

Summary
Genomes are promiscuously transcribed, necessitating mechanisms that facilitate the sorting of RNA for function or destruction. The polyA (pA) tail is one such distinguishing feature, which in the Saccharomyces cerevisiae nucleus is bound by the Nab2p protein, yielding transcript protection. As Nab2p also contacts the main nuclear export factor Mex67p, we asked whether transport kinetics contributes to RNA sorting. Indeed, 3′ end sequencing of newly transcribed pA+ RNAs demonstrates that nuclear depletion of Mex67p elicits their instant and global decay. A similar phenotype is evident upon inactivation of other export factors and proportional to the amount of nuclear pA+ RNA. As RNA expression is partially rescued by Nab2p overexpression, we propose that an export block out-titrates Nab2p onto nuclear-retained pA+ RNA, reducing the pool of Nab2p available to protect new transcripts. More generally, we suggest that nuclear RNA decay, negotiated by Nab2p availability, aids in balancing cellular transcript supply with demand.
Keywords: nuclear export of pA+ RNA, nuclear degradation of pA+ RNA, Mex67p, Nab2p, transcription
Graphical Abstract
Highlights
-
•
An export block triggers nuclear accumulation of pA+ RNA and decay of newly made RNA
-
•
Retained RNA sequesters pA-binding protein Nab2p, preventing protection of new RNA
-
•
Efficient RNA export is essential for cellular mRNA stability
Tudek et al. show that nuclear accumulation of existing pA+ RNA, due to an export block, results in the out-titration of the pA-binding protein Nab2p away from newly synthesized pA+ RNAs. The absence of Nab2p results in transcript decay, which highlights the importance of efficient nuclear export for mRNA stability.
Introduction
RNA polymerase II (RNAPII) transcribes a variety of RNAs with different half-lives, reflecting their cellular utilities. In S. cerevisiae these include mRNAs as well as several types of non-coding RNAs, such as small nuclear/nucleolar (sn/sno) RNAs, cryptic unstable transcripts (CUTs), stable unannotated transcripts (SUTs), and Xrn1p-sensitive unstable transcripts (XUTs) (Wyers et al., 2005, Xu et al., 2009, van Dijk et al., 2011). Transcription termination at sn/snoRNA and CUT transcription units (TUs) is mediated by the Nrd1p/Nab3p/Sen1p (NNS) complex, which recruits the non-processive Trf4p/Air2p/Mtr4p-polyadenylation (TRAMP) complex to stimulate the 3′-5′ exonucleolytic RNA exosome complex for complete decay of CUTs or for the 3′ end maturation of sn/snoRNAs (Porrua and Libri, 2015, Vasiljeva and Buratowski, 2006, Wyers et al., 2005). In contrast, transcription termination and 3′ end processing of most mRNAs, as well as SUTs and XUTs, depend on the cleavage factor I (CFI)/cleavage and polyadenylation factor (CPF) complexes that processively add polyA (pA) tails to RNA 3′ ends, conferring RNA stability (van Dijk et al., 2011, Porrua and Libri, 2015).
In S. cerevisiae, pA tail function is mediated by the dedicated pA-binding proteins (PABPs) Nab2p and Pab1p, which at steady state are nuclear and cytoplasmic, respectively. Nab2p binds to the nascent pA tail of newly synthesized RNA (Batisse et al., 2009, Kelly et al., 2007, Kelly and Corbett, 2009, Hector et al., 2002) and remains bound during nuclear export to the cytoplasm, where it is replaced by Pab1p (Green et al., 2002, Aitchison et al., 1996, Tran et al., 2007). Using the anchor-away (AA) technique (Haruki et al., 2008) to rapidly deplete nuclear Nab2p, we recently demonstrated that its binding to nascent pA tails is essential to protect newly made mRNA from decay by the nuclear exosome. In vitro Nab2p can physically block exosome access to a polyadenylated substrate (Schmid et al., 2015), but whether this is its main mode of action in vivo was not explored. In particular, Nab2p partakes in mRNA nuclear export (Green et al., 2002, Hector et al., 2002, Marfatia et al., 2003, Grant et al., 2008, Iglesias et al., 2010), which might contribute to the timely “escape” of transcripts from the degradative environment of the nucleus.
