

Abstract
Heterochromatin protein 1 (HP1), associated with heterochromatin formation, recognizes an epigenetically repressive marker, trimethylated lysine 9 in histone H3 (H3K9me3), and generally contributes to long-term silencing. How HP1 induces heterochromatin is not fully understood. Recent experiments suggested that not one, but two nucleosomes provide a platform for this recognition. Integrating previous and new biochemical assays with computational modeling, we provide near-atomic structural models for HP1 binding to the dinucleosomes. We found that the dimeric HP1α tends to bind two H3K9me3s that are in adjacent nucleosomes, thus bridging two nucleosomes. We identified, to our knowledge, a novel DNA binding motif in the hinge region that is specific to HP1α and is essential for recognizing the H3K9me3 sites of two nucleosomes. An HP1 isoform, HP1γ, does not easily bridge two nucleosomes in extended conformations because of the absence of the above binding motif and its shorter hinge region. We propose a molecular mechanism for chromatin structural changes caused by HP1.
Introduction
Heterochromatin protein 1 (HP1) is a nonhistone chromosome protein with versatile functions (1, 2, 3, 4), including heterochromatin formation, gene silencing in pericentric heterochromatin, control of gene expression, and stabilization of telomeres (1, 5). As such, HP1 is found in diverse eukaryotic organisms, with the exception of budding yeast. Heterochromatin spread is supposed to rely on the ability of HP1 self-association (6, 7, 8).
Related to its broad range of functions, HP1 has isoforms; for mammals, three isoforms, HP1α, HP1β, and HP1γ, have been identified (2, 4). Despite the apparent similarity in sequences and architectures of the three isoforms, they are known to show different localizations, and their deletions lead to different phenotypes (9, 10, 11). HP1α localizes to heterochromatic regions, whereas HP1β is observed in both heterochromatic and euchromatic regions. HP1γ, but not HP1α and HP1β, associates with actively transcribed genes and plays a role in the transcription elongation (12, 13). Despite HP1’s broad range of functions and properties, little is known about the molecular mechanisms that realize these versatile functions.
HP1 consists of two highly conserved globular domains, the chromo-domain (CD) and chromoshadow-domain (CSD), which are linked by a less-conserved disordered hinge region (HR) of various lengths (Fig. 1 A). The CD is known to recognize the trimethylated lysine 9 in the histone H3 tail (H3K9me3), a major epigenetic marker of heterochromatin (14, 15). The CSD forms homo- or heterodimers with CSDs of the same or different isoforms of HP1 (16, 17). Although HP1 has been considered to function as a dimer (18), the isoform-specific oligomerization of HP1 has also been reported. Fission yeast HP1 (swi6), as well as human HP1α, is dimerized via CD and is tetramerized (18, 19), whereas HP1β has been revealed to be only dimerized via CSD (20). The dimeric CSD also provides a platform for the assembly of other related proteins, thus working as a hub-like protein (21). The hinge region is enriched with positively charged amino acid residues and disordered (22), which makes HP1 a highly flexible molecule. Notably, the affinity of the CD to an H3K9me3 peptide is rather weak, which implicates more collaborative recognition involving other parts of molecules. Indeed, much evidence shows that the hinge region of HP1 binds to DNA in a non-sequence-specific manner, which may facilitate HP1 loading to chromatin (20, 23, 24, 25). Mouse HP1 (CBX1, 3, and 5) binds not only to a methylated histone tail, but also to the histone-fold domain of histone H3 (17).
Figure 1.

Schematic view of HP1 constructs. (A) The HP1 forms the homodimer via CSD (depicted in green) and recognizes the trimethylated H3K9 (shown as “me3”) via the CD (blue). Between the two globular domains, a disordered hinge region is present. (B) A mononucleosome without linker DNA is shown. One CD is bound to K9 of one H3, and the other H3 is termed “A-distal H3.” (C) A mononucleosome with two linker DNAs of 25 bp connected to both ends of nucleosomal DNA is shown. (D) A dinucleosome with linker DNA of 25 bp and one CD bound to K9 in a proximal H3 of nucleosome A (designated as “A-prox-bound”) is shown. An unbound CD is termed “free CD.” The other three H3 tails are labeled. (E) A dinucleosome with one CD bound to K9 in a distal H3 of nucleosome A (“A-dist-bound”) is shown. To see this figure in color, go online.
Although the molecular architecture of HP1 and the domain-specific functions have been relatively well characterized, how the biochemical features of HP1 lead to versatile phenotypes is largely unknown. This is partly due to the limited number of studies concerning the interaction between nucleosomes and HP1 (6, 18, 20, 24, 25, 26, 27, 28, 29). Recently, energy transfer using fluorescently labeled nucleosomes and HP1 has elucidated stochastic HP1-chromatin interaction events (28, 29). Electron microscopy observation shows that HP1 clusters on the nucleosomal array (6). How two CDs of HP1 dimer consequently recognize K9me3 on the chromatin structure is crucial for heterochromatin expansion. However, this point has not been revealed. We found that the HP1α dimer binds to a dinucleosome containing H3K9me3 with a high affinity, but not to a mononucleosome containing H3K9me3 (24). Notably, once linker DNA is attached to both ends of the mononucleosome, it interacts with HP1α with a higher affinity than in the case without the linker DNA. The affinities with HP1α are at the same level between di- and tetranucleosomes with the H3K9me3 marker. These together suggest that the dinucleosome provides a basic platform for the HP1α dimer to bind on. Remarkably, HP1γ does not strongly bind to an extended form of tetranucleosome to which HP1α binds strongly (26). When the tetranucleosome took a more compact form, HP1γ recognized the compact tetranucleosome with a high affinity. This provides us a hint for the phenotype differences among isoforms.
However, biochemical assays alone do not provide high-resolution structural insights. For that purpose, structural biology methods may be desired. Yet, because of the highly disordered nature of HP1 and histone tails, standard structural biology methods have not provided high-resolution information, either. As such, computer simulations are potentially useful to obtain high-resolution dynamical structures. But, for this size of molecule that involves large fluctuations, straightforward atomistic molecular dynamics (MD) simulations are not efficient enough to sample a large conformational space. Given these facts, the mesoscopic Monte Carlo simulation approaches pioneered by Langowski, Schlick and others have opened up structural modeling studies of polynucleosome systems (30, 31, 32, 33, 34, 35). More recently, multiscale MD simulations have also been applied to provide structural and dynamical insights into nucleosomes and their interaction with other proteins (36, 37, 38, 39, 40, 41). Notably, computer simulations for this size of molecule always need to be verified experimentally.
In this study, using multiscale molecular simulations and biochemical assays, we clarified binding modes of HP1 with mono- and dinucleosomes, from which we obtained a near-atomic resolution structural model of the HP1-dinucleosome complex. We also tested the current computational results by biochemical experiments.
Materials and Methods
Biochemical experiments expression and purification of proteins
The cDNA of human wild-type HP1α and γ or mutated HP1 were subcloned from pGEX-6P1, as described by Mishima et al. (26). Full-length and mutagenized HP1 were expressed and purified as described (26). Recombinant histones and H3K9me3 were prepared as described elsewhere (24).
Preparation of dinucleosomes
The 601.2 tandemly repeated DNA templates with 25-bp linker DNA are shown in Fig. S1. Nucleosomes were reconstituted with histone octamers prepared with recombinant histones and the template by a salt-dialysis method (42, 43). The reconstituted dinucleosomes were purified by glycerol density-gradient centrifugation as described (42). The concentrations of dinucleosomes were determined by measuring the absorbance at 260 nm.
Pull-down assay
The binding activity of HP1 to dinucleosomes was determined as described by Mishima et al. (26). Briefly, 80 pmol of glutathione S-transferase (GST), GST-HP1α (wild-type (WT) or mutant (mut)), and GST-HP1γ bound gluthathione Sepharose (GE Healthcare, Little Chalfont, UK) of 10 μL packed volume was mixed with 1 pmol reconstituted nucleosomes in 20 μL of a binding buffer comprising 50 mM NaCl, 0.2 mM dithiothreitol, 0.2 mM phenylmethylsulfonyl fluoride, 1 mM ethylenediaminetetraacetic acid, 0.1% (v/v) Nonidet P-40, 20% (v/v) glycerol, and 20 mM Tris-HCl (pH 7.4). The input, unbound, wash, and bound fractions were subjected to sodium dodecyl sulfate-polyacrylamide gel (SDS-PAGE), and proteins were fluorescently detected by using Lumitein (Biotium, Fremont, CA). The density of histone bands was quantitated with Image Gauge V4.0 software (GE Healthcare).
