

Abstract
Gefitinib is the molecular target drug for advanced non‐small‐cell lung cancer. The primary target of gefitinib is the positive mutation of epidermal growth factor receptor, but it also inhibits cyclin G‐associated kinase (GAK). To reveal the molecular bases of GAK and gefitinib binding, structure analyses were conducted and determined two forms of the gefitinib‐bound nanobody⋅GAK kinase domain complex structures. The first form, GAK_1, has one gefitinib at the ATP binding pocket, whereas the second form, GAK_2, binds one each in the ATP binding site and a novel binding site adjacent to the activation segment C‐terminal helix, a unique element of the Numb‐associated kinase family. In the novel binding site, gefitinib binds in the hydrophobic groove around the activation segment, disrupting the conserved hydrogen bonds for the catalytic activity. These structures suggest possibilities for the development of selective GAK inhibitors for viral infections, such as the hepatitis C virus.
Keywords: complex structures, gefitinib, inhibitor development, novel binding sites, protein kinases
1. Introduction
Protein kinases play important roles in the intracellular signaling pathways that regulate cell functions such as proliferation, differentiation, and survival. In cancer cells, expression of mutated protein kinase genes can cause over‐activation of these intracellular signaling pathways, resulting in promotion of malignant transformation. Therefore, drugs that inhibit the activity of protein kinases are an important means of treating cancers. Kinase inhibitors are categorized as type I inhibitors, which bind to the ATP binding site of the target kinase; type II inhibitors, which bind to the ATP binding site and to a neighboring region; and type III inhibitors, which bind to an allosteric site.1 Type I inhibitors generally have low selectivity, because the amino acid sequence and structure of the ATP binding pocket of kinases have high similarities. In addition to these three types, kinase inhibitors are further classified into subdivisions with more specific criteria.2
Gefitinib (trade name: Iressa) is a type I kinase inhibitor used for the treatment of advanced non‐small‐cell lung cancer that is positive for a mutation in the epidermal growth factor receptor (EGFR) gene.3, 4 The structure of gefitinib has a 4‐anilinoquinazoline skeleton (Figure 1). Although gefitinib is currently in clinical use, serious adverse drug reactions such as interstitial lung disease and respiratory dysfunction have been reported,5 suggesting that gefitinib has unidentified off‐target inhibitory effects.
Figure 1.
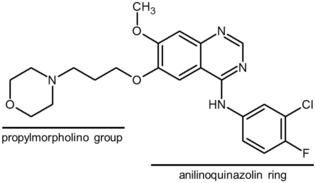
Chemical structure of gefitinib.
In addition to inhibiting signaling through EGFR (IC50=14 nm),6 Brehmer et al. have reported that gefitinib also inhibits cyclin G‐associated kinase (GAK) and receptor‐interacting serine/threonine protein kinase 2 (RICK; also known as RIPK2, RIP2, and CARDIAK) (IC50=90 and 49 nm, respectively), as determined by using a proteomics approach.6 GAK regulates clathrin‐mediated membrane trafficking,7 transcriptional coactivation,8 and mitotic chromosome congression,9 and is also essential for cell growth.10, 11 Knockout mice expressing the kinase‐dead form of GAK develop respiratory dysfunction and die immediately after birth, suggesting that GAK inhibition is one of the underlying causes of the serious adverse drug reactions associated with gefitinib treatment.12 GAK also phosphorylates the μ subunit of adaptor protein 2, which induces rearrangement of its structure; this inhibits adaptor protein‐mediated trafficking in the endocytic pathway,13 which is associated with hepatitis C virus entry into host cells and increased risk of developing Parkinson's disease. Therefore, GAK is a potential therapeutic target for the treatment of hepatitis C and Parkinson's disease.13 Investigations of the structural details of GAK inhibition will contribute not only to understanding the adverse drug reactions, but also to developing selective inhibitors for several severe diseases, such as Parkinson's disease.