mRNA nuclear export is mediated by a set of RNA-binding proteins, which converge at the key Mex67p-Mtr2p heterodimer (Segref et al., 1997, Santos-Rosa et al., 1998). Targeting of Mex67p-Mtr2p to transcripts in vivo is facilitated by RNA-binding adaptor proteins, such as the SR-like protein Npl3p, the Yra1p subunit of the transcription-export (TREX) complex, and Nab2p. Prior to exit through the nuclear pore complex (NPC), Yra1p dissociates (Gilbert and Guthrie, 2004, Iglesias et al., 2010, Hautbergue et al., 2008, Kelly and Corbett, 2009), while the remaining export factors form contacts with nucleoporins (NUPs) of the NPC and stay bound to the mRNA during NPC traversal (Terry and Wente, 2007, Grant et al., 2008). Upon arrival of the messenger ribonucleoprotein (mRNP) at the cytoplasmic side, the Dbp5p helicase is activated by its interaction with the NUP Gle1p (Hodge et al., 1999), which then leads to release of the mRNP into the cytoplasm and relocation of shuttling proteins such as Mex67p-Mtr2p, Npl3p, and Nab2p back into the nucleus (Tran et al., 2007, Lund and Guthrie, 2005).
Interestingly, mutation or depletion of selected mRNA export factors was shown to elicit nuclear exosome-dependent decay of the heat shock (hs)-inducible transcripts HSP104 and SSA4 (Rougemaille et al., 2007, Libri et al., 2002, Assenholt et al., 2008, Jimeno et al., 2002). We therefore wondered whether this phenotype might reflect a mechanistic aspect of the global mRNA decline observed in Nab2p-depleted cells (Schmid et al., 2015). Here, we show that rapid nuclear depletion of Mex67p compromises production of pA+ RNAs by triggering their immediate decay without affecting their transcription. This is a general phenotype triggered by defective nuclear export of pA+ RNA, with the degree of transcript decay following the strength of the export block. Finally, we demonstrate increased binding of Nab2p to pA+ RNA during an export block and that the associated RNA decay phenotype can be partially rescued by Nab2p overexpression. This suggests that nuclear pA+ RNA accumulation depletes the available pool of Nab2p, leaving pA tails of newly produced RNAs unprotected and subject to decay. Our results therefore establish Nab2p as a limiting and essential factor for nuclear mRNA production and highlight the importance of rapid pA+ RNA export for gene expression.
Results
Rapid Nuclear Depletion of Mex67p Globally Inhibits the Net Production of New pA+ RNA
Because of its tight connection to mRNA export factors, we asked whether the previously demonstrated requirement of Nab2p to protect newly produced mRNA from decay (Schmid et al., 2015) might relate to its role in nuclear export. We therefore compared the phenotypes induced by rapid nuclear depletion of Nab2p and Mex67p by taking advantage of the AA system in which the AA-tagged protein of interest is nuclear depleted following rapamycin addition. As previously described, and consistent with the essential nature of these proteins, nuclear depletion of Mex67p or Nab2p resulted in cell death (Figure S1A; Haruki et al., 2008, Schmid et al., 2015). Moreover, loss of nuclear Mex67p caused an instant and dramatic nuclear accumulation of pA+ RNA (Figure 1A), consistent with the central role of the protein in mRNA export (Santos-Rosa et al., 1998, Haruki et al., 2008). In contrast, nuclear depletion of Nab2p caused only modest pA+ RNA accumulation, despite a clear nuclear depletion of the protein (see below), probably because of the presence of other adaptor proteins capable of recruiting Mex67p-Mtr2p. Despite these different pA+ RNA localization phenotypes, the abundance of selected mRNAs was significantly decreased in both Mex67- and Nab2-AA, but not wild-type (WT), cells upon rapamycin treatment (Figure S1B). Some transcripts, such as HHF1, HTA2, RRP6, RPS9A, and TLC1-pA (red-colored series), a precursor of the telomerase component TLC1, were decreased as soon as 10 min after rapamycin addition, whereas others, such as PGK1, PMA1, TDH3, RPL21A, RPL36A (blue-colored series), and NAB2 (orange color) declined at a slower rate, especially in Mex67-AA cells. In contrast, levels of SCR1, a non-adenylated product of RNAPIII, were not downregulated throughout the course of the experiment. Only very few protein-coding transcripts increased in abundance upon Mex67p depletion. However, this is likely due to special mechanistic circumstances (see below), and the overall initial resemblance of results from Mex67- and Nab2-AA cells encouraged us to compare RNA production in the two strains genome-wide.
Figure 1.

