

Abstract
Gastrointestinal and gynecological malignancies disseminate in the peritoneal cavity - a condition known as peritoneal carcinomatosis (PC). Intraperitoneal (IP) administration can be used to improve therapeutic index of anticancer drugs used for PC treatment. Activity of IP anticancer drugs can be further potentiated by encapsulation in nanocarriers and/or affinity targeting with tumor-specific affinity ligands, such as tumor homing peptides. Here we evaluated a novel tumor penetrating peptide, linTT1 (AKRGARSTA), as a PC targeting ligand for nanoparticles. We first demonstrated that the primary homing receptor for linTTl, p32 (or gClqR), is expressed on the cell surface of peritoneal carcinoma cell lines of gastric (MKN-45P), ovarian (SKOV-3), and colon (CT-26) origin, and that peritoneal tumors in mice and clinical peritoneal carcinoma explants express p32 protein accessible from the IP space. Iron oxide nanoworms (NWs) functionalized with the linTT1 peptide were taken up and routed to mitochondria in cultured PC cells. NWs functionalized with linTT1 peptide in tandem with a pro-apoptotic [D(KLAKLAK)2] peptide showed p32-dependent cytotoxicity in MKN-45P, SKOV-3, and CT- 26 cells. Upon IP administration in mice bearing MKN-45P, SKOV-3, and CT-26 tumors, linTT1-functionalized NWs showed robust homing and penetration into malignant lesions, whereas only a background accumulation was seen in control tissues. In tumors, the linTT1-NW accumulation was seen predominantly in CD31-positive blood vessels, in LYVE-1-positive lymphatic structures, and in CD11b-positive tumor macrophages. Experimental therapy of mice bearing peritoneal MKN-45P xenografts and CT-26 syngeneic tumors with IP linTT1-D(KLAKLAK)2-NWs resulted in significant reduction of weight of peritoneal tumors and significant decrease in the number of metastatic tumor nodules, whereas treatment with untargeted D(KLAKLAK)2-NWs had no effect. Our data show that targeting of p32 with linTTl tumor-penetrating peptide improves tumor selectivity and antitumor efficacy of IP pro-apoptotic NWs. P32-directed intraperitoneal targeting of other anticancer agents and nanoparticles using peptides and other affinity ligands may represent a general strategy to increase their therapeutic index.
Keywords: Peptide, p32, Neuropilin-1, Nanomedicine, Iron oxide nanoworms, Pro-apoptotic peptide, Tumor targeting, Peritoneal carcinomatosis, MR imaging
1. Introduction
Gastrointestinal and gynecological malignancies often disseminate in the peritoneal space and trigger severe complications such as bowel obstruction and the formation of ascites. At the time of diagnosis, peritoneal metastases are present in about 50% of gastric, 30% of ovarian, and 40% of colorectal cancer patients [1]. This condition, known as peritoneal carcinomatosis (PC), has a poor prognosis with a median survival of only a few months [2]. PC is a vexing condition as complete surgical removal of disseminated microlesions is impossible and systemic chemotherapy has a limited anticancer effect due to poor vascularization of tumor nodules and the presence of peritoneum- plasma barrier [3,4]. Intraperitoneal (IP) chemotherapy is used to achieve an increased concentration of the anticancer drugs in the peritoneal cavity and to reduce systemic exposure [5]. IP therapy using mildly heated drug solutions, Hyperthermic IntraPeritoneal Chemotherapy (HIPEC), further enhances drug penetration into malignant tissue, and combining HIPEC with cytoreductive surgery results in improved efficacy of the treatment [6–9]. Encapsulation of cytotoxic drugs in nanoformulations has been also shown to improve the anticancer effect of IP chemotherapy [10]. Even with these improvements, many patients develop recurrent disease [11]. Ideally, PC treatment would not rely on specialized, complex equipment and procedures, and would instead be based on smart drugs that are potent, tumor selective, and show minimal peritoneal and systemic toxicities.
Tumor targeting with affinity ligands, such as peptides or antibodies, utilizes malignancy-associated molecular markers in the tumor microenvironment to deliver payloads to cancerous tissues [12,13]. Tumor homing peptides enable delivery of drugs and diagnostic compounds directly into tumors, thereby improving tumor detection and increasing the efficacy of the treatment while reducing side effects [14]. Tumor homing peptides identified by in vivo phage biopanning screens are particularly well suited for NP targeting, as phages used as scaffolds to display random peptides are biological nanoparticles themselves [15]. A series of recent studies have demonstrated the utility of iRGD, a tumor-penetrating peptide, for improved tumor-specific penetration of intraperitoneal drugs and nanoparticles and for enhanced IP chemotherapy in mice [16,17]. iRGD uses as recruitment receptors αν in- tegrins, cell surface molecules commonly upregulated during angiogenic response and in tumor cells, and subsequently activates the transtissue transport (CendR) pathway described below.
A recently identified tumor penetrating peptide TT1 (active both as a disulfide-bridged CKRGARSTC and as linTT1, AKRGARSTA) homes robustly to breast cancer in mouse models and enhances the antitumor potency of therapeutic payloads [18,19]. The primary homing receptor for TT1 family of peptides is p32 (also known as gC1qR), a mitochondrial protein aberrantly expressed on the cell surface of activated malignant and stromal cells in solid tumors, often in hypoxic areas deep in the tumor tissue [20]. TT1 belongs to a novel class of tumor targeting peptides, tumor penetrating C-end Rule (CendR) peptides characterized by a multistep homing and tumor penetration pathway. After binding to p32 TT1 peptide is proteolytically cleaved by a urokinase type plasminogen activator at the second arginine residue (AKRGARSTA) and the processed peptide acquires affinity towards tissue penetration receptor NRP-1 via its C-terminal RGAR CendR motif [19] to trigger vascular exit and tumor penetration [21,22].
Here, we set out to explore potential applications of linTTl peptide as an IP targeting probe to PC lesions. As nanocarriers we used dextran- coated and PEGylated paramagnetic iron oxide nanoworms (NW) - a nanoscale agent extensively validated for peptide-mediated tumor targeting as a drug carrier and a MRI contrast agent [23–30]. Aspect ratio is known to influence performance of iron oxide nanoparticles in biological systems [29]. First, compared to spherical iron oxide nanoparticles, iron oxide nanoworms have extended circulation half-life. Second, the elongated NWs, with their larger surface area, present multiple targeting ligands that can cooperatively interact with cell surfaces, rendering the platform well-suited for affinity targeting. Finally, linearly aggregated 10 cores in IONWs generate improved T2- relaxivity for improved MR imaging [29]. We used intraperitoneal linTTl-functionalized NWs carrying pro-apoptotic D[KLAKLAK]2 effector module [19,31] for experimental therapy on a panel of peritoneal tumors in mice. Our data indicate that linTT1 peptide functionalization greatly improves tumor selectivity of NWs and increases therapeutic efficacy of a pro-apoptotic nanosystem based on the NWs.
2. Materials and methods
2.1. Materials
(K3[Fe(CN)6]), HC1, Nuclear Fast Red solution, Xylene substitute, MTT reagent (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide), isopropanol, Triton-X and Tween-20 were purchased from Sigma-Aldrich, Germany. Phosphate-buffered saline (PBS) was purchased from Lonza (Belgium).
2.2. Peptides and NW preparation
Peptides were synthesized using Fmoc/t-Bu chemistry on a micro- wave assisted automated peptide synthesizer (Liberty, CEM Corporation, NC, USA). Peptides were purified by HPLC using 0.1% TFA in acetonitrile-water mixtures to 90%−95% purity and validated by Q-TOF mass spectral analysis. Fluorescent peptides were synthesized by using 5(6)-carboxyfluorescein (FAM) with 6-aminohexanoic acid spacer attached to the N-terminus of the peptide. The peptides were N-term- inally amidated and had free C-termini.
The NWs were aminated by combining 0.25 ml of 28% aqueous ammonium hydroxide (#44939, Sigma-Aldrich, Germany) with 1 ml of NWs with an iron concentration of 1.3mg/ml and stirring at room temperature for 24 h. Subsequently, the NWs were purified by dialysis against PBS using 20,000 MW cut-off dialysis cassettes (#66030, Thermo Fisher Scientific, USA) at 4 °C for 48 h (adopted from [30]).
NWs coated with peptides were prepared as previously described [29,30]. Briefly, aminated NWs were PEGylated with maleimide- 5KPEG-NHS (JenKem Technology, TX, USA). Peptides were coupled to NWs through a thioether bond between the thiol group of a cysteine residue added to the N-terminus of the peptide and the maleimide on the functionalized particles. Dynamic Light Scattering (DLS; Zetasizer Nano ZS, Malvern Instruments, UK) was used to assess the poly- dispersity and size of NW preparations. Transmission electron microscopy (TEM, Tecnai 10, Philips, Netherlands) was used to image the nanoparticles.
2.3. Cell lines and experimental animals
MKN-45P human gastric cancer cells were isolated from parental MKN-45P cells [32]. SKOV-3 human ovarian carcinoma and CT-26 mouse colon carcinoma cell lines were purchased from ATCC (SKOV-3 ATCC HBT-7; CT-26 ATCC CLR-2638). The cells were cultivated in DMEM (Lonza, Belgium) containing 100 IU/ml of penicillin, streptomycin, and 10% of heat-inactivated fetal bovine serum (GE Healthcare, UK) in 5% CO2 atmosphere. For animal experimentation athymic nude mice were purchased from HSD and Balb/c mice were purchased from Charles River. Animal experimentation protocols were approved by Estonian Ministry of Agriculture, Committee of Animal Experimentation (Project #42).
