz JN, Nuchel J, Niehoff A, Bloch W, Schonborn K, Hayashi S, Kamper M, Brinckmann J, Plomann M, Paulsson M, Krieg T, Zaucke F, Eckes B. COMP-assisted collagen secretion--a novel intracellular function required for fibrosis. J Cell Sci. 2016;129(4):706–16. doi: 10.1242/jcs.180216. [DOI] [PubMed] [Google Scholar]
- 117.Englund E, Canesin G, Papadakos KS, Vishnu N, Persson E, Reitsma B, Anand A, Jacobsson L, Helczynski L, Mulder H, Bjartell A, Blom AM. Cartilage oligomeric matrix protein promotes prostate cancer progression by enhancing invasion and disrupting intracellular calcium homeostasis. Oncotarget. 2017;8(58):98298–98311. doi: 10.18632/oncotarget.21176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Liberti MV, Locasale JW. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem Sci. 2016;41(3):211–8. [Google Scholar]
- 119.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lynch JM, Maillet M, Vanhoutte D, Schloemer A, Sargent MA, Blair NS, Lynch KA, Okada T, Aronow BJ, Osinska H, Prywes R, Lorenz JN, Mori K, Lawler J, Robbins J, Molkentin JD. A thrombospondin-dependent pathway for a protective ER stress response. Cell. 2012;149(6):1257–68. doi: 10.1016/j.cell.2012.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Towbin JA, Lowe AM, Colan SD, Sleeper LA, Orav EJ, Clunie S, Messere J, Cox GF, Lurie PR, Hsu D, Canter C, Wilkinson JD, Lipshultz SE. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA. 2006;296(15):1867–76. doi: 10.1001/jama.296.15.1867. [DOI] [PubMed] [Google Scholar]
- 122.Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB A. American Heart, H.F. Council on Clinical Cardiology, C. Transplantation, C. Quality of, R. Outcomes, G. Functional, G. Translational Biology Interdisciplinary Working, E. Council on, Prevention, Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113(14):1807–16. doi: 10.1161/CIRCULATIONAHA.106.174287. [DOI] [PubMed] [Google Scholar]
- 123.Brancaccio M, Hirsch E, Notte A, Selvetella G, Lembo G, Tarone G. Integrin signalling: the tug-of-war in heart hypertrophy. Cardiovasc Res. 2006;70(3):422–33. doi: 10.1016/j.cardiores.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 124.Svensson L, Aszodi A, Heinegard D, Hunziker EB, Reinholt FP, Fassler R, Oldberg A. Cartilage oligomeric matrix protein-deficient mice have normal skeletal development. Mol Cell Biol. 2002;22(12):4366–71. doi: 10.1128/MCB.22.12.4366-4371.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]