Rapid Nuclear Depletion of Mex67p Results in Global Inhibition of pA+ RNA Net Production
(A) Fluorescent in situ hybridization (FISH) analysis of pA+ RNA using a Cy3-labeled dT18 DNA/LNA probe on fixed WT, Nab2-AA, or Mex67-AA cells subjected to rapamycin treatment for 15 min at 30°C (top). FISH Cy3 images were adjusted to a common display range (defined as the highest and lowest pixel values for the three images) to allow comparison between strain phenotypes. DAPI staining was used to visualize chromatin-rich regions of cell nuclei (bottom).
(B) Schematic workflow of the pA+ RNA 3′ end sequencing experiment. Mex67-AA and Nab2-AA cultures were treated with 4tU for 10 min (“Control (0min)”) or pre-treated with rapamycin for 5 min before 4tU addition (“Depletion (15 min)”). Cells that were not treated with 4tU or rapamycin were used as reference to estimate the background of the 4tU purification (“IP background”). All obtained RNA samples were supplemented with S. pombe spike-in transcripts, biotinylated, and purified on streptavidin beads. Both total and 4tU-purified fractions were subjected to pA+ RNA 3′ end sequencing.
(C) Log2 fold changes (FC) of pA+ RNA net production in 4tU-labeled Mex67-AA (x axis) and Nab2-AA (y axis) cells treated with rapamycin for 15 min and compared with their respective controls (no rapamycin). Three major pA+-containing transcript classes were analyzed: mRNAs, SUTs, and XUTs. Only transcripts purified significantly above background in the control samples are depicted. The number of genes (n), Spearman correlation coefficients (rho), and median (m) values for both datasets are specified on each graph. See also Table S1.
See also Figure S1.
Given the specific impact of Mex67p and Nab2p depletion on polyadenylated RNA, we chose to sequence pA+ RNA 3′ ends from cells subjected to a brief (15 min) rapamycin exposure, minimizing any secondary effects due to protein inactivation. However, because long-lived transcripts synthesized prior to Mex67p or Nab2p nuclear depletion would mask effects on newly synthesized RNA, we also metabolically labeled cells with 4-thiouracil (4tU) 5 min after rapamycin addition to allow the synthesis of new RNA for 10 min before its specific purification (Figure 1B). We calculated fold changes in mRNAs, SUTs, and XUTs levels between the 15 and 0 min depletion time points for both strains. Two fractions were analyzed, the steady-state transcript levels (Figure S1C, “total RNA”) and RNA “net production” within the 10 min labeling period (Figure 1C, “4tU RNA”), understood as the RNA amount deriving from transcription minus any decay. Mex67-AA and Nab2-AA strains were compared on the x and y axes, respectively. Consistent with previous data (Schmid et al., 2015) and single-transcript analysis (Figure S1B), modest but significant declines in mRNA, SUT, and XUT levels were observed in the total RNA fraction upon nuclear depletion of Mex67p or Nab2p (Figure S1C). Importantly, this drop in expression was greatly exacerbated in the 4tU-labeled fractions (Figure 1C).
Although the effects of Nab2p nuclear depletion on mRNA levels and net production were quantitatively weaker than those of the Mex67p depletion, the response triggered was qualitatively similar, with Spearman correlation coefficients of 0.588 between the two total fractions (Figure S1C) and 0.666 between the 4tU-labeled samples (Figure 1C). Changes in SUT and XUT expression correlated less strongly between samples but showed globally similar downregulation. The more robust depletion effect seen for the 4tU-labeled samples validated the quality of the 4tU RNA purification and indicated that the observed phenotypes were caused by an impairment of de novo pA+ RNA net production rather than accelerated decay of pre-existing cytoplasmic transcripts. These conclusions were reinforced by the observation that downregulation of mRNA levels were in agreement with cytoplasmic decay rates considering the total, but not the 4tU-labeled, fractions (Figure S1D). This was the case for both published data (Sun et al., 2012, Presnyak et al., 2015, Miller et al., 2011) and estimates derived from our own data. In other words, a decline in levels of short-lived transcripts could readily be observed in the total fraction, whereas individual transcript half-lives had no relation to the strength of new pA+ RNA net production inhibition. We conclude that nuclear depletion of Mex67p and Nab2p results in a rapid and strong genome-wide inhibition of the net production of transcripts from CFI/CPF-dependent TUs.