GAK is composed of four domains: a kinase domain in the N‐terminus, a PTEN‐like domain, a clathrin‐binding domain, and a J domain at the C‐terminus. The kinase domain belongs to the Numb‐associated kinase (NAK) family that includes MPSK1 (myristoylated and palmitoylated serine/threonine kinase 1), AAK1 (adaptor‐associated kinase 1), and BMP2 (bone morphogenetic protein 2)‐inducible kinase,14 the structures of which have all been solved.15, 16 The active structures of these kinases all have a large α‐helix, including an activation segment C‐terminal helix (ASCH) at the C lobe, which allows for continuous kinase activity without the need for phosphorylation of the activation loop.
Here, we report the crystal structures of two GAK kinase domain⋅gefitinib⋅Nanobody (Nb) complexes. The two structures differ in the number of gefitinib binding sites: both structures possess a common ATP binding site; however, one also possesses a secondary binding site in a segment that is important for the catalytic activity of GAK, suggesting that gefitinib strongly inhibits GAK. The binding mode of gefitinib in the common ATP binding site is similar in GAK and EGFR. Together, these crystal structures provide important information that will be useful for the structure‐based design of drugs with greater selectivity for EGFR and GAK.
2. Results and Discussion
2.1. Structure of GAK Kinase Domain
Initially, we attempted to determine the structure of the GAK kinase domain⋅gefitinib complex; however, the crystal diffracted at a low resolution (ca. 3.5 Å) and we were unable to determine the gefitinib electron density in the structure. Recently, high‐resolution crystal structures of the active and inactive forms of GAK kinase domain in complex with Nbs were reported (Protein Data Bank [PDB] ID: 4C57 [2.55 Å] and 4C59 [2.8 Å], respectively).15 Therefore, in the present study, we used a cell‐free synthesis system17 to synthesize two Nbs and we were able to co‐crystallize GAK kinase domain⋅gefitinib⋅Nb complexes. Consequently, two crystal forms (GAK_1 and GAK_2), which contain the 4C57 type Nb, were obtained and solved by using a molecular replacement method with the first Nb (PDB ID: 4C57) as the search model; these structures were refined to resolutions of 2.80 and 2.50 Å, respectively (see Table S1 and Figure 2). Thus, using Nbs was effective for determining the structure of the GAK kinase domain⋅gefitinib complex.
Figure 2.
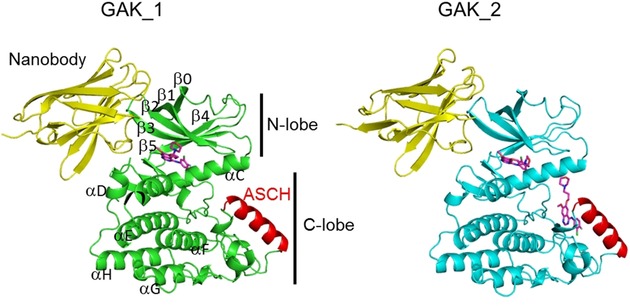
Ribbon diagrams of the overall structures of the GAK kinase domain⋅gefitinib⋅nanobody complexes (GAK_1, green; GAK_2, cyan). Gefitinib molecules are shown as purple stick models. ASCH, activation segment C‐terminal helix.
The two GAK kinase domain structures both have a general bilobal structure, comprising a larger C lobe and a smaller N lobe (Figure 2). Although the complexes crystallized with different crystal packing that included one or two complex molecules in the asymmetric unit, the root mean square deviation (rmsd) value for the common Cα atoms of the GAK kinase domain in both crystal structures was 0.6 Å. A comparison of GAK_1 and the previously reported GAK kinase domain in complex with a Nb (PDB ID: 4C57)15 provided a Cα rmsd of 0.9 Å, indicating that the structures were almost identical.
GAK_1 and GAK_2 adopt an active conformation,2 featuring the DFG‐in and αC‐in configuration, which possesses a salt bridge between Lys69 at a conserved active site in β3 and Glu85 in αC, and an ordered activated segment including ASCH. ASCH is stabilized by hydrogen bonds formed between the conserved amino acids of the catalytic loop and the activation segment (Mg2+‐binding loop, activation loop, P+1 loop, and APE loop). The main difference between the two GAK structures is that they differ in the number of gefitinib molecules that are bound: GAK_1 includes one gefitinib molecule bound to the ATP binding site of the GAK kinase domain, whereas GAK_2 includes two gefitinib molecules—one bound to the ATP binding site and one bound to a binding site located in the C lobe.
