

Abstract
In this paper, we trace the history of current research into the genetic and biochemical mechanisms that underlie folate-preventable neural tube defects (NTDs). The inspired suggestion by Smithells that common vitamins might prevent NTDs ignited a decade of biochemical investigations—first exploring the nutritional and metabolic factors related to NTDs, then onto the hunt for NTD genes. Although NTDs were known to have a strong genetic component, the concept of common genetic variance being linked to disease risk was relatively novel in 1995, when the first folate-related polymorphism associated with NTDs was discovered. The realization that more genes must be involved started a rush to find polymorphic needles in genetic haystacks. Early efforts entailed the intellectually challenging and time-consuming task of identifying and analyzing candidate single nucleotide polymorphisms (SNPs) in folate pathway genes. Luckily, human genome research has developed rapidly, and the search for the genetic factors that contribute to the etiology of human NTDs has evolved to mirror the increased level of knowledge and data available on the human genome. Large-scale candidate gene analysis and genome-wide association studies are now readily available. With the technical hurdles removed, the remaining challenge is to gather a sample large enough to uncover the polymorphisms that contribute to NTD risk. In some respects the real work is beginning. Although moving forward is exciting, it is humbling that the most important result—prevention of NTDs by maternal folic acid supplementation—was achieved years ago, the direct result of Smithells’ groundbreaking studies.
Keywords: MTHFR, folic acid, vitamin B12, homocysteine, candidate genes
Smithells’ Journey to Neural Tube Defect Prevention: A Brief Account
The role of nutrition in the etiology of neural tube defects (NTDs) has been appreciated since the 1960s, when Hibbard and Smithells (1965) suggested a possible link between folate deficiency and NTDs. Smithells et al. (1976) collected blood samples during the first trimester of pregnancy from 900 women in Leeds. In six of these mothers who gave birth to infants with central nervous system (CNS) defects (five with NTDs and one with microcephaly), red cell folate and white blood cell vitamin C levels were significantly lower than in controls. These findings, although based on a small number of CNS defect cases, led the authors to state that they would test the hypothesis that preconceptional vitamin supplementation would prevent CNS defects (Smithells et al., 1976).
The resulting intervention study was pivotal in the story of folic acid and NTDs. Women with a previous history of an NTD birth were recruited into a non-randomized controlled clinical trial of periconceptional multivitamin supplementation. The preliminary results published in 1980 showed that 1 of 178 (0.6%) infants/fetuses of supplemented mothers had an NTD compared to 13 of 260 (5.0%) infants/fetuses of unsupplemented mothers (Smithells et al., 1980). This striking result caused great excitement and led to a lengthy and heated debate in the Lancet and elsewhere on the interpretation and implications of the findings, specifically on the need for a more scientifically rigorous testing of the hypothesis in a randomized controlled trial. These findings were confirmed in further studies by the Smithells team (Smithells et al., 1983, 1989). His work had a profound influence on future research into the etiology and prevention of NTDs and led to the seminal intervention trials that eventually established unequivocally the role of folate (folic acid) in the etiology and prevention of NTDs.
Although not explicitly part of their work, the familial aspect of NTDs was implicit in these early studies. Smithells and his team focused on high-risk families (i.e., those having a previous child with an NTD), because the risk of having a second child with an NTD is roughly 20-fold higher than the population risk. Such familial aggregation suggested that inherited factors played a strong role in the etiology of NTDs. Many years after Smithells’ work, when the debate over the value of folic acid for the prevention of NTDs had been finally settled in Smithells’ favor, advances in genetic technologies allowed scientists to search for genetic polymorphisms associated with NTDs. First tested were genes encoding key proteins in the folate/homocysteine metabolic cycle.
Trials and Afterward
Evidence that folic acid could prevent NTDs came from two main types of studies: (1) observational studies of dietary folate intake and of supplementation with folic acid and (2) interventional studies. The main observational studies examining the effect of periconceptional use of vitamin supplements containing folic acid found, with one exception (Mills et al., 1989), statistically significant protective effects in the range of 35 to 71% (odds ratios [OR], 0.29–0.65) against NTD occurrence (Mulinare et al., 1988; Milunsky et al., 1989; Werler et al., 1993; Shaw et al., 1995). High intakes of dietary folate during the periconceptional period were also shown to be protective (Bower and Stanley, 1989). The nonrandomized controlled trial by Smithells et al. (1980) was the first intervention study to test the efficacy of a multivitamin supplement containing folic acid in preventing pregnancies affected by NTDs, and its key impact has already been described. Later intervention studies included both randomized and nonrandomized controlled trials (Laurence et al., 1981; Medical Research Council, 1991; Czeizel and Dudas, 1992; Kirke et al., 1992), with the strongest evidence for the protective effect of folic acid coming from two randomized controlled trials; the Medical Research Council (MRC) trial on NTD recurrence (Medical Research Council, 1991) and the Hungarian trial on occurrence (Czeizel and Dudas, 1992).
The Czeizel and Dudas (1992) trial compared multivitamins that included 0.8 mg of folic acid versus trace elements that were a virtual placebo in women who did not have a history of prior pregnancies with NTDs. The group that received the multivitamin containing folic acid had no NTD-affected offspring in 2104 pregnancies. The trace element group, in contrast, had six NTD-affected offspring in 2052 pregnancies (p = 0.03). This study answered a key question: could multivitamins containing folic acid prevent NTDs in the general population? However, the study did not show that folic acid was the active agent. The MRC trial randomized women who had a prior affected conceptus to receive folic acid (4 mg), multivitamins, both, or neither (MRC, 1991). The analysis demonstrated that folic acid was highly protective (relative risk [RR], 0.28 [95% confidence interval {CI}, 0.12–0.71]), whereas the other vitamins did not show a significant protective effect (RR, 0.80, [95% CI, 0.32–1.72]). Finally, unequivocal support for the efficacy of a lower dose of folic acid (0.4 mg daily) was obtained from a large nonrandomized intervention study, conducted in two regions of China (Berry et al., 1999). This study also found that the protective effect of folic acid was more marked in the region with a high prevalence at birth of NTD than in the region with lower prevalence.
Several points about the two randomized controlled trials were not appreciated at the time. In the Hungarian study, but not the MRC study, vitamin B12 was present in the vitamin tablets. Thus, a portion of the dramatic protective effect found in that study could have been due to a synergistic effect of vitamin B12 and folic acid. It should be noted that NTD rates in their study population were in the range of 2 per 1000, so that their finding of no NTDs in over two thousand pregnancies showed a greater protective effect than would have been expected, even assuming a 50 to 70% protective effect for folic acid alone. In contrast, the lack of a significant protective effect in the MRC trial group consuming vitamins without folic acid could have been due to the absence of vitamin B12 in the vitamin arm of the study. Considering what we know now, it is also worth mentioning that both the Hungarian and the MRC trials included vitamins B2 and B6 in their supplemented groups.
The Perspective from Ireland: A High-Risk Country
The work of the Smithells’ research team was of special interest in Ireland, where the prevalence at birth of NTDs has traditionally been high. His belief in the protective effect of folate was supported in a randomized intervention trial of NTD recurrence among previously affected Irish mothers, although the trial ended prematurely when the results of the MRC trial were published (Kirke et al., 1992). In 1980, Smithells delivered a lecture on folate and NTDs in Dublin. His observation of significantly lower maternal red cell folate levels in early pregnancy in mothers of NTD cases prompted the suggestion of B12 involvement and led to a collaborative publication, showing that mothers of anencephalic infants had low B12 status (Schorah, et al., 1980). This work and other studies demonstrating lower blood folate levels in mothers of NTD-affected infants compared with mothers of unaffected infants (Smithells et al., 1976; Yates et al., 1987) provided the motivation to study folate biochemist try among Irish families with children affected by NTDs. No case-control differences could be found in serum folate or vitamin B12 in a small study of 32 mothers during an NTD-affected pregnancy and 395 control mothers (Molloy et al., 1985), using blood samples obtained as part of the Irish national rubella screening program. Between 1986 and 1990 we collected 56,049 blood samples from women attending their first antenatal clinic visit in the three major Dublin maternity hospitals at an estimated gestational age of 15 weeks. This large sample bank provided the material for a nested case-control study of 81 women who were carrying NTD-affected fetuses during their pregnancy and 247 women carrying normal fetuses. The study showed that both plasma folate and B12 levels were significantly lower in women who were carrying affected fetuses than in controls (Kirke et al., 1993). Moreover, the B12 and folate effects appeared to be independent, and the data demonstrated the highest risk in women in whom both vitamins were in the lowest quartile. Further analysis showed that the risk of having an NTD-affected child was shown to be strongly related to the mother’s red cell folate level (Daly et al., 1995); decreasing from 6.6 per 1000 births in women whose red cell folates were below 150 ng/ml (340 nmol/l) to 0.8 per 1000 births in women whose red cell folates were over 399 ng/ml (906 nmol/l). One of the most important outcomes was that the study showed that the problem was not just a matter of deficiency; the risk remained elevated until the maternal red cell folate levels were well above the deficiency range. Analysis of blood folate levels in 27 women who were enrolled in the MRC trial and had an NTD affected child were consistent with these results (Wald et al., 1996). All of these biochemical studies pointed to a subtle alteration in folate homeostasis in families with NTDs and supported the hypothesis that specific genetic variants would be associated with interindividual differences in the efficiency of handling metabolites that were directly or indirectly associated with folate pathways, consistent with Garrod’s model of biochemical individuality (Garrod, 1902).