Gel shift assay
GST-HP1 (1, 2, or 4 μM) was mixed with 25 nM of mononucleosomal DNA without linker, of length 147 bp, in the binding buffer. After the incubation, the mixtures were electrophoresed in a 0.7% agarose gel with 0.5× Tris-borate-ethylenediaminetetraacetic acid, and then DNA bands were stained with GelGreen (Biotium) and quantitated with a fluoro-imager, FLA9500 (GE Healthcare, Massachusetts), as described by Mishima et al. (26).
Reference structures for computer modeling
For nucleosome structures, we refer to the x-ray crystallographic structures, for which the Protein Data Bank (PDB) code is 1KX5 (44). Combining the structures with the 25-bp-long standard B-type DNA model, we generated dinucleosome structures. The native structures for the CD and CSD were taken from 3FDT and 3I3C, respectively. The complex structure of the CD with the H3K9me3-containing peptide was also taken from 3FDT (45).
Coarse-grained MD simulations
We briefly describe the simulations method here. We used a total energy function that consists of three terms,
where the first, the second, and the third terms represent potential energies for proteins, DNA, and the interaction between them, respectively.
For proteins, we utilized the atomic interaction-based CG model 2+ (AICG2+) developed by Li et al. (46, 47):
Here, the first term means virtual bonds between adjacent amino acids, and the second term represents the sequence-dependent bond-angle and dihedral-angle potentials defined below. The third and fourth terms are structure-based local potentials giving biases to the native local structures. The fifth term is the native contact potential that binds amino acid pairs that are found in the native structures. The last term is a simple short-ranged repulsion. The third term, , is a sum of the two contributions: the bond-angle potential and the dihedral-angle potential, both of which were obtained via the Boltzmann inversion of the probability distributions in a loop library from the PDB, specifically,
where and are the probability distributions of the corresponding variables estimated from a loop library of PDB structures. See the original work for a more detailed description of the AICG2+ potential (46). For intrinsically disordered regions, we did not include the structure-based term and thus used only the first, the second, and the last terms. For histones, the N-terminal 43, 23, 15, and 33 residues in H3, H4, H2A, and H2B were treated as the intrinsically disordered regions. For HP1, we set residues 1–19, 69–120, and 181–191 as disordered regions.
For DNA, we used the 3SPN.1 model developed by the de Pablo group (48):
See the original work for more details.
For protein-DNA interactions, we defined the following energy function,
where the first term is for the specific interaction imposed between nucleosomal DNA and histones, whereas the second and the third terms are general physical-chemical interactions; the second term is a simple short-range repulsion, and the third term is the Debye-Hückel electrostatic interactions. We chose . The value was determined to be 0.8× the default contact energy for proteins (0.3) for mononucleosomes in the previous work (49). Integer charges were set for each CG particle: +1 for Arg and Lys and −1 for Asp, Glu, and P (the phosphate group). The dielectric constant is set as ε = 78. The Debye screening length is calculated as usual, depending on the in silico ionic strength.
The equation of motion that drives the system is the standard Langevin equation,
where the random noise is the Gaussian white noise with the mean and variance
respectively. is the Boltzmann constant, and T is the temperature set as 300 K. γ is the friction coefficient, and we used a low friction (0.02 in CafeMol unit (50)) to speed up the dynamics. Each MD trajectory contains 108 steps starting from the above-mentioned extended configuration.
For MD, we used the CG MD software CafeMol developed in our group throughout the work (50). This software is freely available at http://www.cafemol.org, and this work is reproducible by CafeMol 2.1. Unless otherwise denoted, we used the default parameter values in CafeMol.
Reverse-mapping from a coarse-grained model to a fully atomistic model
For protein, we constructed a fully atomistic model by a one-to-one threading method of the PHYRE2 modeling server (51). For DNA, we modeled the fully atomistic structure by superposing fully atomistic nucleotides on each coarse-grained nucleotide and replacing the coarse-grained model with the fully atomistic model. Fully atomistic nucleotides (dAMP, dGMP, dTMP, dCTP) were obtained from a B-type DNA structure generated by NAB (52, 53), and we superposed three points (center-of-mass of sugar, center-of-mass of phosphate, N1 or N5 atom) on the corresponding coarse-grained particle. When we modeled the 5′ nucleotide, we used the center-of-mass of sugar of 5′ nucleotide, the N1 or N5 position of the nucleotide, and the center-of-mass of the phosphate of the adjacent nucleotide for superposition.
Fully atomistic MD simulations
MD simulations were conducted by GROMACS 5.1.1 (54, 55, 56). The simulation system was a triclinic box with periodic boundary conditions, including proteins, DNA, 350,048 water molecules, 1031 Na+ ions, and 679 Cl− ions. Ions were added to neutralize the total charge and to set ion concentration to ∼0.1 M. We used the Amber99sb force field with parmbsc0 parameters and the TIP3P water model (57, 58, 59, 60, 61). We used particle mesh Ewald methods (62) for electrostatic interactions with a 1.0-nm short-range cutoff. The cutoff for van der Waals interactions was 1.0 nm. First, we conducted energy minimization by the steepest-descent minimization algorithm. Second, we conducted NVT equilibration at 300 K for 100 ps. Next, we conducted NPT equilibration at 300 K, 1 bar for 1 ns. Finally, the production run was conducted at 300 K, 1 bar for 30 ns. In the equilibration steps, we restrained the position of all heavy atoms. In the NVT equilibration, NPT equilibration, and production run, all covalent bonds were constrained by Linear Constraint Solver (LINCS) (63). For the water molecules, we used SETTLE (64). For the thermostat, we used the velocity rescaling method (65). For the NPT equilibration and production run, the Parrinello-Rahman barostat was used (66).
Analysis
We used the gmx hbond program for counting the number of hydrogen bonds (54, 55). To find atom pairs forming a hydrogen bond, we used the gmx hbond and inspected visually.
Results
HP1α binding to dinucleosome: biochemical experiments
By using the extended dinucleosomes reconstituted in vitro as binding substrates, we have recently reported that HP1α specifically binds to nucleosomes reconstituted with H3K9me3 (24). In this construct, two nucleosome core particles are connected by 20-bp linker DNA. If this linker DNA takes the standard B-type duplex DNA shape with one turn of 10.4 bp, the histone H3 tails on the two nucleosome core particles face almost the same direction (Figs. S1 and S2). To investigate the effect of the rela`tive orientation of two nucleosomes, we made a construct with the length of linker DNA increased by 5 bp. By using the DNA template with 25-bp linker DNA, we have reconstituted dinucleosomes (Fig. S3) and validated the nucleosomes with restriction enzymes and micrococcal nuclease (Fig. S3, B and C). We then examined the interaction between the dinucleosome and the beads on which GST or GST-HP1α bound (Fig. S4). The dinucleosome reconstituted with H3K9me3 selectively bound to GST- HP1α (Fig. 2 A and B). As a control, the dinucleosome could not be recovered on GST bound on beads (Fig. 2 B), irrespective of H3K9 methylation status. The binding activity was comparable to the reported results using dinucleosomes with 20-bp linker DNA (24), suggesting that HP1α did not recognize the relative rotational positioning of core particles. To test a possible artifact of the GST tag, we used Flag-tagged HP1α (F-HP1α), finding that it significantly bound to dinucleosomes with 25-bp linker DNA in H3K9me3 dependent manner (Fig. S5).
Figure 2.
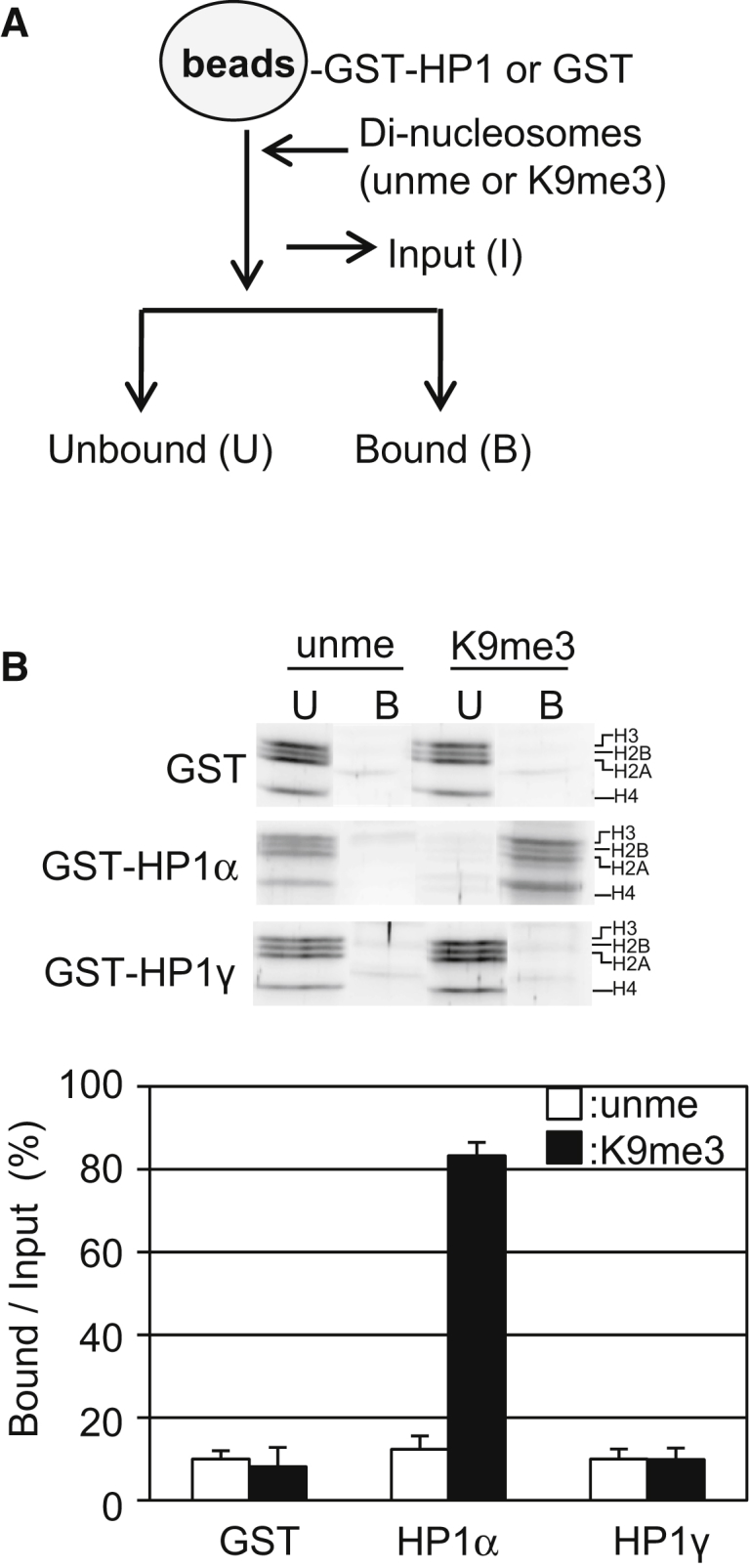
Biochemical assay of HP1 binding to dinucleosomes with 25-bp linker DNA. (A) A schematic illustration of the pull-down assay is given. The number of dinucleosomes bound to GSH Sepharose on which GST-HP1 or GST were anchored were analyzed. (B) HP1 isoform-specific dinucleosome binding activity is shown. Dinucleosomes reconstituted with unmethylated H3 (unme) or H3K9me3(K9me3) were mixed with the beads, and the unbound (U) and bound (B) fractions were separated and then subjected to SDS-PAGE (upper panel). SDS-PAGE data are taken from the whole gel image shown in Fig. S11. The amounts of each histone in the bound fractions over input (%) are shown as mean ± standard error (n = 3) (lower panel).
Modeling the HP1-dinucleosome complex by simulations
Both HP1 and dinucleosomes are highly flexible molecules containing intrinsically disordered regions, and therefore computational modeling is useful to characterize their structural assembly. Yet conventional fully atomistic MD simulations are currently too time-consuming for these large systems. Thus, here we used a coarse-grained (CG) MD approach that we have been developing (46, 50, 67, 68).
In our CG MD approach, each amino acid in proteins is simplified as a single particle located at its Cα position, whereas each nucleotide in DNAs is represented by three particles, one each for phosphate, sugar, and base. For proteins, globular domains for which crystallographic structures are available are biased to their structures with fluctuations (46), whereas disordered tails are treated as flexible chains for which conformations depend on their amino acid sequences (69). We have previously examined and applied this model to several proteins, including protein complexes with DNA (46, 47, 49, 70). DNA was modeled to stabilize the standard B-type DNA with certain bending rigidity (48), which allows it to bend spontaneously as well as upon binding to proteins. At the nucleosome core, interactions between histone cores and nucleosomal DNA were tuned in our previous work (49) and are utilized here. Because HP1 is dimerized via its CSD domain with high affinity, we put attractive interactions between two CSD domains so that HP1 keeps the dimeric form throughout the simulations. Similarly, between the H3K9-trimethylated fragment in H3 tails and the CD of HP1, we put a structure-based attractive interaction so that these interactions are kept throughout the simulations. We note that this attractive interaction is added purely based on experimental knowledge: The CD domain has significant affinity to the H3K9-trimethylated fragment of H3 tails, and there exists a complex crystal structure of a close homolog of the CD domain with the H3K9-trimethylated fragment of H3 tail (45). In our current structural modeling, the strength of this attractive interaction would not affect the overall structure. For other parts, the interactions between HP1 and the dinucleosome are electrostatic interactions and steric repulsions. The time propagation in MD was conducted by the standard Langevin dynamics with the stochastic force.
We begin with a simulation setup of an HP1α dimer bound on a dinucleosome where one of the CDs in the HP1α dimer is bound to an H3 tail, of which K9 is assumed to be trimethylated (Figs. 1 D and 3 A). The other CD is unbound to the H3 tail (termed the free CD). The other three H3K9s are assumed to be unmethylated. (Note that we are interested in the case where all the four H3K9s in the dinucleosome are trimethylated. Here, to estimate the expected time that the free CD finds one of the H3K9, we used unmethylated H3K9s in this simulation. If all the H3K9s were trimethylated in the simulation, the free CD would be bound to one of the H3K9s first met in the simulation and would not sample any more. Assumed here is that the expected time of the free CD to approach the H3K9s is independent of the methylation state of their H3K9s.) For convenience, we designate the nucleosome that binds HP1 as nucleosome A and the other as nucleosome B. Of the two H3 tails in nucleosome A, we first chose the one that is close to the linker DNA for the docking to the CD domain and thus designate this setup as “bound CD on A-proximal H3 tail” (Fig. 3 A).
Figure 3.

Representative trajectories of HP1α on nucleosomes. (A) The construct used is shown. One CD is bound on the trimethylated “A-proximal H3K9,” and the other CD is free at the initial conformation. The other three H3K9s are assumed to be unmethylated. (B) Time courses of the distances from the free CD to three H3K9s are plotted. (C) A close-up view of a time window in (B) is shown, in which two snapshots depicted in (E) occur. (D) For a mononucleosome without linker DNA, the time course of the distance between the free CD and the A-distal H3K9 is shown. To see this figure in color, go online.
With this setup, we performed 10 independent MD simulations with stochastic forces at 300 K with the monovalent ion concentration 100 mM. During the simulations, we monitored distances between the three unmethylated H3K9s and the free CD (Fig. 3 A for the definition of the free CD; for the distances, we used the distance between the Glu52 of HP1 and Lys9 of H3 that are located at the binding sites). For a representative trajectory, the time series of the three distances are plotted in Fig. 3, B and C together with some snapshots in Fig. 3 E (video is available as Video S1). At the initial configuration (Fig. S6), the free CD domain was placed far from any of the three unmethylated H3K9s so that all the distances are large. After a quick decrease in these distances at the initial phase, they fluctuated largely in the range of 50–150 Ǻ. Very rarely, however, we see the free CD approach some of the unmethylated H3K9.
Focusing on these rare approaches of the free CD to the unmethylated H3K9, we obtained histograms of the distances between H3K9 and the free CD at a short-distance range (Fig. 4 A). To avoid possible bias due to an arbitrary initial structure, in each of the 10 trajectories, we only used data obtained in the second half of the stimulation time for statistical analysis. We see in the figure that, of the three unmethylated H3K9s, the free CD preferentially approached the K9 in the proximal H3 tail in nucleosome B (“B-proximal” black curve in Fig. 4 A). Notably, even though the H3K9 of nucleosome A that is distant from the linker DNA (“A-distal”) belongs to the same nucleosome as the H3K9 to which the bound CD is tethered, the free CD did not approach frequently to the “A-distal” H3K9. The third unmethylated H3K9, called “B-distal,” was rarely approached, too.
Figure 4.

Results of HP1α-dinucleosome molecular simulations in which one CD is bound on the trimethylated H3K and the other CD is free. (A) Statistics of the distances between the free CD and unmethylated H3K9s are shown. One CD is bound on the “A-proximal H3K9.” The red arrows show 6.6 Ǻ, corresponding to the distance at the complex crystal structure of the CD with an H3K9-trimethylated H3 tail fragment. (B) The case of “A-distal H3K9” (bottom) is shown. (C) The number of occurrences in which the distance was below 16.5 Ǻ is given. Results with larger and smaller cutoffs are given in Fig. S12. To see this figure in color, go online.
For statistics, we counted the events of approach of the free CD to unmethylated H3K9 within 2.5 × 6.6 = 16.5 Ǻ in the second half of the trajectory data (shown in Fig. 4 C), in which 6.6 Ǻ corresponds to the distance between HP1αE52 and H3K9 at the complex crystal structure (Fig. S7). Clearly, the approach events of the free CD were dominated by the “B-proximal” H3K9.
Next, we changed the trimethylated H3K9 site from the “A-proximal” H3 to the “A-distal” H3 and conducted the same kind of simulations and analysis as above (this setup termed the “bound CD on A-distal H3 tail”). The other three H3K9s are assumed to be unmethylated. Monitoring the three distances from the free CD to the unmethylated H3K9s, we obtained an apparently similar time series as above (Fig. S8). The distances largely fluctuated in the range of 50–150 Ǻ and only rarely decreased so that the free CD could interact with H3K9s. The histogram of the three distances depicted in Fig. 4 B shows that the free CD preferentially approached to “B-distal” H3K9. The approaches to the “A-proximal” and “B-proximal” H3K9s were less frequent. Using the same threshold as above, the number of approach events was the largest for the “B-distal” H3K9 (Figs. 1 E and 4 C). In the same way as above, the approach to the H3K9 that belongs to the same nucleosome as the H3K9 to which the bound CD is tethered was notably rare (“A-proximal”).
We then set up another simulation system in which all the H3K9s in the dinucleosome were trimethylated so that both of the CDs could bind to H3K9s (note that this setup is similar to Fig. 1 D, in which all the H3K9s in H3 tails (purple) are now regarded as trimethylated). We started the simulation with one CD bound on one of H3K9s and the other CD free, observing if and to where the free CD binds.
When the CD was bound on “A-proximal” H3K9 at the initial conformation, we found that, in 9 out of 20 runs, the free CD was bound to the “B-proximal H3K9.” The free CD was bound on the “B-distal H3K9” in 4 runs out of 20. Thus, for 13 of 20 cases, HP1α bridged the two nucleosomes. In only four runs, the free CD bound on the “A-distal H3K9” so that HP1α bound to two of the H3K9s in nucleosome A. For the other three cases, the free CD remained free by the end of simulations. Thus, the results are consistent with the earlier simulation setup in which only one of the H3K9 sites was trimethylated.
In summary, the comparative simulations that started with one CD bound on nucleosome A and the other CD free of binding suggest that the free CD preferentially approaches/binds to the H3K9 of nucleosome B, thus bridging the two nucleosomes.
Modeling HP1-mononucleosome complex by simulations
A previous in vitro experiment showed that HP1α binds to the H3K9 trimethylated dinucleosome more preferentially than its binding to the H3K9 trimethylated mononucleosome (24). Notably, for the case of the mononucleosome, the binding probability markedly increased with the linker DNA attached to both ends of nucleosomal DNA. This suggests roles of nonspecific interaction of HP1α to the naked dsDNA. Here, we exemplified these differences and sought a structural reasoning for them.
For this purpose, we conducted the same kind of simulations for HP1α bound to a mononucleosome with and without linker DNA (Fig. 1, B and C). For the case with the linker DNA, we connected a 25-bp linker DNA to each end of the nucleosomal DNA. In each construct, one H3K9 is trimethylated and one CD is bound at the initial condition. During MD simulations, we observed distances between the free CD and another H3K9 that was assumed to be unmethylated. A representative time course of the distance between the free CD and the unmethylated H3K9 is depicted in Fig. 3 D. Similar to the case of the dinucleosome, the distance fluctuated in the range of 50–150 Ǻ. We performed 10 independent MD runs, each containing 108 MD steps.
The number of approaches of the free CD to the unmethylated H3K9 was very rare for the mononucleosome without the linker DNA (Fig. 4 C). With the linker DNA, the number of approaches to the unmethylated H3K9 increased greatly but was still smaller than that for the case of dinucleosomes. The linker-DNA-dependent binding activity of HP1α to mononucleosomes is consistent with the biochemical analysis reported. With the H3K9 trimethylated dinucleosome, HP1α prefers bridging two nucleosomes. The same binding mode is not available with the mononucleosome, in which HP1α cannot effectively bind to the two H3K9s on a single nucleosome.
Interactions between HP1α and DNA: computational analysis
Although our molecular simulations gave consistent results with biochemical experiments, the underlying mechanisms were not clear yet. Here, we analyze in detail the physical interactions between HP1 and DNA in dinucleosomes. For the simulations of the HP1α-dinucleosome system in which one CD was bound on the trimethylated H3K9 in nucleosome A, we obtained the contact frequencies of every amino acid of HP1 to every nucleotide in the dinucleosome, averaged over snapshots in the second half of all 10 trajectory data (Fig. 5 A). Here, the contact was defined by the approach of the amino acid within 10 Ǻ to one of the nucleotide particles.
Figure 5.

HP1α-DNA interactions in molecular simulations. (A) The horizontal axis indicates the DNA base pair index of dinucleosome, 1–147; nucleosome A, 148–172; and linker DNA, 173–319. The vertical axis means residues in the HP1α dimer, the bottom half for the bound HP1α and the top half for the free HP1α. Contact frequencies are represented by colors (see the right bar for the color definition). (B) The frequency of HP1α binding to every nucleotide in DNA is shown. To see this figure in color, go online.
Clearly, in Fig. 5 A, the major interactions were found in the linker DNA region, with some minor interactions near the dyad in nucleosome A, the position of which is ∼50–75-bp from the 5′ end. The latter’s minor interaction region is located on the entry/exit site of nucleosome structure, which is close to the position of the histone H3 tail.
Among the amino acids in HP1α, five regions showed clear interactions with DNA: the first region, around residue 5, is in the N-terminal tail; the second region, around residue 30, is in the CD; and those around residues 70, 90, and 105 are within the disordered hinge region. Both the sequences KKTKRT at 3–8 and DRRVVK at 27–32 are basic and thus are expected to be attracted to DNA in a non-sequence-specific manner. In these two regions of the two HP1α molecules, only the first monomer showed significant contacts with DNA.
More notable interactions are found in the three segments in the hinge region of HP1α that interacted with DNA; their sequences are KYKKMK at 69–74, SNKRKS at 87–92, and KSKKKR at 102–107, highly positively charged. This strong nonspecific interaction in the hinge region with DNA is consistent with earlier experimental reports: in more detail, of the three segments, the latter two have been investigated based on their high sequence conservation among HP1 isoforms (24, 25, 71). However, the first one, whose sequence is not conserved, has not been characterized before. These segments in the hinge region interacted primarily with the linker DNA, with some minor interactions with the near-dyad region of DNA. Notably, not only the hinge region in the bound HP1α but also that in the free HP1α showed marked interaction with the linker DNA. With these interactions, both of the hinge regions are near the linker DNA, and thus the free HP1α must be directed toward nucleosome B. Naturally, the free CD approaches most frequently to the “B-proximal” H3K9, as we observed in our comparative simulations above. For another construct in which the CD is bound on the distal H3 of nucleosome A, we found rather similar interaction patterns (Fig. 5 B).
Interactions at the N-terminal region KKTKRT at 3–8 with DNA are consistent with the report that the N-terminal region of HP1α enhances the DNA binding activity of the hinge region (25). Recently, it was reported that this segment KKKTKR intramolecularly interacts with the following acidic segment, EDEEE at 15–19, and Tyr20/Val21 in a recombinant HP1α that contains CD and the N-terminal tail (72). In this study, this intramolecular interaction was not observed in the presence of nucleosomes, indicating that KKKTKR dominantly binds to DNA when HP1α binds to nucleosomes. Together, these results suggest that the basic segment contributes to the binding of HP1α with multilayered regulations.
Thus, in summary, when one CD is bound on nucleosome A with the trimethylated H3K9, the preferential approach of the free CD to nucleosome B can be understood by the electrostatic attractions between basic segments of the hinge region of HP1 and the linker DNA. Interactions found in the current simulations are consistent with previous experiments. On top of this, we found what is to our knowledge a new interaction at KYKKMK at 69–74, which will be assessed biochemically below.
Interactions between HP1α and DNA: biochemical assays
Of the three basic segments in the hinge region of HP1, two patches, KKK around residue 105 and KRK around residue 90, have been investigated (24, 25, 71), because the patches are conserved among HP1 isoforms (Fig. S9 A). These two patches are crucial for the nucleosome binding to H3K9me3 in a specific manner in vitro (24) and for the intranuclear localization in vivo (71). Here, we examine the newly found third basic patch around residue 70 suggested in Fig. 5 A, KKYKKMK, which is located at vicinity of the C-terminal end of CD and is specific to HP1α but not to other isoforms.
To verify the possible function of these basic amino acids on the interaction between HP1α and nucleosomes, we prepared several HP1α mutants (Fig. 6 A; Fig. S4) and examined their binding activity (Fig. 6). The binding activity of GST-HP1α 1mut, in which lysine at 68 (K68) was substituted with alanine, to the dinucleosome was significantly reduced (Fig. 6 B) compared with WT HP1α (Fig. 2 B). When two mutations, K68A and K69A, were simultaneously introduced in GST-HP1α (GST-HP1α 2mut), the amount of dinucleosome recovered in the bound fraction was significantly reduced, irrespective of K9 methylation status (Fig. 6 B). The GST-HP1α 3mut in which K68, 69, 71, and 72 were substituted with alanine did not bind to dinucleosomes (Fig. 6 B).
Figure 6.

Biochemical assays of HP1α mutations at basic regions in the hinge region. (A) The amino acid sequence of HP1α is given. The region corresponding to CD is underlined, and the mutation sites (K68, K69, K71, and K72) are shown as bold blue letters. The amino acid sequences of three mutants, 1mut (K68A), 2mut (K68A and K69A), and 3mut (K68A, K69A, K71A, and K72A) are also shown. (B) The binding activities of mutated HP1α to dinucleosomes are shown. The binding activity was examined as described in Fig. 2A. The activity of WT HP1α is taken from Fig. 2B. (C) The DNA binding activities of WT and mutated HP1α are shown. Naked DNA was incubated without protein (lane 1), with GST-WT HP1α (WT) (lanes 2–4), 1mut (lanes 5–7), 2mut (lanes 8–10), 3mut (lanes 11–13), or GST (lanes 14–16). To the DNA, 40 (lanes 2, 5, 8, 11, and 14), 80 (lanes 3, 6, 9, 12, and 15), or 160 (lanes 4, 7, 10, 13, and 16) molar excess of protein was added, and a gel retardation assay was performed. The arrowhead and bracket indicate the position of free DNA and shifted DNA, respectively. (D) The sequence of HP1γ is given. The CD is underlined, and the mutation sites are shown as bold blue letters. The mutated sequence is denoted in red. (E) Dinucleosomes reconstituted with unmethylated H3 (unme) or H3K9me3(K9me3) were mixed with the beads, and the binding assay was performed as described in Fig. 2B. Three unbound (U) and bound (B) fractions were separated and then subjected to SDS-PAGE, and each histone is indicated. The experiments were repeated three times, and the binding activity was summarized in the right panel. The binding activity of GST-HP1γ WT is taken from Fig. 2B. To see this figure in color, go online.
In our previous study, mutations in basic amino acid patches (KRK or KKK) in the middle region of HP1α hinge region reduced the binding activity to naked DNA and to the nucleosome (24). Here, we examined the effect of the above three mutations on the naked-DNA binding activity by gel shift assay (Fig. 6 C). GST-HP1α 1mut showed significant DNA binding activity, but the activity was reduced (Fig. 6 C, lanes 5–7) compared with WT (Fig. 6 C, lanes 2–4). The binding activities of GST-HP1α 2mut and 3mut were below the detection level (lanes 8–10 and 11–13 in Fig. 6 C). In the absence of the GST tag, Flag-HP1α 3mut also did not show the binding activity to dinucleosomes, irrespective of K9 methylation status (Fig. S5). Together, as expected by computer simulation, the basic amino acids existing in the vicinity of the C-terminal region of CD are crucial for the binding to DNA and nucleosomes. Notably, these basic residues are not conserved among HP1 isoforms (Fig. S9 A), suggesting this region could contribute to isoform-specific binding. On the other hand, the basic residues of HP1α are conserved from fish to human (Fig. S9 B), supporting its biological relevance.
It has been reported that K69, K71, K72, K84, K104, and K106 in HP1α are identified to be potentially sumoylated in vitro (73). Maison et al. also reported that the sumoylation promotes initial targeting of HP1α to pericentromeric domains and consequently to seeding further HP1 localization, allowing HP1α to specifically associate with the RNA transcript (73). Accompanying our results, it is suggested that this basic patch could play crucial roles in vivo with multilayered regulations.
HP1γ cannot bridge the dinucleosome without compacting agents
We have reported that HP1γ, in contrast to HP1α, does not bind to extended tetranucleosomes with a linker DNA of 20 bp, even when H3K9 is trimethylated, unless additional compacting agents, such as linker histone H1 or Mg2+, are added (26). Here, using the dinucleosome with 25-bp linker DNA, we examined whether the same discrimination could be found. Indeed, we found that HP1γ did not bind to the dinucleosomes with a linker DNA length of 25 bp, irrespective of H3K9 methylation status (Fig. 2 B).
Next, we computationally addressed how HP1γ binding to the H3K9 trimethylated dinucleosome is different from that of HP1α. We conducted the same kind of simulations as above with HP1γ bound on the dinucleosome. Assuming that one CD can transiently bind to the trimethylated H3K9, we set up the simulation so that, in the same way as the case of HP1α, one CD is bound on the trimethylated K9 in the proximal H3 of nucleosome A, whereas the other CD is free to move. Using the second half of time series data from 10 trajectories, we obtained the histogram of the distances between the free CD and unmethylated H3K9 (Fig. 7, A and B) and the frequency of the approaches within 2.5 × 6.6 = 16.5 Ǻ of the unmethylated H3K9 (Fig. 7 C). Relative to the same plot for HP1α, the free CD of the HP1γ did not come as close to the H3K9 as the case of HP1α. The number of approaches within 16.5 Ǻ was markedly fewer. This was consistent with our previous experimental observation. Without the contact between the free CD and one H3K9, HP1γ transiently bound on the dinucleosome solely by the interaction between one CD and one H3K9 would not maintain this complex stably.
Figure 7.

Comparison between HP1γ and HP1α binding to dinucleosome in molecular simulations. (A) The results of HP1γ-dinucleosome simulations in which one CD is bound on the trimethylated K9 of the A-proximal H3 and the other CD is free are shown. The dashed line is the result of HP1α for comparison. (B) The results when one CD is bound on K9 of the A-distal H3 are given. (C) The number of occurrences in which the distance was below 16.5 Ǻ are given. To see this figure in color, go online.
Seeking mechanisms for this less-frequent approach, we performed the same type of interaction analysis of HP1γ with DNA in dinucleosomes as before. In the results depicted in Fig. S10, we find only two segments in the hinge region of HP1γ interacting with DNA, which is in contrast to the three segments of interactions found in HP1α. Of the three segments in HP1α, the first one, KYKKMK at 69–74, is not present in HP1γ, in which the corresponding sequence is NSQKAGK. This less positively charged segment in HP1γ did not interact with DNA significantly (Fig. S10). Thus, less frequent approaches of the free CD in HP1γ to the unmethylated H3K9 can be attributed partly to the absence of basic residues around 69–74 (number based on HP1α) and partly to the shorter length of the hinge region than in the case of HP1α.
To examine the role of KYKKMK at 69–74 in HP1α further, we constructed a mutant of HP1γ in which NSQKAG at 68–73 was replaced with KKYKKM (Fig. 6 D). The mutated HP1 was expressed and purified as described, and the pull-down assay was performed as described by Materials and Methods (Fig. S4 B). The mutated HP1γ partly recovered the binding activity (Fig. 6 E; Fig. S9), which confirms the importance of this positively charged segment in HP1α for the binding. Notably, the mutated region does not overlap the regions reported by Mishima et al. (26). Thus, it is suggested that this novel hinge region contributes to the binding together with the following hinge regions.
How dinucleosome structures are affected by HP1α
An in vitro assay showed that H3K9-trimethylated polynucleosome arrays become more compact upon addition of HP1 (27). Here, we test whether the binding of HP1α to an H3K9-trimethylated dinucleosome induces compaction or not. We estimated the distance between the centers of two nucleosomes for the three constructs: a dinucleosome without bound HP1, one with one CD of HP1α bound to the H3K9me3 in nucleosome A, and one with two CDs of HP1α bound to the H3K9me3 of nucleosomes A and B. Fig. 8 plots the histograms of the nucleosome-nucleosome distances for the three constructs. First, for all the cases, we find bimodal distributions: a population with a shorter distance contains partial docking of the two nucleosomes, and the other population, with a longer distance, corresponds to more extended conformations without internucleosome interactions (representative snapshots depicted in Fig. 8). We see that, when one CD is bound to nucleosome A, the population of the extended conformation increased by ∼20%. For this case, HP1 often is sandwiched by the two nucleosomes, which may stabilize this extended conformation. For the case in which two CDs of HP1 bridge the two nucleosomes, the population with partial docking of nucleosomes is larger than the case of only one CD bound on nucleosome A and is at a similar level to the case without bound HP1. By the direct bridge by HP1, extended conformations become less probable.
Figure 8.

Distributions of the distance between centers of two nucleosomes in molecular simulations. Dinucleosome without HP1 (green), dinucleosome with one CD of HP1 bound to A-proximal H3K9 (red), and dinucleosome with two CDs of HP1 bound to two nucleosomes (blue) are shown. To see this figure in color, go online.
In summary, we did not observe marked compaction of the dinucleosome upon HP1 binding in our computational modeling. At this moment, this is speculative and should be examined in future by biochemical experiments such as the sedimentation assay. As Azzaz et al. reported that human HP1α dimers promote the interactions in both intra- and internucleosome array (27), HP1 could bridge two chromatin fibers in trans to induce higher-order compaction when the concentration of nucleosomes is high.
Near-atomic structure model of HP1α bound to dinucleosomes
Finally, from a CG model of HP1 bound on dinucleosomes, we constructed an atomic resolution model and conducted a 30-ns-long atomistic MD simulation (a video is available as Video S2). For simplicity, we used unmodified lysine in H3K9; even with this simple modeling, the histone H3 tail kept bound to HP1 stably in this short simulation, and thus it would not affect the overall structures.
In the atomistic MD simulation, a hinge region of HP1 remains bound to DNA. The final structure of the simulation is shown at Fig. 9 A. HRs of HP1 contact the linker DNA. We found that lysine and arginine play important role for hinge-DNA interaction. Some of them form hydrogen bonds (Fig. 9 B), and lysine and arginine are generally near DNA (Fig. 9 A); the representative basic residues involved are K106 and R107, which coincide with experimentally identified key residues. The time course of formation of hydrogen bonds between the hinge region and linker DNA is shown in Fig. 9 C. The number of hydrogen bonds gradually increased. To sum up, HP1α-dinucleosome structure led to electrostatic contact between the hinge region and the linker DNA. These results are consistent with the CG simulation.
Figure 9.

Atomistic MD simulation of HP1 binding to dinucleosome system. (A) The final structure of a 30-ns atomistic MD simulation is shown. DNA is in light gray. Histone tails are in red and the other parts of histone proteins in pink. The CD of HP1 is in light green. The CSD of HP1 is in dark blue or cyan. Arginine and lysine in hinge region (K69-G120) of one of HP1 are in dark green. These regions are also indicated by arrows. The other parts of HP1 are in yellow. (B) A close-up view of interactions between HP1 hinge region and DNA is shown. Hydrogen bonds are shown by cyan dotted lines. Molecules are shown by cartoon model, except around the hydrogen bonding part, which is shown by stick model. For visibility, some important atoms are colored differently. Oxygen atoms are in red and nitrogen in blue. Hydrogen atoms are in white and phosphorus in orange. (C) The number of hydrogen bonds between HP1 hinge region and DNA is given. The hinge region we analyzed is one at a central and upper part in (A), whose arginine and lysine are colored by dark green. To see this figure in color, go online.
Discussion
In this study, employing a hybrid approach of multiscale computational modeling and biochemical experimental assays, we provide near-atomic structural models for HP1 binding to H3K9me3-containing dinucleosome. The previous biochemical assays suggested that the fundamental binding unit of the dimeric HP1 is not a mononucleosome, but a dinucleosome. Consistent with this, using CG molecular simulations, we found that the dimeric HP1α tends to bind two H3K9me3 that are in different nucleosomes, thus bridging two nucleosomes. By computational analysis, we found that this tendency was caused by electrostatic attractions between some positively charged residues in the disordered hinge region of HP1 and the linker DNA. By reverse-mapping from the CG model, we obtained a fully atomic model of the HP1 dimer bound to dinucleosome, showing several hydrogen bonds between the hinge regions and DNA. Biochemical mutation assays confirmed these site-specific interactions. Moreover, consistent with the previous experiment, our computational analysis clarified that HP1γ does not easily bridge two nucleosomes in extended conformations because of the lack of one basic segment in the hinge region and its shorter hinge region.
For the compaction of polynucleosome arrays with more than two nucleosomes, some population of two nonadjacent nucleosomes must be directly bridged by HP1, for which the compaction mechanism would be similar to that seen in the case with two CDs bound to two neighboring nucleosomes on a DNA double strand.
Recently, it has been reported that the phosphorylation on HP1 modulates its binding activity. Phosphorylation of HP1γ by PKA impairs its silencing activity (12), and phosphorylation in the N-terminal region of HP1α by casein kinase2 is crucial for the heterochromatin localization (74) and for its binding specificity (25). In contrast, HP1β phosphorylated at either S89 or S175 does not show compromised chromatin binding activity (75). For analyzing the effect of the posttranslational modification on the HP1 binding, the modeling described in this study could be useful, and this type of study will further elucidate its molecular mechanisms.
As the study was in the review process, Machida et al. reported the structure model of HP1 in complex with the H3K9 trimethylated dinucleosome obtained by cryogenic electron microscopy (cryoEM) (76). In the cryoEM model, homodimeric HP1α, HP1β, and HP1γ all bridge the two H3K9 trimethylated nucleosomes in the same way as we show here for HP1α. In the current work, however, HP1γ did not interact with the extended H3K9 trimethylated dinucleosome significantly. Moreover, the cryoEM structure model shows that the HP1 does not directly interact with the linker DNA, which is in sharp contrast to our model here. Although the molecular systems used and preparations in the two studies are similar, they are significantly different as well; notably, the length of the linker DNA is 15 bp in the cryoEM study and 25 bp in the current study. The cryoEM structure models do not identify all the domains in the HP1 dimer, which makes a detailed argument difficult. Possibly a short linker DNA enables the dimeric HP1 to bridge the two nucleosomes without interacting with the linker DNA. Further studies are required to figure out the effect of the linker DNA length on the interaction of HP1.
The current simulations revealed the dynamic and fragile nature of the HP1α dimer that bridges the dinucleosome. This dynamic view is consistent with a recent report that also appeared during the review process of this article (77).
Author Contributions
S.W., Y.M., M.S., and I.S. performed research and analyzed data. I.S. and S.T. designed the research and wrote the manuscript. M.S. contributed analytic tools.
Acknowledgments
This work was supported by the Ministry of Education, Culture, Sports, Science, and Technology Grants-in-Aid for Scientific Research (grants 15H01351, 16H01303, 25118509, 25251019, 26104517, and 15K14418); the Ministry of Education, Culture, Sports, Science, and Technology Strategic Programs for Innovative Research “Supercomputational Life Science”; and the Japan Agency for Medical Research and Development.
Editor: Tamar Schlick.
Footnotes
Shuhei Watanabe and Yuichi Mishima contributed equally to this work.
Supporting Materials and Methods, twelve figures, and two videos are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(18)30394-1.
Contributor Information
Isao Suetake, Email: hotsuetake@hotmail.com.
Shoji Takada, Email: takada@biophys.kyoto-u.ac.jp.
Supporting Material
This video shows a coarse-grained molecular simulation corresponding to Fig. 3.
This video shows a fully atomistic molecular simulation corresponding to Fig. 9.
References
- 1.Eissenberg J.C., Elgin S.C. HP1a: a structural chromosomal protein regulating transcription. Trends Genet. 2014;30:103–110. doi: 10.1016/j.tig.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nishibuchi G., Nakayama J. Biochemical and structural properties of heterochromatin protein 1: understanding its role in chromatin assembly. J. Biochem. 2014;156:11–20. doi: 10.1093/jb/mvu032. [DOI] [PubMed] [Google Scholar]
- 3.Fanti L., Pimpinelli S. HP1: a functionally multifaceted protein. Curr. Opin. Genet. Dev. 2008;18:169–174. doi: 10.1016/j.gde.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 4.Kwon S.H., Workman J.L. The heterochromatin protein 1 (HP1) family: put away a bias toward HP1. Mol. Cells. 2008;26:217–227. [PubMed] [Google Scholar]
- 5.Canzio D., Larson A., Narlikar G.J. Mechanisms of functional promiscuity by HP1 proteins. Trends Cell Biol. 2014;24:377–386. doi: 10.1016/j.tcb.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hiragami-Hamada K., Soeroes S., Fischle W. Dynamic and flexible H3K9me3 bridging via HP1β dimerization establishes a plastic state of condensed chromatin. Nat. Commun. 2016;7:11310. doi: 10.1038/ncomms11310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eissenberg J.C., Elgin S.C. The HP1 protein family: getting a grip on chromatin. Curr. Opin. Genet. Dev. 2000;10:204–210. doi: 10.1016/s0959-437x(00)00058-7. [DOI] [PubMed] [Google Scholar]
- 8.Grewal S.I., Jia S. Heterochromatin revisited. Nat. Rev. Genet. 2007;8:35–46. doi: 10.1038/nrg2008. [DOI] [PubMed] [Google Scholar]
- 9.Dihazi G.H., Jahn O., Dihazi H. Proteomic analysis of embryonic kidney development: heterochromatin proteins as epigenetic regulators of nephrogenesis. Sci. Rep. 2015;5:13951. doi: 10.1038/srep13951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takada Y., Naruse C., Koseki H. HP1γ links histone methylation marks to meiotic synapsis in mice. Development. 2011;138:4207–4217. doi: 10.1242/dev.064444. [DOI] [PubMed] [Google Scholar]
- 11.Aucott R., Bullwinkel J., Singh P.B. HP1-beta is required for development of the cerebral neocortex and neuromuscular junctions. J. Cell Biol. 2008;183:597–606. doi: 10.1083/jcb.200804041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lomberk G., Bensi D., Urrutia R. Evidence for the existence of an HP1-mediated subcode within the histone code. Nat. Cell Biol. 2006;8:407–415. doi: 10.1038/ncb1383. [DOI] [PubMed] [Google Scholar]
- 13.Vakoc C.R., Mandat S.A., Blobel G.A. Histone H3 lysine 9 methylation and HP1gamma are associated with transcription elongation through mammalian chromatin. Mol. Cell. 2005;19:381–391. doi: 10.1016/j.molcel.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 14.Bannister A.J., Zegerman P., Kouzarides T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–124. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- 15.Lachner M., O’Carroll D., Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001;410:116–120. doi: 10.1038/35065132. [DOI] [PubMed] [Google Scholar]
- 16.Ye Q., Callebaut I., Worman H.J. Domain-specific interactions of human HP1-type chromodomain proteins and inner nuclear membrane protein LBR. J. Biol. Chem. 1997;272:14983–14989. doi: 10.1074/jbc.272.23.14983. [DOI] [PubMed] [Google Scholar]
- 17.Nielsen A.L., Oulad-Abdelghani M., Losson R. Heterochromatin formation in mammalian cells: interaction between histones and HP1 proteins. Mol. Cell. 2001;7:729–739. doi: 10.1016/s1097-2765(01)00218-0. [DOI] [PubMed] [Google Scholar]
- 18.Canzio D., Chang E.Y., Al-Sady B. Chromodomain-mediated oligomerization of HP1 suggests a nucleosome-bridging mechanism for heterochromatin assembly. Mol. Cell. 2011;41:67–81. doi: 10.1016/j.molcel.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamada T., Fukuda R., Sugimoto K. Functional domain structure of human heterochromatin protein HP1(Hsalpha): involvement of internal DNA-binding and C-terminal self-association domains in the formation of discrete dots in interphase nuclei. J. Biochem. 1999;125:832–837. doi: 10.1093/oxfordjournals.jbchem.a022356. [DOI] [PubMed] [Google Scholar]
- 20.Munari F., Soeroes S., Zweckstetter M. Methylation of lysine 9 in histone H3 directs alternative modes of highly dynamic interaction of heterochromatin protein hHP1β with the nucleosome. J. Biol. Chem. 2012;287:33756–33765. doi: 10.1074/jbc.M112.390849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lechner M.S., Schultz D.C., Rauscher F.J., III The mammalian heterochromatin protein 1 binds diverse nuclear proteins through a common motif that targets the chromoshadow domain. Biochem. Biophys. Res. Commun. 2005;331:929–937. doi: 10.1016/j.bbrc.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 22.Maison C., Almouzni G. HP1 and the dynamics of heterochromatin maintenance. Nat. Rev. Mol. Cell Biol. 2004;5:296–304. doi: 10.1038/nrm1355. [DOI] [PubMed] [Google Scholar]
- 23.Meehan R.R., Kao C.F., Pennings S. HP1 binding to native chromatin in vitro is determined by the hinge region and not by the chromodomain. EMBO J. 2003;22:3164–3174. doi: 10.1093/emboj/cdg306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mishima Y., Watanabe M., Suetake I. Hinge and chromoshadow of HP1α participate in recognition of K9 methylated histone H3 in nucleosomes. J. Mol. Biol. 2013;425:54–70. doi: 10.1016/j.jmb.2012.10.018. [DOI] [PubMed] [Google Scholar]
- 25.Nishibuchi G., Machida S., Nakayama J. N-terminal phosphorylation of HP1α increases its nucleosome-binding specificity. Nucleic Acids Res. 2014;42:12498–12511. doi: 10.1093/nar/gku995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mishima Y., Jayasinghe C.D., Suetake I. Nucleosome compaction facilitates HP1γ binding to methylated H3K9. Nucleic Acids Res. 2015;43:10200–10212. doi: 10.1093/nar/gkv841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Azzaz A.M., Vitalini M.W., Shogren-Knaak M.A. Human heterochromatin protein 1α promotes nucleosome associations that drive chromatin condensation. J. Biol. Chem. 2014;289:6850–6861. doi: 10.1074/jbc.M113.512137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kilic S., Bachmann A.L., Fierz B. Multivalency governs HP1α association dynamics with the silent chromatin state. Nat. Commun. 2015;6:7313. doi: 10.1038/ncomms8313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bryan L.C., Weilandt D.R., Fierz B. Single-molecule kinetic analysis of HP1-chromatin binding reveals a dynamic network of histone modification and DNA interactions. Nucleic Acids Res. 2017;45:10504–10517. doi: 10.1093/nar/gkx697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wedemann G., Langowski J. Computer simulation of the 30-nanometer chromatin fiber. Biophys. J. 2002;82:2847–2859. doi: 10.1016/S0006-3495(02)75627-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arya G., Schlick T. Role of histone tails in chromatin folding revealed by a mesoscopic oligonucleosome model. Proc. Natl. Acad. Sci. USA. 2006;103:16236–16241. doi: 10.1073/pnas.0604817103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grigoryev S.A., Arya G., Schlick T. Evidence for heteromorphic chromatin fibers from analysis of nucleosome interactions. Proc. Natl. Acad. Sci. USA. 2009;106:13317–13322. doi: 10.1073/pnas.0903280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ozer G., Luque A., Schlick T. The chromatin fiber: multiscale problems and approaches. Curr. Opin. Struct. Biol. 2015;31:124–139. doi: 10.1016/j.sbi.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luque A., Collepardo-Guevara R., Schlick T. Dynamic condensation of linker histone C-terminal domain regulates chromatin structure. Nucleic Acids Res. 2014;42:7553–7560. doi: 10.1093/nar/gku491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perišić O., Schlick T. Dependence of the linker histone and chromatin condensation on the nucleosome environment. J. Phys. Chem. B. 2017;121:7823–7832. doi: 10.1021/acs.jpcb.7b04917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Voltz K., Trylska J., Smith J. Coarse-grained force field for the nucleosome from self-consistent multiscaling. J. Comput. Chem. 2008;29:1429–1439. doi: 10.1002/jcc.20902. [DOI] [PubMed] [Google Scholar]
- 37.Voltz K., Trylska J., Langowski J. Unwrapping of nucleosomal DNA ends: a multiscale molecular dynamics study. Biophys. J. 2012;102:849–858. doi: 10.1016/j.bpj.2011.11.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Collepardo-Guevara R., Portella G., Orozco M. Chromatin unfolding by epigenetic modifications explained by dramatic impairment of internucleosome interactions: a multiscale computational study. J. Am. Chem. Soc. 2015;137:10205–10215. doi: 10.1021/jacs.5b04086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dans P.D., Walther J., Orozco M. Multiscale simulation of DNA. Curr. Opin. Struct. Biol. 2016;37:29–45. doi: 10.1016/j.sbi.2015.11.011. [DOI] [PubMed] [Google Scholar]
- 40.Winogradoff D., Echeverria I., Papoian G.A. The acetylation landscape of the H4 histone tail: disentangling the interplay between the specific and cumulative effects. J. Am. Chem. Soc. 2015;137:6245–6253. doi: 10.1021/jacs.5b00235. [DOI] [PubMed] [Google Scholar]
- 41.Zhang B., Zheng W., Wolynes P.G. Exploring the free energy landscape of nucleosomes. J. Am. Chem. Soc. 2016;138:8126–8133. doi: 10.1021/jacs.6b02893. [DOI] [PubMed] [Google Scholar]
- 42.Takeshima H., Suetake I., Tajima S. Distinct DNA methylation activity of Dnmt3a and Dnmt3b towards naked and nucleosomal DNA. J. Biochem. 2006;139:503–515. doi: 10.1093/jb/mvj044. [DOI] [PubMed] [Google Scholar]
- 43.Luger K., Rechsteiner T.J., Richmond T.J. Preparation of nucleosome core particle from recombinant histones. Methods Enzymol. 1999;304:3–19. doi: 10.1016/s0076-6879(99)04003-3. [DOI] [PubMed] [Google Scholar]
- 44.Davey C.A., Sargent D.F., Richmond T.J. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 a resolution. J. Mol. Biol. 2002;319:1097–1113. doi: 10.1016/S0022-2836(02)00386-8. [DOI] [PubMed] [Google Scholar]
- 45.Kaustov L., Ouyang H., Arrowsmith C.H. Recognition and specificity determinants of the human cbx chromodomains. J. Biol. Chem. 2011;286:521–529. doi: 10.1074/jbc.M110.191411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li W., Terakawa T., Takada S. Energy landscape and multiroute folding of topologically complex proteins adenylate kinase and 2ouf-knot. Proc. Natl. Acad. Sci. USA. 2012;109:17789–17794. doi: 10.1073/pnas.1201807109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li W., Wang W., Takada S. Energy landscape views for interplays among folding, binding, and allostery of calmodulin domains. Proc. Natl. Acad. Sci. USA. 2014;111:10550–10555. doi: 10.1073/pnas.1402768111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sambriski E.J., Schwartz D.C., de Pablo J.J. A mesoscale model of DNA and its renaturation. Biophys. J. 2009;96:1675–1690. doi: 10.1016/j.bpj.2008.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kenzaki H., Takada S. Partial unwrapping and histone tail dynamics in nucleosome revealed by coarse-grained molecular simulations. PLoS Comput. Biol. 2015;11:e1004443. doi: 10.1371/journal.pcbi.1004443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kenzaki H., Koga N., Takada S. CafeMol: a coarse-grained biomolecular simulator for simulating proteins at work. J. Chem. Theory Comput. 2011;7:1979–1989. doi: 10.1021/ct2001045. [DOI] [PubMed] [Google Scholar]
- 51.Kelley L.A., Mezulis S., Sternberg M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015;10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Case D.A., Darden T.A., Kollman P.A. University of California; San Francisco, CA: 2012. AMBER 13. [Google Scholar]
- 53.Macke T.J., Case D.A. Modeling unusual nucleic acid structures. In: Leontis N.B., SantaLucia J., editors. Molecular Modeling of Nucleic Acids. ACS Symposium Series; 1998. Vol. 682, pp. 379–393. [Google Scholar]
- 54.Abraham M.J., Murtola T., Lindah E. Gromacs: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1:19–25. [Google Scholar]
- 55.Páll S., Abraham M.J., Lindahl E. Tackling exascale software challenges in molecular dynamics simulations with GROMACS. In: Markidis S., Laure E., editors. Solving Software Challenges for Exascale, Lecture Notes in Computer Science. Springer; 2015. Vol. 8759, pp. 3–27. [Google Scholar]
- 56.Bondi A. van der Waals volumes and radii. J. Phys. Chem. 1964;68:441–451. [Google Scholar]
- 57.Guy A.T., Piggot T.J., Khalid S. Single-stranded DNA within nanopores: conformational dynamics and implications for sequencing; a molecular dynamics simulation study. Biophys. J. 2012;103:1028–1036. doi: 10.1016/j.bpj.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hornak V., Abel R., Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sorin E.J., Pande V.S. Exploring the helix-coil transition via all-atom equilibrium ensemble simulations. Biophys. J. 2005;88:2472–2493. doi: 10.1529/biophysj.104.051938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pérez A., Marchán I., Orozco M. Refinement of the AMBER force field for nucleic acids: improving the description of α/γ conformers. Biophys. J. 2007;92:3817–3829. doi: 10.1529/biophysj.106.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 62.Essmann U., Perera L., Pedersen L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- 63.Hess B., Bekker H., Fraaije J. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. doi: 10.1021/ct700200b. [DOI] [PubMed] [Google Scholar]
- 64.Miyamoto S., Kollman P.A. SETTLE - an analytical version of the shake and rattle algorithm for rigid water models. J. Comput. Chem. 1992;13:952–962. [Google Scholar]
- 65.Bussi G., Donadio D., Parrinello M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007;126:014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 66.Parrinello M., Rahman A. Polymorphic transitions in single-crystals - a new molecular-dynamics method. J. Appl. Phys. 1981;52:7182–7190. [Google Scholar]
- 67.Li W., Wolynes P.G., Takada S. Frustration, specific sequence dependence, and nonlinearity in large-amplitude fluctuations of allosteric proteins. Proc. Natl. Acad. Sci. USA. 2011;108:3504–3509. doi: 10.1073/pnas.1018983108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Takada S., Kanada R., Kenzaki H. Modeling structural dynamics of biomolecular complexes by coarse-grained molecular simulations. Acc. Chem. Res. 2015;48:3026–3035. doi: 10.1021/acs.accounts.5b00338. [DOI] [PubMed] [Google Scholar]
- 69.Terakawa T., Takada S. Multiscale ensemble modeling of intrinsically disordered proteins: p53 N-terminal domain. Biophys. J. 2011;101:1450–1458. doi: 10.1016/j.bpj.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Terakawa T., Kenzaki H., Takada S. p53 searches on DNA by rotation-uncoupled sliding at C-terminal tails and restricted hopping of core domains. J. Am. Chem. Soc. 2012;134:14555–14562. doi: 10.1021/ja305369u. [DOI] [PubMed] [Google Scholar]
- 71.Muchardt C., Guilleme M., Yaniv M. Coordinated methyl and RNA binding is required for heterochromatin localization of mammalian HP1α. EMBO Rep. 2002;3:975–981. doi: 10.1093/embo-reports/kvf194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shimojo H., Kawaguchi A., Nishimura Y. Extended string-like binding of the phosphorylated HP1α N-terminal tail to the lysine 9-methylated histone H3 tail. Sci. Rep. 2016;6:22527. doi: 10.1038/srep22527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Maison C., Bailly D., Almouzni G. SUMOylation promotes de novo targeting of HP1α to pericentric heterochromatin. Nat. Genet. 2011;43:220–227. doi: 10.1038/ng.765. [DOI] [PubMed] [Google Scholar]
- 74.Hiragami-Hamada K., Shinmyozu K., Nakayama J. N-terminal phosphorylation of HP1alpha promotes its chromatin binding. Mol. Cell. Biol. 2011;31:1186–1200. doi: 10.1128/MCB.01012-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Munari F., Gajda M.J., Zweckstetter M. Characterization of the effects of phosphorylation by CK2 on the structure and binding properties of human HP1β. FEBS Lett. 2014;588:1094–1099. doi: 10.1016/j.febslet.2014.02.019. [DOI] [PubMed] [Google Scholar]
- 76.Machida S., Takizawa Y., Kurumizaka H. Structural basis of heterochromatin formation by human HP1. Mol. Cell. 2018;69:385–397.e8. doi: 10.1016/j.molcel.2017.12.011. [DOI] [PubMed] [Google Scholar]
- 77.Kilic S., Felekyan S., Fierz B. Single-molecule FRET reveals multiscale chromatin dynamics modulated by HP1α. Nat. Commun. 2018;9:235. doi: 10.1038/s41467-017-02619-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This video shows a coarse-grained molecular simulation corresponding to Fig. 3.
This video shows a fully atomistic molecular simulation corresponding to Fig. 9.