2.2. Gefitinib Binding Affinity for the GAK Kinase Domain
In this study, we conducted a surface plasmon resonance (SPR) analysis to determine the dissociation constant (K D) of gefitinib for the GAK kinase domain. By adopting a 1:1 binding model of the steady‐state affinity analysis, its K D was estimated as 15.4 nm, which is close to the previously reported value.18 Considering the existence of the second binding site in the GAK_2 structure, the SPR data were further analyzed by the kinetic heterogenous ligand model. The analysis implied the presence of two binding sites with high and low affinities: K D1=1.62 nm and K D2=285 nm. These affinities may correspond to the two binding sites in the GAK_2 crystal structure.
Both gefitinib bindings in GAK_2 occur in pockets, and not at the crystal packing interface. Thus, the two crystal forms suggest that conformational variations appear in the different crystals. The GAK activation form is stabilized by Nb, which is located approximately 34 Å apart from ASCH. There is no intermolecular interaction around ASCH. Molecular contacts, such crystal packing, in the other part might influence the ligand affinities at the binding sites through the stability of the activated form. These interaction circumstances may underlie the different binding stoichiometries (1:1 and 1:2) in the different crystal forms (GAK_1 and GAK_2).
2.3. Gefitinib Binding to the ATP Binding Site
The electron densities of gefitinib in the ATP binding pocket of GAK_1 and GAK_2 were clearly observed (Figure 3 a,b). In both structures, the anilinoquinazoline ring of gefitinib is located toward the ATP binding pocket. The chloro group of the aniline ring of gefitinib is oriented to the N lobe. The nitrogen atom of the quinazoline ring forms a hydrogen bond with the backbone amide group of Cys126. The binding of gefitinib with GAK_2 is stabilized by a hydrogen bond network formed among Glu85, Thr123, Phe192, Cys190, and three water molecules. The gefitinib molecule also contacts Leu46, Val54, and Ala67 in the roof of the pocket; Glu124 and Leu125 in the hinge region; and Lys127, Gly128, Gln129, and Leu180 in the floor of the cleft. The interaction manners are summarized in Figure S1 a and S1 b. The anilinoquinazoline ring of gefitinib bound to GAK_1 and GAK_2 superimposes well. A propylmorpholino group was added to improve the pharmacokinetic properties of the inhibitor (Figure 1).19 In GAK_1 and GAK_2, the propylmorpholino group differently extends toward solvent region, and there is no specific interaction with GAK.
Figure 3.
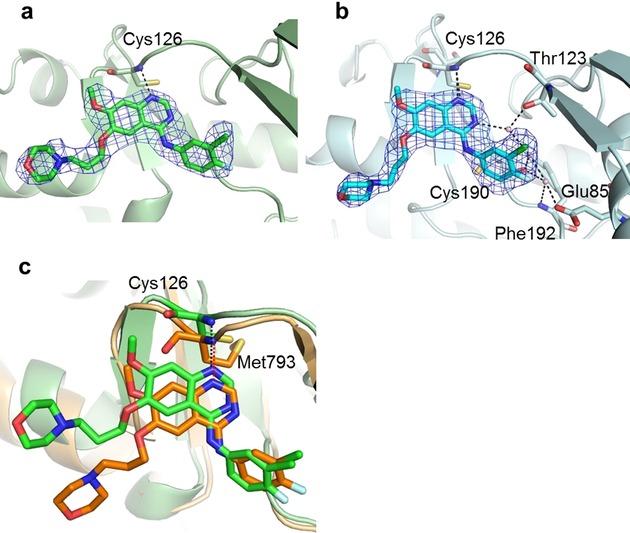
Comparison of gefitinib binding at the GAK ATP binding site. Gefitinib binding at the ATP binding site of a) GAK_1 and b) GAK_2. Gefitinib is shown as a stick model with the backbone carbons in green (GAK_1) and cyan (GAK_2) and the 2 F o−F c electron density map (1σ). c) Superimposed structures of GAK_2 (green ribbon structure) and EGFR (orange ribbon structure). Gefitinib molecules are shown as a green (GAK_2) and orange (EGFR) stick model.
