

Abstract
Uterine serous carcinoma (USC) represents an aggressive histologic subtype of endometrial cancer. It is associated with a poor prognosis, and improved therapies for women battling USCs are greatly needed. ONC201 is an orally bioavailable, first-in-class small molecule that induces tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) independent of p53. ONC201 has demonstrated anti-tumorigenic activity in pre-clinical models of solid tumors through induction of apoptosis and inactivation of the AKT/MAPK pathways. Recent phase I and II clinical trials have shown that ONC201 is well tolerated and may have single agent activity in high grade glioma patients among others. We sought to determine the effects of ONC201 on cell proliferation in USC and identify the mechanisms by which ONC201 inhibits cell growth in this disease. ONC201 inhibited cell proliferation in a dose-dependent manner in ARK1, ARK2 and SPEC-2 cell lines. The anti-proliferative activity of ONC201 in ARK1 and SPEC-2 cells was associated with induction apoptosis independent of p53 via both a TRAIL mediated apoptotic pathway and a mitochondrial apoptosis pathway. Treatment with ONC201 resulted in significant reduction in adhesion and invasion as well as inhibition of the AKT and MAPK pathways. In addition, ONC201 markedly potentiated the anti-tumorigenic effects of paclitaxel in USC cells. Our results suggest that ONC201 has significant anti-proliferative and anti-metastatic effects in USC cells through both induction of apoptosis and inhibition of the AKT and MAPK pathways. ONC201 and paclitaxel are a promising therapeutic combination in USC cells. Thus, ONC201 should be evaluated as a single agent and as a therapeutic partner with paclitaxel in future clinical trials of USC.
Keywords: ONC201, apoptosis, cell proliferation, invasion, uterine serous carcinoma
Introduction
Endometrial cancer is the most common gynecologic malignancy among women with an estimated 63,230 new cases and 11,350 deaths in the United States in 2018 [1]. The majority of women with endometrial cancer present with early-stage disease and the favorable endometrioid histologic subtype. Thus, endometrioid endometrial cancer is associated with an excellent 5-year survival of greater than 85% [2]. Uterine serous carcinoma (USC), however, compromises approximately 10% of endometrial carcinomas, and represents an aggressive histologic subtype of this disease. USC is associated with an increased risk of extrauterine disease and constitutes 40% of all recurrences and deaths from endometrial cancer [3,4]. While the initial treatment of USC includes a combination of surgical staging or cytoreduction and adjuvant chemotherapy, the high recurrence rate makes clear the need for more effective treatment strategies for this deadly disease [5].
ONC201 is a promising first-in-class small molecule that was identified in a phenotypic cell-based screen as an inducer of the tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) in a p53-independent manner [6]. ONC201 causes dual inactivation of the AKT and MAPK signaling pathways, resulting in dephosphorylation of the transcription factor FOXO3a and leading to induction of transcription of TRAIL [6,7]. ONC201 exhibits anti-cancer activity in various advanced solid tumors in pre-clinical cancer models, without significant toxicity in animal cancer models [8]. Unique features of ONC201 are its broad-spectrum activity independent of mutations or tumor type, oral administration, excellent safety profile and ability to penetrate the blood-brain barrier. The FDA has approved ONC201 for phase I/II clinical trials as an anti-tumor therapeutic agent for various human cancers [6]. The first-in-human phase I dose-escalation study has confirmed that ONC201 is exceptionally well tolerated with favorable pharmacokinetics and pharmacodynamics in advanced solid tumors including recurrent endometrial cancer [9]. Recently, another phase I clinical trial has showed that ONC201 is clinically active and well tolerated with oral administration in refractory metastatic endometrial cancer patients [10].
The overall aim of this study was to investigate the anti-tumorigenic and anti-metastatic effects of ONC201 in USC cell lines. Our results demonstrate that ONC201 inhibits cell growth, induces apoptosis, inhibits adhesion and invasion, and increases the sensitivity of tumor cells to paclitaxel, supporting the potential of ONC201 for further evaluation in clinical trials in women with USC.
Materials and methods
Cell culture and reagents
Three USC cell lines, ARK1, ARK2 and SPEC-2, were used for all experiments. The ARK1 and ARK2 cells were maintained in RPMI 1640 medium with 10% fetal bovine serum (FBS). The SPEC-2 cells were maintained in DMEM/F12 with 10% FBS. All medium was supplemented with 100 U/ml of penicillin and 100 ug/ml of streptomycin. The cells were cultured in humidified 5% CO2 at 37°C. ONC201 was obtained from Oncoceutics, Inc. MTT ((3-5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) was purchased from Sigma (St. Louis, MO). All antibodies were purchased from Cell Signaling (Beverly, MA). Enhanced chemiluminescence western blotting detection reagents were purchased from Amersham (Arlington Heights, IL). All other chemicals were purchased from Sigma.
MTT assay
The ARK1 and SPEC-2 cells were plated and grown in 96-well plates at a concentration of 4000 to 8000 cells/well for 24 hours. The cells were subsequently treated with varying doses of ONC201 for 72 hours. MTT (5 mg/ml) was added to the 96-well plates at 5 μl/well, followed by an additional hour of incubation. The MTT reaction was terminated through the addition of 100 μl of DMSO. The results were read by measuring absorption at 570 nm with a microplate reader (Tecan, Morrisville, NC). The effect of ONC201 was calculated as a percentage of control cell growth obtained from DMSO treated cells grown in the same 96-well plates. Each experiment was performed in triplicate to assess for consistency of results.