Homocysteine and Neural Tube Defects
During the same time, the role of homocysteine in the etiology of NTDs was suggested by Steegers-Theunissen in a letter to the New England Journal of Medicine (Steegers-Theunissen et al., 1991). Her group performed a methionine loading test in mothers who previously had affected infants and control subjects. The mothers of the affected children tended to have higher postload total plasma homocysteine, suggesting that they were less able to metabolize homocysteine. The same group later found higher amniotic fluid total homocysteine in NTD case mothers than in controls (Steegers-Theunissen et al., 1995). Mills et al. (1995) studied homocysteine metabolism directly in pregnant mothers who were carrying affected fetuses at the time of investigation with control mothers carrying unaffected fetuses and found significantly higher homocysteine levels in case mothers. The homocysteine findings were precisely what would be expected if functional differences were not restricted to folate. The involvement of vitamin B12 as well as folate reinforced the idea that folic acid prophylaxis was overcoming a metabolic block directly or indirectly related tosome aspect of one-carbon metabolism. It also suggested a candidate gene in which to look for genetic variants. There are only two enzymes in humans that require vitamin B12 (cobalamin). Only one of these, methionine synthase, involves both vitamin B12 and folate. Through this enzyme, vitamin B12 and folate closely interact in the two major metabolic cycles that deal with the intracellular management of one-carbon units (Fig. 1). The methionine synthase axis is central to both the methylation and DNA synthesis aspects of one-carbon metabolism, because folate enters the cell as 5-methyltetrahydrofolate (5-methylTHF) and must release its methyl group through the methionine synthase reaction to be to be retained in the cell. As free tetrahydrofolate, it can then be polyglutamated and can accept one-carbon units from serine, formate, and other sources for use in nucleotide synthesis or generation of 5-methylTHF. Vitamin B12 deficiency, in effect, causes an intracellular functional folate deficiency, and an inadequate (even if not frankly deficient) B12 status might result in an imbalance in the flux of folate-derived one-carbon units through the DNA synthesis and methylation cycles. The finding of independent folate and B12 effects by Kirke et al. (1993), now confirmed in other cohorts in Ireland (Molloy et al., in press) and in several cohorts elsewhere (Suarez et al., 2003; Ray and Blom 2003; Ray et al., 2007), along with higher homocysteine levels in mothers carrying affected fetuses, strongly suggested that the conversion of homocysteine to methionine was a key reaction and that methionine synthase might be involved directly or indirectly in the etiology of NTDs. As discussed in Table 1, the genes encoding methionine synthase or methionine synthase reductase proved not to be the key factors, but the gene encoding the precursor enzyme in the folate pathway, 5,10 methylenetetrahydrofolate reductase (MTHFR), was already making news by 1995.
Figure 1.
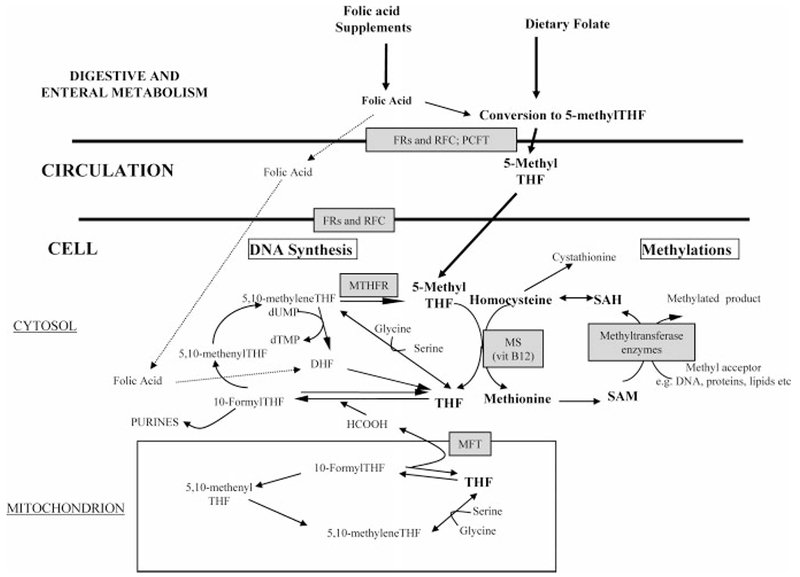
Pathways of one-carbon metabolism. Folate cofactors are used in the distribution of one-carbon units across two distinct metabolic cycles, one relating to the de novo synthesis of DNA and the other involved in providing methyl groups for at least 40 different methyltransferase reactions. In the DNA synthesis cycle, formyl derivatives of tetrahydrofolate (THF) are used in de novo purine biosynthesis. In de novo pyrimidine biosynthesis, 5,10-methyleneTHF is required in the conversion of deoxyuridine monophosphate (dUMP) to deoxythymidine monophosphate (dTMP). Thus folate cofactors play an essential role in cellular proliferation. The THF cofactor pool also provides a continuous supply of methyl groups for biologic methylation reactions via the conversion of homocysteine to methionine, using 5-methylTHF as the methyl donor, with vitamin B12-dependent methionine synthase (MS) transferring the methyl group to homocysteine. (In liver and kidney homocysteine is also remethylated to methionine through an alternative folate-independent pathway involving betaine homocysteine methyltransferase.) Methionine is then converted to S-adenosylmethionine (SAM), the cosubstrate of all methyltransferase enzymes. This cycle is completed by the regeneration of homocysteine from S-adenosylhomocysteine (SAH), the coproduct of the methylation process. Methylenetetrahydrofolate reductase (MTHFR) catalyzes the irreversible conversion of 5,10-methyleneTHF to 5-methylTHF, thereby committing one-carbon units to methylation reactions and away from DNA synthesis. Mitochondrial and cytosolic folate-linked metabolism of serine and formate generate the majority of one-carbon groups for all of these processes. In addition to all of the enzymes involved in folate pathways, the reduced folate carrier (RFC), folate receptors (FRs), proton coupled folate transporter (PCFT), and mitochondrial folate transporter (MFT) play key roles in maintaining folate concentrations within the cell.
Table 1.
