

Abstract
Background
Interstitial lung disease (ILD) is a slowly progressing fatal fibrotic lung disease with a widely variable clinical course and a poor prognosis. Clinicians and patients would benefit from a highly efficient and accurate predictor for ILD. The purpose of this study was to evaluate whether blood biomarkers can predict ILD progression.
Methods
In this study, 85 patients diagnosed as having ILD at the Guangzhou Institute of Respiratory Health participated, including 20 patients with idiopathic pulmonary fibrosis (IPF). During the mean follow-up time of 12 months, every patient was examined during four or five visits in our center. Serum samples were collected at baseline, and after 1, 2, 6, and 12 months and tested for the Klebs von den Lungen-6 (KL-6) concentration. Dynamic fluctuations in this biomarker concentration were examined using a logistic regression model to see if they reflected the progression of ILD.
Results
The baseline levels of serum KL-6 in the ILD patients were significantly increased compared to healthy controls. Serum KL-6 levels were significantly elevated in patients with progression of disease (1,985.2±1,497.8 vs. 1,387.6±1,313.1 µg/mL; P<0.001). Logistic regression revealed sequential changes of KL-6 was a significant predictor of ILD progression in the next follow-up (OR, 2.569; 95% CI, 2.260–2.880; P=0.001), and that sequential changes of KL-6 were significant predictors for the progression of IPF (OR, 3.611; 95% CI, 1.048–12.442; P<0.01). Baseline concentrations were not predictive for ILD or IPF. Univariate Cox analysis showed that KL-6 was significantly associated with survival [relative risk (RR), 1.901; 95% CI, 1.294–2.793; P<0.001], along with other variables.
Conclusions
Serum levels of KL-6 were elevated in ILD patients with severe respiratory function compared to those without. The rate of poor prognosis and mortality was associated with increased biomarker concentrations. Sequential measurements of biomarkers could be valuable in disease monitoring and evaluations in clinical management.
Keywords: Interstitial lung disease (ILD), biomarker, KL-6, prediction
Introduction
Interstitial lung diseases (ILDs) are a heterogeneous group of pulmonary disorders characterized by various patterns of inflammation and fibrosis. A hallmark of these diseases is type II epithelial injury and the abnormal accumulation of extracellular matrix in the lung parenchyma. Approximately two-thirds of ILD cases have an unknown etiology are classified as idiopathic interstitial pneumonia (1-3). The remaining one-third of cases are associated with various environmental or occupational factors including cigarette smoking, aspiration, certain drugs, and radiation exposure, or with connecting tissue diseases (CTDs) (4,5). The disease course varies widely between individuals. In general, idiopathic pulmonary fibrosis (IPF) produces the most rapid deterioration and has a 5-year survival rate of only 80% (2).
Monitoring the activity of ILD with the goal of improved survival depends on knowledge of the pathogenesis of the disease. In general, the dyspnea score, degree of restricted pulmonary function degree, inflammatory and fibrous lesions evident on high-resolution computed tomography (HRCT) scans, lymphocytosis (<20–25%) in bronchoalveolar lavage (BAL) fluid, 6-minute walking distance (6MWD), desaturation, and poor curative effect are reportedly correlated with worse disease outcome (6-11). These parameters are not always sensitive and can require the patient’s cooperation. Furthermore, repeated CT scans results in more radiation exposure. Reliable, less invasive, relatively inexpensive disease-specific biomarkers are needed.
Blood biomarkers can avoid the aforementioned shortcomings and are easy to detect. Klebs von den Lungen-6 (KL-6) is a high-molecular-weight mucin-like glycoprotein that is classified as human MUC1 mucin. It is highly expressed on regenerating type II alveolar cells (12,13). High serum levels of KL-6 have been found in patients with various respiratory diseases, including ILD types of idiopathic interstitial pneumonia, collagen vascular disease-associated interstitial pneumonia, hypersensitivity pneumonia, sarcoidosis, lung cancer, and tuberculosis (14-20). The presence and severity of pulmonary fibrosis also correlates with elevated serum levels of KL-6 (19,20). However, the studies have tended to involve a small number of patients and tested the protein marker at a single time point.
In this study, we hypothesized that the change in the serum concentrations of KL-6 can effectively predict the activities of pulmonary fibrosis. To explore this, we prospectively measured concentrations of two biomarkers in patients experiencing progression of ILD and patients with no progression. The observed relationship between the changes in the concentrations of the serum biomarkers and patient outcome suggests that these markers might be valuable as surrogate clinical measures in monitoring ILD.
Methods
The study was designed as a 12-month, single-center, observational study that was conducted at, Guangzhou Institute of Respiratory Health, Guangzhou, China. Ethical approval for the study was obtained by the Ethical Committee of the First Affiliated Hospital of Guangzhou Medical University (ID Medical ethics 24, No. 17).