Colony formation assay
ARK1 and SPEC-2 cells growing in log phase were seeded in 6 cm dishes (1000 cells/dish) in regular growth media. Cells were allowed to adhere for 24 hours, and then treated with ONC201 for 48 hours. The cells were cultured for 14 days with media changes every third or fourth day. The dishes were stained with 0.5% crystal violet, and colonies were counted under the microscope. Each experiment was repeated three times for consistency of results.
Annexin V assay
Apoptosis was detected with the Annexin V FITC kit (Biolegend, San Diego, CA) on Cellometer (Nexcelom, Lawrence, MA). Briefly, 2 × 105 cells/well were seeded into 6-well plates, incubated overnight and then treated with ONC201 at different doses for 24 hours. The cells were then collected, washed with PBS and resuspended in 100 μl binding buffer. Subsequently, 1 μl of Annexin V-FITC (100 μg/ml) and 0.5 μl of propidium iodide (2 mg/ml) were added in the binding buffer, and the plates placed in the dark for 15 min. The samples were immediately measured by Cellometer. The results were analyzed by FCS4 express software (Molecular Devices, Sunnyvale, CA). All experiments were performed in triplicate to assess for consistency of response.
Caspase 3 activity assay
Cleaved caspase 3 activity was assessed by the Cleaved Caspase 3 Activity Assay kit (Raybiotech, Norcross, GA). After treating the cells with ONC201 in 96-well plates, 100 ul of prepared 1 × detection anti-Cleaved-Caspase-3 loading buffer was added into each well and mixed gently. The plates were then incubated with shaking for 60 min at 37°C, 5% CO2. The ELISA plates were read by measuring absorption at 450 nm. All experiments were performed in triplicate and repeated twice to assess for consistency of response.
siRNA transfection
Transfection of siRNA was performed using p53 siRNA (Santa Cruz Biotechnology Inc., Santa Cruz, CA) and QIAGEN HiperFect Transfection Reagent. In brief, SPEC-2 cells were plated at a density of 2.5 × 105 cells/well in 6-well plates. The transfection complexes containing p53 siRNA and 6 ul HiperFect Transfection Reagent for a 6-well plate were added into the media. A scramble siRNA was used as the negative control. The cells were harvested for western blot and other assays after transfection for 48-72 hours. The impact of siRNA transfection on cell proliferation was evaluated by MTT assay. The experiments were repeated twice in triplicate to assess for consistency of response.
Adhesion assay
Each well in a 96-well plate was coated with 100 μl laminin-1 (10 μg/ml) and incubated at 37°C for 1 hour. The fluid was then aspirated, and 200 μl blocking buffer was added to each well for 45-60 min at 37°C. The wells were washed with PBS, and the plate was allowed to chill on ice. To each well, 2.5 × 103 cells were added with PBS and varying concentrations of ONC201 directly. The plate was then allowed to incubate at 37°C for 2 hours. The medium was then aspirated, and the cells were fixed by directly adding 100 μl of 5% glutaraldehyde and incubating for 30 min at room temperature. Adhered cells were then washed with PBS and stained with 100 μl of 0.1% crystal violet for 30 min. The cells were washed repeatedly with water, and 100 μl of 10% acetic acid was added to each well to solubilize the dye. After 5 min of shaking, the absorbance was measured at 570 nm using a microplate reader from Tecan. All experiments were performed in duplicate to assess for consistency of response.
Invasion assay
Cell invasion assays were performed using 96-well HTS transwells (Corning Life Sciences, Wilmington, NC) coated with 0.5-1 × BME (Trevigen, Gaithersburg, Maryland). Starved (serum free media for 12 hours) ARK1 and SPEC-2 cells (50,000 cells/well) were seeded for 12 hours in the upper chambers of the wells in 50 μl FBS-free medium, and the lower chambers were filled with 150 μl regular medium with different concentrations of ONC201. The plate was incubated for 24 hours at 37°C to allow invasion into the lower chamber. After washing the upper and lower chambers with PBS once, 100 μl calcein AM solution was added into the lower chamber and incubated at 37°C for 30-60 min. The lower chamber plate was measured by the plate reader for reading fluorescence at EX/EM 485/520 nM. All experiments were performed in duplicate to assess for consistency of response.
Wound healing assay
ARK1 and SPEC-2 cells were plated at 2 × 105 cells per well in a 6-well plate and incubated overnight. A uniform wound was created through the cell monolayer using a 20 ul pipette tip. The cells were washed and treated with different concentrations of ONC201 for 48 hours. Pictures were taken at 24 and 48 hours after scratching, and the area of the scratch was analyzed with ImageJ software (National Institutes of Health, Bethesda, MD). Percent closure was measured by comparing to control cells.
Western immunoblotting
The ARK1 and SPEC-2 cells were plated at 2 × 105 cells/well in 6-well plates in their corresponding media and then treated with ONC201 for 24-36 hours. Cell lysates were prepared in RIPA buffer. Equal amounts of protein were separated by gel electrophoresis and transferred onto a PVDF membrane. The membrane was blocked with 5% nonfat dry milk and then incubated with a 1:1000 dilution of primary antibody overnight at 4°C. The membrane was then washed and incubated with a secondary peroxidase-conjugated antibody for 1 hour after washing. Antibody binding was detected using an enhanced chemiluminescence detection system on the Alpha Innotech Imaging System (Protein Simple, Santa Clara, CA). After developing, the membrane was stripped and re-probed using antibodies against β-actin or α-tubulin to confirm equal loading. Intensity for each band was measured and normalized to β-actin or α-tubulin as an internal control. Each experiment was repeated two times to assess for consistency of results.