2.4. Binding Mode of Gefitinib to GAK or EGFR
Six structures of the gefitinib⋅EGFR complex (PDB: 4WKQ, 4I22, 3UG2, 2ITO, 2ITY, and 2ITZ; two with wild‐type and four with mutant EGFR) have been reported.20 EGFR(L858R) and EGFR(G719S) are gefitinib sensitive, whereas the other structures, which have a second mutated T790M, are gefitinib resistant. In the gefitinib⋅EGFR complex, the aniline ring of gefitinib extends into the hydrophobic pocket of the ATP binding cleft, and the nitrogen atom in the quinazoline ring of gefitinib forms only a single hydrogen bond with the backbone amide of Met793 in the hinge region of EGFR.20a The propylmorpholino group is oriented differently in the gefitinib⋅EGFR complex as well as the gefitinib⋅GAK complex. To examine the causes of the serious adverse drug reactions associated with gefitinib administration, the amino acid sequence, overall structure, and binding mode with gefitinib of EGFR(L858R) and GAK_1 were compared. EGFR(L858R), and GAK_1 has a sequence identity of 13.5 % and a structural similarity with an rmsd of 3.36 Å over 210 common Cα atoms and 0.72 Å for the common main chain atoms composing the ATP binding site. Superimposing the two structures (Figure 3 c) revealed that the location of the anilinoquinazoline ring and its hydrogen bond with Cys129 in the GAK kinase domain [corresponding to Met793 in EGFR(L858R)] are conserved. The gefitinib binding pocket region is also similar. However, in EGFR(L858R), a rigid amino acid, Pro794, in the hinge region appears to provide a characteristic structural environment.
2.5. Second Gefitinib Binding Site in the GAK Kinase Domain
A novel second gefitinib binding site was observed in the activation segment of GAK_2 (Figure 4 a). Crystal structures with two binding sites have been reported for MPSK116 and Casein kinase 1 γ1 (PDB ID: 2BUJ and 2CMW, respectively). However, the second binding sites in the structures of these two kinases are at the surface of the N lobe, which is different to the location of the second binding site in GAK_2. In GAK 2, the anilinoquinazoline ring is located parallel to ASCH, and the propylmorpholino group extends toward the αC helix. The anilinoquinazoline ring of gefitinib is surrounded by a hydrophobic environment formed by the side chains of His200, Val214, IIe218, Ser194, Asn221, and Thr222, and the main chain of Gly193. A cation–π interaction is formed between the guanidino group of Arg172 and the quinazoline skeleton of gefitinib, and a stacking interaction is formed between the imidazole ring of His200 and the aniline ring. In addition, hydrogen bonds between Arg172 and Gln244, mediated by two water molecules, stabilize the binding of the second gefitinib molecule to GAK_2. The interaction manners are summarized in Figure S2 a and S2 b. Through the binding of the second gefitinib molecule, ASCH moves toward the solvent region by 3.5 Å compared with in GAK_1, which disrupts the hydrogen bonds between Asp173–Thr223 and Ser194–Asn221 (Figure 4 b). The former hydrogen bond corresponds with the substrate‐induced hydrogen bond between Asp166 and Thr201 in cAMP‐dependent protein kinase.21 Substitution of Thr201 with an alanine residue in cAMP‐dependent protein kinase abolishes its enzyme activity.21 The latter hydrogen bond stabilizes the active conformation by connecting the activation loop and ASCH. These hydrogen bonds are important to anchor the C‐terminal region of the active loop and for the kinase activity.22 Considering the role of these hydrogen bonds, the binding of gefitinib to the second binding site likely further inhibits substrate binding activity.
Figure 4.