Decreased pA+ RNA Net Production in Mex67p- and Nab2p-Depleted Cells Is Due to Increased Transcript Decay
The massive downregulation of pA+ RNA net production in Nab2p and Mex67p nuclear-depleted cells could be due to either a global transcriptional shutdown or rapid decay of newly made RNA. We did not observe any substantial correlation between the degree of 4tU RNA downregulation in Nab2p or Mex67p nuclear-depleted cells and various published transcription estimates (Figure 2A; Mayer et al., 2010, Warfield et al., 2017, Churchman and Weissman, 2011, Milligan et al., 2016), suggesting that the decrease in pA+ RNA net production occurs post-transcriptionally. Nevertheless, to irrevocably address this critical question, we evaluated transcription levels of the affected strains, using a 2 min pulse of 4tU labeling followed by the simultaneous detection of mature pA+ RNA and nascent pA− 3′ ends (Figure 2B), the latter constituting a reliable transcription measure (Schmid et al. 2018 [this issue of Cell Reports]).
Figure 2.

Decreased pA+ RNA Net Production in Mex67p- and Nab2p-Depleted Cells Is Due to Increased Transcript Decay
(A) Spearman correlation coefficient matrix comparing mRNA log2 FC of 15/0 min rapamycin ratios in Mex67- and Nab2-AA cells as in Figures 1C and S1C against various estimates of transcription rates. Transcription estimates were derived from RNAPII NET-seq (Churchman and Weissman 2011), RNAPII-CRAC (Milligan et al., 2016), RNAPII-ChIP-seq (Warfield et al., 2017), and RNAPII-ChIP-tiling (Mayer et al., 2010) data.
(B) Schematic workflow of the pA+/pA− RNA 3′ end sequencing experiment. Cells were treated with 4tU for 2 min. In case of the Mex67-AA- and Nab2-AA-depleted cells, cultures were pre-treated with rapamycin for 13 min before 4tU labeling. Addition of S. pombe spike-in RNAs, biotinylation, and purification of 4tU-labeled RNA were done as described in Figure 1B. Samples were then split in two aliquots, in which one was subjected to pA+ RNA 3′ end sequencing as in Figure 1B, and the other was in vitro polyadenylated and rRNA-depleted prior to pA+ RNA 3′ end sequencing.
(C) Genome Browser view of spike-in normalized pA− and pA+ RNA 3′ end reads derived from 2 min 4tU-labeling experiments of untreated (0′) and 15 min nuclear-depleted (15′) Mex67-AA cells, spanning across the gene-coding strand of PGK1 on chromosome III. Annotations below the tracks show genomic A-rich sites masked in the analysis and boundaries of translated and transcribed regions as previously defined in www.yeastgenome.org and Xu et al. (2009), respectively.
(D) Levels of nascent (“pA− (body)”: mean of pA− signal in the region from gene TSS to 200 bp upstream of TES) and mature (“pA+ (3′ end)”: mean of pA+ signal in the region from 200 bp up- and downstream of TES) mRNAs in 2 min 4tU-labeled samples in Mex67-AA (left) and Nab2-AA (right) cells after 0 (red violins) or 15 (blue violins) min of rapamycin treatment. All signals are shown relative to S. pombe spike-ins, with background subtracted, log2 and length scaled. Levels of transcripts that were not purified above background were set to a pseudocount of 10−6. Violin plots shown are overlaid with box plots depicting the median values with boxes demarcating first and third quartiles of the data. See also Table S2.
See also Figure S2.
Sequence reads mapped across five regions of chromosomes I, III, VI, and VII (Figures 2C and S2A) demonstrated that pA+ 3′ ends from 4tU-labeled samples mainly detected the annotated 3′ ends of the relevant mRNAs, whereas pA− 3′ ends were scattered along the analyzed TUs. Summing pA− reads mapping to protein-coding gene bodies (from transcription start sites [TSSs] to 200 bp upstream of transcript end sites [TESs]) thus yielded a transcription estimate, whereas pA+ reads mapping to gene ends (TES ± 200 bp) yielded an estimate of mature pA+ RNA net production. A subsequent comparison of mRNA gene body pA− 3′ end read densities between rapamycin-treated and untreated Mex67-AA and Nab2-AA cells showed only modest differences (Figures 2C, 2D, and S2A). Consistently, chromatin immunoprecipitation (ChIP) of the RNAPII subunit Rbp3p on selected downregulated genes did not reveal any major changes between control and Mex67p nuclear-depleted cells (Figure S2B, gray series). In contrast, pA+ 3′ ends were robustly downregulated in the 4tU-labeled fractions as a consequence of nuclear depletion of either Mex67p or Nab2p (Figures 2C, 2D, and S2A), demonstrating that mRNA net production was diminished despite virtually unaltered transcription. We therefore conclude that nuclear depletion of Mex67p or Nab2p triggers the rapid decay of newly produced pA+ RNA.