2.4. In vitro binding of nanoworms to recombinant p32 protein
Recombinant hexahistidine-tagged p32 was bacterially expressed and purified as described [18]. For cell-free binding assays, Ni-NTA magnetic agarose beads (Qiagen, Germany) in binding buffer (50 mM Tris pH 7.4, 150 mM NaCl, 0.05% NP40, 5 mM imidazole) were coated with p32 protein (at 15 μg of protein/10 μL beads). Fluorescently labeled NWs were incubated with the p32-coated beads in binding buffer containing 1% BSA at RT for 1 h. Incubation was followed by washes and elution with 400 mM imidazole containing binding buffer. The fluorescence of eluted samples was quantified using a fluorescence plate reader (FlexStation II, Molecular Devices, CA, USA).
2.5. In vitro cell viability assay
Cell viability was assessed by colorimetric assay based on reducing the tetrazolium dye MTT 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenylte- trazolium bromide by NAD(P)H-dependent cellular oxidoreductase to insoluble purple formazan. Briefly, cells were seeded in 96-well plates (10,000 MKN-45P or SKOV-3 cells, and 5000 CT-26 cells) and grown in full medium at 37 °C. At 24 h different concentrations of NWs (3, 10, 30, 100, 300 μg/ml iron) were added to the wells. After 6 h the medium was replaced with a fresh medium, and 24 h after the NW addition, 10 μL of 5 mg/ml MTT reagent in PBS was added. Two h later the medium was removed, the blue formazan crystals were dissolved in 100 μL of isopropanol and the absorbance was measured at 570 nm with a microplate reader (Tecan Austria, Austria).
2.6. Evaluation of cellular binding
For flow cytometry MKN-45P, CT-26, SKOV-3 cells in suspension were incubated with NWs in complete cell culture medium for 1 h The NW-containing solution was removed, cells were washed and analyzed by flow cytometry (Accuri, BD Biosciences, CA, USA). Anti-p32 antibody inhibition was done by pre-incubating the cells in suspension with 20 μg/ml of in-house rabbit polyclonal p32 antibody, followed by NW incubation for 1 h, washes and flow cytometry.
For confocal imaging of FAM-labeled NWs, MKN-45P, CT-26, SKOV- 3 (5 × 104 cells) were seeded on glass coverslips in a 24-well plate. After 24 h, NW samples at 40 μg Fe/well were added to the cells and incubated at 37 °C for 3 h. The cells were washed with PBS, fixed with 4% of paraformaldehyde in PBS pH 7.4 (PFA), co-stained with DAPI, and imaged with fluorescence confocal microscopy (Olympus FV1200MPE, Germany).
For immunofluorescence staining, the cultured cells were incubated with rabbit anti-fluorescein (cat. no. A889, Thermo Fisher Scientific, MA, USA) primary antibody to detect the NWs and with mouse anti- cytochrome-C (cat. no 89918, Thermo Fisher Scientific, MA, USA) to label mitochondria. The images were analyzed using the FV10-ASW 4.2 Viewer image software (Olympus, Germany). Optionally, iron was stained using the Prussian Blue cytochemistry [33] followed by coun- terstaining with Nuclear Fast Red.
2.7. In vivo biodistribution studies
Nude mice were injected IP with 2 × 106 MKN-45P cells or 5 × 106 SKOV-3 cells. Tumors were allowed to develop for 14 days in MKN-45P- injected mice and 4 weeks in SKOV-3-injected mice. Balb/c mice received an IP injection of 2 × 106 CT26 cells 7 days before injecting NW samples. FAM-labeled NWs were injected IP or IV (5 mg/kg Fe) and 5 h later the animals were perfused with 10 ml of PBS. The tumors and organs were excised, snap-frozen in liquid nitrogen, and stored at — 80 °C for further analysis.
2.8. Tissue immunofluorescence and confocal microscopy
For immunofluorescence staining of tissues, 10 μm cryosections were equilibrated at RT, fixed in PFA for 15 min, permeabilized using PBS containing 0.2% Triton-X for 10 min, and blocked in PBS containing 0.05% Tween-20, 5% FBS, 5% BSA, and 5% goat serum (GE Healthcare, UK) for 1 h. The sections were immunostained with rabbit anti-fluorescein (cat. no. A889, Thermo Fisher Scientific, MA, USA), rat anti-mouse CD31, biotin rat anti-mouse CD11b (cat. no. 553370; 557395, BD Biosciences, CA, USA), rat anti-mouse LYVE-1 (cat. no. 14044382, eBioscience, CA, USA), rabbit polyclonal anti-Ki67 (cat. no. NB500–170, Novusbio, UK), and rabbit anti-Cleaved Caspase-3 (Asp 175),(cat. no. 966, Cell Signaling Technology, Inc., MA, USA) as primary antibodies,. Alexa 488-conjugated goat anti-rabbit IgG, Alexa 647-conjugated goat anti-rat IgG, Alexa 546-conjugated goat anti-rabbit IgG (all Invitrogen, Thermo Fisher Scientific, MA, USA) and strepta- vidin Dylight® 550 (Thermo Fisher Scientific, MA, USA) were used as secondary antibodies. Nuclei were counterstained with 10 pg/ml DAPI. The stained tissue sections were examined by fluorescence confocal microscopy using Olympus FV1200MPE instrument, and the images were processed and analyzed using the FV10-ASW 4.2 Viewer image software (Olympus, Germany) and Image J freeware.
2.9. Magnetic resonance imaging
Nude mice bearing MKN-45P IP tumors were injected in- traperitoneally with NWs coated with linTT1 peptide or FAM only (5 mg/kg Fe per injection). The mice were subjected to imaging before injection and 5 h after the NW injection. For each scan with isoflurane (3.5% induction, 1.5–2.0% maintenance) in air/O2 (2:1) and positioned into a MR receiver mouse coil. Fast spin echo T2-weighted iron sensitive MRI scans were acquired using 9.4 T MR system (Bruker, USA). The settings were: TR/TE = 1.8 s/23 ms, slice thickness 0.5 mm.
2.10. Binding of dextran to NWs
Peptide-coated NW samples in complete medium were incubated with 0.3 mg 70-kDa lysine-fixable dextran labeled with Texas red (Molecular Probes, MA, USA) in complete medium at 37 °C for 90 min, followed by washing at 5000 g using 100 kDa Amicon®Ultra Centrifugal Filters (Merck Millipore, Darmstadt, Germany). NWs on the filter were resuspended in complete medium and their fluorescence was measured using a fluorescence plate reader at 595/615. (FlexStation II, Molecular Devices, CA, USA).
2.11. Bystander effect studies in vivo
MKN-45P tumor mice were sequentially injected with 5 mg/kg of NWs and 0.3 mg Texas red-conjugated 70-kDa lysine-fixable dextran (Molecular Probes, MA, USA) in PBS into the abdominal cavity. The volume of injection was in 1 ml. After 90 min, the mice were terminated under deep anesthesia, tissues were excised and processed for immunofluorescence.
2.12. Ex vivo dipping assay on clinical tumor samples
Fresh surgical samples of peritoneal metastases of colon cancer patients were collected under protocols approved by the Ethics Committee of the University of Tartu, Estonia (permit #243/T27). Explants amounting to about 1 cm3 were incubated at 37 °C with NWs (40 μg/ml Fe diluted in DMEM supplemented with 1% of BSA) for 4 h. Next, the explants were washed with PBS, snap-frozen, cryosectioned at 10 μm, and immunostained using rabbit anti-fluorescein primary antibodies, followed by detection with Alexa-546 anti-rabbit secondary antibody (Invitrogen, Thermo Fisher Scientific, MA, USA).
2.13. Experimental tumor therapy
IP MKN-45P tumors were induced in nude mice by IP injection of 1.5 × 106 MKN-45P cells in 500 μL of PBS. Five days after tumor induction, the mice were randomized into 5 groups (8 mice in each group). Starting on day 5, mice were IP injected every other day with linTT1-NW, D(KLAKLAK)2-NW, linTT1-D(KLAKLAK)2-NWs, or non-tar- geted NWs at a dose 5 mg/kg Fe per injection, or with 500 μl of PBS. Body weight was monitored daily and the study was terminated when the body weight of the first animal in the study decreased by 20% compared to the start of the treatment. The mice were perfused with 10 ml of PBS and larger tumors in the IP cavity were weighed, and metastatic nodules smaller than 2 mm in diameter were counted.
Syngeneic peritoneal colon carcinomas were induced by IP injection of 2.5 × 105 CT-26 cells in 500 μL of PBS in Balb/c mice. Four days after tumor induction, the mice were randomized (6 mice in each group) and starting on day 5, mice were injected IP every other day with linTT1-NW, D(KLAKLAK)2-NW, linTT1-D(KLAKLAK)2-NWs, or non- targeted NWs at a dose 5 mg/kg Fe, or with PBS. Thirteen days after tumor induction the animals were perfused with 10 ml of PBS, the volume of peritoneal ascites was measured, and tumor burden was assessed by weighing the tumors and counting tumor nodules as for the MKN-45P model.
2.14. Statistical analysis
Student’s t-test was performed using GraphPad Prism Software (Graphpad, CA, USA) and one-way analysis of variance (ANOVA) and Fisher LSD was performed with Statistica 8 software (StatSoft, OK, USA).