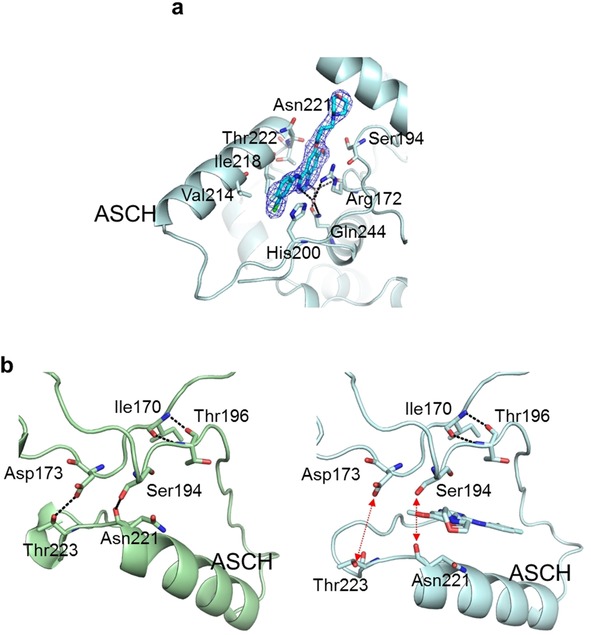
Second gefitinib binding site. a) Location of the second gefitinib binding site with an overlay of the 2 F o−F c electron density map (1σ) of gefitinib. b) Structures of GAK_1 (green ribbon structure; left panel) and GAK_2 (cyan ribbon structure; right panel) around the second binding site. A gefitinib molecule bound to GAK_2 is shown as a stick model.
2.6. Comparison of the Second Binding Sites among NAK Family Kinases
The crystal structures of the NAK family kinases have been determined23 for MPSK1 (PDB: 2BUJ), AAK1 (4WSQ), and BMP2K (4W9X).16 The superimposition of these kinases and GAK_1 produced rmsd values of 2.2 Å (GAK‐MPSK1), 1.8 Å (GAK‐AAK1), and 1.4 Å (GAK‐BMP2K). These structural similarities can be correlated with the sequence conservations: 23.2 % (GAK‐MPSK1), 37.0 % (GAK‐AAK1), and 37.1 % (GAK‐BMPK2).
Although ASCH is a unique motif for the NAK family kinases, the motif length and sequence vary among the kinases (Figure 5). The second gefitinib binding site is present in this region. A comparison among the NAK family kinase structures suggested that hydrophobic contacts might be possible for each kinase, because hydrophobic interactions are achieved with aliphatic side chain moieties. The other interactions in GAK around the second binding site are 1) a cation–π interaction formed between the guanidino group of Arg172 and the quinazoline skeleton of gefitinib, 2) a stacking interaction formed between the imidazole ring of His200 and the aniline ring, and 3) hydrogen bonds between Arg172 and Gln244, mediated by two water molecules (see above). Among these amino acids (Arg172, His200, and Gln244), Arg172 is conserved in the NAK family kinases (Figure 5), whereas His200 and Gln244 are not conserved. Therefore, the stacking interaction and the water‐mediated hydrogen bonds with gefitinib seem to be the selective interactions for GAK against other kinases (MPSK1, AAK1, and BMPK2).
Figure 5.

Sequence alignment of the NAK family kinases. The alignment is shown from the HRD motif to the APE motif.
GAK is reportedly involved in the hepatitis C virus life cycle, and thus an inhibitor of GAK can be considered as an anti‐hepatitis C agent.18 Selective inhibitors for the NAK family kinases to inhibit the ATP binding site have been explored.23 Our results suggest that gefitinib binding at the second site is a selective interaction for both GAK and gefitinib. The second binding site, as well as the ATP binding site, may be a potential target site for GAK‐selective inhibitor development.
3. Conclusions
Here, we solved the crystal structures of two GAK kinase domain⋅gefitinib⋅Nb complexes and discovered a novel second gefitinib binding site in GAK. The two crystal structures revealed that the kinase activity of GAK and EGFR are inhibited by gefitinib through the same inhibitory mechanism. In addition, this is the first time a second binding site near ASCH has been reported, which is unique in the NAK family kinases. Detailed information on the structures of EGFR and GAK is important for the design of highly selective drugs targeting these proteins, as demonstrated by our discovery that gefitinib binding at both the first and second site causes strong inhibition. Although the binding of ligands at secondary sites is yet to be reported for other NAK family kinases, the present results suggest that the second binding site may be a latent compound‐binding site in NAK family kinases. The structural information presented here will be useful to direct in silico screening for the development of novel drugs with greater selectivity for GAK.