Written informed consent was obtained from each participant before enrolment at the time of their diagnosis. Serum samples were prospectively collected at baseline, and at 1, 3, 6, and 12 months. Baseline clinical data were obtained within 3 days of diagnosis or baseline blood sampling. All diagnosed patients were followed-up for at least 1 year. Follow-up examinations included analysis of clinical and laboratory parameters, with prospective analysis of medical records. Mortality data was acquired from the Department of Respiration before the censoring date of 01 June 2015. Measurements of lung function parameters, including forced vital capacity (FVC) and diffusion capacity for carbon monoxide (DLCO) were acquired according to Standardization of spirometry (20). The results were expressed as percentages of normal predicted value at baseline, 1, 3, 6, and 12 months (18,21,22). We defined disease progression based on computed tomography (CT) scan or spirometry changes. The latter changes were defined as the change in pulmonary infiltration or spirometry index changes, change in absolute FVC of 10% (with or without a concomitant change in DLCO), or a change in absolute DLCO of 15% (with or without a concomitant change in FVC). HRCT scans were done using a Somatom Plus S CT unit (Siemens; defined as +128), with thin slices (1-mm) acquired at least every 10 mm. The scans were done at full inspiration from the lung apices to the bases. The HRCT data were reconstructed with a high spatial frequency algorithm and photographed at window widths of 1,500 Hounsfield units and window clinical information. Ground glass opacities (GGO) were characterized by hazy regions of increased lung parenchyma opacity or attenuation, in which vessels could still be seen. Reticular opacities included intralobular reticular opacities and septal line thickening. Bronchial dilatation associated with fibrosis, which is characteristically irregular and cork-screw shaped, was defined as abnormally dilated bronchial divisions with or without bronchial wall thickening, including thickened bronchovascular bundle and traction bronchiectasis. Honeycombing was manifest as air-filled cystic spaces in the lung with readily observable thick-walls that were usually 3 to 10 mm in diameter. The extent of GGO, consolidation, reticular opacities, and honeycombing was scored to the nearest 5% in three zones in each lung—the upper zone (defined as at the aortic arch), the lower zone (defined as the pulmonary veins), and the middle zone (located between the upper zone and the lower zone, i.e., between the aortic arch and the pulmonary veins). The interstitial lesions were divided into inflammatory lesions and fibrosis lesions. The area of the inflammatory lesions was the mean extent of GGO and consolidation in each of the six zones. The area of lung fibrotic lesions was the mean extent of reticular opacities and honeycombing in each of the six zones as scored by two observers. Both observers received training prior to the study to reduce interobserver bias. The observers were evaluated as satisfactory for consistency in visual scoring. Baseline and follow-up images were scored during the trial, and the observers were blinded to the basic and clinical information of the patients and the temporal sequence of the examinations. The lesions were visually classified and scored in a semi-quantitative manner for the extent of interstitial abnormalities (GGO or opacities) as follows: 0 = absent; 1 = <25% involvement; 2 = 25–50% involvement; 3 = 50–75% involvement; and 4 = >75% involvement (20). The CT total score for each patient was calculated by adding the score for each lobe. Finally, we obtained the mean value of the answer forms from two different thoracic radiologists for each patient. The PROFILE study is registered at www.ClinicalTrials.gov as NCT02960672.
Procedures
Blood (3 mL) was drawn from each participant using standardized operating procedures at baseline, 1, 3, 6, and 12 months. Serum was separated by centrifugation and all specimens were stored at −80 °C until assayed. All samples were processed within 2 h. We detected for KL-6 by latex agglutination test (SEKISUI MEDICAL CO, LTD). All assays were conducted in duplicate and the mean values were obtained. Serum levels were evaluated in healthy controls (15 females and 10 males; age range, 21–63 years).
Statistical analyses
Disease progression was defined by death or if decline in FVC >10% or DLCO >15% or more was observed at 12 months. Missing lung function index values were not imputed. Even though 12-month FVC or DLCO data were absent, subjects were still thought to have progressed if ≥10% declines in FVC or DLCO were observed continuously at 6 months. If biomarker data were below the lower limit of detection, values were imputed to be half the lower limit of detection. Values above the upper limit were conservatively imputed as the upper limit of detection.
The statistical evaluation of our data was performed using SPSS 19.0 (SPSS Inc., Chicago, IL, USA; www.spss.com). All values are expressed as the mean ± standard deviation or median for continuous variables, and as percentages for categorical variables. All analyses were two-sided and the level of significance was established at P=0.05. The independent sample t-test was used for comparisons of progressive disease and non-progressive disease. Stata software statistical software version 12.0 (Stata, College Station, TX, USA) was used for some statistical analyses. Multiple logistic regression was applied to identify prognostic factors including variables with significant differences (P=0.05) on university analyses. Survival analysis was done with a Cox regression hazards model to assess the association between continuous variables and overall survival. Forest plot was used to illustrate the differences between independent factors.
Results
General information of subjects
Eighty-five eligible ILD patients were enrolled from March 2013 to September 2015, including 20 IPF patients. The mean age of the subjects was 54 years and 59% were male. The mean pulmonary function parameters [FVC, FEV1, DLCO, and total lung capacity (TLC)] of the subjects were worse than healthy controls (Table 1). No correlations were seen between KL-6 serum levels and smoking habits, age, or body mass index (data not shown). Concerning the distribution in etiology and diagnostic methods of 85 patients with ILD, 28.2% of patients were separately diagnosed based on the surgical lung biopsies or bronchoscopy, 32.9% of patients were diagnosed based on immunological markers and muscle/labial gland biopsy, and 10.6% patients were diagnosed by HRCT (Tables S1,S2).
Table 1. Demographic and background characteristics of patients with ILD.