We then asked which nuclease might be responsible. Examination of new RNA production in a Mex67-AA/rrp6Δ background did not reveal any substantial rescue (Figure S2C). Simultaneous depletion of both exosome nucleases, Rrp6p and Dis3p, resulted in the predicted stabilization of its NEL025c target (Figure S2D) but did not rescue the decay of new hs RNAs under export block conditions (Figure S2E). We then turned to depletion, via the auxin-inducible degron (AID; Morawska and Ulrich, 2013), of one or both of the major yeast 5′-3′ nucleases, the predominantly cytoplasmic Xrn1p and the nuclear Rat1p enzymes. This yielded strains with the predicted stabilization of 3′ extended read-through fragments of two selected mRNAs (Figure S2F). However, when examining total RNA levels from Rat1-AID/Mex67-AA or Rat1-AID/Xrn1-AID/Mex67-AA cells (Figure S2G), only mild rescue of some RNA levels could be detected. We conclude that neither of the tested nucleases is solely responsible for the decay of new RNA. Yet another enzyme might partake in the decay or, perhaps more likely, degradation is performed redundantly by several nuclear decay factors.
Inspection of our data also revealed increased levels of pA− 3′ end signals downstream the TESs of many protein-coding genes in Mex67p nuclear-depleted cells, which was less visible in Nab2p-depleted cells (Figure S2H). This implied the rapid induction of a transcription termination defect upon Mex67p inactivation, which was previously observed on reporter genes (Hammell et al., 2002) and verified by RT-qPCR analysis of 3′ extended RNAs derived from the HHF1, RPL36A, and TDH3 genes (Figure S2I, green series). As 3′ extended transcripts are usually labile (Minvielle-Sebastia et al., 1991, Libri et al., 2002), we considered the possibility that newly made pA+ RNAs in Mex67p-depleted cells were unstable because of this transcription termination defect. However, not all loci showed an increased signal downstream of their TESs, and there was only a marginal negative correlation between the severity of the defect and the extent to which the same mRNA was downregulated (Figures S2H and S2J). This argued against a general causative link between the phenotypes and suggested that transcription termination defects only contribute to RNA decline in selected cases. Still, a termination defect, though of a different type, may explain the aforementioned stabilization of a small set of RNAs. The upregulation of one such target in Mex67p-depleted cells, the NRD1 mRNA (Figure S2I, red bar), was for example likely due to a transcription termination defect at the NNS-responsive region positioned in the 5′ end of the NRD1 gene (Porrua and Libri, 2015). Consistent with this notion, RNAPII ChIP levels increased specifically downstream of the NRD1 gene NNS site upon Mex67p nuclear depletion (Figure S2B, compare NRD1 5′ end with 3′ end). We also observed stabilization of several CUTs (Figure S2I, orange series). This was seemingly not caused by decreased Rrp6p levels, which remained unaffected up to 60 min of Mex67p or Nab2p nuclear depletion (Figure S2K). Instead, analysis of pA− reads revealed increased density downstream of many CUT loci (Figure S2L), consistent with read-through transcription as observed at the NRD1 locus. This suggests that NNS-mediated transcription termination is, to some extent, also perturbed upon Mex67p nuclear depletion (see Discussion).
Decreased mRNA Net Production Correlates with the Severity of the Nuclear Export Block
Previous studies using mutated variants of diverse mRNA export factors found hs RNA expression to be impaired during an export block (Rougemaille et al., 2007, Libri et al., 2002, Assenholt et al., 2008, Jimeno et al., 2002). As this phenotype is reminiscent of what we observed upon Nab2p and Mex67p nuclear depletion, decay of newly made pA+ RNA might be a general phenotype linked to export factor inactivation. To test this prediction, we tagged the Mex67p cofactor Mtr2p, the export adaptors Yra1p and Npl3p, and the NPC-associated RNA helicase Dbp5p with the AA epitope and analyzed the phenotypic consequences of nuclear depletion of these proteins.