3. Results
3.1. LinTT1 -NWs bind to purified p32 protein
We synthesized and characterized NWs as described previously [29,30]. The mean hydrodynamic size of NWs was 88 nm. In TEM images NWs appeared as ~ 100 nm strings of iron cores with a diameter of ~ 30 nm (Fig. 1A). Fluorescent linTT1 and control peptides or only FAM label were coated on the NWs through a thioether bond between the cysteine thiol from the peptide and the maleimide on the functionalized particles, resulting in loading of ~165μmol of peptide/g of iron. To test the effect of linTT1 functionalization on the tropism of NWs, we tested the interaction of linTT1-NWs with immobilized recombinant p32 protein. LinTT1-NWs bound to p32, whereas untargeted NWs, or NWs functionalized with a control RPARPAR peptide, showed only background binding (Fig. 1B). We used RPARPAR as a specificity control because RPARPAR binds to NRP-1, a receptor with specificity somewhat similar to that of p32 (both proteins favor positively charged ligands) [21]. In another control experiment, we observed no binding of linTT1-NWs to recombinant NRP-1, whereas RPARPAR-NW bound specifically to this receptor (Fig. S1). These studies show that linTT1 peptide coated on NWs is accessible for receptor interactions and that linTT1 peptide can be used for selective targeting of NWs to p32.
Fig. 1.
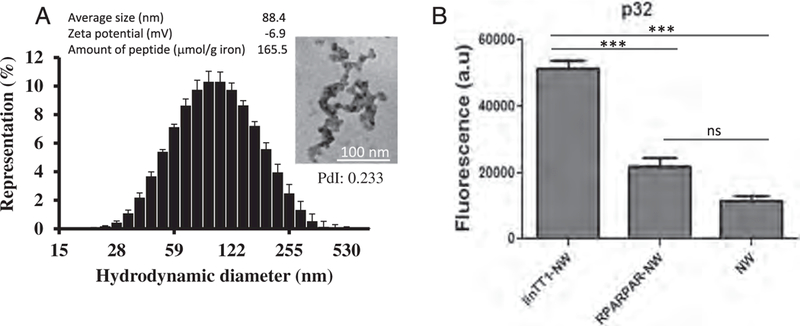
Characterization of iron oxide NWs used in this study. (A) Physicochemical properties of NWs. Size distribution of non-functionalized NWs measured by DLS at 25 C. PdI - polydispersity index. Error bars represent standard deviation (n = 3). TEM image of non-functionalized NWs (scale bar is 100 nm, magnification 160,000 ×). Average diameter calculation was based on DLS (n = 3); zeta potential was measured with NWs functionalized with linTT1 peptide (n = 3); the amount of peptide is given as μmol/g of iron. (B) Binding of FAM-labeled linTT1-NWs, RPARPAR-NWs or control FAM-NWs to recombinant p32. Thirty μg/ml of NWs were incubated with immobilized his-tagged p32 for 1 h, followed by washes to remove unbound NWs and quantification of bound FAM-NWs by spectrometry. Y axis is the NW fluorescence in arbitrary units (a.u.). N = 3; statistical analysis by one-way ANOVA; error bars, mean + SEM, ***p < 0.001.
3.2. LinTTl -NWs bind cultured peritoneal carcinomatosis cells in a p32- dependent manner
The target of linTT1 peptide, p32, is a mitochondrial chaperone that is aberrantly expressed on the cell surface in activated cells [20]. To establish the relevance of the linΤΤ1-ρ32 platform for targeting PC, we first tested the expression of the cell surface p32 on cultured PC cell lines by flow cytometry. Incubation of the live MKN-45P (gastric carcinoma), SKOV-3 (ovarian carcinoma), and CT-26 (colon carcinoma) cells with anti-p32 antibody showed that these cells express p32 at the cell surface (Fig. S2A). We next determined whether the cell surface p32 can be targeted with linTTl-NWs. Flow cytometry demonstrated that linTT1-NW, but not untargeted FAM-coated NW, bound to MKN- 45P, SKOV-3, and CT-26 cells (Fig. 2A). LinTT1-NW interaction with the cells was dependent on the presence of p32, as the pre-incubation of the cells with a blocking anti-p32 antibody inhibited linTT1-NW binding up to 90% (Fig. 2B). Confocal microscopy on cultured cells revealed prominent binding and internalization of linTT1-NW in the MKN-45P, CT-26 and SKOV-3 cells after 3 and 24 h of incubation, whereas NW showed minimal binding (Fig. 2C and Fig. S2B). We confirmed the cellular uptake of linTT1-NWs into MKN-45P and CT-26 cells by staining the cells for the presence of iron using Prussian blue cytochemistry (Fig. S3). These experiments show that linTT1-NWs target p32 expressed on the cell surface of cultured PC cell lines.
Fig. 2.
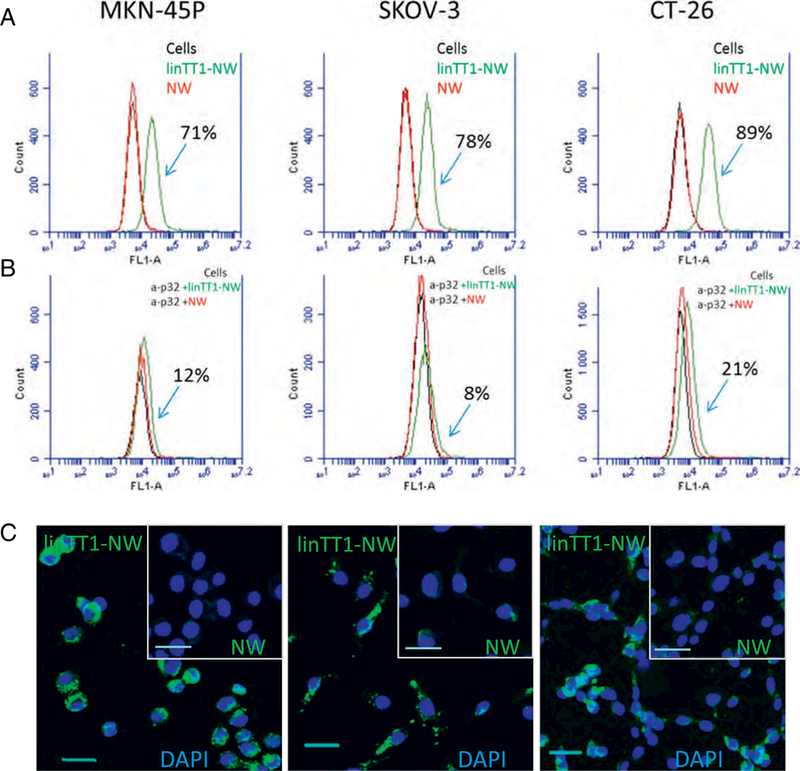
LinTT1-NWs bind to peritoneal carcinomatosis cell lines in a p32-dependent manner. (A) Flow cytometry of MKN-45P, SKOV-3 and CT-26 cells incubated with linTT1-NWs and control FAM labeled NWs. Cells in suspension were incubated with NWs (at 30 μg/ml Fe) for 1 h followed by washes, and flow cytometry analysis. Green line: cells incubated with linTT1- NWs; red line: cells incubated with NWs; black line: cells without NW incubation. (B) Anti-p32 antibody inhibition of NW binding to MKN-45P, SKOV-3 and CT-26 cells. Suspended cells were pre-incubated with 20 μg/ml of p32 antibody, followed by NW incubation for 1 h, washes and flow cytometry. The labeling colour scheme is the same as in A. (C) Fluorescence confocal imaging of cultured adherent MKN-45P, CT-26 and SKOV-3 cells incubated with linTT1-NWs or non-targeted NWs for 3h. Green: NWs; blue: DAPI. Scale bars: 30 μm. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.3. Internalized linTT1-NWs are routed to mitochondria and linTT1-D(KLAKLAK)2-NWs have a cytotoxic effect on MKN-45P cells
In activated cells, p32 pool is present at both on the cell surface and in the mitochondria [31]. Confocal imaging of MKN-45P cells demonstrated that after 3 h of incubation intracellular linTT1-NWs colocalized with cytochrome C, which is a mitochondrial marker (Fig. 3A). The pro- apoptotic D(KLAKLAK)2 peptide is thought to exert its cell-killing activity primarily by destabilizing the mitochondrial membranes [34]. We hypothesized that linTT1 peptide may be well suited for mitochondrial targeting of D(KLAKLAK)2 effector module in the p32-positive PC cells. The two peptides were synthesized as a chimeric peptide for covalent coupling to the NWs through a 5 K-polyethylene glycol (PEG) linker. The final configuration was the following: NW-PEG-Cys-D(KLAKLAK)2- X-AKRGARSTA. The C-terminal positioning of the homing peptide is important for the CendR motif to interact with neuropilin-1 (NRP-1) to trigger cellular uptake and tissue penetration.
Fig. 3.