Statistical analysis
Results for experiments were normalized to the mean of the control and analyzed using the Student t-test. Differences were considered significant if the p value was less than 0.05 (P<0.05) with a confidence interval of 95%.
Results
ONC201 inhibited USC cell growth
The USC cell lines ARK1, ARK2 and SPEC-2 were cultured in medium with various concentrations of ONC201 for 72 hours, and the viability of cultured cells was analyzed using the MTT assay. As shown in Figure 1A, treatment of USC cells with ONC201 dramatically decreased viable cell number in a dose-dependent manner. The mean IC50 values of ONC201 were 6 uM for ARK1, 10 uM for ARK2 and 12 uM for SPEC-2.
Figure 1.

ONC201 inhibited the proliferation of USC cells. The ARK1 and SPEC-2 cells were cultured for 24 hours and then treated with varying concentrations of ONC201 in 96 well plates for 72 hours. Cell proliferation was assessed by MTT assay (A). The ARK1 and SPEC-2 cells were seeded at low density in 6 cm dishes and treated with ONC201 for 48 hours. The cells were cultured for 14 days with medium changes every third or fourth day. Colonies were visualized by crystal violet staining (B). Morphologies of the ARK1 and SPEC-2 cells after treatment of ONC201 for 48 hours (C). The effect of ONC201 on death receptor 5 (DR5) was examined by Western blot analysis. Treatment of ONC201 resulted in a dose-dependent decrease in expression of DR5 protein in both cell lines (D). Each experiment was performed three times.
The colony formation assay is used as an indicator of long-term tumor cell survival. Thus, colony formation assays were performed to investigate the long-term effect of ONC201 on cell growth in the ARK1 and SPEC-2 cell lines. As shown in Figure 1B, the colony-forming ability of ARK1 and SPEC-2 was reduced by 49% and 61%, respectively, after exposure to 10 uM of ONC201 for 2 days and subsequent culture of cells for 14 days. The effects of ONC201 on cellular morphology in both cell lines are shown in Figure 1C. Following ONC201 treatment for 48 hours, SPEC-2 cells shrunk and displayed an elongated shape while control cells were round or oval shape with large nuclei. ONC201 did not change the cell shape in ARK1 cells. Taken together, these results suggest that USC cell lines are sensitive to treatment with ONC201.
Because ONC201 exhibits p53 independent cytotoxicity through TRAIL and DR5 induction in cancer cells, we examined whether ONC201 treatment regulates the expression of DR5 protein in ARK1 and SPEC-2 cells. As shown in Figure 1D, western blotting results showed that ONC201 treatment significantly up-regulated the expression of DR5 protein after 24 hours of treatment in both cell lines.
ONC201 induced apoptosis in USC cells
In order to determine whether the reduction of cell viability by ONC201 was due to increased apoptosis, we detected apoptotic cells by performing an Annexin-V and PI double staining assay. We treated the ARK1 and SPEC-2 cell lines with varying concentrations of ONC201 for 24 hours, and found that the percentage of ARK1 and SPEC-2 cells undergoing apoptosis significantly increased in a dose-dependent manner when compared to controls (Figure 2A). We next treated both cell lines with ONC201 for 24 hours, and found that ONC201 decreased the expression of BCL-1 and MCL-1 and increased cleaved caspase 3, caspase 9 and PARP protein expression in a dose dependent manner. In addition, treatment of ONC201 in both cells also increased cleaved caspase 8 as well as DR5 expression (Figure 2B). Since caspase-8 activation and TRAIL/DR5 expression are characteristic markers of extrinsic apoptotic pathway activation, these data suggest that ONC201 effectively activates both extrinsic and endogenous apoptotic pathways in human USC cells.
Figure 2.

ONC201 induced apoptosis in USC cells. The ARK1 and SPEC-2 cell lines were treated with the varying concentrations of ONC201 (1-100 uM) for 24 hours. Apoptosis was detected using the Annexin-V FITC assay in both cell lines after 24 hours of treatment. ONC201 increased Annexin V expression in the ARK1 and SPEC-2 cells (A). Western blotting shows treatment with ONC201 for 24 hours decreased the expression of MCL-1 and BCL-2, and increased cleaved PARP, caspase 3, caspase 9 and caspase 8 expression in both cell lines (B). Pretreatment with Z-VAD-FMK for 2 hours in ARK1 and SPEC-2 cells resulted in total blocking of ONC201 induced cleaved caspase-3 activity and a significant decrease in the inhibition of ONC201-mediated proliferation in both cell lines (C and D). Western blotting showed siRNA p53 reduced the expression of p53 after 48 hours transfection in the SPEC-2 cells. p53 knockdown did not affect cell proliferation treated by ONC201 compared to control cells (E). Data are shown as mean + SEM of two experiments (*P<0.05). Each experiment was performed three times.
To further evaluate the role of ONC201 on the caspase apoptotic pathways, we used pan-caspase inhibitor (Z-VAD-FMK) to block caspase activity in both cell lines and determined whether the cell proliferation inhibition and caspase-3 activity were changed after ONC201 treatment. Pretreatment with Z-VAD-FMK for 2 hours resulted in total blocking of ONC201 induced cleaved caspase-3 activity and a significant decrease in the inhibition of ONC201-mediated proliferation in both cell lines (Figure 2C and 2D). These results suggest that the caspase cascade dependent pathways are involved in the inhibition of cell proliferation in USC cells treated with ONC201.