Experimental Section
Cloning of GAK
A DNA fragment containing the kinase domain (25–335) of the human GAK gene (OriGene Technologies, USA) was amplified by means of PCR and sub‐cloned into the expression vector pCR2.1TOPO (Life Technologies, USA) with an N‐terminal fusion consisting of an N‐terminal “N11‐tag” (MKDHLIHNHHKHEHAHAEH, affinity tag for nickel resin) and a TEV protease cleavage site. Genes encoding the Nbs were converted from two amino acid sequences (PDB ID: 4C57 and 4C58) and synthesized by Eurofins Genomics (Japan), and the DNA fragments were amplified and sub‐cloned in a similar way to GAK.
Protein Expression and Purification
GAK and the two Nbs were synthesized by using a cell‐free protein synthesis system.24 In the case of the Nbs, 400 μg mL−1 of Escherichia coli disulfide isomerase DsbC and oxidized glutathione (GSSG) (Nacalai Tesque, Japan) was added to the reaction solution to promote the formation of disulfide bonds and optimize the redox conditions.25 The solutions were then applied to a HisTrap column (GE Healthcare, UK) pre‐equilibrated with 20 mm Tris (pH 8.0), 1 m NaCl, 20 mm imidazole, and 10 % glycerol. The samples were eluted with a buffer containing 500 mm imidazole, and the N11‐tag was cleaved with TEV protease at 4 °C overnight in a buffer containing 20 mm Tris (pH 8.0), 500 m NaCl, 20 mm imidazole, and 10 % glycerol. Then, the solutions were applied to a HisTrap column, followed by the elution with a buffer containing 500 mm imidazole, to remove the N11‐tag. The flow‐through fractions (GAK) and the eluted fractions (Nbs) were further purified by using an ion‐exchange column (Resource Q; GE Healthcare, UK) and a size‐exclusion chromatography column (Superdex75; GE Healthcare, UK) in a final buffer containing 20 mm Tris (pH 8.0), 300 mm NaCl, 2 mm DTT, and 10 % glycerol for GAK, and 50 mm Tris (pH 7.5) and 100 mm NaCl for the Nbs. Purified GAK and Nb were mixed in a 1:2 molar ratio, and then the complexes were separated by using a size‐exclusion chromatography column with a buffer containing 50 mm Tris‐HCl (pH 7.5) and 100 mm NaCl. Samples were concentrated to a final concentration of 10 mg mL−1 and stored at −80 °C.
Crystallization and Data Collection
The GAK kinase domain⋅Nb complexes were incubated with 0.5 mm gefitinib (purity >99.9 %, HPLC; Funakoshi, Japan) and 1 % DMSO. To obtain the GAK_1 crystal, GAK kinase domain⋅Nb complex was mixed in a reservoir solution containing a 0.17 m ammonium sulfate, 0.085 m sodium cacodylate trihydrate (pH 6.5), 25.5 % PEG8000, and 15 % glycerol, and co‐crystallized by using the sitting drop method at 20 °C. To obtain the GAK_2 crystal, the GAK kinase domain⋅Nb complex was mixed with a reservoir solution containing a 0.2 m ammonium sulfate, 0.1 m sodium cacodylate trihydrate (pH 6.5), and 15 % PEG8000, and co‐crystallized by using the hanging drop method at 20 °C. The deposited crystals were refined in the same conditions used for the seeding. Both crystals were flash‐frozen in liquid nitrogen with 20 % glycerol as the cryoprotectant.
Structure Determination and Refinement
The diffraction data for the GAK_1 and GAK_2 crystals were collected by using beamline BL32XU at SPring‐8 (Hyogo, Japan) and processed by using the HKL2000,26 XDS,27 and CCP4 software suite.28 Molecular replacement was performed with the Phaser program29 by using the coordinates of Nb and the GAK kinase domain (PDB ID: 4C57) as the search models. The model was built with COOT,30 and refinement was performed with PHENIX software.31 The geometry restraints of gefitinib were generated with the eLBOW module of PHENIX. Ramachandran statistics were calculated with the MolProbity.32 Structural models were drawn by using PyMOL software (the Pymol Molecular Graphics System, Version 1.8, Schrodinger, LLC). Structural comparisons were performed by using the Superpose program in the CCP4 suite.