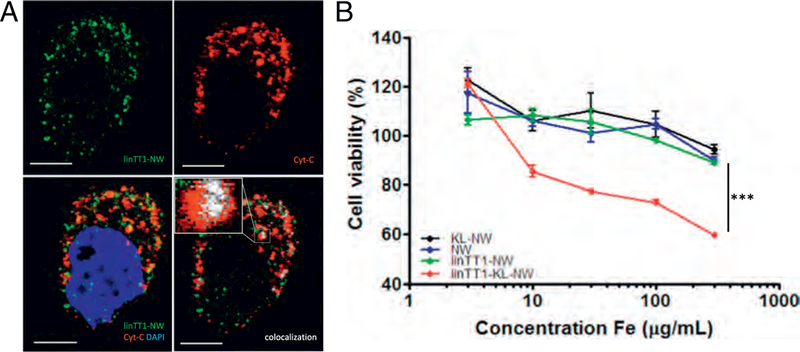
Internalized linTT1-NWs are routed to mitochondria and linTT1-D(KLAKLAK)2-NWs have a cytotoxic effect on MKN-45P cells. (A) Internalized linTT1-NWs colocalize with a mitochondrial marker, cytochrome C. MKN-45P cells were incubated with linTT1-NWs for 3 h, washed, and fixed for immunofluorescence staining. The cells were incubated with rabbit anti-FITC primary antibody to detect the NWs and with mouse anti-cytochrome C to label mitochondria, followed by incubation with anti-mouse Alexa Fluor-546 and anti-rabbit Alexa Fluor 647 secondary antibodies; nuclei stained with DAPI. LinTT1-NW: green; cytochrome C (Cyt-C): red; DAPI: blue; colocalization of FAM and cytochrome C signal: white. Scale bar: 5 μm. (B) Treatment with linTT1-NW coupled to the pro-apoptotic peptide D(KLAKLAK)2, decreases viability of MKN-45P cells. MKN-45P cells were incubated with the indicated NWs over a range of iron concentrations (3, 10, 30, 100, 300 μg/ml), and cell viability was assessed after 6 h incubation by a colorimetric assay based on reducing the tetrazolium dye MTT. KL: D(KLAKLAK)2. Statistical analysis was performed by ANOVA. n = 3; error bars indicate ± SEM; ***p < 0.001, **p < 0.01, *p < 0.05. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The mitochondrial localization of internalized TT1-NW complex (Fig. 3 A) indicated that TT1 peptide is capable of delivering D(KLAKLAK)2 payload to its target site - the mitochondria. The CendR pathway has been partially characterized; the uptake of CendR peptide- coated NPs into cells is an endocytic process that resembles, but is distinct from micropinocytosis [35]. A recent study used a superresolution microscopy to document a direct endosomal-to-mitochon- drial transfer of intraendosomal iron [36]. The dissection of detailed mechanism of mitochondrial translocation of internalized TT1-NWs remains a subject of follow-up studies.
We studied the effect of NWs coated with linTTl-D(KLAKLAK)2 chimeric peptide on viability of cultured MKN-45P, CT-26, and SKOV-3 cells and found that linTTl-D(KLAKLAK)2-NWs reduced the viability of all three PC cell lines, with treatment at the highest concentration, 300 μg Fe/ml (~50 nmol of peptide) causing death of about 55–60% cells (Fig. 3B and Fig. S4). In contrast, control NWs coated with either peptide alone did not have an effect. These observations indicate that the linTT1-NW uptake pathway in PC cells is compatible with in vitro delivery of mitochondrially-acting pro-apoptotic peptide to induce cell death.
3.4. LinTT1-NWs home to peritoneal tumor lesions in vivo
We next studied the biodistribution of linTT1-NWs in mice inoculated with peritoneal MKN-45P tumors. First, two administration routes for targeted particles were compared: systemic IV administration and locoregional IP administration. For both routes, biodistribution of non-targeted NWs was also analyzed and no specific homing to tumors was seen (Fig. 5A and C). Five hours after IP injection of linTT1-NWs, we observed a widespread signal in the tumor tissue (Fig. 4A) and only a low background in the control organs (Fig. 4B). In contrast, IV injected linTT1-NWs gave a tumor signal (Fig. 4A) that was accompanied by a strong non-specific accumulation of linTT1-NWs in control organs, in particular in the liver (Fig. 4B). Quantitative imaging of the mice dosed IP or IV with linTT1-NWs showed that the FAM signal in the tumors was almost twice as high after IP injection, whereas the signal in control tissues was > 3-fold lower after IP administration (Fig. 4C). Based on these data, the IP administration route was used for subsequent studies.
Fig. 5.
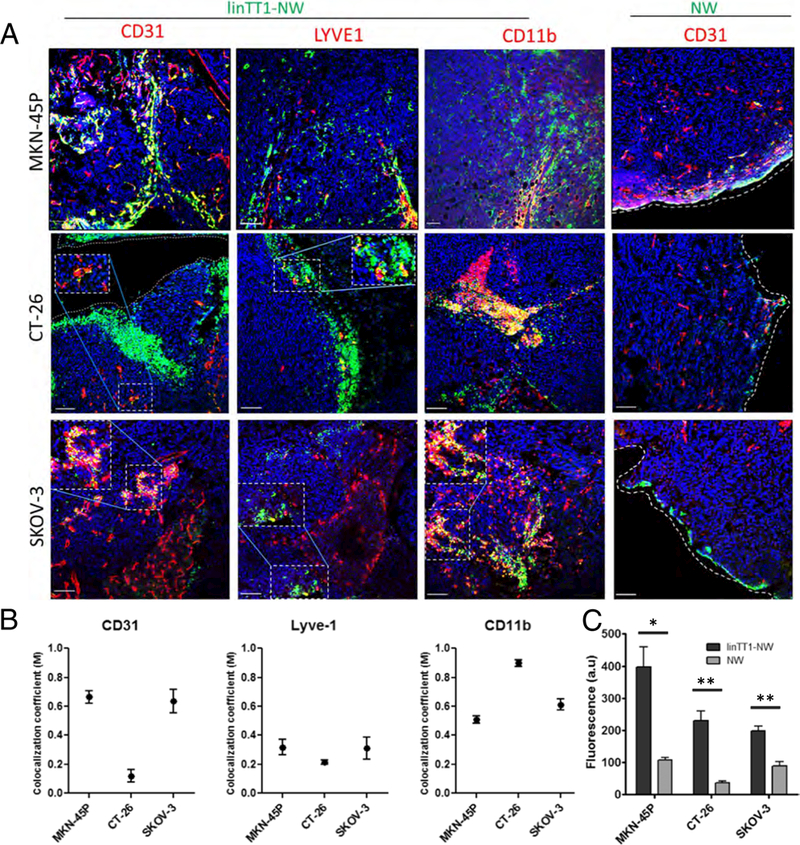
Targeted NW homing in different models of peritoneal carcinomatosis. (A) Confocal Imaging of tumor sections. Mice bearing MKN-45P (upper panel), CT-26 (middle panel) and SKOV-3 (lower panel) tumors were Injected with linTT1-NWs (5 mg iron/kg). Tissues were collected 5 h after IP injection of NWs, and cryosections of tumor tissue were stained with antibodies against CD31 (blood vessels). LYVE-1 (lymphatic vessels) and CD11b (macrophages). Green: NWs; Red: CD31, LYVE-1 or CD11b. Blue: DAPI. Scale bars: 100 μm. (B) Colocalization analysis of linTT1-NWs with CD31, LYVE-1 and CDllb in MKN-45P, CT-26 and SKOV-3 tumor models based on Manders (M) coefficient [39] 0 = no colocalization; 1 = perfect colocalization. Analysis was performed by ImageJ software. Error bars, mean ± SEM (C) Quantification of green fluorescence intensity in the confocal images of tissue sections prepared from MKN-45P, CT-26 and SKOV-3 tumors. Representative fields from multiple sections representing tumors from 3 mice per group are shown. Analysis by Image J, N > 3 mice; Statistical analysis: Student’s t-test; error bars: mean ± SEM; **p < 0.01, *p < 0.05. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 4.
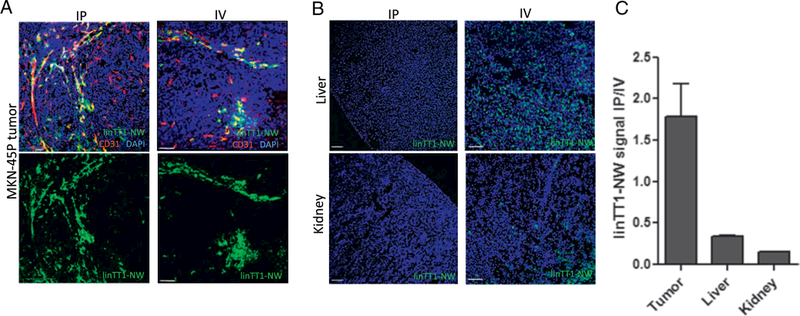
Intraperitoneal linTT1-NWs have improved tumor selectivity over systemically administered NWs. (A) Representative fluorescence confocal images of linTT1-NW at 5 h after IP (first column) or IV injection (second column) demonstrates tumor homing for both routes of administration. Cryosections of tumor tissues were stained with a CD31 antibody to visualize the blood vessels. Green: NWs; Red: CD31, Blue: DAPI. Representative fields from multiple sections (n ≥ 3) prepared from at least 3 tumors are shown. Scale bars: 100 μm. (B) Biodistribution of linTT1-NWs injected IP or IV in non-target organs (liver and kidney) in mice bearing MKN-45P tumors. NWs were injected at a dose of 5 mg/kg, and tissues were collected after 5 h. Blue, DAPI; green: FAM. Scale bars: 100 pm. (C) Quantification of green fluorescence signal in tumor, liver and kidney after IP or IV injection of linTT1-FAM-NWs. Fluorescence signal intensity was quantified by ImageJ software and normalized for tissue area. Representative fields from multiple sections from tumors in three mice are shown. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Cell surface p32 is expressed besides tumor cells also on other activated cells such as vascular and lymphatic endothelial cells and tumor-associated macrophages, especially in hypoxic and nutrient-deprived areas [20,31]. p32 expression level and subcellular localization has also been demonstrated to be affected by the tumor type [20,31]. We next phenotyped the linTT1-NW-positive cells in tumor tissue sections by immunofluorescence staining with a panel of cell type-specific antibodies. In MKN-45P tumors linTT1-NWs showed a colocalization with CD31-positive vascular structures (Fig. 4A and Fig. 5). In addition, overlap was seen with LYVE-1-positive lymphatic vessels and with CD11b-positive macrophages; these cell types were found to be targeted by LyP-1 peptide, the first peptide shown to home to p32 in vivo (Fig. 5) [37,38]. A similar pattern was observed in SKOV-3 ovarian tumor tissue, where linTT1-NWs colocalized with blood vessels and macrophages (Fig. 5), suggesting that a combination of direct penetration from the IP cavity and indirect accumulation via systemic circulation drives tumor accumulation of the linTT1-NWs, as was previously observed for iRGD-coupled polymersomes [16]. In the case of CT-26 tumors, we observed particularly extensive colocalization of linTT1-NWs with CD11b-positive macrophages (Fig. 5).