| Characteristics | ILD patients | Healthy controls | P value |
|---|---|---|---|
| Patients, n | 85 | 20 | – |
| Age, mean ± SD, years | 53.5±10.5 | 51.6±6.5 | 0.39 |
| Male patients, n [%] | 50 [59] | 12 [60] | – |
| Ever smokers, n [%] | 62 [73] | 13 [65] | 0.54 |
| BMI, mean ± SD, kg/m2 | 23.8±3.2 | 23.4±3.8 | 0.62 |
| PFT, %predicted, mean ± SD | |||
| FVC, % | 71.1±17.7 | 84.0±3.3 | <0.01 |
| FEV1, % | 73.8±17.8 | 85.0±1.8 | <0.01 |
| DLCO, % | 49.4±24.3 | 85.0±4.0 | <0.01 |
| TLC, % | 67.4±12.9 | 83.7±2.7 | <0.01 |
| HRCT scan, mean ± SD | |||
| GGO | 19±21 | NA | NA |
| Consolidation | 0±3 | NA | NA |
| Reticulation | 0±11 | NA | NA |
| HC | 0±12 | NA | NA |
| CT Score | 29±20 | NA | NA |
| KL-6, mean ± SD, units/mL | 1,565.2±1,389.0 | 201.8±87.9 | <0.01 |
ILD, interstitial lung disease; BMI, body mass index; PFT, pulmonary function test; FVC, forced vital capacity; FEV1, forced expiratory volume in 1 second; DLCO, diffusion capacity for carbon monoxide; TLC, total lung capacity; HRCT, high resolution CT; GGO, ground glass opacity; HC, honeycombing; KL-6, Klebs von den Lungen-6; NA, normal.
Table S1. The etiology distribution of ILD patients.
| Subtype of ILD | Cases | Proportion (%) | Diagnostic method |
|---|---|---|---|
| IIP | 52 | 61.2 | – |
| IPF | 20 | 23.5 | Surgical lung biopsy, bronchoscopy, HRCT, PFT |
| Possible IPF | 7 | 8.2 | Bronchoscopy, HRCT, PFT |
| Non-IPF | 25 | 29.4 | – |
| NSIP | 18 | 21.2 | – |
| Cellular NSIP | 10 | 11.8 | Surgical lung biopsy, bronchoscopy, HRCT, PFT |
| Fibrotic NSIP | 8 | 9.4 | Surgical lung biopsy, bronchoscopy, HRCT, PFT |
| Others IIP | 7 | 8.2 | Bronchoscopy, HRCT, PFT |
| CTD-ILD | 33 | 38.8 | – |
| PM/DM-ILD | 19 | 22.4 | Muscle biopsy, HRCT, PFT, electromyogram, serum autoantibody test |
| RA-ILD | 3 | 3.5 | Surgical lung biopsy, bronchoscopy, HRCT, PFT, serum autoantibody test |
| Sjogren’s syndrome-ILD | 1 | 1.2 | Labial gland biopsy, HRCT, PFT, serum autoantibody test |
| SLE-ILD | 2 | 2.4 | Bronchoscopy, HRCT, PFT, serum autoantibody test |
| SSc-ILD | 2 | 2.4 | Bronchoscopy, HRCT, PFT, serum autoantibody test |
| Mixed CTD-ILD | 5 | 5.9 | HRCT, PFT, serum autoantibody test |
| Undifferentiated CTD-ILD | 1 | 1.2 | Surgical lung biopsy, HRCT, PFT, serum autoantibody test |
Eight-five cases of ILD patients, 52 cases of IIP patients, including 20 cases of IPF patients, and 33 cases of connective tissue diseases patients, the proportion were 61.2%, 23.5%, and 38.8%. ILD, interstitial lung disease; IIP, idiopathic interstitial pneumonia; IPF, idiopathic pulmonary fibrosis; NSIP, non-specific interstitial pneumonia; CTD, connecting tissue diseases; PM/DM, polymyositis/dermatomyositis; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus; SSc, systemic scleroderma; HRCT, high-resolution computed tomography; PFT, pulmonary function test.
Table S2. The diagnostic methods distribution of ILD patients.
| Subtype of ILD | Surgical lung biopsy (n=24, 28.2%) | Bronchoscopy (n=24, 28.2%) | HRCT (n=9, 10.6%) | Serum autoantibody test + muscle/labial gland biopsy (n=28, 32.9%) |
|---|---|---|---|---|
| IIP | 22 | 28 | 9 | 0 |
| IPF | 11 | 0 | 9 | 0 |
| Possible IPF | 0 | 7 | 0 | 0 |
| Non-IPF | 11 | 21 | 0 | 0 |
| NSIP | 11 | 7 | 0 | 0 |
| Cellular NSIP | 6 | 4 | 0 | 0 |
| Fibrotic NSIP | 5 | 3 | 0 | 0 |
| Others IIP | 0 | 7 | 0 | 0 |
| CTD-ILD | 2 | 3 | 0 | 28 |
| PM/DM-ILD | 0 | 0 | 0 | 19 |
| RA-ILD | 1 | 1 | 0 | 1 |
| Sjogren’s syndrome-ILD | 0 | 0 | 0 | 1 |
| SLE-ILD | 0 | 1 | 0 | 1 |
| SSc-ILD | 0 | 1 | 0 | 1 |
| Mixed CTD-ILD | 0 | 0 | 0 | 5 |
| Undifferentiated | 1 | 0 | 0 | 0 |
28.2% of patients were separately diagnosed based on the surgical lung biopsies or bronchoscopy, 32.9% of patients were diagnosed based on immunological markers and muscle/labial gland biopsy, and 10.6% patients were diagnosed by HRCT. ILD, interstitial lung disease; IIP, idiopathic interstitial pneumonia; IPF, idiopathic pulmonary fibrosis; NSIP, non-specific interstitial pneumonia; CTD, connecting tissue diseases; PM/DM, polymyositis/dermatomyositis; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus; SSc, systemic scleroderma; HRCT, high-resolution computed tomography.
KL-6 distinguishes ILD patients from healthy controls
Baseline concentrations of KL-6 were significantly higher in ILD patients compared with healthy controls (1,565.2±1,389.0 vs. 201.8±87.9 µg/mL, P<0.01) (Table 1).
KL-6 concentration may be associated with disease severity
Patients were grouped as the FVC >50% group and FVC <50% group to analyze the correlation of serum concentrations of biomarkers and different lung function groups. Serum levels of KL-6 were increased in ILD patients with severe respiratory function than those without (996±1,343.3 vs. 1,484.5±1,915.5 µg/mL, P<0.01). A statistically significant correlation in KL-6 concentrations was evident between the two groups with different lung function injury, KL-6 concentrations were significantly increased in the FVC <50% group compared to the FVC >50% group (Figure S1).
Figure S1.
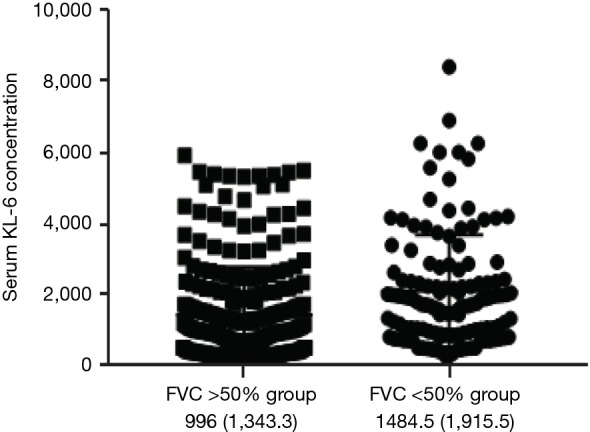
Comparison of serum KL-6 concentration the FVC >50% group and the FVC <50% group. FVC >50% group n=210, FVC<50% group n=122, an independent sample t-test was performed. KL-6 concentrations were significantly higher in the FVC% <50% group compared to FVC% >50% group, P=0.03. FVC, forced vital capacity. KL-6, Klebs von den Lungen-6.
KL-6 concentration in progressive and non-progressive period
KL-6 concentration was highly elevated in progressive ILD compared with the non-progressive period (1,985.2±1,497.8 vs. 1,387.6±1,313.1 µg/mL, P<0.001) (Figure S2).
Figure S2.
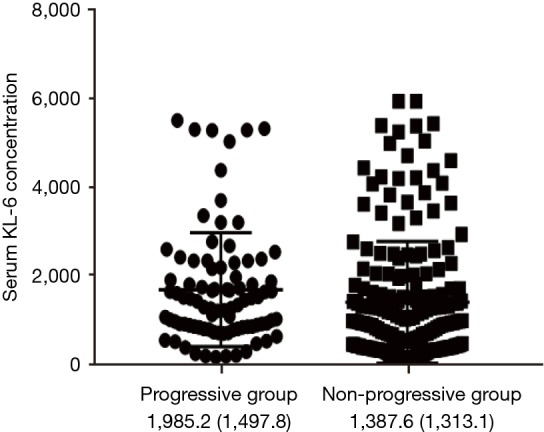
Comparison of serum KL-6 concentration between the progressive group and the non-progressive group. FVC >50% group n=88, FVC <50% group n=244, an independent sample t-test was performed. KL-6 concentration was highly elevated in progressive compared with non-progressive period, P<0.001. “Progressive” meant over 10% decrease in %FVC or over 15% decrease in %DLCO. “Non-progressive” meant within 10% change of %FVC and within 15% change of %DLCO, or increase in %FVC or increase in %DLCO. KL-6, Klebs von den Lungen-6; FVC, forced vital capacity; DLCO, diffusion capacity for carbon monoxide.
Role of baseline levels of KL-6 in the prediction of disease progression
We analyzed the predictive value of baseline biomarkers for IPF and ILD progression. Unexpectedly, IPF or ILD baseline levels of KL-6 could not predict disease deterioration at any stage (P=0.673, 0.606, 0.719, 0.746).
Role of sequential changes of KL-6 in the prediction of disease progression
Receiver operating characteristic analysis was performed to evaluate whether serum KL-6 concentrations were predictive of disease progression. Sequential changes of serum KL-6 concentrations were associated with disease progression. At the cut-off level of ≥759.5 U/mL, serum KL-6 levels obtained a sensitivity of 86.4% and a specificity of 41.7% in predicting disease progression (area under the curve =0.655, P<0.0001) (Figure 1).
Figure 1.
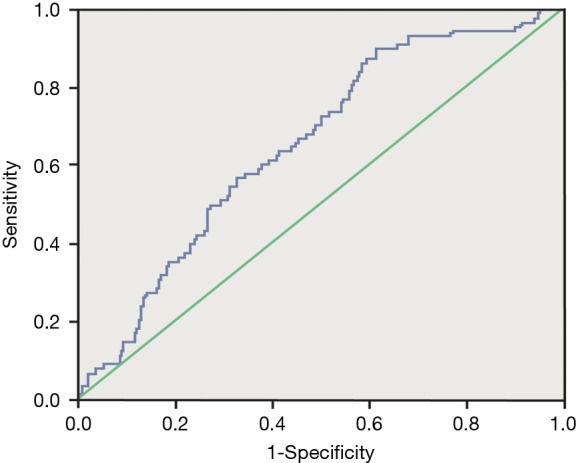
Receiver operating characteristic curve analysis. The curves show the power of initial serum KL-6 for predicting disease progression. At the cut-off level of ≥759.5 U/mL, serum KL-6 levels yielded a sensitivity of 86.4% and a specificity of 41.7% to predict disease progression (AUC =0.655, P<0.0001). KL-6, Klebs von den Lungen-6.
We also defined ΔKL-6 as the difference between the two approaching measurements, and tried to evaluate its predictive value of progression of next follow-up. Using univariate analysis, both ΔKL-6 [odds ratio (OR), 2.57; 95% confidence interval (CI), 2.26–2.88; P=0.001] showed significant predictive value for ILD progression and had better predictive power than any other clinical parameters, as were the age, sex, FVC %predicted, and extent of HRCT (Figure 2). However, in the multivariate model that took into account serum markers, baseline demographics, pulmonary function indexes, and extent of HRCT, serum KL-6 levels could not show significant predictive value for ILD progression when data were adjusted for covariates (P>0.05).
Figure 2.

Predicting value of sequential changes in biomarkers for disease progression in ILD patients assessed by logistic regression model. KL-6, Klebs von den Lungen-6; FVC, forced vital capacity; FEV1, forced expiratory volume in 1 second; DLCO, diffusion capacity for carbon monoxide; TLC, total lung capacity; GGO, ground glass opacities; HC, honeycombing; OR, odds ratio; CI, confidence interval; ILD, interstitial lung disease.
Moreover, especially in IPF, we also tried to demonstrate its predictive value in IPF progression. Using univariate analysis, ΔKL-6 (OR, 2.099; 95% CI, 1.194–3.689; P<0.01) also showed significant predictive value for IPF progression of next follow-up and had better predictive power than any other clinical parameters, as did age, sex, DLCO %predicted, TLC %predicted, and extent of HRCT. Multivariate analysis was done taking into account KL-6, baseline demographics, pulmonary function indexes, and extent of HRCT. Surprisingly, ΔKL-6 (OR, 3.611; 95% CI, 1.05–6.22; P<0.01) and fibrotic score (OR, 1.321; 95% CI, 1.055–1.632; P<0.01) also displayed significant predictive value for IPF progression when data were adjusted for covariates (Figure 3).
Figure 3.

Predicting value of sequential changes in biomarkers for disease progression in IPF patients assessed by logistic regression model. KL-6, Klebs von den Lungen-6; FEV1, forced expiratory volume in 1 second; DLCO, diffusion capacity for carbon monoxide; TLC, total lung capacity; OR, odds ratio; CI, confidence interval; IPF, idiopathic pulmonary fibrosis.
Changes of KL-6 concentrations correlate with disease mortality
We clarified the causes of death of the patients (Table S3). Using Cox regression analysis, KL-6 was the best independent predictor of mortality after adjustment for other covariates (hazard ratio, 1.901; 95% CI, 1.294–2.793; P<0.001). However, only FVC % was a predictor of mortality in multivariate analysis (Figure S3).
Table S3. The causes of death of the six patients of with ILD.
| Causes of death | Cases | Proportion (%) | Type of ILD |
|---|---|---|---|
| Acute exacerbation | 3 | 50 | 2 cases of IPF, 1 cases of PM-ILD |
| Respiratory failure | 1 | 16.7 | 1 cases of IIP (possible IPF) |
| Infections | 1 | 16.7 | 1 cases of IIP (possible IPF) |
| Heart failure | 1 | 16.7 | 1 cases of PM-ILD |
The causes of death were acute exacerbation in 3 (50%) patients, respiratory failure in 1 (16.7%), pulmonary infection in 1 (16.7%) and heart failure in 1 (16.7%). ILD, interstitial lung disease; IIP, idiopathic interstitial pneumonia; IPF, idiopathic pulmonary fibrosis; PM, polymyositis.
Figure S3.

Predicting value for mortality in patients with ILD assessed by Cox regression model. Using Cox regression analysis, KL-6 was the best independent predictor of mortality after adjustment for other covariates (HR, 1.90; 95% CI, 1.29–2.79; P=0.001). Only FVC% is the predictor of mortality in multivariate analysis (n=322). FVC, forced vital capacity; FEV1, forced expiratory volume in 1 second; DLCO, diffusion capacity for carbon monoxide; TLC, total lung capacity; GGO, ground glass opacity; HC, honeycombing; KL-6, Klebs von den Lungen-6; CI, confidence interval; ILD, interstitial lung disease.
Discussion
We demonstrated that changes in concentrations of serum biomarker KL-6 with time was strongly associated with progression of ILD and survival. This single peripheral blood biomarker offered significant predictive value in patients with ILD.
KL-6 was previously implicated in the pathogenesis of ILD, promoting pulmonary fibroblast migration and proliferation, and is significantly expressed in serum, BAL fluid, and lung tissue of ILD patients. These results imply that elevated KL-6 levels in the peripheral blood reflect the pathological characteristics of diffuse alveolar damage in the interstitial microenvironment (21,23). Ishii et al. found that KL-6 serum levels were elevated in 70–100% of patients with ILDs and so could not be used to differentiate patients with IPF from those with non-specific interstitial pneumonia (21). Moreover, higher concentrations of KL-6 may be associated with disease severity, similar to previous reports (18,24). High serum levels of KL-6 have been found to correlate with the severity of pulmonary fibrosis, which could be lowered to the normal range in patients who showed a dramatic clinical improvement during therapy, with the near-absence of lesions in CT scans. In general, our results support the utility of KL-6 as an ILD biomarker and suggest its role in differential diagnosis, assessing disease severity, and monitoring disease progression. However, baseline levels of KL-6 at the time of diagnosis failed to predict deterioration.
KL-6 is a mucinous high-molecular-weight glycoprotein that has recently been revealed as a useful serum biomarker for the prognosis of disease progression and severity. KL-6 is strongly expressed on proliferated type II alveolar epithelial cells. The elevated serum levels of KL-6 in patients with ILDs may be due to an elevation in KL-6 production by increasing permeability following the destruction of alveolar capillaries and results in regenerating of alveolar type II pneumocytes in the affected lung. Detection of serum biomarker may reflect the degree of leakage of molecules from alveolus to the capillaries (25). Previous studies reported KL-6 to be an effective marker in reflecting the severity of pulmonary fibrotic lesions. Yokoyama et al. reported the median survival time in patients with a KL-6 level <1,000 U/mL was twice longer than in patients with KL-6 level >1,000 U/mL (1.5 year) in 27 patients with IPF (26). Elevated serum KL-6 (KL-6 >1,000 U/mL) in IPF patients at the first visit is associated with increased mortality (27). Satoh et al. also reported that the progression of the disease was significantly more rapid in patients with ILDs whose KL-6 levels were 1,000 U/mL or more at the initial measurement than in patients whose KL-6 levels were less than 1,000 U/mL (27). ILDs are a common complication in CTD, with an overall incidence estimated at 15% (15). The role of KL-6 as a serum biomarker to detect disease activity in patients with CTD-ILD has also been reported in polymyositis/dermatomyositis and systemic sclerosis (16,21,28-33). Therefore, KL-6 has been validated as a useful indicator to map the existence of the pulmonary epithelial cell injury that reflects the presence of fibrotic lung lesions accompanied by regenerating epithelial cells, and is a sensitive marker to estimate disease severity. However, KL-6 serum levels are elevated in most patients with ILDs, especially those with severe lung function injury and disease development. However, KL-6 cannot be used to discriminate between IPF and other ILDs (34). Our data show that KL-6 as a single biomarker has a fairly good ability in predicting disease outcome.
A number of biological biomarkers have been reported for survival, baseline FVC, a change of FVC >10%, and a decrement of >25 m in 6MWD, inflammatory and fibrotic lesions on HRCT scans, lymphocytosis in BAL fluid, and poor response to treatment in relation to disease progression and survival, and even CA153 and CA125 might play a more important role in ILD (6-12,35). However, serial changes in lung function parameters failed to predict disease course in a significant number of patients. Measurements of lung function itself are variable in different participants, with differences of approximately 15% between two measurements obtained from healthy individuals (36). Explanations could include measurements obtained with participants’ cooperation, measurements done by different clinicians or lung function laboratories, and confounders like the baseline disease severity. In addition, physiological markers offer no information about disease pathogenesis and have no ability to identify certain molecular subtypes of disease, unlike molecular biomarkers. ILD is a variable disease and can manifest as steady gradual amelioration/recession, with prolonged stability and/or acute deterioration (36). The disease course can be a challenge to predict for clinicians. Thus, biomarker concentration determined at one point in time may not reflect the state of this dynamic disease. Baseline concentrations of a number of serum markers (including KL-6, matrix metalloproteinase 7, chemokine ligand 18, and surfactant protein-D) weakly predict subsequent disease course in IPF (26,37-39). Baseline concentrations are unlikely to be superior to present physiological measurements of disease severity and prediction (40). Many hospitalized patients receive anti-inflammatory and anti-fibrotic therapy. However, not every type of ILD displays a good response to medical treatment and a benign prognosis, such as IPF and fibrotic Non-specific interstitial pneumonia, which continuously progress, even during a series of medical interventions. Measurement of the baseline KL-6 level does not predict the outcome. We believe that the pretreatment values of KL-6 in our patients who lived and those who died were already elevated by deterioration of pre-existing fibrosis. None of these markers has been identified at serially different time points so it is impossible to know precisely whether changes over time mirror the deterioration of pre-existing fibrosis.
The major strength of our project is that we generated a prospective data set collected at pre-specified time intervals during follow-up from the patients with ILD. This is the first study to monitor sequential longitudinal change in biomarker concentrations of each patient and analyze their outcomes of each follow-up in the hospital according to clinical parameters with the aim of identifying biomarkers of disease behavior. We defined ΔKL-6 as the difference between the two approaching measurements, and estimated the OR for progression in patients by changes of the last two follow-up concentrations. Therefore, we manifested those changes in the concentrations of KL-6 that were strongly associated with outcome (progression and survival). The results indicate that elevated KL-6 in the last two follow-ups indicate the elevated rate of disease deterioration. ΔKL-6 was a sensitive and powerful marker in predicting ILD progression. Song et al. also found that three biomarkers (KL-6, matrix metalloproteinase 7, and surfactant protein-D) were statistically significant in disease prediction (P=0.037) (41). However, measurement of all three biomarkers is inconvenient and expensive, and the statistical efficacy of the combination of three biomarkers was much weaker (P=0.037) than our results (P<0.001). Our study indicates that baseline KL-6 concentrations are unlikely to predict disease outcome, which is different from the findings of Song et al. The discrepancy might be due to our small sample size. Further studies with a large number of subjects may show statistical significance.
This study still has several limitations. The study involved a small number of patients and ILD consists of various types of disease. Thus, the subjects from a single specialized center may not be representative of all patients with ILD. Secondly, the majority of the patients had mild and moderate disease, so data are lacking concerning severe ILD. Such patients might die before enrolment, which reduces the power of our study to indicate a relation between biomarkers and prognosis. Thirdly, the study subjects did not receive uniform treatments because the guidelines did not provide clinicians with uniform criteria of treatments until 2014. The patients with different subtypes of ILD received different treatments, including corticosteroid and/or immunosuppressive agents, which are no longer current standard treatments. The effects of pirfenidone and nintedanib on serum KL-6 and other biomarkers and their implications should be investigated in future studies. Finally, even though it was sufficient to reveal the prognostic value of such biomarkers, the follow-up period of this study was short and involved a small number of patients.
Despite these limitations, this is the first biomarker study in ILD of any severity which involves serial serum sample collection at different time points. KL-6 levels that display an elevated trend predict poor prognosis. When sequential increases of KL-6 levels are evident during a 1-year follow-up, clinicians should pay attention to the risk of poor prognosis of patients and additional therapy should be considered.
Acknowledgements
Funding: The study was supported by National Key Research Project-Accurate Medical Research (NO.2016YFC0905701).
Ethical Statement: This study was approved ethically by the First Affiliated Hospital of Guangzhou Medical University (ID Medical ethics 24, No. 17). All patients were notified consent for the use of their tissues for scientific research.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.American Thoracic Society. European Respiratory Society American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med 2002;165:277-304. [DOI] [PubMed] [Google Scholar]
- 2.Bjoraker JA, Ryu JH, Edwin MK, et al. Prognostic significance of histopathologic subsets in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1998;157:199-203. 10.1164/ajrccm.157.1.9704130 [DOI] [PubMed] [Google Scholar]
- 3.Kim DS, Collard HR, King TE., Jr Classification and natural history of the idiopathic interstitial pneumonias. Proc Am Thorac Soc 2006;3:285-92. 10.1513/pats.200601-005TK [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.American Thoracic Society Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am J Respir Crit Care Med 2000;161:646-64. 10.1164/ajrccm.161.2.ats3-00 [DOI] [PubMed] [Google Scholar]
- 5.The diagnosis, assessment and treatment of diffuse parenchymal lung disease in adults. Introduction. Thorax 1999;54 Suppl 1:S1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.King TE, Jr, Tooze JA, Schwarz MI, et al. Predicting survival in idiopathic pulmonary fibrosis: scoring system and survival model. Am J Respir Crit Care Med 2001;164:1171-81. 10.1164/ajrccm.164.7.2003140 [DOI] [PubMed] [Google Scholar]
- 7.Collard HR, King TE, Jr, Bartelson BB, et al. Changes in clinical and physiologic variables predict survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2003;168:538-42. 10.1164/rccm.200211-1311OC [DOI] [PubMed] [Google Scholar]
- 8.Latsi PI, du Bois RM, Nicholson AG, et al. Fibrotic idiopathic interstitial pneumonia: the prognostic value of longitudinal functional trends. Am J Respir Crit Care Med 2003;168:531-7. 10.1164/rccm.200210-1245OC [DOI] [PubMed] [Google Scholar]
- 9.Jegal Y, Dong SK, Shim TS, et al. Physiology is a stronger predictor of survival than pathology in fibrotic interstitial pneumonia. American Journal of Respiratory & Critical Care Medicine 2005;171:639-44. 10.1164/rccm.200403-331OC [DOI] [PubMed] [Google Scholar]
- 10.Lama VN, Flaherty KR, Toews GB, et al. Prognostic value of desaturation during a 6-minute walk test in idiopathic interstitial pneumonia. Am J Respir Crit Care Med 2003;168:1084-90. 10.1164/rccm.200302-219OC [DOI] [PubMed] [Google Scholar]
- 11.Wells AU, Hansell DM, Rubens MB, et al. The predictive value of appearances on thin-section computed tomography in fibrosing alveolitis. Am Rev Respir Dis 1993;148:1076-82. 10.1164/ajrccm/148.4_Pt_1.1076 [DOI] [PubMed] [Google Scholar]
- 12.Nukiwa T. The role of biomarkers in management of interstitial lung disease: implications of biomarkers derived from type II pneumocytes. In: The European Respiratory Monograph. Number 46, December 2009. European Respiratory Society Publications, 2009:47-66. [Google Scholar]
- 13.Kohno N, Kyoizumi S, Awaya Y, et al. New serum indicator of interstitial pneumonitis activity. Sialylated carbohydrate antigen KL-6. Chest 1989;96:68-73. 10.1378/chest.96.1.68 [DOI] [PubMed] [Google Scholar]
- 14.Inoue Y, Nishimura K, Shiode M, et al. Evaluation of serum KL-6 levels in patients with pulmonary tuberculosis. Tuber Lung Dis 1995;76:230-3. 10.1016/S0962-8479(05)80010-3 [DOI] [PubMed] [Google Scholar]
- 15.Bandoh S, Fujita J, Ohtsuki Y, et al. Sequential changes of KL-6 in sera of patients with interstitial pneumonia associated with polymyositis/dermatomyositis. Ann Rheum Dis 2000;59:257-62. 10.1136/ard.59.4.257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kubo M, Ihn H, Yamane K, et al. Serum KL-6 in adult patients with polymyositis and dermatomyositis. Rheumatology (Oxford) 2000;39:632-6. 10.1093/rheumatology/39.6.632 [DOI] [PubMed] [Google Scholar]
- 17.Pardo A, Selman M, Kaminski N. Approaching the degradome in idiopathic pulmonary fibrosis. Int J Biochem Cell Biol 2008;40:1141-55. 10.1016/j.biocel.2007.11.020 [DOI] [PubMed] [Google Scholar]
- 18.Rosas IO, Richards TJ, Konishi K, et al. MMP1 and MMP7 as potential peripheral blood biomarkers in idiopathic pulmonary fibrosis. PLoS Med 2008;5:e93. 10.1371/journal.pmed.0050093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller MR, Hankinson J, Brusasco V, et al. Standardisation of spirometry. Eur Respir J 2005;26:319-38. 10.1183/09031936.05.00034805 [DOI] [PubMed] [Google Scholar]
- 20.Kazerooni EA, Martinez FJ, Flint A, et al. Thin-section CT obtained at 10-mm increments versus limited three-level thin-section CT for idiopathic pulmonary fibrosis: correlation with pathologic scoring. AJR Am J Roentgenol 1997;169:977-83. 10.2214/ajr.169.4.9308447 [DOI] [PubMed] [Google Scholar]
- 21.Ishii H, Mukae H, Kadota J, et al. High serum concentrations of surfactant protein A in usual interstitial pneumonia compared with non-specific interstitial pneumonia. Thorax 2003;58:52-7. 10.1136/thorax.58.1.52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hirasawa Y, Kohno N, Yokoyama A, et al. KL-6, a human MUC1 mucin, is chemotactic for human fibroblasts. Am J Respir Cell Mol Biol 1997;17:501-7. 10.1165/ajrcmb.17.4.2253 [DOI] [PubMed] [Google Scholar]
- 23.Fujisawa T, Suda T, Nakamura Y, et al. Differences in clinical features and prognosis of interstitial lung diseases between polymyositis and dermatomyositis. J Rheumatol 2005;32:58-64. [PubMed] [Google Scholar]
- 24.Inoue Y, Barker E, Daniloff E, et al. Pulmonary epithelial cell injury and alveolar-capillary permeability in berylliosis. Am J Respir Crit Care Med 1997;156:109-15. 10.1164/ajrccm.156.1.9612043 [DOI] [PubMed] [Google Scholar]
- 25.Kinoshita F, Hamano H, Harada H, et al. Role of KL-6 in evaluating the disease severity of rheumatoid lung disease: comparison with HRCT. Respir Med 2004;98:1131-7. 10.1016/j.rmed.2004.04.003 [DOI] [PubMed] [Google Scholar]
- 26.Yokoyama A, Kondo K, Nakajima M, et al. Prognostic value of circulating KL-6 in idiopathic pulmonary fibrosis. Respirology 2006;11:164-8. 10.1111/j.1440-1843.2006.00834.x [DOI] [PubMed] [Google Scholar]
- 27.Satoh H, Kurishima K, Ishikawa H, et al. Increased levels of KL-6 and subsequent mortality in patients with interstitial lung diseases. J Intern Med 2006;260:429-34. 10.1111/j.1365-2796.2006.01704.x [DOI] [PubMed] [Google Scholar]
- 28.Bonella F, Volpe A, Caramaschi P, et al. Surfactant protein D and KL-6 serum levels in systemic sclerosis: correlation with lung and systemic involvement. Sarcoidosis Vasc Diffuse Lung Dis 2011;28:27-33. [PubMed] [Google Scholar]
- 29.Yamane K, Ihn H, Kubo M, et al. Serum levels of KL-6 as a useful marker for evaluating pulmonary fibrosis in patients with systemic sclerosis. J Rheumatol 2000;27:930-4. [PubMed] [Google Scholar]
- 30.Yanaba K, Hasegawa M, Hamaguchi Y, et al. Longitudinal analysis of serum KL-6 levels in patients with systemic sclerosis: association with the activity of pulmonary fibrosis. Clin Exp Rheumatol 2003;21:429-36. [PubMed] [Google Scholar]
- 31.Yanaba K, Hasegawa M, Takehara K, et al. Comparative study of serum surfactant protein-D and KL-6 concentrations in patients with systemic sclerosis as markers for monitoring the activity of pulmonary fibrosis. J Rheumatol 2004;31:1112-20. [PubMed] [Google Scholar]
- 32.Kodera M, Hasegawa M, Komura K, et al. Serum pulmonary and activation-regulated chemokine/CCL18 levels in patients with systemic sclerosis: a sensitive indicator of active pulmonary fibrosis. Arthritis Rheum 2005;52:2889-96. 10.1002/art.21257 [DOI] [PubMed] [Google Scholar]
- 33.Hant FN, Ludwicka-Bradley A, Wang HJ, et al. Surfactant protein D and KL-6 as serum biomarkers of interstitial lung disease in patients with scleroderma. J Rheumatol 2009;36:773-80. 10.3899/jrheum.080633 [DOI] [PubMed] [Google Scholar]
- 34.Zuo F, Kaminski N, Eugui E, et al. Gene expression analysis reveals matrilysin as a key regulator of pulmonary fibrosis in mice and humans. Proc Natl Acad Sci U S A 2002;99:6292-7. 10.1073/pnas.092134099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang T, Zheng XJ, Liang BM, et al. Clinical features of rheumatoid arthritis-associated interstitial lung disease. Sci Rep 2015;5:14897. 10.1038/srep14897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jensen RL, Teeter JG, England RD, et al. Instrument accuracy and reproducibility in measurements of pulmonary function. Chest 2007;132:388-95. 10.1378/chest.06-1998 [DOI] [PubMed] [Google Scholar]
- 37.Richards TJ, Kaminski N, Baribaud F, et al. Peripheral blood proteins predict mortality in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2012;185:67-76. 10.1164/rccm.201101-0058OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prasse A, Probst C, Bargagli E, et al. Serum CC-chemokine ligand 18 concentration predicts outcome in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2009;179:717-23. 10.1164/rccm.200808-1201OC [DOI] [PubMed] [Google Scholar]
- 39.Greene KE, King TE, Jr, Kuroki Y, et al. Serum surfactant proteins-A and -D as biomarkers in idiopathic pulmonary fibrosis. Eur Respir J 2002;19:439-46. 10.1183/09031936.02.00081102 [DOI] [PubMed] [Google Scholar]
- 40.Jenkins RG, Simpson JK, Saini G, et al. Longitudinal change in collagen degradation biomarkers in idiopathic pulmonary fibrosis: an analysis from the prospective, multicentre PROFILE study. Lancet Respir Med 2015;3:462-72. 10.1016/S2213-2600(15)00048-X [DOI] [PubMed] [Google Scholar]
- 41.Song JW, Do KH, Jang SJ, et al. Blood biomarkers MMP-7 and SP-A: predictors of outcome in idiopathic pulmonary fibrosis. Chest 2013;143:1422-9. 10.1378/chest.11-2735 [DOI] [PubMed] [Google Scholar]