Surface Plasmon Resonance
Experiments were conducted on a BIAcoreTM T200 instrument (GE Healthcare Life Sciences). GAK was immobilized on a Sensor Chip CM5, using an Amine Coupling Kit (GE Healthcare Life Sciences). All data were collected in buffer containing 10 mm HEPES (pH 7.3), 150 mm NaCl, 1 mm MgCl2, and 0.005 % surfactant P‐20. Serial concentrations (0–50 μm) of gefitinib were injected, and the responses were measured. The experiments were performed with five sample concentrations, in triplicate. Dissociation constants (KD) were computed by fitting to a 1:1 interaction model in the steady‐state affinity analysis and a heterogeneous ligand model in the kinetic analysis, using the Biacore software, BIAevaluation (GE Healthcare Life Sciences). The stoichiometry was calculated based on the theoretical Rmax value, where the theoretical Rmax = MWA/MWL × RL × SM (MW: molecular weight, A: analyte, L: ligand, RL: immobilization level of ligand in RU, SM: stoichiometry).
Accession Codes
The coordinates and structure factors of the final models of the Nb⋅GAK kinase domain complexes were deposited in the PDB (PDB IDs: 5Y7Z and 5Y80).
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Mio Inoue and Ken Ishii for preparing the plasmids. We also thank Drs. Shun‐ichi Sekine and Toshifumi Fujii, and the beamline staff at SPring‐8 for their assistance during data collection. This research was supported by the Platform Project for Supporting in Drug Discovery and Life Science Research (Platform for Drug Discovery, Informatics and Structural Life Science) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT).
N. Ohbayashi, K. Murayama, M. Kato-Murayama, M. Kukimoto-Niino, T. Uejima, T. Matsuda, N. Ohsawa, S. Yokoyoma, H. Nojima, M. Shirouzu, ChemistryOpen 2018, 7, 713.
References
- 1. Dar A. C., Shokat K. M., Annu. Rev. Biochem. 2011, 80, 769–795. [DOI] [PubMed] [Google Scholar]
- 2. R. Roskoski, Jr. , Pharmacological Res. 2016, 103, 26–48. [DOI] [PubMed] [Google Scholar]
- 3.AstraZeneca, London, UK; http://www.astrazeneca.com.
- 4. Araki T., Yashima H., Shimizu K., Aomori T., Hashita T., Kaira K., Nakamura T., Yamamoto K., Clin. Med. Insights. Oncol. 2012, 6, 407–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jiang H., Jpn. J. Clin. Oncol. 2009, 39, 137–150. [DOI] [PubMed] [Google Scholar]
- 6. Brehmer D., Greff Z., Godl K., Blencke S., Kurtenbach A., Weber M., Muller S., Klebl B., Cotten M., Keri G., Wissing J., Daub H., Cancer Res. 2005, 65, 379–382. [PubMed] [Google Scholar]
- 7. Eisenberg E., Greene L. E., Traffic 2007, 8, 640–646. [DOI] [PubMed] [Google Scholar]
- 8. Ray M. R., Wafa L. A., Cheng H., Snoek R., Fazli L., Gleave M., Rennie P. S., Int. J. Cancer 2006, 118, 1108–1119. [DOI] [PubMed] [Google Scholar]
- 9. Shimizu H., Nagamori I., Yabuta N., Nojima H., J. Cell Sci. 2009, 122, 3145–3152. [DOI] [PubMed] [Google Scholar]
- 10. Lee D. W., Zhao X., Yim Y. I., Eisenberg E., Greene L. E., Mol. Biol. Cell 2008, 19, 2766–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Olszewski M. B., Chandris P., Park B. C., Eisenberg E., Greene L. E., Traffic 2014, 15, 60–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tabara H., Naito Y., Ito A., Katsuma A., Sakurai M. A., Ohno S., Shimizu H., Yabuta N., Nojima H., PloS one 2011, 6, e26034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang G., Pan J., Chen S. D., Prog. Neurobiol. 2012, 98, 207–221. [DOI] [PubMed] [Google Scholar]