Iron oxide nanoparticles generate hypointense areas in T2-weighted magnetic resonance imaging (MRI) [40]. Magnetic relaxivity of iron oxide in MRI is further increased with elongated NWs compared to spherical iron oxide nanoparticles [29]. To investigate the potential of linTT1-targeted NWs as a tumor detection and imaging agent, we injected mice bearing MKN-45P tumors IP with linTT1-coated or control NW and subjected the mice to MRI scans 5 h later. The tumors in mice injected with linTT1-NW showed hypointense regions, whereas untargeted NW produced no detectable signal decrease under the same imaging conditions (Fig. 6A and B). Post-MRI ex vivo fluorescence imaging of tumors and control organs confirmed selective accumulation of linTT1-NW in tumor tissue (Fig. S5).
Fig. 6.
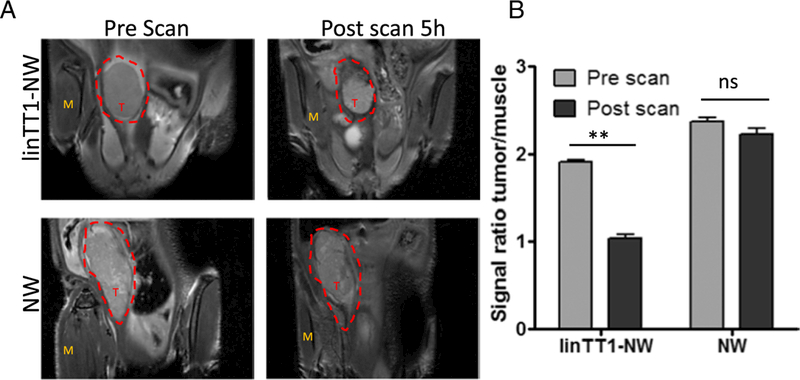
Tumor imaging with linTT1-NWs. T2-weighted magnetic resonance images of mice bearing IP MKN-45P tumors. The mice were injected intraperitoneally with linTT1-coated or control FAM-coated NWs (5 mg/kg of iron). (A) Coronal images of the tumors were acquired using 9.4 T Bruker MR system (TR/TE =1.8 s/ 23 ms; slice thickness 0.5 mm) before NW injection (pre-scan) and 5 h after NW injection (post scan). T, tumor; M, muscle (B) linTT1-NWs produced significant hypointensities in the tumor tissue whereas control NWs gave no signal. The signal intensity was normalized to the adjacent muscle tissue. Five images per time point were analyzed. Statistical analysis: Student’s t-test; error bars: mean +SEM; **p ˂ 0.01.
The linTT1 peptide belongs to the family of tumor penetrating peptides. An important feature of this group of targeting ligands is their ability to cause bystander effect-increased target accumulation of coadministered payloads. Prototypic member of the family, iRGD, increases accumulation and activity of IP coadministered payloads, including anticancer drugs [41–44]. We hypothesized that interaction of the truncated form of the linTT1 with NRP-1 might trigger the transtissue transport pathway for co-administered compounds [22]. To determine if IP linTT1peptide coupled to NW has such activity, we co-administered linTT1-NWs (or iRGD-NWs as a positive control) with 70 kDa fluorescently-labeled dextran, collected the tissues 90 min later, and studied the biodistribution of the dextran in tissue sections. We observed a significant increase in tumor dextran accumulation with linTT1-NWs and iRGD-NWs, whereas only a minimal signal was present when dextran was injected alone (Fig. 7). This accumulation was not due to binding of the free dextran on the peptide-targeted NWs, as the free dextran did not form a complex with the NWs during in vitro incubation (Fig. S7).
Fig. 7.
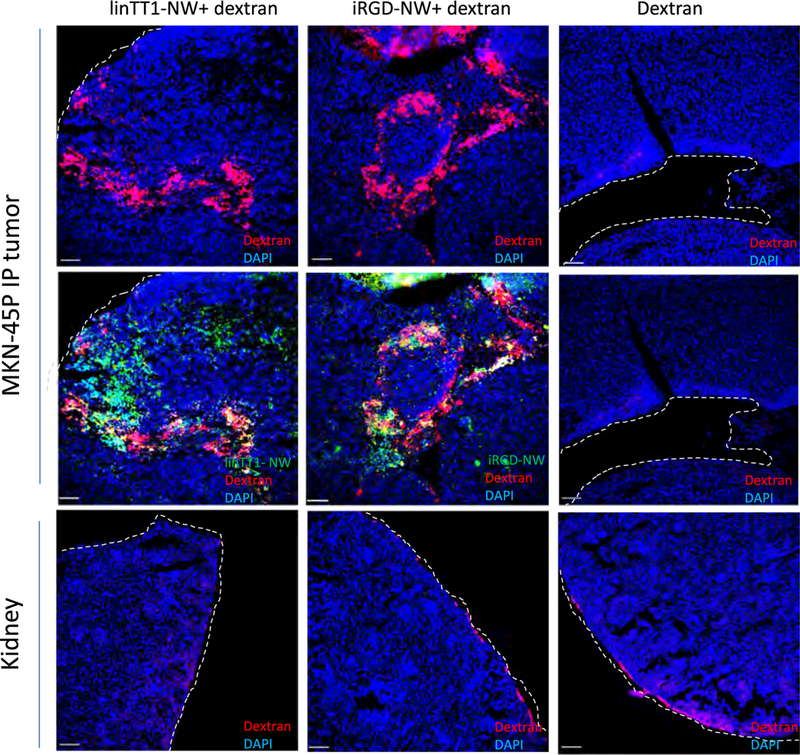
IP linTT1-NWs increase MKN-45P tumor accumulation of coadministered 70 kDa dextran. Mice bearing disseminated MKN-45P tumors were injected IP with the indicated NW formulations (5 mg/kg Fe) and 0.3 mg Lys-Dextran 70 kDa in 1 ml PBS. After 90 min, the mice were perfused and tissues processed for confocal imaging.
To explore the translational relevance of the linTT1-NW platform, we assessed the binding and penetration of targeted NWs on fresh surgical explants of peritoneal human colon cancer. LinTT1-NWs showed binding and higher fluorescence in human colon cancer tissue explants during a short-term culture, whereas control NWs only weakly labeled the surface of the explants (Fig. S6).
These optical and MR imaging data show that IP-administered, linTT1-functionalized NWs accumulate in PC lesions in mouse models of PC and in clinical carcinoma explants.
3.5. IP linTT1-D(KLAKLAK)2-NWs show therapeutic efficacy on mouse models of PC
Accumulation of linTT1-targeted NWs in PC lesions and p32-de- pendent in vitro cytotoxicity of linTT1-D(KLAKLAK)2-NWs in PC cell lines prompted us to evaluate the suitability of linTT1-NW platform for the therapy of peritoneally disseminated MKN-45P and CT-26 tumors in mice.
We used the same treatment regimen previously used in systemic therapy of glioblastomas D(KLAKLAK)2-NWs targeted with different peptides given every other day at a NW dose of 5 mg/kg Fe) [27], but gave the injections IP. In the MKN-45P tumor model, the treatment with linTTl-D(KLAKLAK)2-NWs resulted in a significant decrease in tumor burden (weight of peritoneal malignant tissue) and in significant reduction in the number of tumor nodules (Fig. 8A and B). The control NW treatments did not differ from the vehicle control.
Fig. 8.
