

Abstract
Psoriasis vulgaris is a common, heterogeneous, chronic inflammatory skin disease characterized by thickened, red, scaly plaques and systemic inflammation. Psoriasis is also associated with multiple comorbid conditions, such as joint destruction, cardiovascular disease, stroke, hypertension, metabolic syndrome, and chronic kidney disease. The discovery of interleukin-17-producing T helper cells in a mouse model of autoimmunity transformed our understanding of inflammation driven by T lymphocytes and the subsequent development of disease like psoriasis. Under the regulation of interleukin-23, T cells that produce high levels of interleukin-17 create a self-amplifying, feed-forward inflammatory response in keratinocytes that drives the development of thickened skin lesions infiltrated with a mixture of inflammatory cell populations. Recently, the FDA approved multiple highly effective psoriasis therapies that disrupt interleukin-17 (secukinumab, ixekizumab, and brodalumab) and interleukin-23 (guselkumab and tildrakizumab) signaling in the skin, thus leading to a major paradigm shift in the way that psoriatic disease is managed.
INTRODUCTION
Psoriasis vulgaris is a common, heterogeneous disease associated with the spontaneous development of inflammatory skin lesions and systemic inflammation (1). The systemic inflammation associated with psoriasis results in an increased risk for death (2, 3) and significant psoriasis-associated comorbidities such as psoriatic arthritis, cardiovascular disease, stroke, metabolic syndrome (obesity, hypertension, dyslipidemia, and diabetes), chronic kidney disease, gastrointestinal disease, mood disorder, and malignancy (4). For the last three decades, patients with moderate-to-severe psoriasis have been treated with phototherapy or a variety of systemic medications including methotrexate, cyclosporine, and targeted monoclonal antibodies such as tumor necrosis factor-α (TNFα) antagonists. However, a substantial proportion of psoriasis patients have disease that is inadequately treated with these first-generation systemic therapies either because of a primary response failure or gradual loss of efficacy. Patients also occasionally discontinue therapy due to treatment-related adverse events or comorbid conditions.
The discovery of the roles for interleukin-17 (IL-17) and IL-23 on the development of psoriatic disease has led to substantial increases in our understanding of the pathogenic immune events in psoriasis and has led to a paradigm shift in the treatment of this condition. Over the last few years, we have witnessed the FDA approval of several inhibitors of the IL-23/IL-17 signaling axis for the treatment of psoriasis (Table 1). These new biologic therapies have proven to be highly effective and result in dramatic improvements in approximately 80–90% of psoriasis patients. The unprecedented success of selective IL-17 and IL-23 antagonists for the treatment of psoriasis underscores the essential nature of these cytokines in the pathogenesis of this chronic inflammatory condition. In this article, we provide an overview of the major laboratory findings that led to the discovery of the dominant role of the IL-23/IL-17 axis in psoriatic disease and the subsequent development and clinical testing of novel IL-17 and IL-23 antagonists.
Table 1:
Summary of phase III clinical trial data of FDA-approved medications for the treatment of moderate-to-severe plaque psoriasis and psoriatic arthritis.
| Outcome | Etanercept (50 mg)a | Adalimumab (40 mg)b | Infliximab (5 mg/kg)c | Certolizumab (200 or 400 mg)d | Ustekinumab (45 or 90 mg)e | Secukinumab (300 mg)f | Ixekizumab (80 mg)g | Brodalumab (210 mg)h | Guselkumab (100 mg)i | Tildrakizumab (100 mg)j |
| PASI75 | 54–61% | 59–70% | 60–82% | 75–83% | 51–79% | 63–88% | 60–94% | 84–88% | 83–91% | 73–80% |
| PASI90 | 28–38% | 42–49% | 39–58% | 44–55% | 49–60% | 49–71% | 50–88% | 73–82% | 60–80% | 52–56% |
| PASI100 | 10–11% | 22% | 21% | 13–19% | 21–30% | 40–41% | 28–53% | 50–58% | 24–44% | 23–24% |
| Etanercept (25 mg) | Adalimumab (40 mg) | Infliximab (5 mg/kg) | Certolizumab (200 mg) | Ustekinumab (45 or 90 mg) | Secukinumab (150 mg) | Ixekizumab (80 mg) | Brodalumab (140 or 210 mg) | Guselkumab (100 mg) | Tildrakizumab (multiple doses) | |
| ACR20 | 55% | 57–65% | 54% | 64% | 42–44% | 50–51% | 48–62% | In progress | In progress | In phase 2 |
| ACR50 | 40% | 39–43% | 41% | 44% | 17–25% | 35% | 33–47% | In progress | In progress | In phase 2 |
| ACR75 | 10% | 23–27% | 27% | 28% | 0–12% | 19–21% | 12–34% | In progress | In progress | In phase 2 |
Abbreviations: PASI, Psoriasis Area and Severity Index; ACR, American College of Rheumatology.
Clinical trial data is based on phase III clinical trials. All PASI assessments occurred at week 24 unless otherwise indicated. All
Etanercept: PASI values for a dosage of 50 mg biweekly. ACR values for a dosage of 25 mg biweekly.
Adalimumab: PASI and ACR values for a dosage of 40mg every other week.
Infliximab: PASI values for a dosage of 5 mg/kg at weeks 0, 2, and 6. ACR values for a dosage of 5 mg/kg at weeks 0, 2, 6, 14, and 22.
Certolizumab pegol: PASI assessments at week 16 for a dosage of 400 mg every other week or 400 mg at weeks 0, 2, and 4, followed by 200 mg every other week. ACR values for a dosage of 400 mg at weeks 0, 2, and 4, followed by 200 mg every other week.
Ustekinumab: PASI assessments at week 24 or 28 depending on study. PASI and ACR values at a dosage of 45 mg or 90 mg at weeks 0 and 4, followed by every 12 weeks.
Secukinumab: PASI values for a dosage of 300 mg every week for 4–5 weeks, followed by every four weeks. ACR values for initial dosages of 10 mg/kg at weeks 0, 2, and 4, followed by 150 mg every 4 weeks.
Ixekizumab: PASI values for a starting dose of 160 mg, followed by 80 mg every other week. At week 12, dosages adjusted to 80 mg every four weeks. ACR values for a starting dose of 160 mg, followed by 80 mg every other week.
Brodalumab: PASI values for a dosage of 210 mg q2weeks, with an additional dose at week 1.
Guselkumab: PASI values at a dosage of 100 mg at weeks 0 and 4, followed by every 8 weeks.
Tildrakizumab: PASI assessments at week 28 for a dosage of 100 mg at weeks 0, 4, and 16.
DISCOVERY OF IL-23 AND THE TH17 LINEAGE
As definitive proof emerged showing that psoriasis was a T cell mediated condition (5), subsequent work focused on the identification and characterization of the various T cell subsets found in the skin and blood. The initial characterization of specific T cell populations was determined by the cytokines produced by specific lymphocyte subsets. For example, CD4+ type 1 T helper (Th1) cells were defined by their production of interferon-γ (IFNγ), whereas Th2 cells were associated with the production of IL-4, IL-5, and IL-13. These “polar” T lymphocyte subsets were frequently used as a method of categorizing autoimmune and inflammatory diseases based on their differential expression of Th1 vs. Th2 cells. In this way, psoriasis was primarily considered to be a predominant Th1 condition due to a strong IFNγ signature found in lesional tissues (6). Therefore, early disease models of psoriasis suggested that IFNγ in cooperation with IL-12, a master regulator of Th1 cell development and differentiation, could be the principal driver of this disease (7, 8).
In the late 1990s, laboratory advances and structure-based alignment tools led to the discovery of novel cytokines related to IL-12. These “gene hunting” technologies identified a new dimeric cytokine that was structurally similar to IL-12 in that it shared its p40 subunit, but also contained a unique p19 subunit (9). The pairing of these p40 and p19 subunits resulted in a novel heterodimeric cytokine that was later named IL-23. Given the structural similarities between IL-23 and IL-12, as well as their concomitant production by activated phagocytic and dendritic cells (DCs), it was reasonable to assume that IL-23 could also regulate the differentiation of the Th1 cell subset. However, it was shown that IL-23 stimulated the production of IL-17 from a subset of CD4+ T cells that were negative for IFNγ and IL-4, a finding that challenged the conventional Th1-Th2 classification system (10).
Definitive proof for a novel IL-17-producing subset of T helper cells (Th17) under the regulation of IL-23 came as a result of studies involving the experimental autoimmune encephalomyelitis (EAE) murine model. Much like human psoriasis, the EAE murine model was initially thought to be mediated by the Th1 cell subset since the disease phenotype of these mice was prevented by the absence (Il12p40−/−) or neutralization of the p40 subunit (11, 12). However, the idea that the EAE model was driven primarily by aberrant Th1 signaling was incongruous with the finding that mice deficient in IFNγ and signal transducer and activator of transcription 1 (STAT1) signaling still developed an autoimmune disease phenotype (13, 14). Subsequent work using mice deficient in IL-12, IL-23, or both found that the EAE model was critically dependent on IL-23, which regulates the differentiation of IL-17 and IL-22-producing subsets of CD4+ lymphocytes (12). A more severe autoimmune phenotype was observed in mice deficient in IL-12 compared to wild-type controls (12) suggesting a potential protective role for the IL-12/Th1 axis in this particular mouse model. Additionally, it was also shown that IL-23 did not stimulate IL-17 production in Th1 and Th2-polarized subsets. These findings challenged the conventional Th1-Th2 classification system and broadened our understanding of the various effector T cell lineages, such as the Th17 subset.
Following the discovery of the Th17 lineage, investigators sought to determine whether this lymphocyte subset was associated with human diseases such as psoriasis. Consistent with findings from the EAE mouse model, psoriatic plaques were found to have upregulation of IL-17-producing lymphocytes (15, 16) as well as the p40 and p19 subunits that make functional IL-23 (17). In contrast, the p35 subunit of IL-12 was not increased in psoriatic tissues, indicating that IL-23 rather than IL-12 was the primary regulator of the pathogenic Th17 cells found in psoriasis lesions. IL-22-producing cells were also found in the cellular infiltrate of psoriatic plaques, leading to the further characterization of the distinct Th22 subset found in humans. IL-17 and IL-22 were shown to induce a number of psoriasis-related genes (e.g. the S100 proteins), while IFNγ had no appreciable effect on these molecules (18). Thus, investigators began to incorporate the presence of multiple T lymphocyte subsets (i.e. Th1, Th17, and Th22) into updated models of psoriasis based on the idea that the sum of genes activated in psoriasis were produced by distinct actions of “polar” T cell cytokines on keratinocytes and other tissue-resident cells (19).
A CURRENT IMMUNE MODEL OF PSORIASIS
Psoriasis is typified by the presence of large, erythematous, scaly plaques commonly found on the scalp, trunk, and extensor surfaces of affected patients. Histologically, psoriatic skin lesions are characterized by hyperproliferative keratinocytes and a mixed cellular infiltrate in the epidermal and dermal layers of the skin. The initiation of psoriasis lesions begins with immune activation in susceptible individuals following environmental stimuli (e.g. exogenous trauma or infection) and/or loss of immune tolerance via the recognition of three recently described psoriasis autoantigens (e.g. LL37/cathelicidin, ADAMTSL5, and PLA2G4D-generated neolipid antigens) (20–22). Psoriatic autoantigens are then presented to CD4+ or CD8+ T lymphocytes by DC subsets that include TNFα/iNOS-producing (TIP)-DC (BDCA-1 negative), resident DC (BDCA-1 and CD11c positive), and/or mature DC populations. TNFα and IL-23 secreted by these activated DCs drive the polarization and clonal expansion of CD4+ and CD8+ IL-17 and IL-22-producing T cells (subsequently referred to as T17 and T22 cells, respectively), leading to the production of considerable amounts of IL-17 and IL-22 in psoriatic plaques (Figure 1).
Figure 1. Current pathogenic model of psoriasis.
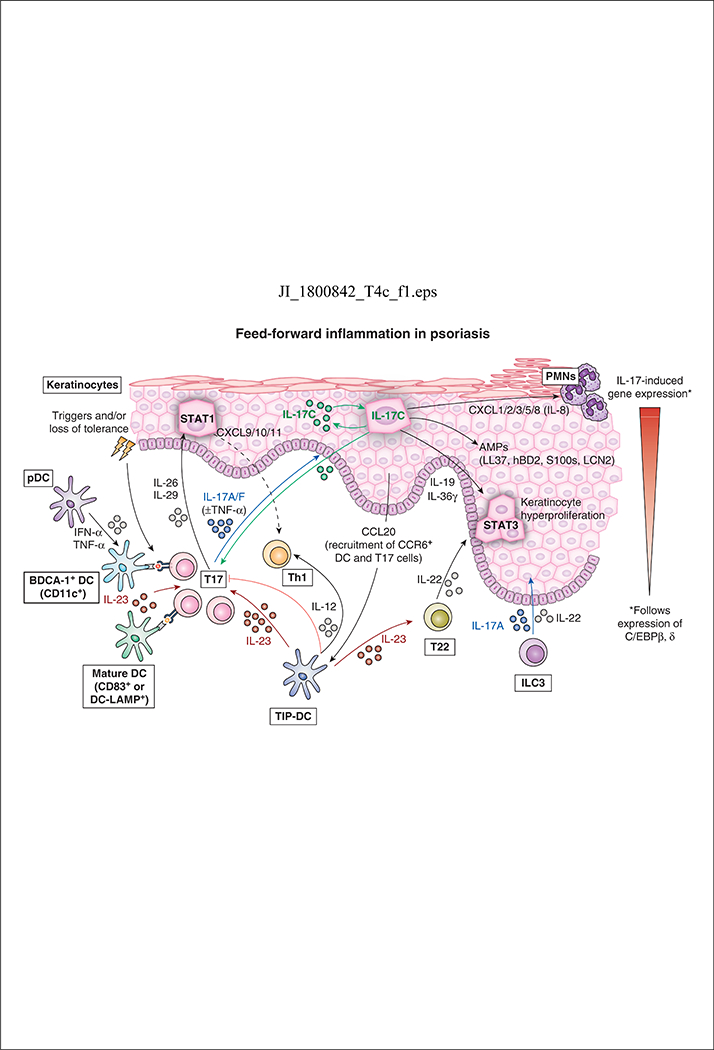
Psoriasis initiation begins with environmental triggers and/or loss of tolerance, which leads to activation of plasmacytoid dendritic cells (pDCs) and IL-23-producing dermal DCs. Several pro-inflammatory DC cell populations present psoriasis autoantigens and trigger T17 cell polarization and clonal expansion. Activated T17 cells produce key cytokines including IL-17, IL-26, IL-29, and TNFα that act on epidermal keratinocytes to promote a feed-forward inflammatory response in skin. IL-17 acting alone or synergistically with TNFα induces expression of psoriasis-related genes in keratinocytes leading to epidermal hyperplasia and the production of antimicrobial peptides (e.g. hBD2, S100s, and LL37/cathelicidin). Keratinocyte-derived CCR20 recruits CCR6+ cells (IL-23-producing TIP-DCs and IL-17-producing T cells) to further promote inflammation. CXCL1/2/3/5/8 are also produced by keratinocytes and recruit neutrophils and macrophages into inflamed skin. IL-23 promotes the clonal expansion and differentiation of T22 cells that produce IL-22, which works together with IL-19/IL-36γ to alter the terminal differentiation and proliferation of keratinocytes. IL-12 produced by activated DCs as well as keratinocyte-derived CXCL9/10/11 promotes the influx of Th1 cells into lesional psoriatic skin. Type 3 innate lymphoid cells (ILC3) in the skin also produce IL-17 and IL-22, which contribute to the development of skin inflammation.
Activated T17 cells in the skin produce several cytokines, including IL-17 (IL-17A/IL-17F), TNFα, IL-26, and IL-29. IL-17, the major effector cytokine in psoriasis, acts alone or synergistically with TNFα to induce the expression and release of many psoriasis-related proteins from keratinocytes, including hBD2, LCN2, S100 proteins, and LL37/cathelicidin (23). The effects of IL-17 on epidermal keratinocytes produce a feed-forward inflammatory response by activating CCAAT enhancer–binding protein (C/EBPβ and δ), STAT1, and nuclear factor κB (NF-κB), which amplify the primary signals driving the development of mature psoriatic plaques (24). IL-22, IL-19, and IL-36 in response to IL-17 contribute to the development of epidermal hyperplasia that gives the skin a thickened, scaly appearance with retained nuclei (parakeratosis) seen histologically. Additionally, the production of keratinocyte-derived antimicrobial peptides (AMPs), such as LL37/cathelicidin, enhances innate immunity and protects the skin from cutaneous infection, whereas the upregulation of CCL20 and CXCL1/2/3/5/8 promote the influx of various immune cell populations (e.g. neutrophils, macrophages, DCs, and CCR6+ cells) into inflamed skin (25–27). Keratinocytes also synthesize IL-17C in response to IL-17A/F and this cytokine amplifies many psoriasis-associated genes that can also stimulate T17 cells to increase IL-17A/F production. Myeloid DCs, which produce significant amounts of IL-23, also promote the differentiation of T17 cells that produce a positive feedback loop that sustains the IL-23/IL-17 signaling pathway in psoriasis lesions.
The production of IL-26 and IL-29 by activated T17 cells also drives STAT1 signaling in keratinocytes, leading to the subsequent release of proinflammatory chemokines CXCL9/10/11 from keratinocytes which recruit Th1 cells into lesional skin (28, 29). Inflammatory TIP-DCs in lesional skin also secrete IL-12 and contribute to the recruitment of Th1 cells in psoriatic plaques. Both keratinocyte-driven chemotaxis and DC-driven T cell polarization/clonal expansion likely contribute to the abundant infiltration of Th1 cells found in psoriasis skin lesions. Of note, infiltration of Th1 cells in psoriatic plaques likely contributes little to the pathogenesis of this condition as demonstrated by the limited clinical efficacy of IFNγ blockade in patients with psoriasis (30), while the IL-12/Th1 axis is potentially suppressive of the IL-23/T17 axis that drives psoriasiform hyperplasia (12).
Innate immunity and psoriasis: neutrophils, innate lymphoid cells, and natural killer cells
The dysregulation of DCs and skin-derived AMPs in psoriatic skin underscore the importance of the innate immune system in this inflammatory disease. As a result, scientists have sought to better understand the specific actions of innate immune cell populations in the initiation and maintenance of psoriasis skin lesions. Neutrophils, innate lymphoid cells (ILCs), and natural killer (NK) cells are among the innate immune cell populations that may contribute to the pathogenesis of psoriasis.
Neutrophils have long been recognized as a common immune cell infiltrate in lesional psoriatic skin, which are recruited into the stratum corneum during early psoriasis and form aggregates known as Munro’s microabscesses. The recruitment of neutrophils into psoriatic skin is likely driven by the increase of TNFα and IL-17 signaling in lesional psoriatic skin, which triggers keratinocytes to secrete neutrophil-recruiting factors such as CXCL-1/2/3/5/8 (26). In turn, neutrophils can secrete a variety of pro-inflammatory signals, including reactive oxygen species, IL-17, LL-37, and the release of neutrophil extracellular traps or NETs (31). However, the role of neutrophils in the pathogenesis of psoriasis is not entirely clear since many psoriatic plaques and murine models of skin inflammation lack Munro’s microabscesses, and neutrophils are not the predominant cellular source of IL-17 in the skin (32–34).
ILCs are another innate immune cell population that may play a pathogenic role in skin disease. ILCs are a rare population of immune cells in healthy skin and blood that are characterized by the lack of antigen specific T or B-cell receptors. Their primary function in the skin is to form a protective barrier in epithelial tissues (e.g. mucosal skin) by initiating a non-specific immune response to skin damage, environmental exposures, or microorganisms. ILCs can be categorized into 3 major groups characterized by transcription factor expression and cytokine production, respectively: Type 1 (T-bet/Th1), Type 2 (RORα and GATA3/Th2), and Type 3 (RORγt/Th17 and Th22) (35). The role of Type 3 ILC (ILC3) cells in psoriatic disease is of particular interest due to the upregulation of this group in psoriasis skin and blood (36, 37) as well as their expression of IL23R, response to IL-23 and IL-1β signaling, and ability to produce IL-23 and IL-17A cytokines. However, despite the association between ILC3s and factors implicated in the IL-23/T17 signaling pathway, no studies have clearly defined the relative contribution of IL-17 by ILC3s versus other cellular sources of IL-17 in the skin of psoriasis patients. Further, the clinical significance and impact of targeted, systemic therapies on ILC3s in the skin has not been determined in a large cohort of plaque psoriasis patients.
Natural killer (NK), the prototypical ILC, and NKT cells have also been implicated in psoriasis. NK cells are capable of producing cytokines central to the pathogenesis of psoriasis, including IFNγ, TNFα, and IL-22 (38, 39). In vitro experiments suggest a potential contributory role for NKT cells in psoriasis since the incubation of NKT cells with CD1d-expressing keratinocytes led to increases in IFNγ production (40, 41). Studies also involving the engraftment of either allogenic normal or pre-psoriatic skin onto severe combined immunodeficiency (SCID) mice, followed by the injection of NKT cells from psoriasis patients, led to the development of psoriatic plaques (41, 42). However, the exact role of NK and NKT cells in psoriasis and extent of their involvement in the pathogenesis of the disease has yet to be determined.
NOVEL IL-17 AND IL-23 ANTAGONISTS FOR THE TREATMENT OF PSORIASIS
The recognition of psoriasis as an auto-inflammatory disease heralded the use of systemic biological agents for the treatment of this condition. By deciphering the immune axes activated in psoriasis, greater specificity and targeting of pathogenic cytokines became possible (Table 1). Among the first biologics used for the treatment of psoriasis were antagonists against TNFα, a broad-acting pro-inflammatory cytokine secreted by a variety of immune cells including macrophages and monocytes. The production of IFNα and TNFα by plasmacytoid DCs in psoriatic skin drives the production of IL-23 by myeloid DCs and the subsequent activation of IL-17-producing lymphocytes. TNFα also acts synergistically with IL-17 to upregulate the expression of inflammatory genes implicated in psoriasis (24).
TNFα antagonists: etanercept, adalimumab, infliximab, and certolizumab
Etanercept, adalimumab, infliximab, and certolizumab received FDA-approval for moderate-to-severe plaque psoriasis in 2004, 2005, 2006, and 2018, respectively. Phase 3 clinical trials for these agents revealed that ~50–80% of all treated plaque psoriasis patients achieved a 75% improvement in the psoriasis area and severity index (PASI75) compared to placebo (43–51). All of these anti-TNFα agents have also proven to be effective at reducing inflammation in the joints of patients with psoriatic arthritis. Approximately 50–65% of patients achieve a 20% improvement in their American College of Rheumatology (ACR20) score after 24 weeks. The efficacy of this drug class appears to be significantly less than their observed benefits in the skin. The primary mechanism by which TNFα antagonists result in disease improvement in treated psoriasis patients is most likely due to its indirect effect on IL-17 signaling via the regulation of IL-23 production from myeloid or CD11c+ DCs. The exact mechanism for the benefits in the joints are not entirely understood.
Common adverse events associated with TNFα antagonists include increased infection risk, reactivation of tuberculosis, injection site reactions, and possibly lymphoma. Worsening of pre-existing congestive heart failure and the development of demyelinating disease have also been reported; thus, the use of TNFα antagonists should be avoided in these patient populations. Interestingly, TNFα inhibitors have also been implicated in the development of paradoxical psoriasiform and eczematous-like cutaneous eruptions that are characterized by a strong type I IFN (IFNα and IFNβ) molecular signature that is inconsistent with the diagnosis of psoriasis or atopic dermatitis (52–54).
IL-12/23 p40 subunit antagonist: ustekinumab
As a human monoclonal antibody against the shared p40 subunit of the IL-12 and IL-23 cytokines, ustekinumab inhibits both the Th1 and T17 signaling pathways that are upregulated in psoriasis. In phase 3 clinical trials (55, 56), ustekinumab demonstrated a PASI75 of 50–80%. It subsequently received FDA approval for the treatment of moderate-to-severe plaque psoriasis in 2009. In the head-to-head phase 3 ACCEPT trial (57), ustekinumab showed superior efficacy to etanercept over a 12-week period, with 70% of patients achieving a PASI75 response with ustekinumab compared to 57% of those receiving etanercept. Ustekinumab also demonstrated efficacy in the treatment of psoriatic arthritis (58, 59), with 40% of patients achieving an ACR20 response after 24 weeks. Resultantly, ustekinumab received FDA-approval for psoriatic arthritis in 2013.
The common adverse events reported with ustekinumab include headache, upper respiratory tract infection, nasopharyngitis, arthralgias, and infections (55, 56), with a comparable safety profile to that of etanercept (57). Rare cases of non-melanoma skin cancers and reversible posterior leukoencephalopathy syndrome have also been reported in patients treated with ustekinumab for psoriasis (60).
IL-17 antagonists: secukinumab, ixekizumab, and brodalumab
While secukinumab and ixekizumab are human monoclonal antibodies against IL-17A, brodalumab antagonizes the IL-17A receptor (IL-17RA) and disrupts signaling of IL-17A, IL-17C, IL-17F, and IL-17A/F heterodimers, giving it potential advantages over selective IL-17A inhibition with secukinumab or ixekizumab. In phase 3 clinical trials (61–65), 30–60% of patients treated with IL-17 inhibitors demonstrated a PASI100 response, showcasing the importance of IL-17 in the pathogenesis of plaque psoriasis. Head-to-head clinical trials of secukinumab versus ustekinumab (66), ixekizumab versus etanercept (61, 62), and brodalumab versus ustekinumab (64) have demonstrated the superior efficacy of the IL-17 antagonists. Secukinumab and ixekizumab have also been FDA-approved for the treatment of psoriatic arthritis based on phase 3 trial results showing 50–60% of patients achieving an ACR20 response after 24 weeks (67–70). Phase 3 results for the use of brodalumab in patients with psoriatic arthritis are pending.
Given similarities in their mechanisms of action, IL-17 antagonists exhibit comparable safety profiles in clinical trials with nasopharyngitis, upper respiratory tract infections, mucocutaneous candidiasis, transient neutropenia, and injection site reactions being the most common adverse effects. Mucocutaneous candidiasis seen with IL-17 inhibition or genetic deficiencies (e.g. chronic mucocutaneous candidiasis) (71) reflects the innate, protective role of this cytokine against microbial pathogens on the skin. Secukinumab and brodalumab were also tested for clinical efficacy in inflammatory bowel disease (IBD) patients without comorbid inflammatory skin disease, and these trials reported some worsening of gastrointestinal symptoms with IL-17 blockade, suggesting use with caution in psoriasis patients with comorbid gastrointestinal disease (72, 73). Importantly, four patients in the AMAGINE 1 and 2 trials committed suicide during the treatment period prompting investigations into a possible association between brodalumab and increased suicidality. This resulted in a black box warning for brodalumab mandating surveillance under the Risk Evaluation and Mitigation Strategy (REMS) program. However, an analysis of more than 4,000 patients treated with brodalumab (9161 patient-years of exposure) did not support a causal relationship between brodalumab and psychiatric adverse events (74).
IL-23 p19 subunit antagonists: guselkumab, tildrakizumab, risankizumab, and mirikizumab
With the discovery of IL-23 as the “master regulator” of T17 cells, several antagonists against the p19 subunit of IL-23 are currently being investigated for the treatment of psoriasis, including guselkumab, tildrakizumab, risankizumab, and mirikizumab. In contrast to ustekinumab, this class of medication targets IL-23 by inhibiting the p19 subunit without disrupting the IL-12 signaling pathway.
Guselkumab was the first selective IL-23 antagonist to receive FDA approval for moderate-to-severe plaque psoriasis in July 2017. In March 2018, tildrakizumab received FDA approval for the same indication. The results of phase 3 clinical trials have indicated that more than 50% of treated patients reach a PASI90 response (75–77), with head-to-head trials of guselkumab versus ustekinumab (78) and tildrakizumab versus etanercept (75) demonstrating superiority of these two biologics. Guselkumab and tildrakizumab are not currently approved for the treatment of psoriatic arthritis.
The most common adverse events associated with the use of guselkumab and tildrakizumab include upper respiratory tract infections, nasopharyngitis, and headaches (75–77). Rates of urticaria and mucocutaneous candidiasis were infrequent and comparable to healthy control subjects. Moreover, no new cases or exacerbation of IBD occurred in the tildrakizumab trials. The impact of IL-23 blockade on the gastrointestinal system or IBD is not fully understood and is under investigation. Follow-up studies investigating the long-term efficacy and safety of guselkumab are needed.
Several other agents that selectively target IL-23 are currently undergoing clinical testing. These other agents include risankizumab (BI 655066) and mirikizumab (LY3074828). These two agents are humanized monoclonal antibodies against the p19 subunit of IL-23 and are in various stages of clinical testing for psoriasis, psoriatic arthritis, and/or IBD. Risankizumab is in phase II testing for psoriatic arthritis and asthma, as well as phase III testing for plaque psoriasis and Crohn’s disease. Finally, mirikizumab is in phase II testing for plaque psoriasis, Crohn’s disease, and ulcerative colitis.
FUTURE PERSPECTIVES AND OPEN QUESTIONS
The ability to selectively target the IL-23/T17 signaling axis in psoriasis has resulted in a number of highly efficacious systemic therapies with excellent safety profiles. As a result of these novel, selective therapies, as many as 80–90% of treated patients are experiencing dramatic improvements in their psoriatic disease. Clinicians, however, continue to encounter subsets of psoriasis patients (e.g. palmoplantar psoriasis, psoriatic arthritis) with severe or recalcitrant disease that does not improve following treatment with novel biologic therapies. The current inability to predict which psoriasis patients will respond to specific therapies often results in a frustrating cycle of medication trial and error for the patient and clinician.
The variable clinical responses to specific systemic therapies and the recalcitrant psoriasis patient population highlight some important open questions in psoriasis research. One promising area of ongoing translational research is the development and testing of bispecific antibodies which would allow for the simultaneous targeting of two molecules with a single antibody (79). For example, early proof-of-concept studies comparing the clinical efficacy of bimekizumab, a humanized IgG antibody that neutralizes IL-17A and IL-17F, suggest that dual blockade may have advantages over therapies that inhibit IL-17A alone (80). Phase II clinical trials for bimekizumab in the treatment of psoriasis were recently completed; however, study results will not be available until later this year.
In a recent phase II clinical trial of 166 patients with moderate-to-severe plaque psoriasis (81), investigators compared the clinical efficacy of risankizumab (antagonist of the p19 subunit of IL-23) against ustekinumab (antagonist of the p40 subunit that is common to both IL-12 and IL-23). This head-to-head trial allowed for the direct comparison of selective IL-23 blockade versus dual inhibition of IL-12 and IL-23 in the treatment of plaque psoriasis. Interestingly, selective blockade of IL-23 with risankizumab showed clinical superiority over inhibition of IL-12 and IL-23 via ustekinumab. This finding was somewhat of a surprise given that both drugs effectively inhibit IL-23. It also demonstrates the complexity of inflammation driven by IL-23 where there is a family of dimeric cytokines which can alternatively share subunits to create 6 distinct cytokines with differing pro- and anti-inflammatory properties (Figure 2).
Figure 2. Extended IL-12 Cytokine Family.
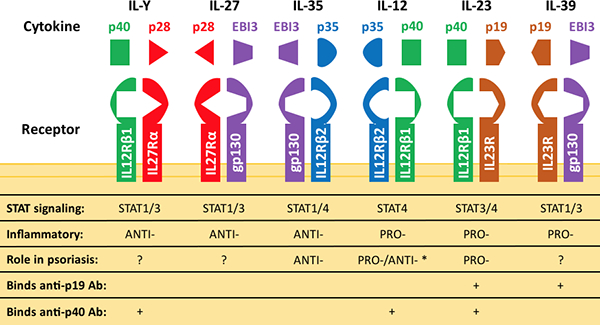
The IL-12 family, which includes IL-12, IL-23, IL-27, IL-35, IL-39, and IL-Y, is made up of heterodimeric cytokines that bind to their respective receptors to mediate inflammation. The different subunits forming each cytokine, as well as their corresponding receptor chains, STAT signaling pathways, and functional roles in psoriasis, are highlighted. Some cytokines can have opposing effects in the skin depending on the biological context, as indicated by an asterisk (*). The specific roles of IL-Y, IL-27, IL-35, and IL-39 in psoriatic disease have not been systematically studied
Using the imiquimod-induced mouse model of psoriasiform dermatitis, which is dependent on the induction of IL-23 and T-cells producing IL-17, Kulig et al. (82) showed that mice lacking the p35 subunit of IL-12 (Il12a−/−) or the IL-12-specific receptor subunit (Il12rb2−/−) developed significantly increased skin inflammation, similar to an earlier finding in the EAE model of multiple sclerosis, in which IL-12 knockout led to worsening inflammation (14). Thus, dual blockade of IL-12 and IL-23 with ustekinumab might lead to lesser disease improvement compared to single blockade of IL-23, as suggested by clinical trial outcomes. Note that an antibody against the p19 subunit can also theoretically neutralize IL-39 (Figure 2), which is another proinflammatory cytokine likely expressed in psoriatic skin. Additionally, blockade of IL-23 through the inhibition of the p19 subunit would also allow IL-12 and IL-Y to maintain their beneficial anti-inflammatory effects on T17-centered inflammation in the skin. Further studies are needed to systematically explore the individual roles of the extended IL-12 cytokine family, which includes IL-12, IL-23, IL-27, IL-35, IL-39, and IL-Y, in psoriasis, as there are reports that the individual subunits of this family (with the exception of p35) are all up-regulated in psoriasis lesions.
One major obstacle hampering the elucidation of the role of the IL-12 cytokine family and the mechanisms of other signaling pathways in psoriasis is the lack of a single animal model that recapitulates the complexity of this inflammatory skin condition in humans. While more than 40 murine models of psoriasis have been proposed to date, each of these models have significant drawbacks. The advantages and disadvantages of the three most common model types used (i.e. inducible, genetically engineered, and xenotransplantation) have been reviewed in detail elsewhere (83–85). In short, these limitations are primarily due to 1) the methods by which inflammation in the skin or joints are induced and evaluated, 2) the inherent immune system and genetic background of the mice, or 3) the human donor tissue variability and technical challenges associated with xenotransplantation (84). Still, the most faithful model of psoriasis may be xenotransplantation of non-lesional psoriatic skin onto the AGR129 mouse background – mice which lack type I and II IFN receptors in addition to being RAG-2−/− (86). The AGR129 murine model has been used to demonstrate the importance of IL-23 in the generation of psoriasis and has also shown that T-cell infiltration into and within the epidermis is essential for disease induction (86, 87). This model also demonstrated that IL-23 inhibition could prevent psoriatic disease induction before clinical studies showed efficacy of this approach. Nevertheless, continued efforts to develop a robust murine model that mimics the genetic perturbations and immune dysregulation observed in human disease is crucial if we are to advance our understanding of immunologic mechanisms driving psoriasis.
CONCLUSIONS
The discovery of the IL-23/T17 signaling pathway and significant therapeutic advances made over the last two decades have made psoriasis one of the most effectively treated chronic inflammatory conditions in all of medicine. It serves as a model of how meticulous research discoveries directly led to the development and approval of highly effective, targeted psoriasis therapies. As our understanding of the etiology and immunologic signals driving psoriatic disease continues to expand, we look forward with optimism towards more effective anti-psoriatic medications and improved treatment strategies for patients with severe or recalcitrant disease
ACKNOWLEDGEMENTS
JEH would like to acknowledge support from The National Psoriasis Foundation. TCC would like to thank the Eighth Core Laboratory, Department of Medical Research, National Taiwan University Hospital, for their support.
JEH was supported in part by The National Psoriasis Foundation/USA Early Career Research Grant. JEH and JGK are supported in part by The Rockefeller University CTSA award grant UL1TR001866 and KL2TR001865 from the National Center for Advancing Translational Sciences (NCATS), National Institutes of Health (NIH) Clinical and Translational Science Award (CTSA) program.
ABBREVIATIONS
- ACR20
American College of Rheumatology 20
- C/EBPβ and δ
CCAAT enhancer–binding protein
- DCs
dendritic cells
- EAE
experimental autoimmune encephalomyelitis
- IBD
inflammatory bowel disease
- IFNγ
interferon-γ
- IL
interleukin
- NF-κB
nuclear factor κB
- PASI
psoriasis area and severity index
- SCID
severe combined immunodeficiency
- STAT1
signal transducer and activator of transcription 1
- Th
T helper
- TNFα
tumor necrosis factor-α
- TIP-DC TNFα
iNOS-producing dendritic cell
REFERENCES
- 1.Rachakonda TD, Schupp CW, and Armstrong AW. 2014. Psoriasis prevalence among adults in the United States. J Am Acad Dermatol 70: 512–516. [DOI] [PubMed] [Google Scholar]
- 2.Gelfand JM, Troxel AB, Lewis JD, Kurd SK, Shin DB, Wang X, Margolis DJ, and Strom BL. 2007. The risk of mortality in patients with psoriasis: results from a population-based study. Arch Dermatol 143: 1493–1499. [DOI] [PubMed] [Google Scholar]
- 3.Takeshita J, Grewal S, Langan SM, Mehta NN, Ogdie A, Van Voorhees AS, and Gelfand JM. 2017. Psoriasis and comorbid diseases: Implications for management. J Am Acad Dermatol 76: 393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takeshita J, Grewal S, Langan SM, Mehta NN, Ogdie A, Van Voorhees AS, and Gelfand JM. 2017. Psoriasis and comorbid diseases: Epidemiology. J Am Acad Dermatol 76: 377–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gottlieb SL, Gilleaudeau P, Johnson R, Estes L, Woodworth TG, Gottlieb AB, and Krueger JG. 1995. Response of psoriasis to a lymphocyte-selective toxin (DAB389IL-2) suggests a primary immune, but not keratinocyte, pathogenic basis. Nat Med 1: 442–447. [DOI] [PubMed] [Google Scholar]
- 6.Lew W, Bowcock AM, and Krueger JG. 2004. Psoriasis vulgaris: cutaneous lymphoid tissue supports T-cell activation and “Type 1” inflammatory gene expression. Trends Immunol 25: 295–305. [DOI] [PubMed] [Google Scholar]
- 7.Austin LM, Ozawa M, Kikuchi T, Walters IB, and Krueger JG. 1999. The majority of epidermal T cells in Psoriasis vulgaris lesions can produce type 1 cytokines, interferon-gamma, interleukin-2, and tumor necrosis factor-alpha, defining TC1 (cytotoxic T lymphocyte) and TH1 effector populations: a type 1 differentiation bias is also measured in circulating blood T cells in psoriatic patients. J Invest Dermatol 113: 752–759. [DOI] [PubMed] [Google Scholar]
- 8.Yawalkar N, Karlen S, Hunger R, Brand CU, and Braathen LR. 1998. Expression of interleukin-12 is increased in psoriatic skin. J Invest Dermatol 111: 1053–1057. [DOI] [PubMed] [Google Scholar]
- 9.Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, Vega F, Yu N, Wang J, Singh K, Zonin F, Vaisberg E, Churakova T, Liu M, Gorman D, Wagner J, Zurawski S, Liu Y, Abrams JS, Moore KW, Rennick D, de Waal-Malefyt R, Hannum C, Bazan JF, and Kastelein RA. 2000. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 13: 715–725. [DOI] [PubMed] [Google Scholar]
- 10.Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, and Gurney AL. 2003. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem 278: 1910–1914. [DOI] [PubMed] [Google Scholar]
- 11.Constantinescu CS, Wysocka M, Hilliard B, Ventura ES, Lavi E, Trinchieri G, and Rostami A. 1998. Antibodies against IL-12 prevent superantigen-induced and spontaneous relapses of experimental autoimmune encephalomyelitis. J Immunol 161: 5097–5104. [PubMed] [Google Scholar]
- 12.Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, Zurawski S, Wiekowski M, Lira SA, Gorman D, Kastelein RA, and Sedgwick JD. 2003. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 421: 744–748. [DOI] [PubMed] [Google Scholar]
- 13.Ferber IA, Brocke S, Taylor-Edwards C, Ridgway W, Dinisco C, Steinman L, Dalton D, and Fathman CG. 1996. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE). J Immunol 156: 5–7. [PubMed] [Google Scholar]
- 14.Bettelli E, Sullivan B, Szabo SJ, Sobel RA, Glimcher LH, and Kuchroo VK. 2004. Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J Exp Med 200: 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lowes MA, Kikuchi T, Fuentes-Duculan J, Cardinale I, Zaba LC, Haider AS, Bowman EP, and Krueger JG. 2008. Psoriasis vulgaris lesions contain discrete populations of Th1 and Th17 T cells. J Invest Dermatol 128: 1207–1211. [DOI] [PubMed] [Google Scholar]
- 16.Haider AS, Lowes MA, Suarez-Farinas M, Zaba LC, Cardinale I, Khatcherian A, Novitskaya I, Wittkowski KM, and Krueger JG. 2008. Identification of cellular pathways of “type 1,” Th17 T cells, and TNF- and inducible nitric oxide synthase-producing dendritic cells in autoimmune inflammation through pharmacogenomic study of cyclosporine A in psoriasis. J Immunol 180: 1913–1920. [DOI] [PubMed] [Google Scholar]
- 17.Lee E, Trepicchio WL, Oestreicher JL, Pittman D, Wang F, Chamian F, Dhodapkar M, and Krueger JG. 2004. Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J Exp Med 199: 125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wolk K, Witte E, Wallace E, Docke WD, Kunz S, Asadullah K, Volk HD, Sterry W, and Sabat R. 2006. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: a potential role in psoriasis. Eur J Immunol 36: 1309–1323. [DOI] [PubMed] [Google Scholar]
- 19.Lowes MA, Bowcock AM, and Krueger JG. 2007. Pathogenesis and therapy of psoriasis. Nature 445: 866–873. [DOI] [PubMed] [Google Scholar]
- 20.Cheung KL, Jarrett R, Subramaniam S, Salimi M, Gutowska-Owsiak D, Chen YL, Hardman C, Xue L, Cerundolo V, and Ogg G. 2016. Psoriatic T cells recognize neolipid antigens generated by mast cell phospholipase delivered by exosomes and presented by CD1a. J Exp Med 213: 2399–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arakawa A, Siewert K, Stohr J, Besgen P, Kim SM, Ruhl G, Nickel J, Vollmer S, Thomas P, Krebs S, Pinkert S, Spannagl M, Held K, Kammerbauer C, Besch R, Dornmair K, and Prinz JC. 2015. Melanocyte antigen triggers autoimmunity in human psoriasis. J Exp Med 212: 2203–2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lande R, Botti E, Jandus C, Dojcinovic D, Fanelli G, Conrad C, Chamilos G, Feldmeyer L, Marinari B, Chon S, Vence L, Riccieri V, Guillaume P, Navarini AA, Romero P, Costanzo A, Piccolella E, Gilliet M, and Frasca L. 2014. The antimicrobial peptide LL37 is a T-cell autoantigen in psoriasis. Nat Commun 5: 5621. [DOI] [PubMed] [Google Scholar]
- 23.Wang CQ, Akalu YT, Suarez-Farinas M, Gonzalez J, Mitsui H, Lowes MA, Orlow SJ, Manga P, and Krueger JG. 2013. IL-17 and TNF synergistically modulate cytokine expression while suppressing melanogenesis: potential relevance to psoriasis. J Invest Dermatol 133: 2741–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hawkes JE, Chan TC, and Krueger JG. 2017. Psoriasis pathogenesis and the development of novel targeted immune therapies. The Journal of allergy and clinical immunology 140: 645–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harper EG, Guo C, Rizzo H, Lillis JV, Kurtz SE, Skorcheva I, Purdy D, Fitch E, Iordanov M, and Blauvelt A. 2009. Th17 cytokines stimulate CCL20 expression in keratinocytes in vitro and in vivo: implications for psoriasis pathogenesis. J Invest Dermatol 129: 2175–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chiricozzi A, Guttman-Yassky E, Suarez-Farinas M, Nograles KE, Tian S, Cardinale I, Chimenti S, and Krueger JG. 2011. Integrative responses to IL-17 and TNF-alpha in human keratinocytes account for key inflammatory pathogenic circuits in psoriasis. J Invest Dermatol 131: 677–687. [DOI] [PubMed] [Google Scholar]
- 27.Homey B, Dieu-Nosjean MC, Wiesenborn A, Massacrier C, Pin JJ, Oldham E, Catron D, Buchanan ME, Muller A, deWaal Malefyt R, Deng G, Orozco R, Ruzicka T, Lehmann P, Lebecque S, Caux C, and Zlotnik A. 2000. Up-regulation of macrophage inflammatory protein-3 alpha/CCL20 and CC chemokine receptor 6 in psoriasis. J Immunol 164: 6621–6632. [DOI] [PubMed] [Google Scholar]
- 28.Stephen-Victor E, Fickenscher H, and Bayry J. 2016. IL-26: An Emerging Proinflammatory Member of the IL-10 Cytokine Family with Multifaceted Actions in Antiviral, Antimicrobial, and Autoimmune Responses. PLoS Pathog 12: e1005624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolk K, Witte K, Witte E, Raftery M, Kokolakis G, Philipp S, Schonrich G, Warszawska K, Kirsch S, Prosch S, Sterry W, Volk HD, and Sabat R. 2013. IL-29 is produced by T(H)17 cells and mediates the cutaneous antiviral competence in psoriasis. Sci Transl Med 5: 204ra129. [DOI] [PubMed] [Google Scholar]
- 30.Harden JL, Johnson-Huang LM, Chamian MF, Lee E, Pearce T, Leonardi CL, Haider A, Lowes MA, and Krueger JG. 2015. Humanized anti-IFN-gamma (HuZAF) in the treatment of psoriasis. J Allergy Clin Immunol 135: 553–556. [DOI] [PubMed] [Google Scholar]
- 31.Lin AM, Rubin CJ, Khandpur R, Wang JY, Riblett M, Yalavarthi S, Villanueva EC, Shah P, Kaplan MJ, and Bruce AT. 2011. Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J Immunol 187: 490–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chiricozzi A, Romanelli P, Volpe E, Borsellino G, and Romanelli M. 2018. Scanning the Immunopathogenesis of Psoriasis. International journal of molecular sciences 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schon MP, Broekaert SM, and Erpenbeck L. 2017. Sexy again: the renaissance of neutrophils in psoriasis. Experimental dermatology 26: 305–311. [DOI] [PubMed] [Google Scholar]
- 34.Yamanaka K, Yamagiwa A, Akeda T, Kondo M, Kakeda M, Habe K, Imafuku S, Sano S, and Mizutani H. 2017. Neutrophils are not the dominant interleukin-17 producer in psoriasis. The Journal of dermatology 44: e170–e171. [DOI] [PubMed] [Google Scholar]
- 35.Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie AN, Mebius RE, Powrie F, and Vivier E. 2013. Innate lymphoid cells--a proposal for uniform nomenclature. Nat Rev Immunol 13: 145–149. [DOI] [PubMed] [Google Scholar]
- 36.Teunissen MBM, Munneke JM, Bernink JH, Spuls PI, Res PCM, Te Velde A, Cheuk S, Brouwer MWD, Menting SP, Eidsmo L, Spits H, Hazenberg MD, and Mjosberg J. 2014. Composition of innate lymphoid cell subsets in the human skin: enrichment of NCR(+) ILC3 in lesional skin and blood of psoriasis patients. J Invest Dermatol 134: 2351–2360. [DOI] [PubMed] [Google Scholar]
- 37.Villanova F, Flutter B, Tosi I, Grys K, Sreeneebus H, Perera GK, Chapman A, Smith CH, Di Meglio P, and Nestle FO. 2014. Characterization of innate lymphoid cells in human skin and blood demonstrates increase of NKp44+ ILC3 in psoriasis. J Invest Dermatol 134: 984–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cella M, Fuchs A, Vermi W, Facchetti F, Otero K, Lennerz JK, Doherty JM, Mills JC, and Colonna M. 2009. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature 457: 722–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ottaviani C, Nasorri F, Bedini C, de Pita O, Girolomoni G, and Cavani A. 2006. CD56brightCD16(−) NK cells accumulate in psoriatic skin in response to CXCL10 and CCL5 and exacerbate skin inflammation. European journal of immunology 36: 118–128. [DOI] [PubMed] [Google Scholar]
- 40.Bonish B, Jullien D, Dutronc Y, Huang BB, Modlin R, Spada FM, Porcelli SA, and Nickoloff BJ. 2000. Overexpression of CD1d by keratinocytes in psoriasis and CD1d-dependent IFN-gamma production by NK-T cells. J Immunol 165: 4076–4085. [DOI] [PubMed] [Google Scholar]
- 41.Nickoloff BJ, Bonish B, Huang BB, and Porcelli SA. 2000. Characterization of a T cell line bearing natural killer receptors and capable of creating psoriasis in a SCID mouse model system. Journal of dermatological science 24: 212–225. [DOI] [PubMed] [Google Scholar]
- 42.Nickoloff BJ, Wrone-Smith T, Bonish B, and Porcelli SA. 1999. Response of murine and normal human skin to injection of allogeneic blood-derived psoriatic immunocytes: detection of T cells expressing receptors typically present on natural killer cells, including CD94, CD158, and CD161. Arch Dermatol 135: 546–552. [DOI] [PubMed] [Google Scholar]
- 43.Papp KA, Tyring S, Lahfa M, Prinz J, Griffiths CE, Nakanishi AM, Zitnik R, van de Kerkhof PC, and Melvin L. 2005. A global phase III randomized controlled trial of etanercept in psoriasis: safety, efficacy, and effect of dose reduction. The British journal of dermatology 152: 1304–1312. [DOI] [PubMed] [Google Scholar]
- 44.Tyring S, Gordon KB, Poulin Y, Langley RG, Gottlieb AB, Dunn M, and Jahreis A. 2007. Long-term safety and efficacy of 50 mg of etanercept twice weekly in patients with psoriasis. Archives of dermatology 143: 719–726. [DOI] [PubMed] [Google Scholar]
- 45.Tyring S, Gottlieb A, Papp K, Gordon K, Leonardi C, Wang A, Lalla D, Woolley M, Jahreis A, Zitnik R, Cella D, and Krishnan R. 2006. Etanercept and clinical outcomes, fatigue, and depression in psoriasis: double-blind placebo-controlled randomised phase III trial. Lancet (London, England) 367: 29–35. [DOI] [PubMed] [Google Scholar]
- 46.Leonardi CL, Powers JL, Matheson RT, Goffe BS, Zitnik R, Wang A, and Gottlieb AB. 2003. Etanercept as monotherapy in patients with psoriasis. The New England journal of medicine 349: 2014–2022. [DOI] [PubMed] [Google Scholar]
- 47.Reich K, Nestle FO, Papp K, Ortonne JP, Evans R, Guzzo C, Li S, Dooley LT, and Griffiths CE. 2005. Infliximab induction and maintenance therapy for moderate-to-severe psoriasis: a phase III, multicentre, double-blind trial. Lancet (London, England) 366: 1367–1374. [DOI] [PubMed] [Google Scholar]
- 48.Gottlieb AB, Evans R, Li S, Dooley LT, Guzzo CA, Baker D, Bala M, Marano CW, and Menter A. 2004. Infliximab induction therapy for patients with severe plaque-type psoriasis: a randomized, double-blind, placebo-controlled trial. Journal of the American Academy of Dermatology 51: 534–542. [DOI] [PubMed] [Google Scholar]
- 49.Menter A, Tyring SK, Gordon K, Kimball AB, Leonardi CL, Langley RG, Strober BE, Kaul M, Gu Y, Okun M, and Papp K. 2008. Adalimumab therapy for moderate to severe psoriasis: A randomized, controlled phase III trial. Journal of the American Academy of Dermatology 58: 106–115. [DOI] [PubMed] [Google Scholar]
- 50.Gottlieb AB, Blauvelt A, Thaci D, Leonardi CL, Poulin Y, Drew J, Peterson L, Arendt C, Burge D, and Reich K. 2018. Certolizumab Pegol for the Treatment of Chronic Plaque Psoriasis: Results through 48 Weeks from Two Phase 3, Multicenter, Randomized, Double-Blinded, Placebo-Controlled Studies (CIMPASI-1 and CIMPASI-2). J Am Acad Dermatol. [DOI] [PubMed] [Google Scholar]
- 51.Lebwohl M, Blauvelt A, Paul C, Sofen H, Weglowska J, Piguet V, Burge D, Rolleri R, Drew J, Peterson L, and Augustin M. 2018. Certolizumab Pegol for the Treatment of Chronic Plaque Psoriasis: Results Through 48 Weeks of a Phase 3, Multicenter, Randomized, Double-Blinded, Etanercept- and Placebo-Controlled Study (CIMPACT). J Am Acad Dermatol. [DOI] [PubMed] [Google Scholar]
- 52.Joyau C, Veyrac G, Dixneuf V, and Jolliet P. 2012. Anti-tumour necrosis factor alpha therapy and increased risk of de novo psoriasis: is it really a paradoxical side effect? Clinical and experimental rheumatology 30: 700–706. [PubMed] [Google Scholar]
- 53.Stoffel E, Maier H, Riedl E, Bruggen MC, Reininger B, Schaschinger M, Bangert C, Guenova E, Stingl G, and Brunner PM. 2017. Analysis of anti-TNF-induced skin lesions reveals strong Th1 activation with some distinct immunological characteristics. Br J Dermatol. [DOI] [PubMed] [Google Scholar]
- 54.Conrad C, Di Domizio J, Mylonas A, Belkhodja C, Demaria O, Navarini AA, Lapointe AK, French LE, Vernez M, and Gilliet M. 2018. TNF blockade induces a dysregulated type I interferon response without autoimmunity in paradoxical psoriasis. Nat Commun 9: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leonardi CL, Kimball AB, Papp KA, Yeilding N, Guzzo C, Wang Y, Li S, Dooley LT, Gordon KB, and P. s. investigators. 2008. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1). Lancet 371: 1665–1674. [DOI] [PubMed] [Google Scholar]
- 56.Papp KA, Langley RG, Lebwohl M, Krueger GG, Szapary P, Yeilding N, Guzzo C, Hsu MC, Wang Y, Li S, Dooley LT, Reich K, and P. s. investigators. 2008. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2). Lancet 371: 1675–1684. [DOI] [PubMed] [Google Scholar]
- 57.Griffiths CE, Strober BE, van de Kerkhof P, Ho V, Fidelus-Gort R, Yeilding N, Guzzo C, Xia Y, Zhou B, Li S, Dooley LT, Goldstein NH, Menter A, and A. S. Group. 2010. Comparison of ustekinumab and etanercept for moderate-to-severe psoriasis. N Engl J Med 362: 118–128. [DOI] [PubMed] [Google Scholar]
- 58.McInnes IB, Kavanaugh A, Gottlieb AB, Puig L, Rahman P, Ritchlin C, Brodmerkel C, Li S, Wang Y, Mendelsohn AM, Doyle MK, and P. S. Group. 2013. Efficacy and safety of ustekinumab in patients with active psoriatic arthritis: 1 year results of the phase 3, multicentre, double-blind, placebo-controlled PSUMMIT 1 trial. Lancet 382: 780–789. [DOI] [PubMed] [Google Scholar]
- 59.Ritchlin C, Rahman P, Kavanaugh A, McInnes IB, Puig L, Li S, Wang Y, Shen YK, Doyle MK, Mendelsohn AM, Gottlieb AB, and P. S. Group. 2014. Efficacy and safety of the anti-IL-12/23 p40 monoclonal antibody, ustekinumab, in patients with active psoriatic arthritis despite conventional non-biological and biological anti-tumour necrosis factor therapy: 6-month and 1-year results of the phase 3, multicentre, double-blind, placebo-controlled, randomised PSUMMIT 2 trial. Ann Rheum Dis 73: 990–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gottlieb A, and Narang K. 2013. Ustekinumab in the treatment of psoriatic arthritis: latest findings and clinical potential. Ther Adv Musculoskelet Dis 5: 277–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gordon KB, Blauvelt A, Papp KA, Langley RG, Luger T, Ohtsuki M, Reich K, Amato D, Ball SG, Braun DK, Cameron GS, Erickson J, Konrad RJ, Muram TM, Nickoloff BJ, Osuntokun OO, Secrest RJ, Zhao F, Mallbris L, Leonardi CL, U.-S. Group, U.-S. Group, and U.-S. Group 2016. Phase 3 Trials of Ixekizumab in Moderate-to-Severe Plaque Psoriasis. N Engl J Med 375: 345–356. [DOI] [PubMed] [Google Scholar]
- 62.Griffiths CE, Reich K, Lebwohl M, van de Kerkhof P, Paul C, Menter A, Cameron GS, Erickson J, Zhang L, Secrest RJ, Ball S, Braun DK, Osuntokun OO, Heffernan MP, Nickoloff BJ, Papp K, Uncover, and U.−. investigators. 2015. Comparison of ixekizumab with etanercept or placebo in moderate-to-severe psoriasis (UNCOVER-2 and UNCOVER-3): results from two phase 3 randomised trials. Lancet 386: 541–551. [DOI] [PubMed] [Google Scholar]
- 63.Langley RG, Elewski BE, Lebwohl M, Reich K, Griffiths CE, Papp K, Puig L, Nakagawa H, Spelman L, Sigurgeirsson B, Rivas E, Tsai TF, Wasel N, Tyring S, Salko T, Hampele I, Notter M, Karpov A, Helou S, Papavassilis C, E. S. Group, and F. S. Group. 2014. Secukinumab in plaque psoriasis--results of two phase 3 trials. N Engl J Med 371: 326–338. [DOI] [PubMed] [Google Scholar]
- 64.Lebwohl M, Strober B, Menter A, Gordon K, Weglowska J, Puig L, Papp K, Spelman L, Toth D, Kerdel F, Armstrong AW, Stingl G, Kimball AB, Bachelez H, Wu JJ, Crowley J, Langley RG, Blicharski T, Paul C, Lacour JP, Tyring S, Kircik L, Chimenti S, Callis Duffin K, Bagel J, Koo J, Aras G, Li J, Song W, Milmont CE, Shi Y, Erondu N, Klekotka P, Kotzin B, and Nirula A. 2015. Phase 3 Studies Comparing Brodalumab with Ustekinumab in Psoriasis. N Engl J Med 373: 1318–1328. [DOI] [PubMed] [Google Scholar]
- 65.Papp KA, Reich K, Paul C, Blauvelt A, Baran W, Bolduc C, Toth D, Langley RG, Cather J, Gottlieb AB, Thaci D, Krueger JG, Russell CB, Milmont CE, Li J, Klekotka PA, Kricorian G, and Nirula A. 2016. A prospective phase III, randomized, double-blind, placebo-controlled study of brodalumab in patients with moderate-to-severe plaque psoriasis. Br J Dermatol 175: 273–286. [DOI] [PubMed] [Google Scholar]
- 66.Blauvelt A, Reich K, Tsai TF, Tyring S, Vanaclocha F, Kingo K, Ziv M, Pinter A, Vender R, Hugot S, You R, Milutinovic M, and Thaci D. 2017. Secukinumab is superior to ustekinumab in clearing skin of subjects with moderate-to-severe plaque psoriasis up to 1 year: Results from the CLEAR study. Journal of the American Academy of Dermatology 76: 60–69.e69. [DOI] [PubMed] [Google Scholar]
- 67.McInnes IB, Mease PJ, Kirkham B, Kavanaugh A, Ritchlin CT, Rahman P, van der Heijde D, Landewe R, Conaghan PG, Gottlieb AB, Richards H, Pricop L, Ligozio G, Patekar M, Mpofu S, and F. S. Group. 2015. Secukinumab, a human anti-interleukin-17A monoclonal antibody, in patients with psoriatic arthritis (FUTURE 2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 386: 1137–1146. [DOI] [PubMed] [Google Scholar]
- 68.Mease PJ, McInnes IB, Kirkham B, Kavanaugh A, Rahman P, van der Heijde D, Landewe R, Nash P, Pricop L, Yuan J, Richards HB, Mpofu S, and F. S. Group. 2015. Secukinumab Inhibition of Interleukin-17A in Patients with Psoriatic Arthritis. N Engl J Med 373: 1329–1339. [DOI] [PubMed] [Google Scholar]
- 69.Mease PJ, van der Heijde D, Ritchlin CT, Okada M, Cuchacovich RS, Shuler CL, Lin CY, Braun DK, Lee CH, Gladman DD, and S.-P. S. Group. 2017. Ixekizumab, an interleukin-17A specific monoclonal antibody, for the treatment of biologic-naive patients with active psoriatic arthritis: results from the 24-week randomised, double-blind, placebo-controlled and active (adalimumab)-controlled period of the phase III trial SPIRIT-P1. Ann Rheum Dis 76: 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nash P, Kirkham B, Okada M, Rahman P, Combe B, Burmester GR, Adams DH, Kerr L, Lee C, Shuler CL, Genovese M, and S.-P. S. Group. 2017. Ixekizumab for the treatment of patients with active psoriatic arthritis and an inadequate response to tumour necrosis factor inhibitors: results from the 24-week randomised, double-blind, placebo-controlled period of the SPIRIT-P2 phase 3 trial. Lancet 389: 2317–2327. [DOI] [PubMed] [Google Scholar]
- 71.Puel A, Cypowyj S, Bustamante J, Wright JF, Liu L, Lim HK, Migaud M, Israel L, Chrabieh M, Audry M, Gumbleton M, Toulon A, Bodemer C, El-Baghdadi J, Whitters M, Paradis T, Brooks J, Collins M, Wolfman NM, Al-Muhsen S, Galicchio M, Abel L, Picard C, and Casanova JL. 2011. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science (New York, N.Y.) 332: 65–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, Higgins PD, Wehkamp J, Feagan BG, Yao MD, Karczewski M, Karczewski J, Pezous N, Bek S, Bruin G, Mellgard B, Berger C, Londei M, Bertolino AP, Tougas G, Travis SP, and Secukinumab G in Crohn’s Disease Study. 2012. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut 61: 1693–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Targan SR, Feagan B, Vermeire S, Panaccione R, Melmed GY, Landers C, Li D, Russell C, Newmark R, Zhang N, Chon Y, Hsu YH, Lin SL, and Klekotka P. 2016. A Randomized, Double-Blind, Placebo-Controlled Phase 2 Study of Brodalumab in Patients With Moderate-to-Severe Crohn’s Disease. Am J Gastroenterol 111: 1599–1607. [DOI] [PubMed] [Google Scholar]
- 74.Lebwohl MG, Papp KA, Marangell LB, Koo J, Blauvelt A, Gooderham M, Wu JJ, Rastogi S, Harris S, Pillai R, and Israel RJ. 2018. Psychiatric adverse events during treatment with brodalumab: Analysis of psoriasis clinical trials. J Am Acad Dermatol 78: 81–89 e85. [DOI] [PubMed] [Google Scholar]
- 75.Reich K, Papp KA, Blauvelt A, Tyring SK, Sinclair R, Thaci D, Nograles K, Mehta A, Cichanowitz N, Li Q, Liu K, La Rosa C, Green S, and Kimball AB. 2017. Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE 1 and reSURFACE 2): results from two randomised controlled, phase 3 trials. Lancet 390: 276–288. [DOI] [PubMed] [Google Scholar]
- 76.Blauvelt A, Papp KA, Griffiths CE, Randazzo B, Wasfi Y, Shen YK, Li S, and Kimball AB. 2017. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the continuous treatment of patients with moderate to severe psoriasis: Results from the phase III, double-blinded, placebo- and active comparator-controlled VOYAGE 1 trial. J Am Acad Dermatol 76: 405–417. [DOI] [PubMed] [Google Scholar]
- 77.Reich K, Armstrong AW, Foley P, Song M, Wasfi Y, Randazzo B, Li S, Shen YK, and Gordon KB. 2017. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the treatment of patients with moderate to severe psoriasis with randomized withdrawal and retreatment: Results from the phase III, double-blind, placebo- and active comparator-controlled VOYAGE 2 trial. J Am Acad Dermatol 76: 418–431. [DOI] [PubMed] [Google Scholar]
- 78.Langley RG, Tsai TF, Flavin S, Song M, Randazzo B, Wasfi Y, Jiang J, Li S, and Puig L. 2018. Efficacy and safety of guselkumab in patients with psoriasis who have an inadequate response to ustekinumab: results of the randomized, double-blind, phase III NAVIGATE trial. Br J Dermatol 178: 114–123. [DOI] [PubMed] [Google Scholar]
- 79.Torres T, Romanelli M, and Chiricozzi A. 2016. A revolutionary therapeutic approach for psoriasis: bispecific biological agents. Expert Opin Investig Drugs 25: 751–754. [DOI] [PubMed] [Google Scholar]
- 80.Glatt S, Baeten D, Baker T, Griffiths M, Ionescu L, Lawson ADG, Maroof A, Oliver R, Popa S, Strimenopoulou F, Vajjah P, Watling MIL, Yeremenko N, Miossec P, and Shaw S. 2017. Dual IL-17A and IL-17F neutralisation by bimekizumab in psoriatic arthritis: evidence from preclinical experiments and a randomised placebo-controlled clinical trial that IL-17F contributes to human chronic tissue inflammation. Ann Rheum Dis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Papp KA, Blauvelt A, Bukhalo M, Gooderham M, Krueger JG, Lacour JP, Menter A, Philipp S, Sofen H, Tyring S, Berner BR, Visvanathan S, Pamulapati C, Bennett N, Flack M, Scholl P, and Padula SJ. 2017. Risankizumab versus Ustekinumab for Moderate-to-Severe Plaque Psoriasis. N Engl J Med 376: 1551–1560. [DOI] [PubMed] [Google Scholar]
- 82.Kulig P, Musiol S, Freiberger SN, Schreiner B, Gyulveszi G, Russo G, Pantelyushin S, Kishihara K, Alessandrini F, Kundig T, Sallusto F, Hofbauer GF, Haak S, and Becher B. 2016. IL-12 protects from psoriasiform skin inflammation. Nat Commun 7: 13466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gudjonsson JE, Johnston A, Dyson M, Valdimarsson H, and Elder JT. 2007. Mouse models of psoriasis. J Invest Dermatol 127: 1292–1308. [DOI] [PubMed] [Google Scholar]
- 84.Hawkes JE, Adalsteinsson JA, Gudjonsson JE, and Ward NL. 2018. Research Techniques Made Simple: Murine Models of Human Psoriasis. J Invest Dermatol 138: e1–e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hawkes JE, Gudjonsson JE, and Ward NL. 2017. The Snowballing Literature on Imiquimod-Induced Skin Inflammation in Mice: A Critical Appraisal. J Invest Dermatol 137: 546–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Boyman O, Hefti HP, Conrad C, Nickoloff BJ, Suter M, and Nestle FO. 2004. Spontaneous development of psoriasis in a new animal model shows an essential role for resident T cells and tumor necrosis factor-alpha. J Exp Med 199: 731–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tonel G, Conrad C, Laggner U, Di Meglio P, Grys K, McClanahan TK, Blumenschein WM, Qin JZ, Xin H, Oldham E, Kastelein R, Nickoloff BJ, and Nestle FO. 2010. Cutting edge: A critical functional role for IL-23 in psoriasis. J Immunol 185: 5688–5691. [DOI] [PMC free article] [PubMed] [Google Scholar]