Given that p53 serves as an important molecular target in USC, we also assessed whether knockdown of p53 by siRNA would alter cellular sensitivity to ONC201 in SPEC-2 cells with mutant p53 [11]. As shown in Figure 2E, p53 siRNA dramatically decreased p53 protein expression in SPEC-2. Treatment of p53 siRNA transfected cells with different concentrations of ONC201 for 72 hours did not change sensitivity to ONC201 when compared with the control. In agreement with prior studies in other cancers, these results suggest that ONC201 inhibits cell proliferation in USC cells in a p53 independent manner [12].
ONC201 inhibited adhesion and invasion in USC cells
In order to determine the effect of ONC201 on the invasive ability of USC cells, laminin adhesion, wound healing and transwell invasion assays were employed. Incubation of the ARK1 and SPEC-2 cells with ONC201 (1, 50 and 100 uM) for 2 hours showed significant inhibition of cell adhesion (Figure 3A). Exposure to ONC201 at concentrations ranging from 1-100 uM for 4 hours significantly suppressed invasiveness of the cells compared to vehicle-treated control cells as determined by the transwell invasion assay (Figure 3B). Inhibition of cell adhesion and invasion was dose-dependent in both cell lines. To examine the effect of ONC201 on motility in USC cells, we used a scratch wound healing assay to measure the extent of cell migration into the scratched area. ARK1 cells were almost confluent at 72 hours after scratching, whereas ONC201 significantly slowed down cell migration into the “wounded” area in both cells after 72 hours of treatment (Figure 3C).
Figure 3.

ONC201 decreased adhesion and invasion in USC cells. The ARK1 and SPEC-2 cell lines were cultured for 24 hours and then treated with ONC201 in a laminin-coated 96 well plate for 2 hours to assess adhesion (A) or a BME coated 96 transwell plate for 4 hours to assess invasion (B), respectively. The data represents relative inhibition in each cell line. Wound healing assay showed that ONC201 increased wound width in both cells in a dose-dependent manner (C). VEGF, Snail, Slug, E-Cadherin and N-cadherin were measured by Western blotting in cell lysates after 24 hours exposure to ONC201 (D). Each experiment was performed three times (*P<0.05, **P<0.01).
Since adhesion and invasion of cancer cells are mediated by a variety of membrane proteins as well as modulation of cytoskeletal assembly, we further analyzed the effect of ONC201 on the epithelial-mesenchymal transition (EMT) and angiogenesis in USC cells. The expression of cadherins, Slug, Snail and VEGF were assessed by western blotting analysis. After 24 hours of treatment, ONC201 induced the expression of E-cadherin and Slug, and reduced N-cadherin, Snail and VEGF expression in ARK1 and SPEC-2 cells (Figure 3D). These results further support the role of ONC201 in the inhibition of adhesion, invasion and angiogenesis in USC cells.
Effect of ONC201 on the AMPK/AKT/mTOR and MAPK pathways
Given deregulation of the P13K/AKT pathway in USC tumors and induction of TRAIL by ONC201 via Akt and ERK inactivation [8,13,14], we investigated whether these pathways were involved in the anti-proliferative effects of ONC201 in USC cells. Western blotting showed that phosphorylation of AKT, S6 and p42/44 was repressed by ONC201 in a dose dependent manner after 24 hours of treatment in both cell lines (Figure 4). Meanwhile, ONC201 also increased phosphorylation of AMPK in the ARK1 and SPEC-2 cells. Together, these data suggest that ONC201 exerts its anti-tumorigenic activity via activation of AMPK and inhibition of the AKT/mTOR and MAPK signaling pathways in USC cells.
Figure 4.
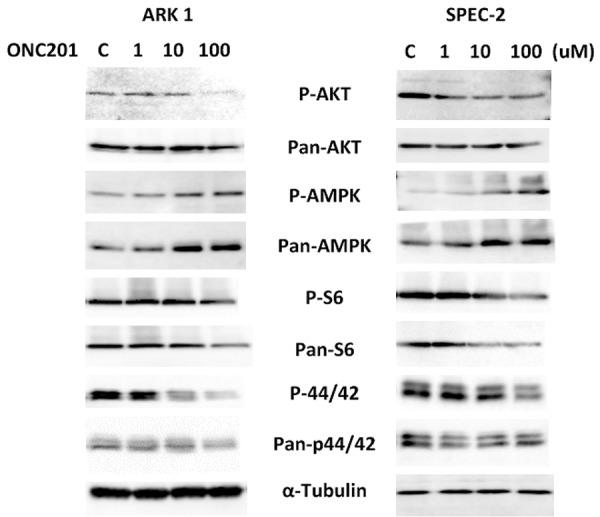
Effect of ONC201 on AMPK/AKT and MAPK pathways in USC cells. The ARK1 and SPEC-2 cells were treated with ONC201 at different doses for 24 hours. Phosphorylated-p42/44, phosphorylated-AKT, phosphorylated-AMPK and phosphorylated-pS6 were assessed by Western blotting. ONC201 inhibited downstream targets of the AKT and MAPK pathways in both cell lines and increased AMPK activation in the ARK1 and SPEC-2 cells. Each experiment was performed two times.
ONC201 synergized with paclitaxel in USC cells
ONC201 has been previously shown to synergize with paclitaxel in non-small cell lung cancer cell lines and nude mouse xenografts [8]. Thus, we assessed the effect of ONC201 on sensitivity to paclitaxel in ARK1 and SPEC-2 cells. The antagonism or synergism of the combinations was evaluated using Chou-Talalay method [15]. Both cell lines were pre-treated with a low concentration (0.5 uM) of ONC201 for 24 hours and then combined with serial dilutions of paclitaxel for 48 hours. MTT assays demonstrated that the combination of 0.5 uM of ONC201 with paclitaxel resulted in synergistic inhibitory effects in both cell lines at the majority of dose combinations (CI<1, Figure 5A-C). These results suggest that treatment of ONC201 increases the sensitivity of the USC cells to paclitaxel.
Figure 5.
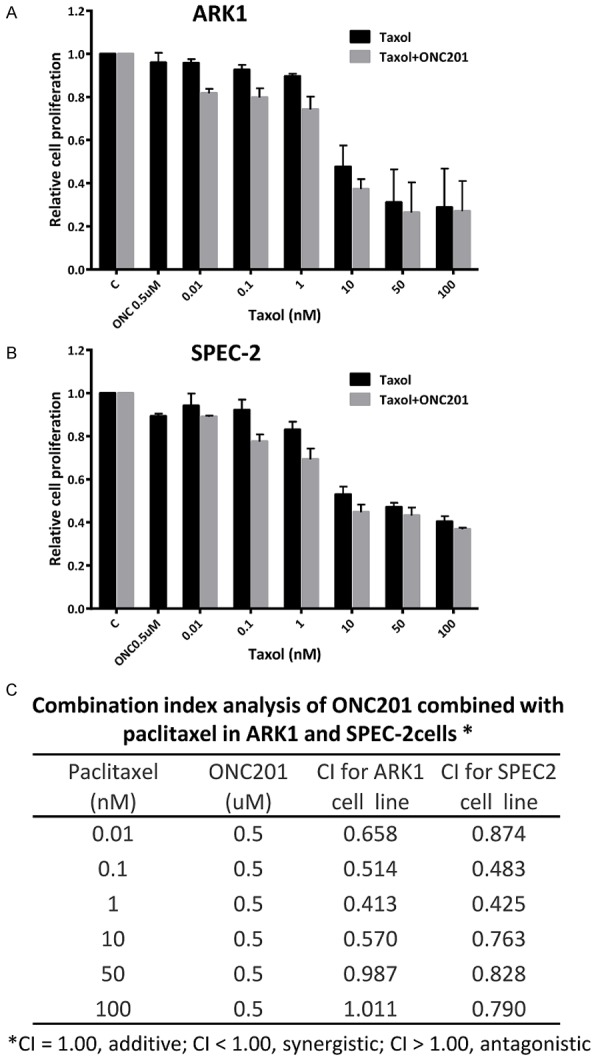
ONC201 synergized with paclitaxel in USC cells. The ARK1 (A) and SPEC-2 (B) cells were pretreated with ONC201 (0.5 uM) for 24 hours and then treated with different concentrations of paclitaxel for 48 hours. MTT assays showed that ONC201 increased sensitivity to paclitaxel in both cell lines. Combination index (CI) was calculated by Chou-Talalay method (C) supported by CompuSyn.
Discussion
Given the aggressiveness of USC, there remains a great need for improved therapies in women battling this cancer. In this study, we found that ONC201 exhibits anti-tumorigenic activity in human USC cells through inhibition of cell proliferation as well as induction of p53-independent apoptosis. In parallel to ONC201’s anti-proliferative effects, ONC201 treatment was associated with inhibition of the AKT/mTOR and MAPK pathways and activation of both TRAIL/DR5/caspase 8 and caspase 9/3, and PARP mediated apoptotic pathways in USC cells. Intriguingly, ONC201 significantly reduced the ability of adhesion and invasion through mediation of cadherins and their associated transcription factors. Thus, our study provides a strong rationale for the application of ONC201 as an anti-tumorigenic and anti-metastatic agent in USC.
The mechanism of action of ONC201’s inhibition of cancer cell proliferation is an area of active study. The anti-tumorigenic activity of ONC201 was initially reported to be associated with an early-stage integrated stress response (ISR) activation and a late stage inactivation of AKT and ERK pathways. This ultimately resulted in induction of TRAIL and DR5 and cell death through apoptosis in a p53 independent and a cell-type dependent manner [6,16]. Dephosphorylated FOXO3a, a master regulator of the TRAIL gene promoter, triggers the induction of TRAIL and DR5 expression, which activates the extrinsic apoptotic pathway and consequently cause caspase-8 dependent apoptosis in ONC201 treated-solid tumor cells [8,17]. Recent studies found that the anti-tumorigenic activity of ONC201 did not require FOXO3a-dependent transcription of TRAIL or caspase 8 activation in hematological malignancies, rather the intrinsic apoptotic pathway was significantly activated in response to ONC201 [7,12,18]. Overexpression of BCL-2 protected against ONC201-induced apoptosis and inhibition of BCL-2 expression synergistically increased apoptosis induced by ONC201 in these cells [7]. Interestingly, ONC201 exhibited potent cytotoxic and cytostatic activities through both extrinsic (TRAIL/caspase-8-dependent) and mitochondrial (caspase-9-dependent) apoptotic pathways in pancreatic cancer cells [19]. In agreement with previous studies, we found that treatment with ONC201 reduced BCL-2 expression and induced DR5, cleaved caspase 8, cleaved caspase 9, cleaved caspase 3 and PARP expression in USC cells. Inhibition of caspases activation by Z-VAD-FMK significantly attenuated ONC201-induced survival loss and apoptosis. These findings indicate that ONC201 inhibits USC cells growth through both a TRAIL dependent and a TRAIL independent manner, suggesting that the mechanism of action of ONC201 may be tissue- or cancer type-specific [16,18].
Recent whole-exome sequencing data finds that USC has a unique molecular signatures distinct from most endometrioid endometrial cancers, including overexpression of HER2 and mutations in TP53 as well as mutations in the PIK3CA/AKT/mTOR and cyclin E/FBXW7 pathways [13,20]. The overexpression of HER2 results in the phosphorylation of tyrosine kinase residues and constitutively activated PIK3CA/AKT/mTOR and RAS/RAF/MEK pathways, which are vital for regulating cell proliferation, apoptosis resistance, differentiation, metabolism, invasion and survival [13]. The complexity and function of the cellular signaling network in USC cells opens the possibility of actively targeting multiple pathways, which might lead to superior anti-tumor activity. Several PI3K/AKT/mTOR inhibitors and anti-HER2 therapies are currently under evaluation in clinical trials against a variety of human cancers, including USC [21]. In the current study, we showed that ONC201 simultaneously reduced phosphorylation of AKT and p42/44 and induced AMPK activation in USC cells in vitro, suggesting that the effects of ONC201 on cell growth may be intricately linked to the inhibition of PIK3CA/AKT/mTOR and RAS/RAF/MEK pathways in USC cells.
USC is commonly found to have lymphovascular space invasion and lymph node involvement, even in apparently early stage disease. Cell adhesion and invasion are early steps in metastasis which involve tumor cell degradation of the extracellular matrix (ECM) and resultant cell migration [15]. TRAIL signaling exerts regulatory effects on cancer cell adhesion, migration and invasion. TRAIL has been shown to significantly inhibit liver metastasis in three separate tumor models [22]. TRAIL-resistant breast cancer cells have enhanced invasiveness and induce the epithelial to mesenchymal transition (EMT) by down-regulation of PTEN [23]. Recently, Wagner et al found that the migration and invasion capabilities of cancer cells were significantly attenuated by ONC201, even in TRAIL-resistant cancer cells [24]. Our results also confirm the ability of ONC201 to reduce adhesion and invasion in USC cells, as measured by the transwell assay and wound healing assay as well as decreased expression of Snail and VEGF, and increased expression of E-cadherin. However, the underlying biological mechanisms by which ONC201 affected EMT and VEGF remain areas for further investigation.
Cytotoxic chemotherapy with a combination of paclitaxel and carboplatin is the standard adjuvant therapy for advanced stage and recurrent USC. Low concentrations of TRAIL significantly enhanced the lethality of paclitaxel against human cancer cells, and the combination of TRAIL and paclitaxel resulted in a significantly improved anti-tumorigenic effect compared with either TRAIL or paclitaxel alone in in nude mice bearing glioblastoma xenografts [25]. Furthermore, paclitaxel also markedly sensitized TRAIL-mediated apoptosis in TRAIL-resistant gastric cancer cells and animal models through activation of the mitochondrial apoptotic pathway, upregulation of TRAIL receptors and inactivation of the MAPK pathway [26]. It has been reported that ONC201 has broad synergistic potential with chemotherapeutics and other small molecules in a variety of solid tumors and hematologic malignancies including pancreatic cancer, myeloma, hepatocellular carcinoma, lymphoma and non-small cell lung cancer [16,27,28]. Synergistic interactions between ONC201 and paclitaxel have been observed in non-small cell lung cancer cells and non-small cell lung cancer xenografts in nude mice [8]. After treating the USC cells with varying concentrations of paclitaxel in combination with ONC201, we found that low concentrations of ONC201 increased sensitivity of the cells to paclitaxel in both cell lines (CI<1). Given the unique pharmacokinetic (PK) and pharmacodynamic (PD) profiles of ONC201, the combination of ONC201 and paclitaxel is an attractive potential therapeutic strategy for USC. Further work on the potential application of this combination in clinical trials is certainly warranted [6,9,29,30].
In conclusion, ONC201 causes significant inhibition of USC cell proliferation and invasion through activation of TRAIL/DR5/caspase 8 and mitochondrial apoptotic pathways as well as modulation of AMPK/AKT/mTOR and MAPK pathways. Furthermore, the combination of ONC201 and paclitaxel has a synergistic inhibitory effect in USC cells. Our recent work finds that oral administration of ONC201 showed a significant reduction in tumor growth and serum VEGF production in the absence of significant toxicities or side effects in a transgenic mouse model of endometrial adenocarcinoma (Data not shown). Given that ONC201 has been well-tolerated and has shown preliminary efficacy in solid tumor clinical trials [9,10,29,30], our results provide a strong rationale for the investigation of ONC201 as a single agent and in combination with paclitaxel in USC clinical trials.
Acknowledgements
This work was generously supported by V Foundation Translational Award and Beijing health system high-level health personnel training program fund (2014-3-073).
Disclosure of conflict of interest
None.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.McGunigal M, Liu J, Kalir T, Chadha M, Gupta V. Survival differences among uterine papillary serous, clear cell and grade 3 endometrioid adenocarcinoma endometrial cancers: a national cancer database analysis. Int J Gynecol Cancer. 2017;27:85–92. doi: 10.1097/IGC.0000000000000844. [DOI] [PubMed] [Google Scholar]
- 3.de Jonge MM, Mooyaart AL, Vreeswijk MP, de Kroon CD, van Wezel T, van Asperen CJ, Smit VT, Dekkers OM, Bosse T. Linking uterine serous carcinoma to BRCA1/2-associated cancer syndrome: a meta-analysis and case report. Eur J Cancer. 2017;72:215–225. doi: 10.1016/j.ejca.2016.11.028. [DOI] [PubMed] [Google Scholar]
- 4.Sagae S, Susumu N, Viswanathan AN, Aoki D, Backes FJ, Provencher DM, Vaughan M, Creutzberg CL, Kurzeder C, Kristensen G, Lee C, Kurtz JE, Glasspool RM, Small W Jr. Gynecologic Cancer InterGroup (GCIG) consensus review for uterine serous carcinoma. Int J Gynecol Cancer. 2014;24:S83–89. doi: 10.1097/IGC.0000000000000264. [DOI] [PubMed] [Google Scholar]
- 5.Boruta DM, Gehrig PA, Fader AN, Olawaiye AB. Management of women with uterine papillary serous cancer: a Society of Gynecologic Oncology (SGO) review. Gynecol Oncol. 2009;115:142–153. doi: 10.1016/j.ygyno.2009.06.011. [DOI] [PubMed] [Google Scholar]
- 6.Allen JE, Kline CL, Prabhu VV, Wagner J, Ishizawa J, Madhukar N, Lev A, Baumeister M, Zhou L, Lulla A, Stogniew M, Schalop L, Benes C, Kaufman HL, Pottorf RS, Nallaganchu BR, Olson GL, Al-Mulla F, Duvic M, Wu GS, Dicker DT, Talekar MK, Lim B, Elemento O, Oster W, Bertino J, Flaherty K, Wang ML, Borthakur G, Andreeff M, Stein M, El-Deiry WS. Discovery and clinical introduction of first-in-class imipridone ONC201. Oncotarget. 2016;7:74380–74392. doi: 10.18632/oncotarget.11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ishizawa J, Kojima K, Chachad D, Ruvolo P, Ruvolo V, Jacamo RO, Borthakur G, Mu H, Zeng Z, Tabe Y, Allen JE, Wang Z, Ma W, Lee HC, Orlowski R, Sarbassov dos D, Lorenzi PL, Huang X, Neelapu SS, McDonnell T, Miranda RN, Wang M, Kantarjian H, Konopleva M, Davis RE, Andreeff M. ATF4 induction through an atypical integrated stress response to ONC201 triggers p53-independent apoptosis in hematological malignancies. Sci Signal. 2016;9:ra17. doi: 10.1126/scisignal.aac4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Allen JE, Krigsfeld G, Mayes PA, Patel L, Dicker DT, Patel AS, Dolloff NG, Messaris E, Scata KA, Wang W, Zhou JY, Wu GS, El-Deiry WS. Dual inactivation of Akt and ERK by TIC10 signals Foxo3a nuclear translocation, TRAIL gene induction, and potent antitumor effects. Sci Transl Med. 2013;5:171ra117. doi: 10.1126/scitranslmed.3004828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stein MN, Bertino JR, Kaufman HL, Mayer T, Moss R, Silk A, Chan N, Malhotra J, Rodriguez L, Aisner J, Aiken RD, Haffty BG, DiPaola RS, Saunders T, Zloza A, Damare S, Beckett Y, Yu B, Najmi S, Gabel C, Dickerson S, Zheng L, El-Deiry WS, Allen JE, Stogniew M, Oster W, Mehnert JM. First-in-human clinical trial of oral ONC201 in patients with refractory solid tumors. Clin Cancer Res. 2017;23:4163–4169. doi: 10.1158/1078-0432.CCR-16-2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lorna Rodriguez-Rodriguez JMM, Silk AW, Chan N, Malhotra J, Aisner J, Saunders T, Yu BN, Dickerson S, Tarapore R, Allen JE, Stogniew M, Oster W, Kaufman H, Haffty BG, Bertino JR, Stein MN. Clinical activity of the selective DRD2 antagonist ONC201, an imipridone, in metastatic endometrial cancer (mEC) J. Clin. Oncol. 2017;35:5592–5592. [Google Scholar]
- 11.Kalogera E, Roy D, Khurana A, Mondal S, Weaver AL, He X, Dowdy SC, Shridhar V. Quinacrine in endometrial cancer: repurposing an old antimalarial drug. Gynecol Oncol. 2017;146:187–195. doi: 10.1016/j.ygyno.2017.04.022. [DOI] [PubMed] [Google Scholar]
- 12.Tu YS, He J, Liu H, Lee HC, Wang H, Ishizawa J, Allen JE, Andreeff M, Orlowski RZ, Davis RE, Yang J. The Imipridone ONC201 induces apoptosis and overcomes chemotherapy resistance by up-regulation of bim in multiple myeloma. Neoplasia. 2017;19:772–780. doi: 10.1016/j.neo.2017.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Menderes G, Clark M, Santin AD. Novel targeted therapies in uterine serous carcinoma, an aggressive variant of endometrial cancer. Discov Med. 2016;21:293–303. [PubMed] [Google Scholar]
- 14.Mahdi H, Xiu J, Reddy SK, DeBernardo R. Alteration in PI3K/mTOR, MAPK pathways and Her2 expression/amplification is more frequent in uterine serous carcinoma than ovarian serous carcinoma. J Surg Oncol. 2015;112:188–194. doi: 10.1002/jso.23993. [DOI] [PubMed] [Google Scholar]
- 15.Kilgore J, Jackson AL, Clark LH, Guo H, Zhang L, Jones HM, Gilliam TP, Gehrig PA, Zhou C, Bae-Jump VL. Buformin exhibits anti-proliferative and anti-invasive effects in endometrial cancer cells. Am J Transl Res. 2016;8:2705–2715. [PMC free article] [PubMed] [Google Scholar]
- 16.Talekar MK, Allen JE, Dicker DT, El-Deiry WS. ONC201 induces cell death in pediatric non-Hodgkin’s lymphoma cells. Cell Cycle. 2015;14:2422–2428. doi: 10.1080/15384101.2015.1054086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kline CL, Van den Heuvel AP, Allen JE, Prabhu VV, Dicker DT, El-Deiry WS. ONC201 kills solid tumor cells by triggering an integrated stress response dependent on ATF4 activation by specific eIF2alpha kinases. Sci Signal. 2016;9:ra18. doi: 10.1126/scisignal.aac4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Endo Greer Y, Lipkowitz S. ONC201: stressing tumors to death. Sci Signal. 2016;9:fs1. doi: 10.1126/scisignal.aad7955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Q, Wang H, Ran L, Zhang Z, Jiang R. The preclinical evaluation of TIC10/ONC201 as an anti-pancreatic cancer agent. Biochem Biophys Res Commun. 2016;476:260–266. doi: 10.1016/j.bbrc.2016.05.106. [DOI] [PubMed] [Google Scholar]
- 20.Fader AN, Santin AD, Gehrig PA. Early stage uterine serous carcinoma: management updates and genomic advances. Gynecol Oncol. 2013;129:244–250. doi: 10.1016/j.ygyno.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 21.Black JD, English DP, Roque DM, Santin AD. Targeted therapy in uterine serous carcinoma: an aggressive variant of endometrial cancer. Womens Health (Lond) 2014;10:45–57. doi: 10.2217/whe.13.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takeda K, Hayakawa Y, Smyth MJ, Kayagaki N, Yamaguchi N, Kakuta S, Iwakura Y, Yagita H, Okumura K. Involvement of tumor necrosis factor-related apoptosis-inducing ligand in surveillance of tumor metastasis by liver natural killer cells. Nat Med. 2001;7:94–100. doi: 10.1038/83416. [DOI] [PubMed] [Google Scholar]
- 23.Wang H, Xu C, Kong X, Li X, Kong X, Wang Y, Ding X, Yang Q. Trail resistance induces epithelial-mesenchymal transition and enhances invasiveness by suppressing PTEN via miR-221 in breast cancer. PLoS One. 2014;9:e99067. doi: 10.1371/journal.pone.0099067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wagner J, Kline CL, Ralff MD, Lev A, Lulla A, Zhou L, Olson GL, Nallaganchu BR, Benes CH, Allen JE, Prabhu VV, Stogniew M, Oster W, El-Deiry WS. Preclinical evaluation of the imipridone family, analogs of clinical stage anti-cancer small molecule ONC201, reveals potent anti-cancer effects of ONC212. Cell Cycle. 2017;16:1790–1799. doi: 10.1080/15384101.2017.1325046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dorsey JF, Mintz A, Tian X, Dowling ML, Plastaras JP, Dicker DT, Kao GD, El-Deiry WS. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and paclitaxel have cooperative in vivo effects against glioblastoma multiforme cells. Mol Cancer Ther. 2009;8:3285–3295. doi: 10.1158/1535-7163.MCT-09-0415. [DOI] [PubMed] [Google Scholar]
- 26.Li L, Wen XZ, Bu ZD, Cheng XJ, Xing XF, Wang XH, Zhang LH, Guo T, Du H, Hu Y, Fan B, Ji JF. Paclitaxel enhances tumoricidal potential of TRAIL via inhibition of MAPK in resistant gastric cancer cells. Oncol Rep. 2016;35:3009–3017. doi: 10.3892/or.2016.4666. [DOI] [PubMed] [Google Scholar]
- 27.Lev A, Lulla AR, Wagner J, Ralff MD, Kiehl JB, Zhou Y, Benes CH, Prabhu VV, Oster W, Astsaturov I, Dicker DT, El-Deiry WS. Anti-pancreatic cancer activity of ONC212 involves the unfolded protein response (UPR) and is reduced by IGF1-R and GRP78/BIP. Oncotarget. 2017;8:81776–81793. doi: 10.18632/oncotarget.20819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Allen JE, Prabhu VV, Talekar M, van den Heuvel AP, Lim B, Dicker DT, Fritz JL, Beck A, El-Deiry WS. Genetic and pharmacological screens converge in identifying FLIP, BCL2, and IAP proteins as key regulators of sensitivity to the TRAIL-inducing anticancer agent ONC201/TIC10. Cancer Res. 2015;75:1668–1674. doi: 10.1158/0008-5472.CAN-14-2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Edwards H, Ge Y. ONC201 shows promise in AML treatment. Cell Cycle. 2018:1. doi: 10.1080/15384101.2017.1421035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arrillaga-Romany I, Chi AS, Allen JE, Oster W, Wen PY, Batchelor TT. A phase 2 study of the first imipridone ONC201, a selective DRD2 antagonist for oncology, administered every three weeks in recurrent glioblastoma. Oncotarget. 2017;8:79298–79304. doi: 10.18632/oncotarget.17837. [DOI] [PMC free article] [PubMed] [Google Scholar]