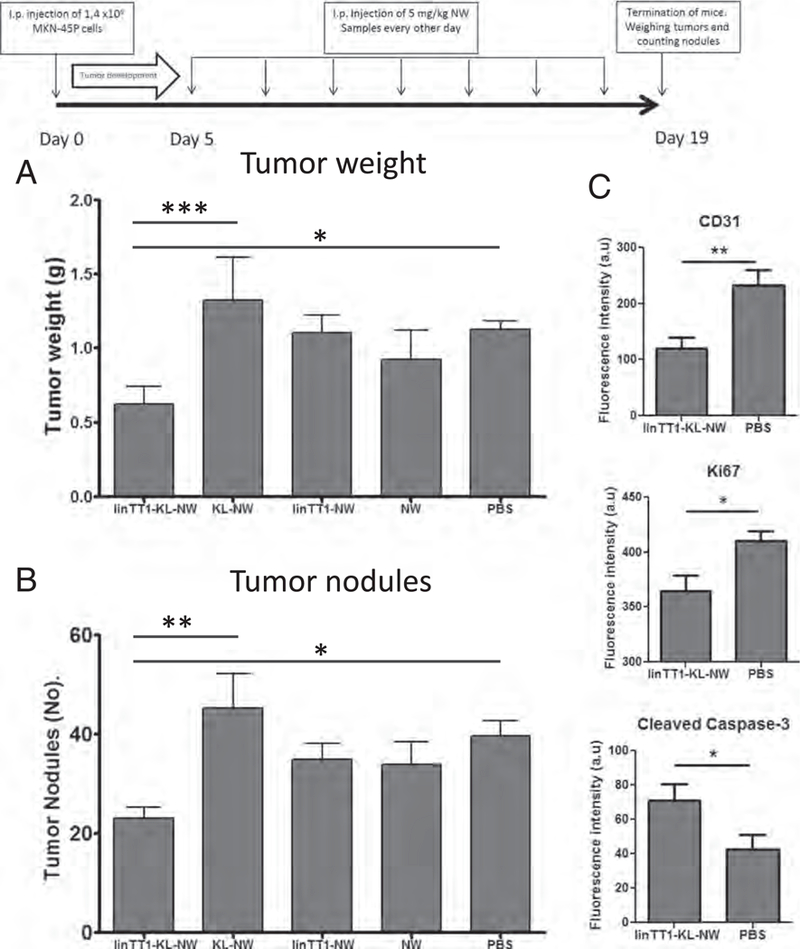
Experimental therapy of mice bearing peritoneal MKN-45P tumors. (A) Mice bearing disseminated IP MKN-45P tumors were injected IP every other day during two weeks with the indicated NW formulations (5 mg/kg Fe). (B) Total tumor weight and (C) number of peritoneal tumor nodules after treatment are shown. KL: D(KLAKLAK)2 apoptotic peptide coupled to NWs. N=8 mice in each group in both panels A and B. Statistical analysis was performed by one-way ANOVA; error bars, mean+ SEM. (D) Quantification of the fluorescence intensity from CD31 (blood vessels), Ki67 (proliferating cells) and cleaved caspase-3 (apoptotic cells) staining. Image J software was used, and 8–10 confocal images were analyzed per group. Statistical analysis: Student's t-test; error bars, mean ± SEM; ***p ˂ 0.001. **p < 0.01, *p < 0.05.
Immunofluorescent staining of post-treatment tumor tissue sections showed a significant decrease in the number of CD31 positive blood vessels in the linTT1-D(KLAKLAK)2-NW group compared with the control group (Fig. 8C and Fig. S8). There were also fewer Ki67-positive proliferating cells in these tumors (Fig. 8C and Fig. S8), and the count of apoptotic caspase-3 positive cells was increased (Fig. 8C). These results suggest that the anti-tumor activity of linTTl-D(KLAKLAK)2-NW is due to a combination of decreased proliferation of cells, enhanced apoptosis, and reduced number of blood vessels in the tumor tissue.
CT-26 syngeneic tumors are highly aggressive, with death ensuing already at 14 days after tumor induction. Consequently, we started experimental therapy on the day 4 after tumor inoculation in this model. On day 13 the experiment was terminated, and the tumor volume, ascites and metastatic nodules were measured. We did not see any significant differences between the treatment groups in total tumor weight, but there was a trend towards reduced tumor burden in linTT1-D(KLAKLAK)2-NW group (Fig. S9A). One of the features of the CT-26 model is the presence of abundant ascitic fluid. The ascites volume was significantly lower in the linTT1-D(KLAKLAK)2-NW group compared to PBS group (Fig. S9B). These studies show that linTT1peptide targeting improves therapeutic efficacy of an IP-administered nanosystem.
4. Discussion
IP therapy for the treatment of PC is a rapidly growing niche that is explored by numerous preclinical and clinical studies. Compared to free drugs, IP-administered nanoformulated cancer drugs may have favorable pharmacokinetic and biodistribution profiles [45], and at least 2 nano-therapeutics, Abraxane™ and Nanotax™ are being clinically evaluated in PC clinical trials [46,47]. Our study was designed to evaluate the effect of functionalizing NPs with the p32-binding linTT1 peptide on the tumor targeting and antitumor efficacies of drug-loaded NPs upon IP administration. Our data indicate that linTT1 functionalization increases tumor selectivity of IP-administered nanoparticles and that this selectivity translates into improved anticancer response to the therapeutic NPs.
The PEGylated iron oxide NWs we used as the scaffold for the targeted nanotherapeutic are a versatile dual-use NPs, optimized for systemic in vivo targeting as a carrier of anticancer agents and as an MRI contrast agent [23,27,29,30]. The high aspect ratio of the NWs is thought to allow more efficient targeting with affinity ligands than is possible with equivalent spherical NPs [29,48]. Moreover, NWs have an increased tissue residence time upon extravasation than spherical NPs.
The linTT1 peptide has been previously validated as a tumor specific peptide. Significantly, the affinity of linTT1 for its target receptor, p32 (aka gC1qR), is lower than that of similar peptides, but it is particularly effective in causing tumor penetration of NPs [18,19]. The reason may be that the avidity effect of the multivalent presentation on NPs does not become so high that it would elicit the so-called “binding- site barrier”, which can hinder tissue penetration of compounds with a high affinity for their receptors [49,50].
The p32 protein has been used as a target molecule in systemic treatment of tumors [19,31,51,52], but this is the first study on PC. We show that a number of cell lines representing tumors that commonly present as PC express p32 at levels suitable for peptide-based tumor targeting. The binding of linTT1-coated NPs to surface p32 of these cells resulted in cellular internalization into p32-expressing PC cell lines in vitro and in tumor accumulation in vivo. Importantly, the internalized NPs were associated with mitochondria, as has been shown previously for a related peptide [28]. This is important because the drug payload we used acts on mitochondria, causing apoptosis. Thus, our targeting system directs the NPs into the tumor and all the way to the appropriate subcellular target.
Our data indicate that for targeting peritoneal tumor lesions, IP administration of linTT1-NWs is superior to the systemic route, with improved tumor/background tissues ratio. Our study demonstrates that non-targeted NWs are taken up in organs that non-specifically capture all NPs (liver and spleen) or excrete NP degradation products (kidney), as has been demonstrated for systemic route [19]. Thus, despite the fact that the NWs are smaller than the openings of lymphatic stomata at the peritoneal barrier (> 500 nm) [53], IP-injected NWs appear to have intraperitoneal residence time sufficient to allow direct IP tumor targeting. The linTT1-coated NWs reached the PC tumor nodules via the blood vessels, but clearly where also able to penetrate into nodules in a circulation independent manner. This was particularly clearly demonstrated by the penetration of linTT1-NWs into human tumor explants ex vivo. Another important implication from this result is that human PC should also be targetable with linTTl. The NW platform used in the current study does not allow quantitative biodistribution analysis of the injected particles. We will use peptide-targeted silver nanoparticle cargo and ICP-MS analysis in the follow-up biodistribution studies.
The statistically significant decrease in the count of tumor nodules we observed in PC mice treated with LinTT1-D(KLAKLAK)2-NWs is important because IP (micro)metastases that remain after de-bulking surgery and adjuvant chemotherapy are a main cause of treatment failure [54–56]. In CT-26 tumor model, which is prone to ascites fluid accumulation, we observed a significant reduction of the volume of ascites following treatment with the NWs. Buildup of malignant ascites involves interference of tumor cells on the peritoneal surface with normal venous and lymphatic drainage and increased vascular permeability. An ability of treatment to reduce ascites formation could be of high clinical importance, as ascites buildup is one of the main causes of PC morbidity [57,58].
Clinical cancers are notoriously heterogenous and it is likely that not all clinical PC cases are susceptible to p32-targeted interventions. MR imaging of linTT1-targeted NW may provide a convenient companion diagnostic tool to establish the sensitivity of the particular clinical case to p32 targeted therapies. Importantly, we also demonstrated that IP linTTl-NWs trigger significant bystander effect and increase tumor retention of coadministered 70 kDa dextran. This is an important observation that suggests that in the context of PC, linTT1-NW may act simultaneously as a drug carrier, imaging agent, and an adjuvant to increase uptake of coadministered drugs and/or imaging agents. The therapeutic potentiation of anticancer drugs by linTT1-NW will be a subject for the follow-up studies.
Our present report on linTT1-mediated targeting of the PC is a sequel to our recently published studies on tumor homing iRGD peptide for the targeting of peritoneal cancer [16,17]. Both iRGD and linTT1 peptides belong to a novel class of tumor-targeting peptides, tumor penetrating peptides, defined by the presence of cryptic R/KXXR/K C- end Rule (CendR) motif (iRGD: CRGDKGPDC, linTT1: AKRGARSTA) that is activated by proteolytic cleavage to enable NRP-1 binding and activation of CendR cell- and tissue penetration pathway [59]. iRGD and linTT1use different primary receptors for initial tumor recruitment: iRGD binds first to angiogenic integrins overexpressed on tumor cells and on angiogenic endothelial cells [60]; linTT1 peptide recognizes first cell surface p32 - a mitochondrial protein aberrantly displayed on the surface of activated tumor cells and cells in tumor stroma (macrophages, lymphatic endothelial cells, endothelial cells) [18]. Thus, linTT1 and iRGD have a differing and likely complementary targeting landscapes and can be potentially used in combination for synergistic targeting of the peritoneal tumors - a testable hypothesis for the follow-up studies.
LinTT1 peptide is a member of a family of p32-targeting ligands identified in our laboratory over the years. LyP-1 peptide, the first peptidic ligand of p32, was identified by in vivo phage display screening for peptides that target lymphatic vessels [38]. LinTT1 peptide was identified using biopanning on purified p32 protein in a cell free system, and was found to have about 10-fold improved affinity compared to LyP-1 [18]. In vitro, linTT1 peptide is cleaved by a tumor- associated protease, urokinase type plasminogen activator (uPA), to convert it from p32-binding peptide to NRP-1 binding CendR peptide (C/AKRGAR↓STC/A, cleavage site indicated by arrow, CendR motif underlined [19]. The relevance of uPA processing for in vivo tissue penetrative targeting of linTT1 peptide is under active investigation. Finally, we have used high-throughput screens of chemical libraries to identify low molecular weight compound leads that target linTT1- binding site on p32 and used the compounds to make nanoparticles that home to p32-positive tumors in vivo [18]. These different p32 ligands are likely to have differing molecular interaction profiles that result in distinct in vivo targeting profiles of the PC lesions.
In summary, functionalization of therapeutic iron oxide NWs with the linTT1 peptide potentiates their tumor selectivity and anti-tumor activity upon IP administration. In a panel of mouse models of PC and on clinical tumor explants linTT1-NW showed efficient tumor accumulation that was not detected with untargeted nanoworms. These observations establish linΤΤ1/ρ32 targeting paradigm for peritoneally disseminated carcinomas and encourage future pre-clinical and clinical studies to validate the system for the treatment of PC.
Supplementary Material
Acknowledgments
We thank Dr. Joji Kitayama for kind gift of MKN-45P cells, Indrek Heinla for help with MRI, Dr. Olav Tammik for colon carcinoma tissue sample, and Rein Laiverik for help with characterization of the NWs.
Funding
This work was supported by the European Union through the European Regional Development Fund (Project No. 2014–2020.4.01.15– 0012), by EMBO Installation grant #2344, European Research Council (281910) starting grant GLIOMADDS from European Regional Development Fund and Wellcome Trust International Fellowship WT095077MA (TT). US National Cancer Institute support was provided by grants CA167174 (to KNS), CA188883 and R44CA183287 (ER), and Cancer Center Support grant CA30199 to the Sanford Burnham Prebys Medical Discovery Institute.
Footnotes
Conflict of interest
T. Teesalu, K.N. Sugahara, and E. Ruoslahti are shareholders and officers of DrugCendR Inc., and E. Ruoslahti is a shareholder of EnduRx Inc. These companies have certain rights to the peptides discussed in the article.
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.jconrel.2017.06.005.
References
- [1].Goodman MD, McPartland S, Detelich D, Saif MW, Chemotherapy for in- traperitoneal use: a review of hyperthermic intraperitoneal chemotherapy and early post-operative intraperitoneal chemotherapy, J. Gastrointest. Oncol. 7 (2016) 45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Coccolini F, Gheza F, Lotti M, Virzi S, Iusco D, Ghermandi C, Melotti R, Baiocchi G, Giulini SM, Ansaloni L, Catena F, Peritoneal carcinomatosis, World J. Gastroenterol. 19 (2013) 6979–6994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Jacquet P, Sugarbaker PH, Peritoneal-plasma barrier, Cancer Treat. Res. 82 (1996) 53–63. [DOI] [PubMed] [Google Scholar]
- [4].Kitayama J, Intraperitoneal chemotherapy against peritoneal carcinomatosis: current status and future perspective, Surg. Oncol. 23 (2014) 99–106. [DOI] [PubMed] [Google Scholar]
- [5].Sugarbaker PH, Treatment of peritoneal carcinomatosis from colon or appendiceal cancer with induction intraperitoneal chemotherapy, Cancer Treat. Res. 82 (1996) 317–325. [DOI] [PubMed] [Google Scholar]
- [6].Bakrin N, Classe JM, Pomel C, Gouy S, Chene G, Glehen O, Hyperthermic intraperitoneal chemotherapy (HIPEC) in ovarian cancer, J. Vise Surg. 151 (2014) 347–353. [DOI] [PubMed] [Google Scholar]
- [7].Armstrong DK, Bundy B, Wenzel L, Huang HQ, Baergen R, Lele S, Copeland LJ, Walker JL, Burger RA, Gynecologic Oncology Group, Intraperitoneal cisplatin and paclitaxel in ovarian cancer, N. Engl. J. Med. 354 (2006) 34–43. [DOI] [PubMed] [Google Scholar]
- [8].Ceelen WP, Flessner MF, Intraperitoneal therapy for peritoneal tumors: biophysics and clinical evidence, Nat. Rev. Clin. Oncol. 7 (2010) 108–115. [DOI] [PubMed] [Google Scholar]
- [9].Baumgartner JM, Kwong TG, Ma GL, Messer K, Kelly KJ, Lowy AM, A novel tool for predicting major complications after cytoreductive surgery with hyperthermic intraperitoneal chemotherapy, Ann. Surg. Oncol. 23 (2016) 1609–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Simon-Gracia L, Hunt H, Scodeller PD, Gaitzsch J, Braun GB, Willmore AA, Ruoslahti E, Battaglia G, Teesalu T, Paclitaxel-loaded polymersomes for enhanced intraperitoneal chemotherapy, Mol. Cancer Ther. (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Van Oudheusden TR, Grull H, Dankers PY, De Hingh IH, Targeting the peritoneum with novel drug delivery systems in peritoneal carcinomatosis: a review of the literature, Anticancer Res. 35 (2015) 627–634. [PubMed] [Google Scholar]
- [12].Ruoslahti E, Peptides as targeting elements and tissue penetration devices for nanoparticles, Adv. Mater. (Deerfield Beach, Fla.) 24 (2012) 3747–3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ruoslahti E, Bhatia S, Sailor M, Targeting of drugs and nanoparticles to tumors, J. Cell Biol. 188 (2010) 759–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ruoslahti E, Tumor penetrating peptides for improved drug delivery, Adv. Drug Deliv. Rev. (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Teesalu T, Sugahara K, Ruoslahti E, Mapping of vascular ZIP codes by phage display, Methods Enzymol. 503 (2012) 35–56. [DOI] [PubMed] [Google Scholar]
- [16].Simon-Gracia L, Hunt H, Scodeller P, Gaitzsch J, Kotamraju VR, Sugahara KN, Tammik O, Ruoslahti E, Battaglia G, Teesalu T, iRGD peptide conjugation potentiates intraperitoneal tumor delivery of paclitaxel with polymersomes, Biomaterials 104 (2016) 247–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sugahara KN, Scodeller P, Braun GB, de Mendoza TH, Yamazaki CM, Kluger MD, Kitayama J, Alvarez E, Howell SB, Teesalu T, Ruoslahti E, Lowy AM, A tumor-penetrating peptide enhances circulation-independent targeting of peritoneal carcinomatosis, J. Control. Release 212 (2015) 59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Paasonen L, Sharma S, Braun GB, Kotamraju VR, Chung TD, She ZG, Sugahara KN, Yliperttula M, Wu B, Pellecchia M, Ruoslahti E, Teesalu T, New p32/Gc1qR ligands for targeted tumor drug delivery, Chembiochem 17 (2016) 570–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sharma S, Kotamraju VR, Molder T, Tobi A, Teesalu T, Ruoslahti E, Tumor- penetrating nanosystem strongly suppresses breast tumor growth, Nano Lett. 17 (2017) 1356–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Fogal V, Zhang L, Krajewski S, Ruoslahti E, Mitochondrial/cell-surface protein p32/gClqR as a molecular target in tumor cells and tumor stroma, Cancer Res. 68 (2008) 7210–7218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Teesalu T, Sugahara K, Kotamraju V, Ruoslahti E, C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration, Proc. Natl. Acad. Sci. U. S. A. 106 (2009) 16157–16162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sugahara K, Teesalu T, Karmali P, Kotamraju V, Agemy L, Greenwald D, Ruoslahti E, Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs, Science (New York, N.Y.) 328 (2010) 1031–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Simberg D, Duza T, Park J, Essler M, Pilch J, Zhang L, Derfus A, Yang M, Hoffman R, Bhatia S, Sailor M, Ruoslahti E, Biomimetic amplification of nanoparticle homing to tumors, Proc. Natl. Acad. Sci. U. S. A. 104 (2007) 932–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Simberg D, Park JH, Karmali PP, Zhang WM, Merkulov S, McCrae K, Bhatia SN, Sailor M, Ruoslahti E, Differential proteomics analysis of the surface heterogeneity of dextran iron oxide nanoparticles and the implications for their in vivo clearance, Biomaterials 30 (2009) 3926–3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Inturi S, Wang G, Chen F, Banda NK, Holers VM, Wu L, Moghimi SM, Simberg D, Modulatory role of surface coating of superparamagnetic iron oxide nanoworms in complement opsonization and leukocyte uptake, ACS Nano 9 (2015) 10758–10768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Alberici L, Roth L, Sugahara K, Agemy L, Kotamraju V, Teesalu T, Bordignon C, Traversari C, Rizzardi G, Ruoslahti E, De novo design of a tumor-penetrating peptide, Cancer Res. 73 (2013) 804–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Agemy L, Sugahara K, Kotamraju V, Gujraty K, Girard O, Kono Y, Mattrey R, Park J, Sailor M, Jimenez A, Cativiela C, Zanuy D, Sayago F, Aleman C, Nussinov R, Ruoslahti E, Nanoparticle-induced vascular blockade in human prostate cancer, Blood 116 (2010) 2847–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Agemy L, Friedmann-Morvinski D, Kotamraju V, Roth L, Sugahara K, Girard O, Mattrey R, Verma I, Ruoslahti E, Targeted nanoparticle enhanced proapoptotic peptide as potential therapy for glioblastoma, Proc. Natl. Acad. Sci. U. S. A. 108 (2011) 17450–17455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Park JH, von Maltzahn G, Zhang L, Schwartz MP, Ruoslahti E, Bhatia SN, Sailor MJ, Magnetic iron oxide nanoworms for tumor targeting and imaging, Adv. Mater. (Deerfield Beach, Fla.) 20 (2008) 1630–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Park JH, von Maltzahn G, Zhang L, Derfus AM, Simberg D, Harris TJ, Ruoslahti D, Bhatia SN, Sailor MJ, Systematic surface engineering of magnetic nanoworms for in vivo tumor targeting, Small 5 (2009) 694–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Agemy L, Kotamraju VR, Friedmann-Morvinski D, Sharma S, Sugahara KN, Ruoslahti E, Proapoptotic peptide-mediated cancer therapy targeted to cell surface p32, Mol. Ther. 21 (2013) 2195–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Koga A, Aoyagi K, Imaizumi T, Miyagi M, Shirouzu K, Comparison between the gastric cancer cell line MKN-45 and the high-potential peritoneal dissemination gastric cancer cell line MKN-45P, Kurume Med. J. 58 (2011) 73–79. [DOI] [PubMed] [Google Scholar]
- [33].van Tilborg GA, Cormode DP, Jarzyna PA, van der Toorn A, van der Pol SM, van Bloois L, Fayad ZA, Storm G, Mulder WJ, de Vries HE, Dijkhuizen RM, Nanoclusters of iron oxide: effect of core composition on structure, biocompatibility, and cell labeling efficacy, Bioconjug. Chem. 23 (2012) 941–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ellerby HM, Arap W, Ellerby LM, Kain R, Andrusiak R, Rio GD, Krajewski S, Lombardo CR, Rao R, Ruoslahti E, Bredesen DE, Pasqualini R, Anti-cancer activity of targeted pro-apoptotic peptides, Nat. Med. 5 (1999) 1032–1038. [DOI] [PubMed] [Google Scholar]
- [35].Pang HB, Braun GB, Friman T, Aza-Blanc P, Ruidiaz ME, Sugahara KN, Teesalu T, Ruoslahti E, An endocytosis pathway initiated through neuropilin-1 and regulated by nutrient availability, Nat. Commun. 5 (2014) 4904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Das A, Nag S, Mason AB, Barroso MM, Endosome-mitochondria interactions are modulated by iron release from transferrin, J. Cell Biol. 214 (2016) 831–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Fogal V, Richardson A, Karmali P, Scheffler I, Smith J, Ruoslahti E, Mitochondrial p32 protein is a critical regulator of tumor metabolism via main- tenance of oxidative phosphorylation, Mol. Cell. Biol. 30 (2010) 1303–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Laakkonen P, Porkka K, Hoffman J, Ruoslahti E, A tumor-homing peptide with a targeting specificity related to lymphatic vessels, Nat. Med. 8 (2002) 751–755. [DOI] [PubMed] [Google Scholar]
- [39].Manders EM, Verbeek F, Aten J, Measurement of co-localization of objects in dual-colour confocal images, J. Microsc. 169 (1993) 375–382. [DOI] [PubMed] [Google Scholar]
- [40].McAteer MA, Sibson NR, von Zur Muhlen C, Schneider JE, Lowe AS, Warrick N, Channon KM, Anthony DC, Choudhury RP, In vivo magnetic resonance imaging of acute brain inflammation using microparticles of iron oxide, Nat. Med. 13 (2007) 1253–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Schmithals C, Koberle V, Korkusuz H, Pleli T, Kakoschky B, Augusto EA, Ibrahim AA, Arencibia JM, Vafaizadeh V, Groner B, Korf HW, Kronenberger B, Zeuzem S, Vogl TJ, Waidmann O, Piiper A, Improving drug penetrability with iRGD leverages the therapeutic response to sorafenib and doxorubicin in hepatocellular carcinoma, Cancer Res. 75 (2015) 3147–3154. [DOI] [PubMed] [Google Scholar]
- [42].Deng C, Zhang Q, Fu Y, Sun X, Gong T, Zhang Z, Coadministration of oligomeric hyaluronic acid-modified liposomes with tumor-penetrating peptide-iRGD enhances the antitumor efficacy of doxorubicin against melanoma, ACS Appl. Mater. Interfaces (2017). [DOI] [PubMed] [Google Scholar]
- [43].Deng C, Jia M, Wei G, Tan T, Fu Y, Gao H, Sun X, Zhang Q, Gong T, Zhang Z, Inducing optimal antitumor immune response through coadministering iRGD with pirarubicin loaded nanostructured lipid carriers for breast cancer therapy, Mol. Pharm. 14 (2017) 296–309. [DOI] [PubMed] [Google Scholar]
- [44].Akashi Y, Oda T, Ohara Y, Miyamoto R, Kurokawa T, Hashimoto S, Enomoto T, Yamada K, Satake M, Ohkohchi N, Anticancer effects of gemcitabine are enhanced by co-ad ministered iRGD peptide in murine pancreatic cancer models that over- expressed neuropilin-1, Br. J. Cancer 110 (2014) 1481–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Dakwar GR, Shariati M, Willaert W, Ceelen W, De Smedt SC, Remaut K, Nanomedicine-based intraperitoneal therapy for the treatment of peritoneal carcinomatosis - mission possible? Adv. Drug Deliv. Rev. (2016). [DOI] [PubMed] [Google Scholar]
- [46].Williamson SK, Johnson GA, Maulhardt HA, Moore KM, McMeekin DS, Schulz TK, Reed GA, Roby KF, Mackay CB, Smith HJ, Weir SJ, Wick JA, Markman M, diZerega GS, Baltezor MJ, Espinosa J, Decedue CJ, A phase I study of intraperitoneal nanoparticulate paclitaxel (Nanotax(R)) in patients with peritoneal malignancies, Cancer Chemother. Pharmacol. 75 (2015) 1075–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Cristea MC, Synold TW, Frankel PH, Rivkin SE, Pharmacologic advantage (PA) of intraperitoneal (IP) nab-paclitaxel in patients with advanced malignancies primarily confined to the peritoneal cavity. http://meetinglibrary.asco.org/content/152193-156, (2015) (accessed 11.01.17).
- [48].Wang G, Inturi S, Serkova NJ, Merkulov S, McCrae K, Russek SE, Banda NK, Simberg D, High-relaxivity superparamagnetic iron oxide nanoworms with de¬creased immune recognition and long-circulating properties, ACS Nano 8 (2014) 12437–12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].van Osdol W, Fujimori K, Weinstein JN, An analysis of monoclonal antibody distribution in microscopic tumor nodules: consequences of a “binding site barrier”, Cancer Res. 51 (1991) 4776–4784. [PubMed] [Google Scholar]
- [50].Thurber GM, Schmidt MM, Wittrup KD, Antibody tumor penetration: transport opposed by systemic and antigen-mediated clearance, Adv. Drug Deliv. Rev. 60 (2008) 1421–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Park JH, von Maltzahn G, Xu MJ, Fogal V, Kotamraju VR, Ruoslahti E, Bhatia SN, Sailor MJ, Cooperative nanomaterial system to sensitize, target, and treat tumors, Proc. Natl. Acad. Sci. U. S. A. 107 (2010) 981–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Karmali P, Kotamraju V, Kastantin M, Black M, Missirlis D, Tirrell M,Ruoslahti E, Targeting of albumin-embedded paclitaxel nanoparticles to tumors, Nanomedicine 5 (2009) 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Tsai M, Lu Z, Wang J, Yeh TK, Wientjes MG, Au JL, Effects of carrier on disposition and antitumor activity of intraperitoneal paclitaxel, Pharm. Res. 24 (2007) 1691–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Sugarbaker PH, Update on the prevention of local recurrence and peritoneal me- tastases in patients with colorectal cancer, World J. Gastroenterol. 20 (2014) 9286–9291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Sugarbaker PH, Improving oncologic outcomes for colorectal cancer at high risk for local-regional recurrence with novel surgical techniques, Expert Rev. Gastroenterol. Hepatol. 10 (2016) 205–213. [DOI] [PubMed] [Google Scholar]
- [56].Braam GJ, van Oudheusden TR, de Hingh IH, Nienhuijs SW, Boerma D, Wiezer MJ, van Ramshorst B, Patterns of recurrence following complete cytoreductive surgery and hyperthermic intraperitoneal chemotherapy in patients with peritoneal carcinomatosis of colorectal cancer, J. Surg. Oncol. 109 (2014) 841–847. [DOI] [PubMed] [Google Scholar]
- [57].Cavazzoni E, Bugiantella W, Graziosi L, Franceschini MS, Donini A, Malignant ascites: pathophysiology and treatment, Int. J. Clin. Oncol. 18 (2013) 1–9. [DOI] [PubMed] [Google Scholar]
- [58].Randle RW, Swett KR, Swords DS, Shen P, Stewart JH, Levine EA, Votanopoulos KI, Efficacy of cytoreductive surgery with hyperthermic intraperitoneal chemotherapy in the management of malignant ascites, Ann. Surg. Oncol. 21 (2014) 1474–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Teesalu T, Sugahara KN, Ruoslahti E, Tumor-penetrating peptides, Front. Oncol. 3 (2013) 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Sugahara K, Teesalu T, Karmali P, Kotamraju V, Agemy L, Girard O, Hanahan D, Mattrey R, Ruoslahti E, Tissue-penetrating delivery of compounds and nanoparticles into tumors, Cancer Cell 16 (2009) 510–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.