Most Commonly Studied Polymorphisms in Folate and Vitamin B12 Genes in Relation to Neural Tube Defects in Humans
| Gene | Enzyme | Association with NTDs | Reference |
|---|---|---|---|
| MTHFR | 5,10-Methylene tetrahydrofolate reductase | >30 association studies worldwide; significant risk factor for NTDs in some populations; suggestion of stronger association among non-Hispanic whites | van der Put et al., 1995; Whitehead et al., 1995; Shields et al., 1999 |
| 677C→T | A 222V | Important cause of low folate status | Botto and Yang, 2000; Kirke et al., 2004; Amorim et al., 2007 |
| MTHFR 1298A→C | As above, E429A | In linkage with 677C→T; no demonstrated risk associations that are independent of 677C→T | van der Put et al., 1998; Stegmann et al., 1999; Barber et al., 2000; Botto and Yang, 2000; De Marco et al., 2002; Parle-McDermott et al., 2003a; Relton et al., 2004a |
| MTR 2756A →G | Methionine synthase D919G | No independent association; may interact with other genes as a maternal risk factor | Brody et al., 1999; Christensen et al., 1999; Johanning et al., 2000; Doolin et al., 2002; Zhu et al., 2003 |
| MTRR 66A→G | Methionine synthase reductase I22M | Several studies show case or maternal risk associations and possible interactive effects with low B12 or other genes | Wilson et al., 1999; Zhu et al., 2003; Relton et al., 2004b; O’Leary et al., 2005b; van der Linden et al., 2006; Candito et al 2008 |
| MTHFD1 1958G→A | Trifunctional C1 synthase R653Q | Maternal risk factor for neural tube defects; no reported change in folate status | Brody et al., 2002; De Marco et al., 2006; Parle-McDermott et al., 2006 |
| SHMT1 1420C→T | Serine hydroxymethyl-transferase L474F | No reported risk association with NTD | Heil et al., 2001; Relton et al., 2004a; Relton et al., 2004b |
| RFC1 80G→A | Reduced folate carrier H27N | Some studies report increased risk of NTD but larger studies are negative; possible interaction with maternal nutrient intake | Shaw et al., 2002; De Marco et al., 2003; Morin et al., 2003a; Vieira et al., 2005; O’Leary et al., 2006; Shang et al., 2008 |
| FRα, FRβ, FRγ Several SNPs | Folate receptors | No reported risk associations with NTD or biochemical changes | Barber et al., 1998; Heil et al., 1999; Trembath et al., 1999; O’Leary et al., 2003 |
| GCPII 1561C →T | Folyl-γ-glutamate carboxypeptidase H475Y | One report of significant maternal risk was not confirmed in two other studies; contradictory results in relation to phenotype | Afman et al., 2003b; Devlin et al., 2000; Morin et al., 2003a; Relton et al., 2003 |
| TSER (Promoter enhancer region) | Thymidylate synthase (28 bp double or triple repeat) | Possible risk factor for NTDs in some ethnic groups but not confirmed; uncertainty about the identity of the functional polymorphism within the repeat; conflicting reports of effects on plasma folate levels | Mandola et al., 2003; Volcik et al., 2003; Wilding et al., 2004 |
| CBS 844ins68 | Cystathionine β synthase | No risk association with NTD | Ramsbottom et al., 1997; Morrison et al., 1998; Speer et al., 1999; Richter et al., 2001; de Franchis et al., 2002; Afman et al., 2003a; Boyles et al., 2006 |
| TCII 776C→G | Transcobalamin II R259P | Significant association in a small study of NTD mothers but no risk association in larger studies; however, several studies show changes in transcobalamin II concentrations in serum or in amniotic fluid of NTD affected mothers | Pietrzyk and Bik-Multanowski, 2003; Ray and Blom, 2003; Swanson et al., 2005; Boyles et al., 2006 |
| DHFR Intron 1 | Dihydrofolate reductase (19 bp deletion) | Conflicting reports of risk association and protection | Johnson et al., 2004; Parle-McDermott et al., 2007; van der Linden et al., 2007 |
| BHMT 742G→A | Betaine-homocysteine methyltransferase R239Q | Conflicting effects in three studies; suggestion of protection in one study with small sample size; no effect in another; risk association in a third, but no correction for multiple gene testing | Morin et al., 2003b; Zhu et al., 2005; Boyles et al., 2006 |
MTHFR 677C→T As the First Genetic Factor Associated with Neural Tube Defects
MTHFR is the second key enzyme that exerts central control of folate metabolic pathways. This enzyme catalyzes the essentially irreversible conversion of 5,10-methyleneTHF to 5-methylTHF and thereby channels one-carbon units away from purine and pyrimidine synthesis and into the provision of methyl groups for S-adenosylmethionine (SAM) mediated methylation reactions. Kang et al. (1988) had previously shown that humans carried different isoforms of MTHFR. One particular form of the enzyme was thermolabile (Kang et al., 1988, 1991). Frosst et al. (1995) were first to clone this enzyme, describe the 677C→T (A222V) polymorphism in MTHFR, and demonstrate that the “T” (valine containing) allele was the thermolabile form of the enzyme. They also reported that individuals homozygous for the T allele had mildly increased plasma homocysteine concentrations. It is now known that there is a wide heterogeneity in the worldwide distribution of the polymorphism, ranging from a homozygous genotype frequency of approximately 0.20 to 0.36 in Mexican and southern European populations to 0.12 in northern Europeans and <0.01 among African groups. (Pepe et al., 1998; Mutchinick et al., 1999; Botto and Yang, 2000; Esfahani et al., 2003; Wilcken et al., 2003; Kirke et al., 2004; Gueant-Rodriguez et al., 2006). The variant is associated with reduced plasma and red cell folate status, suggesting an increased requirement for folate (Molloy et al., 1997). Furthermore, the T variant results in an enzyme that binds its cofactor flavin adenine dinucleotide (FAD) with lower affinity than the C variant (Guenther et al., 1999; Yamada et al., 2001) and can be stabilized by addition of the cofactor FAD or by addition of folate (Yamada et al., 2001; Pejchal et al., 2006). The validity of the in vitro model is supported by the observation that the elevated homocysteine seen in individuals homozygous for the TT variant can be partially attributed to riboflavin status and can be reversed by riboflavin intervention (McNulty et al., 2002, 2006), although the relative importance of cellular folate status in stabilizing the enzyme as compared with riboflavin status has still to be established.
Shortly after the publication of Frosst et al. (1995), Whitehead et al. (1995) and van der Put et al. (1995) reported an association between the TT variant and increased risk for NTD. In a later study of Irish NTD families, using the largest cohort of ethnically homogeneous NTD case family trios worldwide (218 full trios), Shields et al. (1999) reported that the MTHFR 677 TT genotype was associated with an OR of 2.57 (CI, 1.48–4.45; p = 0.0005) in a case-versus-control analysis and contributed an estimated 12% of the population-attributable risk in the Irish population. In that study, log-linear analysis of case and maternal effects within parental triads showed that the critical genetic determinant of risk was the genotype of the developing embryo, and maternal TT genotype played no more than a modest additional role. With an expanded DNA sample base of 397 cases from the same population and 855 randomly selected population based controls, Kirke et al. (2004) later demonstrated that the CT genotype also contributed significant NTD risk and estimated that the CT genotype was responsible for at least as many NTDs as the TT genotype (population-attributable risk 14.9% vs. 11.3% in the TT genotype), because of the much higher proportion of CT heterozygotes in the population
The 677C→T polymorphism in MTHFR has been more thoroughly explored than other folate enzymes in relation to NTDs. A human genome (HuGe) review containing a metaanalysis of existing studies (Botto and Yang, 2000) concluded that both embryonic and maternal TT genotype are equivalent factors in determining risk, with a pooled OR of 1.8 (95% CI, 1.4–2.2) for the case and 2.0 (95% CI, 1.5–2.8) for mothers. In this analysis, the maternal effect was calculated in a mother-versus-control analysis, without consideration of case genotype. However, such an analysis may not be entirely appropriate, because the mother shares one T allele with the embryo and therefore the frequency of the T allele would inevitably be higher in the mother if the case TT genotype contributes directly to NTD risk (Weinberg et al., 1998). In this respect, a more recent study of 175 American Caucasian NTD cases and their families supported the finding of case rather than maternal status as the major contributing factor (Rampersaud et al., 2003). It must also be said that in some populations, this polymorphism has not been found to confer risk for NTDs. Low power is likely to be a contributing factor in some studies, but a wide variation in the frequency of TT genotype among different ethnic groups is probably an additional factor. There may also be complex interactions between nutrient status and prevalence of the variant in certain populations (Gueant-Rodriguez et al., 2006; Amorim et al., 2007) that could influence the effect of the polymorphism on NTD risk. Furthermore, there may be differences in effect according to the severity or location of the NTD lesion (Johanning et al., 2000), a circumstance that might confer intrinsic bias in studies in which case sample availability is restricted to certain lesions.
Although it is biologically plausible that the MTHFR 677C→T polymorphism would be a functional risk factor for NTDs, because it leads to low folate status and elevated plasma homocysteine (Molloy et al., 1998), it is not known precisely how the MTHFR 677C→T polymorphism functions to confer this risk in the embryo. The modest contribution of the genotype to population risk indicates that it is a low penetrance factor because the majority of TT individuals do not have NTDs. Nevertheless, the critical position of the enzyme at a metabolic axis, where one-carbon units are committed to either DNA synthesis or methylation reactions, allows for mechanisms in which either cellular proliferation or methyltransferase activity leading to posttranslation modification of proteins, gene silencing, and cell signaling processes may all be invoked. Mothers who are homozygotes for the TT variant might confer risk by limiting the supply of folate to the developing embryo or by exposing the embryo to higher homocysteine concentrations.
As noted previously, the 677C→T polymorphism has received most attention in terms of NTD risk: however, other polymorphisms within the MTHFR gene have also been described and tested for association with NTDs (O’Leary et al., 2005a), the most widely investigated of these being the 1298A→C polymorphism with NTDs. This SNP is in strong linkage disequilibrium with the 677C→T variant, a fact that is not taken into consideration in some reports. In general, no significant effects of the mutation on NTD risk have been found that were independent of the 677C→T polymorphism (van der Put et al., 1998; Stegmann et al., 1999; Parle-McDermott et al., 2003a). A summary of other key published association studies of the 677C→T polymorphism and the 1298A→C polymorphism with NTDs is given in Table 1.
After MTHFR: Candidate SNPs in Folate Linked Genes Become a Hot Research Topic
Neural tube closure is a complex developmental process, involving many layers of molecular processes (Detrait et al., 2005). There are numerous ways in which the pathways of folate metabolism may be disturbed and potentially result in abnormal closure of the neural tube. Although such folate-responsive factors are still not understood, despite more than one decade of research, the hypothesis that folic acid supplementation works by overcoming a metabolic block in folate-related processes either in the mother or in the developing embryo remains the consensus. We now know a lot more about factors that influence embryonic development; it is clear that although DNA synthesis is an essential feature that can be influenced by folate or vitamin B12, the factors that trigger developmental changes, such as cell signaling events that lead to differential gene expression and activation of apoptotic pathways, are partially controlled by methylation reactions, including DNA, histone, and other protein methylations. All of these methylations are likely to be sensitive to both folate and vitamin B12 and support the notion that vitamin B12 and folate may act synergistically in prevention of NTDs.
After the initial investigations of MTHFR, a wider search for folate genes associated with NTD risk began. Many groups started to collect genetic material from NTD case families and controls and embarked on a candidate gene approach to identify folate-related genetic risk factors based on the current background knowledge of the metabolism of folate and related nutrients. The primary candidates were genes whose products participated directly in folate metabolic pathways and formed the main focus for immediate genetic analysis. Secondary candidate genes were more indirectly involved and tended to be included as new information arose in the literature regarding their potential for being associated with a possible folate-related phenotype or NTD risk. In general, the expectation was that low-penetrance genetic risk factors would be manifested primarily in the offspring or the mother, and paternal genetic involvement would mainly be in their transmission of the risk allele to the developing embryo. However, it was also accepted that analysis of paternal genes would provide a useful control group in the specific case of a polymorphism that confers increased risk on the mother, but not on the embryo that bears the defect.
At the outset of these studies, the newly emerging genetic databases (e.g., dbSNP, HapMap) were beginning to provide a wealth of polymorphic variants on nearly all of the folate related candidate genes. However, genotyping techniques were slow and limited; therefore, it was necessary to generate criteria that would prioritize the polymorphisms to test (the candidate SNP approach). For example, a polymorphism with a known biochemical phenotype or that altered the expression level of a gene would be high priority, as would a polymorphism resulting in a nonsynonymous change, particularly if it altered a highly conserved amino acid in the protein or changed the category (i.e., aliphatic to aromatic) or functional property of an amino acid. The next level of priority would be whether the polymorphism occurred in the coding region and was likely to affect codon usage and translational efficiency or perhaps occurred in a noncoding region that might affect gene transcription, such as those within the 5’ and 3’ untranslated regions or within a noncoding region that might affect mRNA splicing. In those early studies, repeat polymorphisms such as microsatellites residing in close proximity to a candidate gene were generally of lowest priority, but might be considered if it was likely that they might indicate linkage disequilibrium (LD) with an unknown functional variant.
The statistical methodology for the analysis of such disease association studies has also been an important factor in the study outcomes. Two analysis techniques have been recommended: (1) a primary analysis in which the prevalence of a particular polymorphism in the case population is compared with that in the nonaffected control population of a similar genetic background, using a log-linear approach (Weinberg et al., 1998; Wilcox et al., 1998), and (2) a secondary analysis testing for the preferential transmission of a particular allele from heterozygous parents to cases, using a transmission disequilibrium test (Spielman and Ewens, 1996). Because only transmissions from heterozygous parents are informative in the latter analysis, this approach requires large numbers of family triads (offspring, mother, and father).
Because of these complexities, there are clear advantages to performing such studies in an area with a high genetic homogeneity and a high prevalence of NTDs underlying a high genetic predisposition to the condition. Such criteria are met in Ireland, (Hill et al., 2000; Scott et al., 1990), where our collaborative group was able to collect DNA and demographic information on more than 550 Irish families affected by NTDs, including 445 complete triads (offspring, mother, father) and 110 incomplete triads.
Despite technical limitations, considerable progress was made by researchers in the field, using the available polymorphism analysis techniques. Table 1 gives details of NTD association studies involving high priority SNPs in over a dozen primary candidate genes in a number of populations worldwide. It is probably not surprising that the outcomes of these efforts have been inconsistent, because of the underlying low penetrance of the genetic effects, differences in population genetic and environmental susceptibility, low sample size, and often poorly matched controls in study design (Mitchell et al., 2004). In the large, ethnically homogeneous Irish cohort described previously, no important risk polymorphisms were identified in the gene encoding the most plausible biologic candidate, methionine synthase, or in its activating enzyme, methionine synthase reductase (Brody et al., 1999; O’Leary et al., 2005b). In addition, none were identified in the genes encoding the human folate receptor β gene (O’Leary et al., 2003), the reduced folate carrier, (O’Leary et al., 2006), cystathionine β-synthase (Ramsbottom et al., 1997), methylmalonyl CoA mutase (Parle-McDermott et al., 2003a), transcobalamin II (Swanson et al., 2005), or dihydrofolate reductase (Parle-McDermott et al., 2007). However, a 1958G→ A polymorphism in the 10-formylTHF synthetase region of the cytosolic MTHFD1 gene, which encodes the trifunctional C1-synthase enzyme, was found to be a maternal risk factor for NTDs (Brody et al., 2002). Overall, there was an excess of “QQ” homozygotes in the mothers of children with NTD compared with controls (OR, 1.52 [1.16–1.99]; p = 0.003). The effect was later confirmed in a second NTD cohort (Parle-McDermott et al., 2006), and the variant was also shown to be a risk factor for other maternal complications of pregnancy (Parle-McDermott et al., 2005a, b).
From One to Many: The Move from Single Variants to Candidate Genes and Pathways
Although effective, the testing of single candidate genes for NTDs becomes problematic once the top 5 to 10 biologically plausible candidate polymorphisms have been tested, because the next tier of candidates may have an order of magnitude more members. In addition, whereas the number of polymorphisms in a gene predicted to effect function (functional SNPs) is small, such a list does not include variants whose function is unknown or variants whose identity has yet to be discovered. This list of unsurveyed variants would include variants in the 5′ regulatory region that might increase or decrease gene expression, variants in the 3′ region that might increase or decrease the stability of the untranslated mRNA, insertions, deletions, repeats, and copy-number polymorphisms.
Fortunately, as discussed below, recent advances in genotyping technology and bioinformatics have progressively increased the number of variants that can be tested in a single experiment. Accordingly, the search for genetic polymorphisms in the homocysteine/folate pathway that contribute to NTDs has moved from a candidate SNP to a candidate gene approach. By taking advantage of the LD in the human genome, panels of SNPs are now being used to capture all the variation in a gene. The number of SNPs required to “mark” a particular gene depends upon the physical size and the structure LD present in human populations. The International HapMap Project (http://www.hapmap.org) has produced LD maps for several major geographic populations. For example, the LD map of the Caucasian population can be used to accurately predict the LD pattern in the Irish population. These LD data can be used to select “tagging” SNPs to represent any gene in the genome, as recently demonstrated for TP53 in relation to NTDs (Pangilinan et al., 2008). Coupled with technology that allows the assay of more than 1500 SNPs simultaneously, our group recently designed a single experiment capable of surveying more than 60 candidate genes. Similar types of experiments are ongoing in other NTD research laboratories.
Biochemical Hypotheses Take a Back Seat: The Prospect for Genome-Wide Association Studies
Candidate gene studies are limited by our ability to produce a list of candidates. The biology of normal and indeed abnormal function is so complex and so poorly understood that one can easily see how alteration in the function of a gene with no apparent link to folate pathways could play a role in NTDs. When our lack of knowledge of pathologic mechanisms is coupled with a search for genes, we cannot ignore one startling observation: nearly one third of all human genes have no known function. Most are conserved through evolution, indicating that nature has assigned important functions to these unannotated genes (http://www.ncbi.nlm.nih.gov/COG/).
How do we escape only knowing what we already know? Fortunately, this tautology can be solved. In addition to having a map of the human genome, we also have a detailed map of where genomic sequences differ between individuals. This variation map, known as the HapMap, can be used to scan the genome for genes associated with specific diseases (Manolio et al., 2008). Results of genome wide association studies are already causing great scientific interest, (Baker 2008; Couzin and Kaiser, 2007), with many studies within the past two years showing associations of specific genomic regions with common diseases such as cardiovascular disease (McPherson et al., 2007; Helgadottir et al., 2007), stroke (Bilguvar et al., 2008), diabetes (Cooper et al., 2008; Zeggini et al., 2008), lung cancer (Wang et al., 2008; McKay et al., 2008), and breast cancer (Easton et al., 2007). Other such studies have examined genetic determinants of biomarkers, and a recent publication reported a strong association of a genetic locus with plasma vitamin B12 concentration (Hazra et al., 2008). The technical details of these studies are covered elsewhere (Maresso et al., 2008; McCarthy et al., 2008). In practice, the method allows investigators to forget biology. By using 500,000 to 1 million SNPs, every region in the genome can be screened for association with NTDs. Why has this not yet been done for NTDs? The method relies on being able to detect a higher frequency of specific variants in cases compared to controls. Because so many markers are tested, one expects that some frequencies will be skewed by chance. Two steps are required to protect against these chance findings. The first is to use as large a sample size as possible. Even for a trait with a strong genetic component such as NTDs, it would be wise to start with approximately 2000 cases (mothers or affected children, or both) and 2000 controls. Samples of this size have sufficient power to detect some of the genes associated with NTDs. To demonstrate universal applicability and to avoid false positives, it is wise to retest the genes found in genome scans in a second population. Is this type of study feasible? No one group has obtained a sufficient number of samples to perform such an experiment. However, the study would be possible if all the major groups studying NTDs decided to join their efforts and pool their samples. In all likelihood, the results of such a study would identify new genes that in turn could illuminate new areas of folate biochemistry. We believe that Professor Smithells would be excited to know that his seminal discovery would someday by followed by a study of the entire genome, and that such a genetic investigation might definitively answer the question of how folate prevents NTDs.
Acknowledgments
This study was supported by the Health Research Board, Ireland, and the intramural research programs of the Eunice Kennedy Shriver National Institute of Child Health and Human Development and the National Human Genome Research Institute, National Institutes of Health, Bethesda, Maryland.
REFERENCES
- Afman LA, Lievers KJ, Kluijtmans LA, et al. 2003a. Gene-gene interaction between the cystathionine beta-synthase 31 base pair variable number of tandem repeats and the methylenetetrahydrofolate reductase 677C > T polymorphism on homocysteine levels and risk for neural tube defects. Mol Genet Metab 78:211–5. [DOI] [PubMed] [Google Scholar]
- Afman LA, Trijbels FJ, Blom HJ. 2003b. The H475Y polymorphism in the glutamate carboxypeptidase II gene increases plasma folate without affecting the risk for neural tube defects in humans. J Nutr 133:75–77. [DOI] [PubMed] [Google Scholar]
- Amorim MR, Lima MA, Castilla EE, et al. 2007. Non-Latin European descent could be a requirement for association of NTDs and MTHFR variant 677C > T: a meta-analysis. Am J Med Genet A 143A:1726–1732. [DOI] [PubMed] [Google Scholar]
- Baker M 2008. Genome studies: genetics by numbers. Nature 451:516–518. [DOI] [PubMed] [Google Scholar]
- Barber R, Shalat S, Hendricks K, et al. 2000. Investigation of folate pathway gene polymorphisms and the incidence of neural tube defects in a Texas hispanic population. Mol Genet Metab 70:45–52. [DOI] [PubMed] [Google Scholar]
- Barber RC, Shaw GM, Lammer EJ, et al. 1998. Lack of association between mutations in the folate receptor-alpha gene and spina bifida. Am J Med Genet 76:310–317. [PubMed] [Google Scholar]
- Berry RJ, Li Z, Erickson JD, et al. 1999. Prevention of neural-tube defects with folic acid in China. N Engl J Med 341:1485–1490. [DOI] [PubMed] [Google Scholar]
- Bilguvar K, Yasuno K, Niemela M, et al. 2008. Susceptibility loci for intracranial aneurysm in European and Japanese populations. Nat Genet. 40:1472–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botto LD, Yang Q. 2000. 5,10-Methylenetetrahydrofolate reductase gene variants and congenital anomalies: a HuGE review. Am J Epidemiol 151:862–877. [DOI] [PubMed] [Google Scholar]
- Bower C, Stanley FJ. 1989. Dietary folate as a risk factor for neural-tube defects: evidence from a case-control study in Western Australia. Med J Aust 150:613–619. [DOI] [PubMed] [Google Scholar]
- Boyles AL, Billups AV, Deak KL, et al. 2006. Neural tube defects and folate pathway genes: family-based association tests of gene-gene and gene-environment interactions. Environ Health Perspect 114:1547–1552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody LC, Baker PJ, Chines PS, et al. 1999. Methionine synthase: high-resolution mapping of the human gene and evaluation as a candidate locus for neural tube defects. Mol Genet Metab 67:324–333. [DOI] [PubMed] [Google Scholar]
- Brody LC, Conley M, Cox C, et al. 2002. A polymorphism, R653Q, in the trifunctional enzyme methylenetetrahydrofolate dehydrogenase/methenyltetrahydrofolate cyclohydrolase/formyltetrahydrofolate synthetase is a maternal genetic risk factor for neural tube defects: report of the Birth Defects Research Group. Am J Hum Genet 71:1207–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candito M, Rivet R, Herbeth B, et al. 2008. Nutritional and genetic determinants of vitamin B and homocysteine metabolisms in neural tube defects: a multicenter case-control study. Am J Med Genet A 146A:1128–1133. [DOI] [PubMed] [Google Scholar]
- Christensen B, Arbour L, Tran P, et al. 1999. Genetic polymorphisms in methylenetetrahydrofolate reductase and methionine synthase, folate levels in red blood cells, and risk of neural tube defects. Am J Med Genet 84:151–157. [DOI] [PubMed] [Google Scholar]
- Cooper JD, Smyth DJ, Smiles AM, et al. 2008. Meta-analysis of genomewide association study data identifies additional type 1 diabetes risk loci. Nat Genet 40:1399–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couzin J, Kaiser J. 2007. Genome-wide association. Closing the net on common disease genes. Science 316:820–822. [DOI] [PubMed] [Google Scholar]
- Czeizel AE, Dudas I. 1992. Prevention of the first occurrence of neuraltube defects by periconceptional vitamin supplementation. N Engl J Med 327:1832–1835. [DOI] [PubMed] [Google Scholar]
- Daly LE, Kirke PN, Molloy A, et al. 1995. Folate levels and neural tube defects. Implications for prevention. JAMA 274:1698–1702. [DOI] [PubMed] [Google Scholar]
- de Franchis R, Botto LD, Sebastio G, et al. 2002. Spina bifida and folaterelated genes: a study of gene-gene interactions. Genet Med 4:126–130. [DOI] [PubMed] [Google Scholar]
- De Marco P, Calevo MG, Moroni A, et al. 2002. Study of MTHFR and MS polymorphisms as risk factors for NTD in the Italian population. J Hum Genet 47:319–324. [DOI] [PubMed] [Google Scholar]
- De Marco P, Calevo MG, Moroni A, et al. 2003. Reduced folate carrier polymorphism (80A→G) and neural tube defects. Eur J Hum Genet 11:245–252. [DOI] [PubMed] [Google Scholar]
- De Marco P, Merello E, Calevo MG, et al. 2006. Evaluation of a methylenetetrahydrofolate-dehydrogenase 1958G>A polymorphism for neural tube defect risk. J Hum Genet 2006; 51:98–103. [DOI] [PubMed] [Google Scholar]
- Detrait ER, George TM, Etchevers HC, et al. 2005. Human neural tube defects: developmental biology, epidemiology, and genetics. Neurotoxicol Teratol 27:515–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin AM, Ling EH, Peerson JM, et al. 2000. Glutamate carboxypeptidase II: a polymorphism associated with lower levels of serum folate and hyperhomocysteinemia. Hum Mol Genet 9:2837–2844. [DOI] [PubMed] [Google Scholar]
- Doolin MT, Barbaux S, McDonnell M, et al. 2002. Maternal genetic effects, exerted by genes involved in homocysteine remethylation, influence the risk of spina bifida. Am J Hum Genet 71:1222–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easton DF, Pooley KA, Dunning AM, et al. 2007. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 447:1087–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esfahani ST, Cogger EA, Caudill MA. 2003. Heterogeneity in the prevalence of methylenetetrahydrofolate reductase gene polymorphisms in women of different ethnic groups. J Am Diet Assoc 103:200–207. [DOI] [PubMed] [Google Scholar]
- Frosst P, Blom HJ, Milos R, et al. 1995. A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nat Genet 10:111–113. [DOI] [PubMed] [Google Scholar]
- Garrod AE. 1902. The incidence of alkaptonuria: A study of chemical individuality. Lancet ii:616–1620. [Google Scholar]
- Gueant-Rodriguez RM, Gueant JL, Debard R, et al. 2006. Prevalence of methylenetetrahydrofolate reductase 677T and 1298C alleles and folate status: a comparative study in Mexican, West African, and European populations Am J Clin Nutr 83:701–707. [DOI] [PubMed] [Google Scholar]
- Guenther BD, Sheppard CA, Tran P, et al. 1999. The structure and properties of methylenetetrahydrofolate reductase from Escherichia coli suggest how folate ameliorates human hyperhomocysteinemia. Nat Struct Biol 6:359–365. [DOI] [PubMed] [Google Scholar]
- Hazra A, Kraft P, Selhub J, et al. , 2008. Common variants of FUT2 are associated with plasma vitamin B12 levels. Nat Genet. 40:1160–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heil SG, van der Put NM, Trijbels FJ, et al. 1999. Molecular genetic analysis of human folate receptors in neural tube defects. Eur J Hum Genet 7:393–396. [DOI] [PubMed] [Google Scholar]
- Heil SG, Van der Put NM, Waas ET, et al. 2001. Is mutated serine hydroxymethyltransferase (SHMT) involved in the etiology of neural tube defects? Mol Genet Metab 73:164–172. [DOI] [PubMed] [Google Scholar]
- Helgadottir A, Thorleifsson G, Manolescu A, et al. 2007. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science 316:1491–1493. [DOI] [PubMed] [Google Scholar]
- Hibbard ED, Smithells RW. 1965. Folic acid metabolism and human embryopathy. Lancet 285:1254. [Google Scholar]
- Hill EW, Jobling MA, Bradley DG. 2000. Y-chromosome variation and Irish origins. Nature 404:351–352. [DOI] [PubMed] [Google Scholar]
- http://www.ncbi.nlm.nih.gov/COG/2008 Clusters of Orthologous Groups -Phylogenetic classification of proteins encoded in complete genomes. Accessed October 2008.
- Johanning GL, Tamura T, Johnston KE, et al. 2000. Comorbidity of 5,10-methylenetetrahydrofolate reductase and methionine synthase gene polymorphisms and risk for neural tube defects. J Med Genet 37:949–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WG, Stenroos ES, Spychala JR, et al. 2004. New 19 bp deletion polymorphism in intron-1 of dihydrofolate reductase (DHFR): a risk factor for spina bifida acting in mothers during pregnancy? Am J Med Genet A 124A:339–345. [DOI] [PubMed] [Google Scholar]
- Kang SS, Wong PW, Susmano A, et al. 1991. Thermolabile methylenetetrahydrofolate reductase: an inherited risk factor for coronary artery disease. Am J Hum Genet 48:536–545. [PMC free article] [PubMed] [Google Scholar]
- Kang SS, Zhou J, Wong PW, et al. 1988. Intermediate homocysteinemia: a thermolabile variant of methylenetetrahydrofolate reductase. Am J Hum Genet 43:414–421. [PMC free article] [PubMed] [Google Scholar]
- Kirke PN, Daly LE, Elwood JH. 1992. A randomised trial of low dose folic acid to prevent neural tube defects. Arch Dis Child 67:1442–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirke PN, Mills JL, Molloy AM, et al. 2004. Impact of the MTHFR C677T polymorphism on risk of neural tube defects: case-control study. BMJ 328:1535–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirke PN, Molloy AM, Daly LE, et al. 1993. Maternal plasma folate and vitamin B12 are independent risk factors for neural tube defects. QJM 86:703–708. [PubMed] [Google Scholar]
- Laurence KM, James N, Miller MH, et al. 1981. Double-blind randomized controlled trial of folate treatment before conception to prevent recurrence of neural-tube defects. Br Med J (Clin Res Ed) 282:1509–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandola MV, Stoehlmacher J, Muller-Weeks S, et al. 2003. A novel single nucleotide polymorphism within the 50 tandem repeat polymorphism [PubMed] [Google Scholar]
- of the thymidylate synthase gene abolishes USF-1 binding and alters transcriptional activity. Cancer Res 63:2898–2904. [PubMed] [Google Scholar]
- Manolio TA, Brooks LD, Collins FS. 2008. A HapMap harvest of insights into the genetics of common disease. J Clin Invest 118:1590–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maresso K, Broeckel U. 2008. Genotyping platforms for mass-throughput genotyping with SNPs, including human genome-wide scans. Adv Genet 60:107–139. [DOI] [PubMed] [Google Scholar]
- McCarthy MI, Abecasis GR, Cardon LR, et al. 2008. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet 9:356–369. [DOI] [PubMed] [Google Scholar]
- McKay JD, Hung RJ, Gaborieau V, et al. 2008. Lung cancer susceptibility locus at 5p15.33. Nat Genet 40:1404–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNulty H, Dowey le RC, Strain JJ, et al. 2006. Riboflavin lowers homocysteine in individuals homozygous for the MTHFR 677C->T polymorphism. Circulation 113:74–80. [DOI] [PubMed] [Google Scholar]
- McNulty H, McKinley MC, Wilson B, et al. 2002. Impaired functioning of thermolabile methylenetetrahydrofolate reductase is dependent on riboflavin status: implications for riboflavin requirements. Am J Clin Nutr 76:436–441. [DOI] [PubMed] [Google Scholar]
- McPherson R, Pertsemlidis A, Kavaslar N, et al. 2007. A common allele on chromosome 9 associated with coronary heart disease. Science 316:1488–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills JL, McPartlin JM, Kirke PN, et al. 1995. Homocysteine metabolism in pregnancies complicated by neural-tube defects. Lancet 345:149–151. [DOI] [PubMed] [Google Scholar]
- Mills JL, Rhoads GG, Simpson JL, et al. 1989. The absence of a relation between the periconceptional use of vitamins and neural-tube defects. National Institute of Child Health and Human Development Neural Tube Defects Study Group. N Engl J Med 321:430–435. [DOI] [PubMed] [Google Scholar]
- Milunsky A, Jick H, Jick SS, et al. 1989. Multivitamin/folic acid supplementation in early pregnancy reduces the prevalence of neural tube defects. JAMA 262:2847–2852. [DOI] [PubMed] [Google Scholar]
- Mitchell LE, Adzick NS, Melchionne J, et al. 2004. Spina bifida. Lancet 364:1885–1895. [DOI] [PubMed] [Google Scholar]
- Molloy AM, Daly S, Mills JL, et al. 1997. Thermolabile variant of 5,10-methylenetetrahydrofolate reductase associated with low red-cell folates: implications for folate intake recommendations. Lancet 349:1591–1593. [DOI] [PubMed] [Google Scholar]
- Molloy AM, Kirke P, Hillary I, et al. 1985. Maternal serum folate and vitamin B12 concentrations in pregnancies associated with neural tube defects. Arch Dis Child 60:660–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molloy AM, Kirke PN, Troendle JF et al. Maternal vitamin B12 status and risk of neural tube defects in a high prevalence, unfortified population Pediatrics (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molloy AM, Mills JL, Kirke PN, et al. 1998. Low blood folates in NTD pregnancies are only partly explained by thermolabile 5,10-methylenetetrahydrofolate reductase: low folate status alone may be the critical factor. Am J Med Genet 78:155–159. [PubMed] [Google Scholar]
- Morin I, Devlin AM, Leclerc D, et al. 2003a. Evaluation of genetic variants in the reduced folate carrier and in glutamate carboxypeptidase II for spina bifida risk. Mol Genet Metab 79:197–200. [DOI] [PubMed] [Google Scholar]
- Morin I, Platt R, Weisberg I, et al. 2003b. Common variant in betaine-homocysteine methyltransferase (BHMT) and risk for spina bifida. Am J Med Genet A 119A:172–176. [DOI] [PubMed] [Google Scholar]
- Morrison K, Papapetrou C, Hol FA, et al. 1998. Susceptibility to spina bifida: an association study of five candidate genes. Ann Hum Genet 62:379–396. [DOI] [PubMed] [Google Scholar]
- Medical Research Council. 1991. Prevention of neural tube defects: results of the Medical Research Council Vitamin Study. MRC Vitamin Study Research Group. Lancet 338:131–137. [PubMed] [Google Scholar]
- Mulinare J, Cordero JF, Erickson JD, et al. 1988. Periconceptional use of multivitamins and the occurrence of neural tube defects. JAMA 260:3141–5. [PubMed] [Google Scholar]
- Mutchinick OM, Lopez MA, Luna L, et al. 1999. High prevalence of the thermolabile methylenetetrahydrofolate reductase variant in Mexico: a country with a very high prevalence of neural tube defects. Mol Genet Metab 68:461–467. [DOI] [PubMed] [Google Scholar]
- O’Leary VB, Mills JL, Kirke PN, et al. 2003. Analysis of the human folate receptor beta gene for an association with neural tube defects. Mol Genet Metab 79:129–133. [DOI] [PubMed] [Google Scholar]
- O’Leary VB, Mills JL, Parle-McDermott A, et al. 2005a. Screening for new MTHFR polymorphisms and NTD risk. Am J Med Genet A 138A:99–106. [DOI] [PubMed] [Google Scholar]
- O’Leary VB, Mills JL, Pangilinan F, et al. 2005b. Analysis of methionine synthase reductase polymorphisms for neural tube defects risk association. Mol Genet Metab 85:220–227. [DOI] [PubMed] [Google Scholar]
- O’Leary VB, Pangilinan F, Cox C, et al. 2006. Reduced folate carrier polymorphisms and neural tube defect risk. Mol Genet Metab 87:364–369. [DOI] [PubMed] [Google Scholar]
- Pangilinan F, Geiler K, Dolle J, et al. 2008. Construction of a high resolution linkage disequilibrium map to evaluate common genetic variation in TP53 and neural tube defect risk in an Irish population. Am J Med Genet A 146A:2617–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parle-McDermott A, Kirke PN, Mills JL, et al. 2006. Confirmation of the R653Q polymorphism of the trifunctional C1-synthase enzyme as a maternal risk for neural tube defects in the Irish population. Eur J Hum Genet 14:768–772. [DOI] [PubMed] [Google Scholar]
- Parle-McDermott A, Mills JL, Kirke PN, et al. 2003a. Analysis of the MTHFR 1298A→C and 677C→T polymorphisms as risk factors for neural tube defects. J Hum Genet 48:190–193. [DOI] [PubMed] [Google Scholar]
- Parle-McDermott A, McManus EJ, Mills JL, et al. 2003b. Polymorphisms within the vitamin B12 dependent methylmalonyl-coA mutase are not risk factors for neural tube defects. Mol Genet Metab 80:463–468. [DOI] [PubMed] [Google Scholar]
- Parle-McDermott A, Mills JL, Kirke PN, et al. 2005a. MTHFD1 R653Q polymorphism is a maternal genetic risk factor for severe abruption placentae. Am J Med Genet A 132A:365–368. [DOI] [PubMed] [Google Scholar]
- Parle-McDermott A, Pangilinan F, Mills JL, et al. 2005b. A polymorphism in the MTHFD1 gene increases a mother’s risk of having an unexplained second trimester pregnancy loss. Mol Hum Reprod 11:477–480. [DOI] [PubMed] [Google Scholar]
- Parle-McDermott A, Pangilinan F, Mills JL, et al. 2007. The 19-bp deletion polymorphism in intron-1 of dihydrofolate reductase (DHFR) may decrease rather than increase risk for spina bifida in the Irish population. Am J Med Genet A 143A:1174–1180. [DOI] [PubMed] [Google Scholar]
- Pejchal R, Campbell E, Guenther BD, et al. 2006. Structural perturbations in the Ala → Val polymorphism of methylenetetrahydrofolate reductase: how binding of folates may protect against inactivation. Biochemistry 45:4808–4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepe G, Camacho Vanegas O, Giusti B, et al. 1998. Heterogeneity in world distribution of the thermolabile C677T mutation in 5,10-methylenetetrahydrofolate reductase. Am J Hum Genet 63:917–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrzyk JJ, Bik-Multanowski M. 2003. 776C>G polymorphism of the transcobalamin II gene as a risk factor for spina bifida. Mol Genet Metab 80:364. [DOI] [PubMed] [Google Scholar]
- Rampersaud E, Melvin EC, Siegel D, et al. 2003. Updated investigations of the role of methylenetetrahydrofolate reductase in human neural tube defects. Clin Genet 63:210–214. [DOI] [PubMed] [Google Scholar]
- Ramsbottom D, Scott JM, Molloy A, et al. 1997. Are common mutations of cystathionine beta-synthase involved in the aetiology of neural tube defects? Clin Genet 51:39–42. [DOI] [PubMed] [Google Scholar]
- Ray JG, Blom HJ. 2003. Vitamin B12 insufficiency and the risk of fetal neural tube defects. QJM 96:289–95. [DOI] [PubMed] [Google Scholar]
- Ray JG, Wyatt PR, Thompson MD, et al. , 2007. Vitamin B12 and the risk of neural tube defects in a folic-acid-fortified population. Epidemiology 18:362–366. [DOI] [PubMed] [Google Scholar]
- Relton CL, Wilding CS, Jonas PA, et al. , 2003. Genetic susceptibility to neural tube defect pregnancy varies with offspring phenotype. Clin Genet 64:424–428. [DOI] [PubMed] [Google Scholar]
- Relton CL, Wilding CS, Laffling AJ, et al. 2004a. Low erythrocyte folate status and polymorphic variation in folate-related genes are associated with risk of neural tube defect pregnancy. Mol Genet Metab 81:273–281. [DOI] [PubMed] [Google Scholar]
- Relton CL, Wilding CS, Pearce MS, et al. 2004b. Gene-gene interaction in folate-related genes and risk of neural tube defects in a UK population. J Med Genet 41:256–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter B, Stegmann K, Roper B, et al. 2001. Interaction of folate and homocysteine pathway genotypes evaluated in susceptibility to neural tube defects (NTD) in a German population. J Hum Genet 46:105–109. [DOI] [PubMed] [Google Scholar]
- Schorah CJ, Smithells RW, Scott J. 1980. Vitamin B12 and anencephaly. Lancet 1:880. [DOI] [PubMed] [Google Scholar]
- Scott JM, Kirke PN, Weir DG. 1990. The role of nutrition in neural tube defects. Annu Rev Nutr 10:277–295. [DOI] [PubMed] [Google Scholar]
- Shang Y, Zhao H, Niu B, et al. 2008. Correlation of polymorphism of MTHFRs and RFC-1 genes with neural tube defects in China. Birth Defects Res A Clin Mol Teratol 82:3–7. [DOI] [PubMed] [Google Scholar]
- Shaw GM, Lammer EJ, Zhu H, et al. 2002. Maternal periconceptional vitamin use, genetic variation of infant reduced folate carrier (A80G), and risk of spina bifida. Am J Med Genet 108:1–6. [DOI] [PubMed] [Google Scholar]
- Shaw GM, Schaffer D, Velie EM, et al. 1995. Periconceptional vitamin use, dietary folate, and the occurrence of neural tube defects. Epidemiology 6:219–226. [DOI] [PubMed] [Google Scholar]
- Shields DC, Kirke PN, Mills JL, et al. 1999. The ‘‘thermolabile’’ variant of methylenetetrahydrofolate reductase and neural tube defects: An evaluation of genetic risk and the relative importance of the genotypes of the embryo and the mother. Am J Hum Genet 64:1045–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smithells RW, Nevin NC, Seller MJ, et al. 1983. Further experience of vitamin supplementation for prevention of neural tube defect recurrences. Lancet 1:1027–1031. [DOI] [PubMed] [Google Scholar]
- Smithells RW, Sheppard S, Schorah CJ. 1976. Vitamin deficiencies and neural tube defects. Arch Dis Child 51:944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smithells RW, Sheppard S, Schorah CJ, et al. 1980. Possible prevention of neural-tube defects by periconceptional vitamin supplementation. Lancet 1:339–340. [DOI] [PubMed] [Google Scholar]
- Smithells RW, Sheppard S, Wild J, et al. 1989. Prevention of neural tube defect recurrences in Yorkshire: final report. Lancet 2:498–499. [DOI] [PubMed] [Google Scholar]
- Speer MC, Nye J, McLone D, et al. 1999. Possible interaction of genotypes at cystathionine beta-synthase and methylenetetrahydrofolate reductase (MTHFR) in neural tube defects. NTD Collaborative Group. Clin Genet 56:142–144. [DOI] [PubMed] [Google Scholar]
- Spielman RS, Ewens WJ. 1996. The TDT and other family-based tests for linkage disequilibrium and association. Am J Hum Genet 59:983–989. [PMC free article] [PubMed] [Google Scholar]
- Steegers-Theunissen RP, Boers GH, Trijbels FJ, et al. 1991. Neural-tube defects and derangement of homocysteine metabolism. N Engl J Med 324:199–200. [DOI] [PubMed] [Google Scholar]
- Steegers-Theunissen RP, Boers GH, Blom HJ, et al. 1995. Neural tube defects and elevated homocysteine levels in amniotic fluid. Am J Obstet Gynecol 172:1436–1441. [DOI] [PubMed] [Google Scholar]
- Stegmann K, Ziegler A, Ngo ET, et al. 1999. Linkage disequilibrium of MTHFR genotypes 677C/T-1298A/C in the German population and association studies in probands with neural tube defects (NTD). Am J Med Genet 87:23–29. [PubMed] [Google Scholar]
- Suarez L, Hendricks K, Felkner M, Gunter E. 2003. Maternal serum B12 levels and risk for neural tube defects in a Texas-Mexico border population. Ann Epidemiol 13:81–88. [DOI] [PubMed] [Google Scholar]
- Swanson DA, Pangilinan F, Mills JL, et al. 2005. Evaluation of transcobalamin II polymorphisms as neural tube defect risk factors in an Irish population. Birth Defects Res A Clin Mol Teratol 73:239–244. [DOI] [PubMed] [Google Scholar]
- Trembath D, Sherbondy AL, Vandyke DC, et al. 1999. Analysis of select folate pathway genes, PAX3, and human T in a Midwestern neural tube defect population. Teratology 59:331–341. [DOI] [PubMed] [Google Scholar]
- van der Linden IJ, den Heijer M, Afman LA, et al. 2006. The methionine synthase reductase 66A>G polymorphism is a maternal risk factor for spina bifida. J Mol Med 84:1047–1054. [DOI] [PubMed] [Google Scholar]
- van der Linden IJ, Nguyen U, Heil SG, et al. 2007. Variation and expression of dihydrofolate reductase (DHFR) in relation to spina bifida. Mol Genet Metab 91:98–103. [DOI] [PubMed] [Google Scholar]
- van der Put NM, Steegers-Theunissen RP, Frosst P, et al. 1995. Mutated methylenetetrahydrofolate reductase as a risk factor for spina bifida. Lancet 346:1070–1071. [DOI] [PubMed] [Google Scholar]
- van der Put NM, Gabreels F, Stevens EM, et al. 1998. A second common mutation in the methylenetetrahydrofolate reductase gene: an additional risk factor for neural-tube defects? Am J Hum Genet 62:1044–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira AR, Murray JC, Trembath D, et al. 2005. Studies of reduced folate carrier 1 (RFC1) A80G and 5,10-methylenetetrahydrofolate reductase (MTHFR) C677T polymorphisms with neural tube and orofacial cleft defects. Am J Med Genet A 135:220–223. [DOI] [PubMed] [Google Scholar]
- Volcik KA, Shaw GM, Zhu H, et al. 2003. Associations between polymorphisms within the thymidylate synthase gene and spina bifida. Birth Defects Res A Clin Mol Teratol 67:924–928. [DOI] [PubMed] [Google Scholar]
- Wald NJ, Hackshaw AD, Stone R, et al. 1996. Blood folic acid and vitamin B12 in relation to neural tube defects. Br J Obstet Gynaecol 103:319–324. [DOI] [PubMed] [Google Scholar]
- Wang Y, Broderick P, Webb E, et al. 2008. Common 5p15.33 and 6p21.33 variants influence lung cancer risk. Nat Genet 40:1407–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg CR, Wilcox AJ, Lie RT. 1998. A log-linear approach to case-parent-triad data: assessing effects of disease genes that act either directly or through maternal effects and that may be subject to parental imprinting. Am J Hum Genet 62:969–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werler MM, Shapiro S, Mitchell AA. 1993. Periconceptional folic acid exposure and risk of occurrent neural tube defects. JAMA 269:1257–1261. [PubMed] [Google Scholar]
- Whitehead AS, Gallagher P, Mills JL, et al. 1995. A genetic defect in 5,10 methylenetetrahydrofolate reductase in neural tube defects. QJM 88:763–766. [PubMed] [Google Scholar]
- Wilcken B, Bamforth F, Li Z, et al. 2003. Geographical and ethnic variation of the 677C>T allele of 5,10 methylenetetrahydrofolate reductase (MTHFR): findings from over 7000 newborns from 16 areas world wide. J Med Genet 40:619–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox AJ, Weinberg CR, Lie RT. 1998. Distinguishing the effects of maternal and offspring genes through studies of ‘‘case-parent triads”. Am J Epidemiol 148:893–901. [DOI] [PubMed] [Google Scholar]
- Wilding CS, Relton CL, Sutton MJ, et al. 2004. Thymidylate synthase repeat polymorphisms and risk of neural tube defects in a population from the northern United Kingdom. Birth Defects Res A Clin Mol Teratol 70:483–485. [DOI] [PubMed] [Google Scholar]
- Wilson A, Platt R, Wu Q, et al. 1999. A common variant in methionine synthase reductase combined with low cobalamin (vitamin B12) increases risk for spina bifida. Mol Genet Metab 67:317–323. [DOI] [PubMed] [Google Scholar]
- Yamada K, Chen Z, Rozen R, et al. 2001. Effects of common polymorphisms on the properties of recombinant human methylenetetrahydrofolate reductase. Proc Natl Acad Sci USA 98:14853–14858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates JR, Ferguson-Smith MA, Shenkin A, et al. 1987. Is disordered folate metabolism the basis for the genetic predisposition to neural tube defects? Clin Genet 31:279–287. [DOI] [PubMed] [Google Scholar]
- Zeggini E, Scott LJ, Saxena R, et al. 2008. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet 40:638–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Wicker NJ, Shaw GM, et al. 2003. Homocysteine remethylation enzyme polymorphisms and increased risks for neural tube defects. Mol Genet Metab 78:216–221. [DOI] [PubMed] [Google Scholar]
- Zhu H, Curry S, Wen S, et al. 2005. Are the betaine-homocysteine methyltransferase (BHMT and BHMT2) genes risk factors for spina bifida and orofacial clefts? Am J Med Genet A 135A:274–277. [DOI] [PubMed] [Google Scholar]