- 14. Manning G., Whyte D. B., Martinez R., Hunter T., Sudarsanam S., Science 2002, 298, 1912–1934. [DOI] [PubMed] [Google Scholar]
- 15. Chaikuad A., Keates T., Vincke C., Kaufholz M., Zenn M., Zimmermann B., Gutierrez C., Zhang R. G., Hatzos-Skintges C., Joachimiak A., Muyldermans S., Herberg F. W., Knapp S., Muller S., Biochem. J. 2014, 459, 59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Eswaran J., Bernad A., Ligos J. M., Guinea B., Debreczeni J. E., Sobott F., Parker S. A., Najmanovich R., Turk B. E., Knapp S., Structure 2008, 16, 115–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kigawa T., Yabuki T., Matsuda N., Matsuda T., Nakajima R., Tanaka A., Yokoyama S., J. Struct. Funct. Genomics 2004, 5, 63–68. [DOI] [PubMed] [Google Scholar]
- 18. Kovackova S., Chang L., Bekerman E., Neveu G., Barouch-Bentov R., Chaikuad A., Heroven C., Sala M., De Jonghe S., Knapp S., Einav S., Herdewijn P., J. Med. Chem. 2015, 58, 3393–3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Barker A. J., Gibson K. H., Grundy W., Godfrey A. A., Barlow J. J., Healy M. P., Woodburn J. R., Ashton S. E., Curry B. J., Scarlett L., Henthorn L., Richards L., Bioorg. Med. Chem. Lett. 2001, 11, 1911–1914. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Yun C. H., Boggon T. J., Li Y., Woo M. S., Greulich H., Meyerson M., Eck M. J., Cancer Cell 2007, 11, 217–227; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20b. Yoshikawa S., Kukimoto-Niino M., Parker L., Handa N., Terada T., Fujimoto T., Terazawa Y., Wakiyama M., Sato M., Sano S., Kobayashi T., Tanaka T., Chen L., Liu Z. J., Wang B. C., Shirouzu M., Kawa S., Semba K., Yamamoto T., Yokoyama S., Oncogene 2013, 32, 27–38; [DOI] [PubMed] [Google Scholar]
- 20c. Gajiwala K. S., Feng J., Ferre R., Ryan K., Brodsky O., Weinrich S., Kath J. C., Stewart A., Structure 2013, 21, 209–219. [DOI] [PubMed] [Google Scholar]
- 21. Yang J., Ten Eyck L. F., Xuong N. H., Taylor S. S., J. Mol. Biol. 2004, 336, 473–487. [DOI] [PubMed] [Google Scholar]
- 22. Nolen B., Taylor S., Ghosh G., Mol. Cell 2004, 15, 661–675. [DOI] [PubMed] [Google Scholar]
- 23. Sorrell F. J., Szklarz M., Abdul Azeez K. R., Elkins J. M., Knapp S., Structure 2016, 24, 401–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Takanori Kigawa T. M., Yabuki T., Yokoyama S., Bacterial Cell-Free Synstem for Highly Efficient Protein Synthesis, Wiley-VCH, Weinheim, 2008. [Google Scholar]
- 25. Matsuda T., Watanabe S., Kigawa T., Biochem. Biophys. Res. Commun. 2013, 431, 296–301. [DOI] [PubMed] [Google Scholar]
- 26. Otwinowski M. W., Methods Enzymol. 1997, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- 27. Kabsch W., Acta Crystallogr. Sect. D 2010, 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Project Collaborative Computational, Acta Crystallogr. Sect. D 1994, 50, 760–763.15299374 [Google Scholar]
- 29. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J., J. Appl. Crystallogr. 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Emsley P., Cowtan K., Acta Crystallogr. Sect. D 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- 31. Adams P. D., Afonine P. V., Bunkoczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., Acta Crystallogr. Sect. D 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen V. B., Arendall, 3rd W. B., Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C., Acta Crystallogr. Sect. D 2010, 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary