

Abstract
Triple negative breast cancer (TNBC) accounts for about 10-15% of all breast cancers. It is a heterogeneous disease, characterized by early relapse, aggressive behavior, and poor prognosis, when compared to other breast cancer subtypes. Interestingly, most of the heat shock protein 90 (Hsp90) client proteins are oncoproteins, and some are closely related to the key factors that promote the progression of TNBC. Anacardic acid (AA), which is commonly seen in natural plants of Anacardiaceae, exhibits potent Hsp90 ATPase inhibition activity. In this study, the anticancer effects of AA on TNBC MDA-MB-231 cells were investigated. The results of our study showed that AA inhibited cell proliferation, induced G0/G1-phase cell cycle arrest, suppressed cell invasion and migration, and induced apoptosis in the MDA-MB-231 cells. Regulation of the key Hsp90-dependent tumor-related molecules or endoplasmic reticulum stress (ERS) related molecules, such as GRP78, Hsp70, CDK-4, MMP-9, Bcl-2, and Mcl-1 by AA may be related to these effects. Taken together, our results suggest that AA shows potential as a possible new drug for therapy of TNBC.
Keywords: Anacardic acid, TNBC, Hsp90, ERS, anticancer effect
Introduction
Triple negative breast cancer (TNBC) is defined by the lack of estrogen receptors (ERs), progesterone receptors (PRs), and by human epidermal growth factor receptor 2 (HER2)-negative status. TNBC accounts for about 10-15% of all breast cancers and is associated with a higher mortality rate, compared to other subtypes [1].
In recent years, several targeted treatment options have been explored for patients with TNBC, such as poly (ADP ribose) polymerase (PARP) inhibitors, angiogenesis inhibitors, EG-FR-targeted agents, mTOR inhibitors, src tyrosine kinase inhibitors and androgen receptor inhibitors. However, efficacy data and results are limited and the only available treatment options are surgery, chemo and/or radiotherapy.
The molecular chaperone heat shock protein 90 (Hsp90) is responsible for maintaining the correct folding and stability of many signaling proteins, and is emerging as an important target in cancer therapeutics [2,3]. Hsp90 is often overexpressed in a range of cancers, including breast cancer, and high levels of Hsp90 have been associated with poor prognosis in breast cancer patients [4]. Hsp90 inhibition can trigger proteasomal degradation of multiple oncoproteins, thereby reducing cancer cell proliferation, survival, invasion, and angiogenesis, hence promoting apoptosis [5-7]. In particular, HSP90 levels are markedly increased in tumors and high HSP90 expression is associated with a negative prognosis in breast cancer [8].
Bark of plants belonging to the family Anacar-diaceae, are frequently used in treating gastric ulcer, gastritis and gastric cancer in Mexico [9]. Anacardic acid (6-pentadecylsalicylic acid, AA) (Figure 1A), a natural compound, isolated from the traditional medicine Amphipterygium adstringens or commonly seen in plants of Anacar-diaceae, is an important composition of cashew, ginkgo leaf and fruit, and it has been reported to possess antibacterial and anticancer effects [10-12]. Additionally, AA has a number of roles, including the inhibition of lipid synthesis, enzyme activity such as lipoxygenase, prostaglandin endoperoxide synthase and histone acetyltransferase, expression of nuclear factor-κB as well as the activation of aurora kinase A [13-17]. Li H et al. [18] also showed that AA exhibited the inhibition of Hsp90 ATPase activity. However, the molecular mechanisms have not been well understood. The aim of this study was to investigate the effects of AA on cell proliferation, cell cycle, invasion, migration and apoptosis in MDA-MB-231 cells, and to explore its preliminary mechanism.
Figure 1.

Inhibitory effect of AA on cell viability in MDA-MB-231 cells. A. Chemical structure of AA; B. Dose- and time-response curve of the effects of AA on cell viability in MDA-MB-231 cells. Cells were treated with DMSO or variant concentrations of AA for 24 h, 48 h, and 72 h. The cell viability was measured by an MTT assay.
Materials and methods
Reagents and antibodies
Dulbecco’s modified Eagle’s medium (DMEM), trypsin, and fetal bovine serum (FBS) were obtained from Gibco (Grand Island, NY). 3-(4,5-Dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium bromide (MTT), cocktail of protaease inhibitors, and Propidium iodide (PI) were purchased from Sigma (USA). Mitochondrial membrane potential (ΔΨm) assay kit (JC-1) was obtained from Beyotime Institute of Biotechnology (China). Anacardic acid, anti-β-actin, anti-GRP78, anti-HSP70, anti-E-cadherin, anti-Vimentin, and secondary antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-Bcl-2, anti-Mcl-1 (myeloid cell leukemia-1), and anti-MMP-9 antibodies were purchased from Beijing Biosynthesis Biotechnology Co (China). Anti-cas-pase-3 monoclonal antibodies were obtained from Abcam (Cambridge, UK). Anti-CDK-4 was obtained from Proteintech (USA). Anti-Hsp-90 was obtained from Stressgen (USA). Transwell boyden chamber system was purchased from Corning Life Sciences (NY). Matrigel was purchased from BD Biosciences (Bedford, MA, USA).
Cell culture
Human breast cancer MDA-MB-231 cell was obtained from Shanghai Cell Bank (China). Cells were grown in DMEM, supplemented with 10% FBS, penicillin (100 units/ml), streptomycin (100 units/ml), and HEPES (25 mM). All cells were maintained in the presence of 5% CO2 at 37°C.
Cell viability assay
The cell viability was determined by the MTT assay [19]. 1 × 104 cells were plated in 96-well microtiter plates and treated with various concentration of Anacardic acid for 24 h, 48 h, and 72 h. At the end of each time point, 15 μl of MTT (5 mg/ml in PBS) were added to each well incubated at 37°C for 4 h. After 4 h, the MTT solution was removed, and 150 μl DMSO were added to each well to dissolve the formazan crystals. The plate was further incubated at room temperature for 10 min, and the absorbance (A) of the wells was determined using a plate reader at a test wavelength of 570 nm.
Colony formation assay
Cells were cultured in 6-well plates (1 × 104 cells/well) overnight. The medium was then exchanged with fresh medium containing AA. The plates were incubated under cell culture conditions for another 8 days, at which point the medium was removed and the cells were washed twice with PBS and fixed with paraformaldehyde for 10 min at -20°C. The colonies were stained with 2% crystal violet for 10 min, washed with double-distilled water and dried at room temperature, before being counted.
Determination of apoptotic cells (PI staining)
MDA-MB-231 cells were seeded at 1 × 105 cells/well in 6-well cell culture plates and allowed to reach exponential growth for 24 h before treatment. Whole-cell lysates from MDA-MB-231 cells were treated with various concentration of AA for 24 h/48 h using PI staining and then evaluated using flow cytometry. Quantitative analysis of subG1 cells were carried out in a BD Accuri C6 flow cytometer using the Cell Quest software.
Mitochondrial membrane potential (ΔΨm) assay using fluorescent microscope
The cells were seeded at 2 × 105 cells/well in 12-well culture plates and allowed to attach overnight before treatment. Changes in ΔΨm after different treatments were evaluated by staining with the cationic dye tetrechloro-tetraethylbenzimidazol carbocyanine iodide (JC-1) according to the manufacturer’s instruction (Beyotime Institute of Biotechnology, China). After the incubation, the dye was aspirated from the plates, and the plates were washed three times with 1 × JC-1 buffer and examined with an invert fluorescent microscope (Olym-pus IX71, Japan) using both red and green channels.
Cell invasion assay
The invasion assay was performed using a 24-well cell culture plate with 8.0 μm pore membrane inserts. The membrane undersurface was coated with 50 μl Matrigel with serum free medium for 30 min at 37°C. MDA-MB-231 cells were starved in serum-free medium overnight, and 5 × 104 cells were resuspended in 100 μl serum-free medium and placed in the upper chambers. The lower well of each chamber was filled with 800 μl of DMEM supplemented with 10% FBS and incubated for 36 h. Reagents added to the upper surface of the membrane were removed by cotton buds, and the cells on the lower chamber were inubated with paraformaldehyde in PBS buffer and stained with 0.1% crystal violet. Five visual fields were randomly selected for each insert and photographed under a light microscope at 200 × magnification. The number of invasive cells was then counted and analyzed to determine statistically significant differences. Each condition was assayed in triplicate, the experiments were performed independently at least three times, and the results are expressed as the number of cells/field. A one-way analysis of variance was used to determine significant differences.
Cell migration assay
Migration assay was performed using a 24-well cell culture plate with 8.0 μm pore membrance inserts without Matrigel. 5 × 104 MDA-MB-231 cells were added to the upper wells, and the chambers were incubated for 36 h at 37°C. The lower chamber was filled with 800 μl 10% FBS as the chemoattractant. After 36 h in normoxic conditions the cells that had migrated were stained and photographed under a light microscope at 200 × magnification. The number of migratory cells was then counted and analyzed to determine statistically significant differences. Each condition was assayed in triplicate, the experiments were performed independently at least three times, and the results are expressed as the number of cells/field. A one-way analysis of variance was used to determine significant differences.
Wound healing assay
Cell migration was assessed using a wound healing assay. Cells were plated in 6-well plates (Corning Life Sciences) at 5 × 105 cells/well and allowed to grow to 90% confluence. The cells were scraped with a sterile micropipette tip to create a gap of standard width. To remove non-adherent cells, the plates were rinsed gently with medium twice prior to incubation. The wound closure was monitored for 36 h at × 100 magnification. The wound areas were observed under an inverted microscope and imaged at the appropriate fields to calculate the healing percentages. Each experiment was performed in triplicate.
Western blot analysis
The cells were plated in 6-well culture plate at a density of 5 × 105 cells per well. After incubation with drugs, the cell were harvested, washed twice in PBS, and lysed in lysis buffer [50 mM Tris-HCl pH = 7.4, 1% Triton X-100, 150 mM NaCl, and a cocktail of protaease inhibitors (Sigma)]. Lysetes were loaded onto 10%~15% SDS-PAGE and transferred to a PVDF membrance. The membranes were blocked with 5% skim milk, and were incubated with primary antibodies overnight at 4°C. Detection was performed using secondary antibody for 1 h at room temperature and an ECL detection system (Bio-Rad, CA, USA).
Statistical analysis
All experiments were repeated at least three times. Data were presented as the Mean ± SD. Statistical comparisons between Anacardic acid treatment groups and control were carried out using one-way analysis of variance (ANOVA) followed by Dunnett t-tests. Differences were considered significant at P < 0.05, and in here denoted as *. All statistical analyses were performed using SPSS 19.0 software (Chicago, IL, USA).
Results
AA exhibit anti-proliferative effect in MDA-MB-231 cells
In the present study, we have used MDA-MB-231 cancer cells, reported to be a highly metastatic, human TNBC cell line. Previous studies have revealed that AA exerts anticancer effects in various carcinomas [20,21].
To understand anticancer activity of AA on TNBC cells, we analyzed the anti-proliferative effect of AA on MDA-MB-231 cells, after treatment with increasing doses of the compound (0-100 μM) for specified time courses (24 h, 48 h, and 72 h). As shown in Figure 1B, AA showed significant anti-proliferative activity on MDA-MB-231 cells in a dose- and time-dependent manner, with an IC50 value of 19.7 μM at 24 h, after treatment. Similar to the MTT assays, the data-analyzed colony formation assays, which also showed that AA inhibited cell growth at low doses (Figure S1).
AA induces cell cycle arrest of MDA-MB-231 cells
As shown in Figure 2A, with increasing concentrations of AA treatment, the change of Hsp90 protein was not obvious. Previously, AA also showed strong yeast Hsp90 ATPase inhibition activity (IC50, 82.5 μM) [18].
Figure 2.

Effects of AA on the expression of Hsp90 and AA induces cell cycle arrest of MDA-MB-231 cells. A. Whole-cell lysates from MDA-MB-231 cells treated with vehicle or various concentration of AA for 24 h, were subjected to western blot analysis. B. Representative flow cytometry histograms of apoptosis. MDA-MB-231 cells were treated with: (a) Vehicle, (b) 25 μM of AA, (c) 50 μM of AA, and (d) 100 μM of AA for 24 h, respectively. Apoptosis was measured by the propidium iodide (PI) method using flow cytometry. C. Representative flow cytometry histograms of cell cycle. MDA-MB-231 cells were treated with: (a) Vehicle, (b) 25 μM of AA, (c) 50 μM of AA, and (d) 100 μM of AA for 24 h, respectively. D. Cell cycle distribution expressed as percentage of control. Data are presented as mean ± SD of triplicates. AA arrested cells in the G0/G1-phase. The differences among the four treatments were analysed by Dunnett t-tests (*P < 0.05 vs control).
According to the flow cytometric apoptosis detection by PI single staining method, the corresponding share of sub-G1 cells phase ratio represents the apoptosis rate of each group. After 24 h, the rate of apoptosis (Proportion of subG1 cells) in the control was 2.5%, while the apoptotic rate was 2.6%, 3.1%, and 13.2% for the cells treated with 25, 50, or 100 μM of AA, respectively (as shown in Figure 2B). AA exerts a very mild effect on promoting apoptosis. However, AA arrests the cell cycle in G0/G1 phase. The results of flow cytometric analysis showed that the percentage of G0/G1 phase of MDA-MB-231 cells increased after treatment with different concentrations of AA for 24 h. The percentage of cells in G0/G1 phase in the control was 51.1 ± 1.47%, while the rate was 56.8 ± 2.57%, 62.2 ± 3.81%, and 70.7 ± 2.01% for the cells treated with 25, 50, or 100 μM of AA, respectively (*P < 0.05 vs control) (Figure 2C, 2D). No increase in S or G2/M peak was observed in MDA-MB-231 cells.
AA changes the expression of GRP78 and Hsp70 involved in the potential triggering of ERS in MDA-MB-231 cells
Previous studies have shown that AA is a potent inducer of ERS [12,22,23]. Induction of ERS by AA was supported by a dose- and time-dependent increase in expression of the ER signaling downstream molecules, such as GRP78/BiP, phosphorylated eIF2α, ATF4 and CHOP, in both HepG2 and U266 cell lines. Moreover, AA suppressed HepG2 xenograft tumor growth, associated with increased ERS in vivo [24].
A number of cancer-associated proteins have been identified as Hsp90 clients, including GRP78 and Hsp70 proteins, to name but two [7,24]. In order to clarify the cause of the above-mentioned changes by AA, we examined the expression of the relevant proteins and observed that, both GRP78 and Hsp70 proteins gradually increased (Figure 3). This indirectly reflects that AA triggers ERS in MDA-MB-231 cells.
Figure 3.
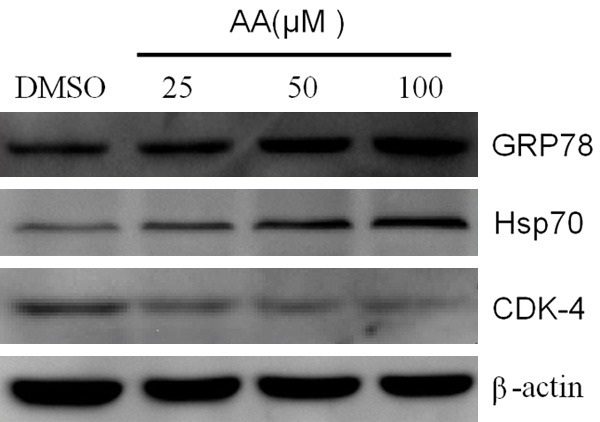
Effects of AA on the expression of GRP78, Hsp70 and CDK-4. Whole-cell lysates from MDA-MB-231 cells treated with vehicle or various concentration of AA for 24 h, were subjected to western blot analysis.
AA induces cell cycle arrest via down-regulation of CDK-4 in MDA-MB-231 cells
Western blot analysis demonstrated that classical client protein, cyclin-dependent kinase CDK-4, was significantly downregulated in the presence of AA (Figure 3). The results showed that AA may induce cell cycle arrest via down-regulation of CDK-4.
AA changes the expression of oncoproteins involved in the invasion and migration of MDA-MB-231 cells
We examined the effects of AA on the invasion and migration of MDA-MB-231 cells. Low concentrations of AA did not have a significant effect on cell death, but suppressed cell invasion and migration. As shown in Figure 4, with increasing concentrations of AA treatment, invasion of cells gradually decreased (*P < 0.05 vs control). Next, we exa 0.05 vs control). Next, we examined the effect of AA on cell migration using the Transwell cell migration assay and wound-healing assays. The results showed that, with increasing concentrations of AA, the migration inhibition rate also increased (Figure 5 and Figure S2).
Figure 4.

AA suppressed the invasion of MDA-MB-231 cells. A. The invasion of MDA-MB-231 cells was suppressed following exposure to vehicle or various concentrations of AA. Using AA untreated cells as the controls, five visual fields were randomly selected for each insert and photographed under a light microscope at 200 × magnification; B. Quantification of invasion of MDA-MB-231 cells inhibited by AA. The differences among the four treatments were analysed by Dunnett t-tests (*P < 0.05 vs control).
Figure 5.

AA suppressed the migration of MDA-MB-231 cells. A. The migration of MDA-MB-231 cells was suppressed following exposure to vehicle or various concentrations of AA. Using AA untreated cells as the controls, five visual fields were randomly selected for each insert and photographed under a light microscope at 200 × magnification; B. Quantification of migration of MDA-MB-231 cells inhibited by AA. The differences among the four treatments were analysed by Dunnett t-tests (*P < 0.05 vs control).
To determine whether there was any change in MMP-9 or Vimentin and E-cadherin in AA-treated cells, we investigated the expression of these proteins. As shown in Figure 6, MMP-9 and Vimentin protein expression was gradually down-regulated with increasing concentrations of AA, meanwhile E-cadherin protein gradually increased.
Figure 6.

AA changes the expression of oncoproteins involved in the invasion and migration potential of MDA-MB-231 cells. Whole-cell lysates from MDA-MB-231 cells treated with vehicle or various concentration of AA for 36 h, were subjected to western blot analysis.
AA induces apoptosis in MDA-MB-231 cells
To clarify whether AA induced apoptosis, MDA-MB-231 cells were treated with AA. After 48 h, the rate of apoptosis in the control was 1.3%, while the apoptotic rate was 8.7%, 35.5%, and 49.0% for the cells treated with 25, 50, or 100 μM of AA, respectively (Figure 7A). One of the early critical events in apoptosis is the loss/disruption of the mitochondrial membrane potential (Δψm) in the cells, which eventually causes the initiation and activation of apoptotic cascades. We sought to determine whether AA treatment had any effect on the Δψm in MDA-MB-231 cells using JC-1 staining. The results in Figure S3 show that compared with the control group the AA-treated cells exhibited reduction in red fluorescence, but increased signals in green fluorescence, indicating a reduction in Δψm, and that AA induced apoptosis in MDA-MB-231 cells only to a small extent, which was mediated by the mitochondria. Induction of apoptosis was preceded by cell cycle arrest.
Figure 7.

AA induces apoptosis in MDA-MB-231 cells and effects of AA on the expression of apoptosis-related proteins. A. Representative flow cytometry histograms of apoptosis. MDA-MB-231 cells were treated with: (a) Vehicle, (b) 25 μM of AA, (c) 50 μM of AA, and (d) 100 μM of AA for 48 h, respectively. B, C. Whole-cell lysates from MDA-MB-231 cells treated with vehicle or various concentration of AA for 48 h, were subjected to western blot analysis.
We next examined whether apoptosis related proteins (Bcl-2, Mcl-1, and Caspase 3), were regulated by AA. As shown in Figure 7B, with increasing concentrations of AA, the expression of anti-apoptotic proteins, Bcl-2 and Mcl-1, was gradually down-regulated. In addition, the Caspase 3 protein cleavage fragment becomes more obvious and results in a dose-dependent cleavage, generating processed caspase-3 (Figure 7C).
Discussion
Our results showed that AA induces cell cycle arrest via down-regulation of CDK-4 and triggers ERS in MDA-MB-231 cells. A previous study has shown that treatment of HD-LM2 cells with 17-AAG predominantly induced G0-G1 cell cycle arrest, which was associated with down-regulation of several cell cycle regulatory proteins, including CDK-4, CDK-6, polo-like kinase 1 (PLK1), cyclin B1 and, to a lesser extent, cyclin D1. Inhibition of HSP90 function by 17-AAG showed a time- and dose-dependent growth inhibition of Hodgkin’s lymphoma cell lines. Moreover, 17-AAG induced cell cycle arrest and apoptosis, which were associated with a decrease in CDK-4, CDK-6, and PLK1 [25]. Currently, three CDK4/6 inhibitors have been tested in clinical breast cancer trials [26].
In spite of early detection and novel therapeutic interventions, metastatic breast cancer is still the leading cause of cancer death in women. Moreover, the poor prognosis of TNBC is linked to the early metastatic spread of the disease that occurs within 2 or 3 years of diagnosis. Many studies have associated this invasive property to the epithelial-mesenchymal transition (EMT) which is a crucial event during cancer metastasis [27,28]. EMT is characterized by a morphological change in the epithelial cells that lose their differentiated phenotype and acquire an invasive mesenchymal one. During EMT, TNBC cells lose epithelial cell markers, such as EGFR, epithelial cadherin (E-cadherin) and show an enhanced expression of mesenchymal cell markers, such as vimentin and neuronal cadherin (N-cadherin) [29,30]. Cadherin switches, such as E-cadherin to N-cadherin, are classic representative markers of the EMT process [31]. Many studies have examined the possible role of EMT in breast cancer. Vimentin is a key regulator of breast cancer cell migration and a marker for mesenchymal subtype, characteristic of cancer cells that have undergone EMT. Expression of vimentin is related to the reduced expression of E-cadherin and upregulation of N-cadherin [32]. Numerous studies have linked aberrant expression of E-cadherin with the development of metastases in breast and other cancers [33]. Recently, Lee et al. suggested that Embelin induced the expression of E-cadherin and inhibited the expression of N-cadherin and vimentin in MDA-MB-231 cells. Thus, Embelin could regulate breast cancer metastasis through modulation of EMT markers [34].
Matrix metalloproteinases, especially MMP-9, play an important role in tumor invasion and metastasis. The imbalance between MMPs and their inhibitors may facilitate tumor progression [7]. In addition, MMP-9 levels in tumor tissue, as well as in serum, plasma, and urine, are significantly elevated in patients with breast cancer [35,36].
Our experiments confirmed that AA can significantly suppress invasion and migration of MDA-MB-231 cells, and its mechanism may be related to the down-regulation of MMP-9 and vimentin protein, whilst increasing the expression of E-cadherin protein. AA suppresses EMT levels in MDA-MB-231 cells, thereby suppressing cell invasion and migration.
For instance, Puttananjaiah S et al. demonstrated that exposure of MCF-7 cells to AA resulted in upregulation of epithelial marker Ecadherin with a concomitant decrease in the expression of mesenchymal markers, Twist and Snail, besides exhibiting a strong anti-migratory and anti-invasive activity. AA functions as a potent EMT inhibitor by targeting the VEGF signaling pathway [37].
Furthermore, it was demonstrated that the antitumor activity of AA and its role in suppressing metastasis was regulated by the reversal of EMT.
Tan J et al. [38] found that AA inhibited cell proliferation, induced apoptosis and caspase-3/9 activities, as well as Bax protein expression, in prostatic cancer. They demonstrated that AA induces cell apoptosis in prostatic cancer through autophagy, by ER stress/DAPK3/Akt signaling pathway. Furthermore, AA can synergize TRAIL induced apoptosis, through the upregulation of death receptors and downregulation of anti-apoptotic proteins in cancer context [39]. Gény C et al. [40] showed that AA acts as a modulator of Bcl-xL/Bak and Mcl-1/Bid interactions. As an HSP90 client protein, Bcl-2 is reduced, as anticipated, by AUY922 [41]. Our results also showed that AA induces apoptosis in MDA-MB-231 cells.
Findings of our study indicate that AA treatment results in decreased viability, cell cycle arrest, increased apoptosis, and suppression of metastatic potential in TNBC cell line, MDA-MB-231. The changes observed in multiple key Hsp90-dependent, tumor-related molecules (such as GRP78, Hsp70, CDK-4 protein and Mcl-1, Bcl-2 protein etc.) regulated by AA, may be related to these effects. In summary, this study supports the notion that AA has certain anticancer activity, and deserves further investigation; AA may provide the basis for the development of rational drug combinations for treating TNBC.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31401533, 81500071, 81502042) and a grant from Affiliated Hospital of Jiangnan University (FYYB201801).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Brewster A, Chavez-MacGregor M, Brown P. Epidemiology, biology, and treatment of triple-negative breast cancer in women of African ancestry. Lancet Oncol. 2014;15:e625–634. doi: 10.1016/S1470-2045(14)70364-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sankhala K, Mita M, Mita A, Takimoto C. Heat shock proteins: a potential anticancer target. Curr Drug Targets. 2011;12:2001–2008. doi: 10.2174/138945011798829339. [DOI] [PubMed] [Google Scholar]
- 3.Whitesell L, Lindquist S. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 4.Cheng Q, Chang J, Geradts J, Neckers L, Haystead T, Spector N, Lyerly H. Amplifi cation and high-level expression of heat shock protein 90 marks aggressive phenotypes of human epidermal growth factor receptor 2 negative breast cancer. Breast Cancer Res. 2012;14:R62. doi: 10.1186/bcr3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chiosis G, Vilenchik M, Kim J, Solit D. Hsp90: the vulnerable chaperone. Drug Discov Today. 2004;9:881–888. doi: 10.1016/S1359-6446(04)03245-3. [DOI] [PubMed] [Google Scholar]
- 6.Mosser D, Morimoto R. Molecular chaperones and the stress of oncogenesis. Oncogene. 2004;23:2907–2918. doi: 10.1038/sj.onc.1207529. [DOI] [PubMed] [Google Scholar]
- 7.Zhao Q, Wu C, Lee J, Zhao S, Li H, Huo Q, Ma T, Zhang J, Hong Y, Liu H. Anticancer effects of the Hsp90 inhibitor 17-demethoxy-reblas-tatin in human breast cancer MDA-MB-231 cells. J Microbiol Biotechnol. 2014;24:914–920. doi: 10.4014/jmb.1311.11052. [DOI] [PubMed] [Google Scholar]
- 8.Pick E, Kluger Y, Giltnane J, Moeder C, Camp R, Rimm D, Kluger H. High HSP90 expression is associated with decreased survival in breast cancer. Cancer Res. 2007;67:2932–2937. doi: 10.1158/0008-5472.CAN-06-4511. [DOI] [PubMed] [Google Scholar]
- 9.Peng C, Zhu J, Sun H, Huang X, Zhao W, Zheng M, Liu L, Tian J. Inhibition of histone H3K9 acetylation by anacardic acid can correct the over-expression of Gata4 in the hearts of fetal mice exposed to alcohol during pregnancy. PLoS One. 2014;9:e104135. doi: 10.1371/journal.pone.0104135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi J, Jeong S, Ku C, Sathishkumar M, Lee J, Mun S, Kim S. Antibacterial activity of hydroxyalkenyl salicylic acids from sarcotesta of Ginkgo biloba against vancomycin-resistant enterococcus. Fitoterapia. 2009;80:18–20. doi: 10.1016/j.fitote.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 11.Sukumari-Ramesh S, Singh N, Jensen M, Dhandapani K, Vender J. Anacardic acid induces caspase-independent apoptosis and radiosensitizes pituitary adenoma cells. J Neurosurg. 2011;114:1681–1690. doi: 10.3171/2010.12.JNS10588. [DOI] [PubMed] [Google Scholar]
- 12.Seong Y, Shin P, Yoon J, Yadunandam A, Kim G. Induction of the endoplasmic reticulum stress and autophagy in human lung carcinoma A549 cells by anacardic acid. Cell Biochem Biophys. 2014;68:369–377. doi: 10.1007/s12013-013-9717-2. [DOI] [PubMed] [Google Scholar]
- 13.Irie J, Murata M, Homma S. Glycerol-3-phosphate dehydrogenase inhibitors, anacardic acids, from ginkgo biloba. Biosci Biotechnol Biochem. 1996;60:240–243. doi: 10.1271/bbb.60.240. [DOI] [PubMed] [Google Scholar]
- 14.Grazzini R, Hesk D, Heininger E, Hildenbrandt G, Reddy C, Cox-Foster D, Medford J, Craig R, Mumma R. Inhibition of lipoxygenase and prostaglandin endoperoxide synthase by anacardic acids. Biochem Biophys Res Commun. 1991;176:775–780. doi: 10.1016/s0006-291x(05)80252-9. [DOI] [PubMed] [Google Scholar]
- 15.Kishore A, Vedamurthy B, Mantelingu K, Agrawal S, Reddy B, Roy S, Rangappa K, Kundu T. Specifi c small-molecule activator of Aurora kinase A induces autophosphorylation in a cell-free system. J Med Chem. 2008;51:792–797. doi: 10.1021/jm700954w. [DOI] [PubMed] [Google Scholar]
- 16.Sun Y, Jiang X, Chen S, Price B. Inhibition of histone acetyltransferase activity by anacardic acid sensitizes tumor cells to ionizing radiation. FEBS Lett. 2006;580:4353–4356. doi: 10.1016/j.febslet.2006.06.092. [DOI] [PubMed] [Google Scholar]
- 17.Sung B, Pandey M, Ahn K, Yi T, Chaturvedi M, Liu M, Aggarwal B. Anacardic acid (6-nonadecyl salicylic acid), an inhibitor of histone acetyltransferase, suppresses expression of nuclear factor-kappaB-regulated gene products involved in cell survival, proliferation, invasion, and inflammation through inhibition of the inhibitory subunit of nuclear factor-kappaBalpha kinase, leading to potentiation of apoptosis. Blood. 2008;111:4880–4891. doi: 10.1182/blood-2007-10-117994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H, Nie L, Huo Q, Zhao S, Ma T, Wu C, Liu H. [Effect of anacardic acid, a Hsp90 inhibitor, on proliferation, invasion and migration of breast cancer MDA-MB-231 cells] Nan Fang Yi Ke Da Xue Xue Bao. 2015;35:355–359. [PubMed] [Google Scholar]
- 19.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 20.Wu Y, He L, Zhang L, Chen J, Yi Z, Zhang J, Liu M, Pang X. Anacardic acid (6-pentadecylsalicylic acid) inhibits tumor angiogenesis by targeting Src/FAK/Rho GTPases signaling pathway. J Pharmacol Exp Ther. 2011;339:403–411. doi: 10.1124/jpet.111.181891. [DOI] [PubMed] [Google Scholar]
- 21.Seong Y, Shin P, Kim G. Anacardic acid induces mitochondrial-mediated apoptosis in the A549 human lung adenocarcinoma cells. Int J Oncol. 2013;42:1045–1051. doi: 10.3892/ijo.2013.1763. [DOI] [PubMed] [Google Scholar]
- 22.Huang H, Hua X, Liu N, Li X, Liu S, Chen X, Zhao C, Lan X, Yang C, Dou Q, Liu J. Anacardic acid induces cell apoptosis associated with induction of ATF4-dependent endoplasmic reticulum stress. Toxicol Lett. 2014;228:170–178. doi: 10.1016/j.toxlet.2014.05.012. [DOI] [PubMed] [Google Scholar]
- 23.Dong X, Liao Y, Liu N, Hua X, Cai J, Liu J, Huang H. Combined therapeutic effects of bortezomib and anacardic acid on multiple myeloma cells via activation of the endoplasmic reticulum stress response. Mol Med Rep. 2016;14:2679–2684. doi: 10.3892/mmr.2016.5533. [DOI] [PubMed] [Google Scholar]
- 24.Georgakis G, Li Y, Rassidakis G, Martinez-Valdez H, Medeiros L, Younes A. Inhibition of heat shock protein 90 function by 17-allylamino-17-demethoxy-geldanamycin in Hodgkin’s lymphoma cells down-regulates Akt kinase, dephosphorylates extracellular signal-regulated kinase, and induces cell cycle arrest and cell death. Clin Cancer Res. 2006;12:584–590. doi: 10.1158/1078-0432.CCR-05-1194. [DOI] [PubMed] [Google Scholar]
- 25.de Groot A, Kuijpers C, Kroep J. CDK4/6 inhibition in early and metastatic breast cancer: a review. Cancer Treat Rev. 2017;60:130–138. doi: 10.1016/j.ctrv.2017.09.003. [DOI] [PubMed] [Google Scholar]
- 26.Rangel M, Bertolette D, Castro N, Klauzinska M, Cuttitta F, Salomon D. Developmental signaling pathways regulating mammary stem cells and contributing to the etiology of triple-negative breast cancer. Breast Cancer Res Treat. 2016;156:211–226. doi: 10.1007/s10549-016-3746-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Creighton C, Gibbons D, Kurie J. The role of epithelial-mesenchymal transition programming in invasion and metastasis: a clinical perspective. Cancer Manag Res. 2013;5:187–195. doi: 10.2147/CMAR.S35171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kashiwagi S, Yashiro M, Takashima T, Nomura S, Noda S, Kawajiri H, Ishikawa T, Wakasa K, Hirakawa K. Signifi cance of E-cadherin expression in triple-negative breast cancer. Br J Cancer. 2010;103:249–255. doi: 10.1038/sj.bjc.6605735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagaraju G, Long T, Park W, Landry J, Taliaferro-Smith L, Farris A, Diaz R, El-Rayes B. Heat shock protein 90 promotes epithelial to mesenchymal transition, invasion, and migration in colorectal cancer. Mol Carcinog. 2015;54:1147–1158. doi: 10.1002/mc.22185. [DOI] [PubMed] [Google Scholar]
- 30.Banyard J, Bielenberg D. The role of EMT and MET in cancer dissemination. Connect Tissue Res. 2015;56:403–413. doi: 10.3109/03008207.2015.1060970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lehtinen L, Ketola K, Mäkelä R, Mpindi J, Viitala M, Kallioniemi O, Iljin K. High-throughput RNAi screening for novel modulators of vimentin expression identifi es MTHFD2 as a regulator of breast cancer cell migration and invasion. Oncotarget. 2013;4:48–63. doi: 10.18632/oncotarget.756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frixen U, Behrens J, Sachs M, Eberle G, Voss B, Warda A, Löchner D, Birchmeier W. E-cadherin-mediated cell-cell adhesion prevents invasiveness of human carcinoma cells. J Cell Biol. 1991;113:173–185. doi: 10.1083/jcb.113.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gamallo C, Palacios J, Suarez A, Pizarro A, Navarro P, Quintanilla M, Cano A. Correlation of E-cadherin expression with differentiation grade and histological type in breast carcinoma. Am J Pathol. 1993;142:987–993. [PMC free article] [PubMed] [Google Scholar]
- 34.Lee H, Ko J, Baek S, Nam D, Lee S, Lee J, Yang W, Um J, Kim S, Shim B, Ahn K. Embelin inhibits invasion and migration of MDA-MB-231 breast cancer cells by suppression of CXC chemokine receptor 4, matrix metalloproteinases-9/2, and epithelial-mesenchymal transition. Phytother Res. 2016;30:1021–1032. doi: 10.1002/ptr.5612. [DOI] [PubMed] [Google Scholar]
- 35.Wu Z, Wu Q, Yang J, Wang H, Ding X, Yang F, Xu X. Prognostic signifi cance of MMP-9 and TIMP-1 serum and tissue expression in breast cancer. Int J Cancer. 2008;122:2050–2056. doi: 10.1002/ijc.23337. [DOI] [PubMed] [Google Scholar]
- 36.Zhao S, Ma W, Zhang M, Tang D, Shi Q, Xu S, Zhang X, Liu Y, Song Y, Liu L, Zhang Q. High expression of CD147 and MMP-9 is correlated with poor prognosis of triple-negative breast cancer (TNBC) patients. Med Oncol. 2013;30:335. doi: 10.1007/s12032-012-0335-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shilpa P, Kaveri K, Salimath B. Anti-metastatic action of anacardic acid targets VEGF-induced signalling pathways in epithelial to mesenchymal transition. Drug Discov Ther. 2015;9:53–65. doi: 10.5582/ddt.2014.01042. [DOI] [PubMed] [Google Scholar]
- 38.Tan J, Jiang X, Yin G, He L, Liu J, Long Z, Jiang Z, Yao K. Anacardic acid induces cell apoptosis of prostatic cancer through autophagy by ER stress/DAPK3/Akt signaling pathway. Oncol Rep. 2017;38:1373–1382. doi: 10.3892/or.2017.5841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harsha Raj M, Yashaswini B, Rössler J, Salimath B. Combinatorial treatment with anacardic acid followed by TRAIL augments induction of apoptosis in TRAIL resistant cancer cells by the regulation of p53, MAPK and NFκβ pathways. Apoptosis. 2016;21:578–593. doi: 10.1007/s10495-016-1223-8. [DOI] [PubMed] [Google Scholar]
- 40.Gény C, Rivière G, Bignon J, Birlirakis N, Guittet E, Awang K, Litaudon M, Roussi F, Dumontet V. Anacardic acids from knema hookeriana as modulators of Bcl-xL/Bak and Mcl-1/Bid Interactions. J Nat Prod. 2016;79:838–844. doi: 10.1021/acs.jnatprod.5b00915. [DOI] [PubMed] [Google Scholar]
- 41.Cohen-Saidon C, Carmi I, Keren A, Razin E. Antiapoptotic function of Bcl-2 in mast cells is dependent on its association with heat shock protein 90beta. Blood. 2006;107:1413–1420. doi: 10.1182/blood-2005-07-2648. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.