Rapamycin addition to Mtr2-AA, Yra1-AA, and Dbp5-AA cells resulted in their lethality, whereas Npl3-AA cells were severely growth impaired (Figure S3A), as expected (Bossie et al., 1992, Portman et al., 1997, Hodge et al., 1999, Santos-Rosa et al., 1998). We next compared the pA+ RNA localization profiles of the AA strains after 15, 40, and 70 min of rapamycin treatment (Figures 3A and 3B). Depletion of Mtr2p and Dbp5p yielded a “Mex67-AA-like” effect with severe nuclear pA+ RNA accumulation (6- to 9-fold) 15 min post-rapamycin addition. In contrast, loss of nuclear Yra1p resulted in more modest pA+ RNA accumulation and depletion of Npl3p did not yield a detectable phenotype, despite severely diminished nuclear Npl3p levels (Figure S3B). Interestingly, all mutant-specific pA+ RNA localization profiles were essentially established already after 15 min of rapamycin treatment, reaching a plateau thereafter (Figure 3B). Thus, a mutant-specific steady-state concentration of nuclear pA+ RNAs is established rapidly, likely reflecting the extent of the export defect. In order to evaluate a possible connection to mRNA synthesis, we employed 4tU labeling to measure the net production of selected mRNAs and found it to be decreased to largely equal extents in Nab2-, Mex67-, Mtr2-, and Dbp5-AA cells after 15 min depletion (Figure 3C). Despite a longer (30 min) nuclear depletion, loss of Yra1p yielded a more modest phenotype, whereas Npl3p depletion only very mildly impaired mRNA net production. We conclude that depletion of all tested export factors resulted in decreased mRNA net production, which, with an apparent exception of Nab2p-depleted cells, appeared related to the degree of the nuclear pA+ RNA export phenotype.
Figure 3.

Decreased mRNA Net Production Correlates with the Severity of the Nuclear Export Block
(A) FISH analysis of pA+ RNA as in Figure 1A on fixed WT, Npl3-, Yra1-, Mtr2-, Dbp5-, Mex67-, and Nab2-AA cells subjected to rapamycin for 15 min at 30°C. Images were adjusted and stained with DAPI as in Figure 1A. WT, Mex67-AA, and Nab2-AA cells were from the same experiment shown in Figure 1A.
(B) Quantification of nuclear pA+ RNA signals of AA cells from (A) incubated for 0, 15, 40, or 70 min with rapamycin. Values are shown relative to mean values of non-rapamycin-treated cells and corrected for background. Error bars at individual time points indicate SDs calculated from nuclear pA+ RNA levels of a minimum of 800 cells counted for each strain. Error bars of mean values indicate SDs on the mean accumulation of pA+ RNA over the time course of each protein depletion.
(C) RT-qPCR analysis of selected 4tU RNAs of cells from (A). RNA was harvested after 10 min of 4tU labeling. All values were normalized to S. pombe spike-in, background-subtracted, and displayed relative to respective non-rapamycin-treated samples. Nab2-, Mex67-, Mtr2-, and Dbp5-AA cells were treated with rapamycin for 15 min, whereas Yra1- and Npl3-AA cells were subjected to rapamycin for 30 min to ensure complete nuclear depletion of these proteins. The mean value of all tested RNAs is indicated in red. Error bars indicate SDs from three to nine biological replicates. Error bars on mean values indicate SD from means of all tested transcripts. qPCR results were analyzed using a two-tailed paired t test for the comparisons indicated in the figure (see STAR Methods details for p value thresholds). Mean RNA production rate for Mex67-AA was calculated from nine replicate samples, six of which are depicted in Figure 4D.
(D) Relationship between levels of nuclear accumulation of pA+ RNA (x axis) and the mean of HSP104 and SSA4 RNA expression (y axis) in the indicated mutants strains relative to the WT control. Single repeats for each strain are shown separately. The mean of replicates from the individual strains is indicated and labeled and a power function has been fitted to the dataset.
See also Figure S3.
In an attempt to strengthen this relationship, we compared the nuclear pA+ RNA accumulation and new mRNA net production profiles in a panel of mutants affecting NPC function. A WT strain and the mex67-5i mutant (Jimeno et al., 2002) were taken as reference points and compared with NPC mutants nup116Δ, nup133Δ, nup60Δ, rat7-1, and gle1-37 (Strahm et al., 1999, Jimeno et al., 2002, Lund and Guthrie, 2005, Terry and Wente, 2007, Doye et al., 1994, Gorsch et al., 1995), of which the latter two are directly implicated in Dbp5p function (Hodge et al., 1999). Moreover, we included the nuclear basket deletion mutants mlp1Δ and mlp2Δ (Strambio-de-Castillia et al., 1999) in the analysis. Because most of these aberrations are permissive at normal growth temperature (25°C–30°C), we assayed nuclear pA+ RNA accumulation after incubation at 38°C for 12 min and compared this with the expression of the HSP104 and SSA4 hs-inducible RNAs. This revealed a tight correlation between the analyzed nuclear RNA accumulation and mRNA net production phenotypes (Figures 3D and S3C). Strains with modest 1.2- to 1.4-fold increases of nuclear pA+ RNA (mlp1Δ, mlp2Δ, and nup60Δ), compared with WT, showed only a mild (∼30%) drop in hs RNA expression, whereas cells showing more than 2-fold increases in nuclear pA+ RNA (nup116Δ, nup133Δ, rat7-1, and gle1-37) exhibited RNA expression decreases of ∼80%, similar to mex67-5 cells (Figure 3D). We conclude that the severe downregulation of new RNA net production correlates well, albeit in a non-linear manner, with the degree of nuclear pA+ RNA accumulation.
Nab2p Is Sequestered on Nuclear-Retained RNA in Export-Deficient Cells, and Excess Nab2p Can Alleviate mRNA Downregulation
Given the strikingly similar mRNA net production phenotypes of cells debilitated in nuclear export and depleted of nuclear Nab2p, which causes only a mild export defect, we wondered whether Nab2p activity might be compromised in export-deficient conditions. Immunofluorescence (IF) and ChIP analysis of Nab2p demonstrated that its predominantly nuclear localization was not altered, and the amount of chromatin-bound Nab2p was not decreased upon Mex67p nuclear depletion (Figures S4A and S4B). Nevertheless, we wondered whether Nab2p might be sequestered on pA+ RNAs accumulating in the nucleus during an export block and hence be unavailable to yield protection to newly made transcripts. To test this idea, we purified RNAs associated with Nab2p from ± rapamycin-treated Mex67-AA cells, which had been UV cross-linked with 0, 300, or 700 J/cm2. Purified Nab2p and its associated P32-labeled RNA were then resolved by SDS-PAGE (Figure 4A) and both total co-purified RNA (Figure 4A, lanes 1–7) and the RNaseA/T1-resistant pool directly linked to Nab2p (Figure 4A, lanes 8–14) were quantified (Figures 4B and 4C, respectively). Nab2p purified from rapamycin-treated and export-blocked cells associated with significantly more RNA than the Nab2p isolated from the control condition. Moreover, Nab2p cross-linking attained the maximum efficiency at lower energies during the export block, reflecting the increased amount of binding sites due to nuclear pA+ RNA accumulation. These results therefore suggest that Nab2p is indeed sequestered by RNAs accumulating in nuclei. Following this conclusion, it is likely that Mex67p and Nab2p act cooperatively to prevent nuclear decay of pA+ RNA. To test this assumption further, we attempted to co-deplete Mex67p and Nab2p from the nucleus by simultaneously AA-tagging these factors. This resulted in some redistribution of Nab2p from the nucleus (Figure S4C), which was incomplete, presumably because both regular Nab2p export and ribosome export, required for the AA system, depend on nuclear Mex67p (Iglesias et al., 2010, Yao et al., 2007). The pA+ RNA accumulation phenotype upon Mex67p/Nab2p double depletion was comparable with that of Mex67p-depleted cells (Figure S4D), and a parallel 4tU-labeling experiment demonstrated that the double depletion did not impair mRNA net production more than nuclear depletion of Mex67p alone (Figure 4D, compare RNA series 2 and 3). Together this indicates that Mex67p and Nab2p act in the same pathway.
Figure 4.

Nab2p Is Sequestered on Nuclear-Retained RNA in Export-Deficient Cells and Excess Nab2p Can Alleviate mRNA Downregulation
(A) SDS-PAGE of eluted samples from an RNA immunoprecipitation (IP) experiment in which endogenous Nab2p was immunoprecipitated from Mex67-AA cells without (“rapamycin –”) or treated with rapamycin for 30 min (“rapamycin +”). Before RNA IP, cells were not subjected to crosslinking (“0 J/cm2”), moderately cross-linked (“300 J/cm2,” ∼2 min cross-linking time), or strongly cross-linked (“700 J/cm2,” ∼5 min cross-linking time). Mock samples were negative control IPs using beads without anti-Nab2p antibody. Shown are IP eluates without (lanes 1–7) or with (lanes 8–14) prior RNaseA/T1 treatment. Top: western blotting analysis of Nab2p levels; bottom: autoradiogram of P32-labeled SDS-PAGE gel (representative example of three independent repeats).
(B) Quantification of the P32-labeled RNA signal from lanes 1–7 in (A). Background was estimated and subtracted on the basis of the mock sample. Single points represent values obtained from three individual repeats. Error bars represent SDs.
(C) Quantification of P32-labeled RNA signal within the Nab2p contained band of lanes 8–14 from (A). Values were normalized as in Figure 4B. Error bars represent SDs from three independent repeats, and statistical significance was tested using a two-tailed paired t test.
(D) RT-qPCR analysis of selected 10 min 4tU-labeled RNAs purified from cells treated for 15 min with rapamycin as in Figure 3C. RNA series 2–4 show new RNA production from control Mex67-AA cells grown in glucose (series 3), which have been simultaneously depleted for Nab2p (Mex67-AA/Nab2-AA) (series 2) or containing a Nab2p expressing plasmid (Mex67-AA p2μ_NAB2) (series 4). Values derive from a minimum of four biological replicates, and error bars indicate SDs. Values for RNA series 3 were also included in the mean shown in Figure 3C. RNA series 5–9 show new RNAs from Mex67-AA cells containing an empty (series 5) or a pPgal_Nab2 (series 6–9) plasmid and incubated for 2.5, 5, 6, or 8 hr in galactose. Values are a mean of two biological replicates for the pPgal_Nab2 containing samples and an average of single replicates of all galactose incubation times for the empty plasmid sample. Error bars indicate median absolute deviation.
(E) Western blotting analysis of Nab2p levels in non-rapamycin-treated Mex67-AA cells transformed with either an empty 2μ pRS426 plasmid (−) or its counterpart (pNAB2) expressing wild-type Nab2p (+). Rpb3p served as a loading control.
(F) Western blotting analysis showing Nab2p and Rpb3p levels in cells containing a NAB2 gene under control of a galactose-inducible promoter (pPgal_Nab2) in glucose (GLU) condition and at the indicated times after galactose (GAL) addition. The image was edited to show only one of the repeats performed in parallel.
See also Figure S4.
Previous in vitro experiments demonstrated that Nab2p can protect pA+ RNA from decay independent of cofactors (Schmid et al., 2015). Hence, lack of Mex67p, or other ways of compromising pA+ RNA nuclear export, likely impairs the function of endogenous Nab2p indirectly, in line with the idea that Nab2p is sequestered on accumulating pA+ RNA. If this is the case, providing excess Nab2p should alleviate mRNA decay in Mex67-AA cells, and we therefore examined the effect of Nab2p overexpression in vivo. Introducing the entire NAB2 locus on a high-copy 2μ plasmid translated only into a roughly 2- to 3-fold increase of Nab2p levels (Figure 4E), probably reflecting auto-regulatory mechanisms counteracting its overexpression (Roth et al., 2009). Even so, 4tU-labeling analysis demonstrated that net production of selected mRNAs was on average 2-fold more efficient upon such Nab2p overexpression (Figure 4D, compare RNA series 3 and 4). To increase Nab2p overexpression even further, we placed the NAB2 ORF behind a galactose-inducible promoter, which attained its full capacity within 6 hr of galactose induction (Figure 4F). Prolonged Nab2p overexpression revealed to be toxic to cell growth (Figure S4E), and we therefore induced Nab2p expression for 2.5, 5, 6, and 8 hr before monitoring its effect on new RNA production, which again revealed a partial rescue (Figure 4D, compare series 5 with 6, 7, 8, and 9). Even though the rescue was still not complete, we note that it was on average more robust than when expressing Nab2p from the 2μ plasmid (Figure S4F). These results collectively support the notion that nuclear Nab2p levels are limiting in Mex67p-depleted cells and thus insufficient to support normal mRNA net production.
Discussion
The compromised net production of new pA+ RNA, resulting from export inhibition, occurs without a significant decrease in cellular transcription levels, demonstrating an extensive decay of new RNAs in these conditions. The phenotype is robust regardless of the cause of the imposed nuclear export block and does not depend on initial transcription levels of the affected loci. Together, this suggests a post-transcriptional mechanism preventing excessive accumulation of nuclear pA+ RNA and that timely mRNA export is important, not only for protein production but also to prevent nuclear decay.
The decline in new pA+ RNA net production is tightly related to the amount of nuclear-retained pA+ RNA. Hence, even slight increases in nuclear pA+ RNA lead to significantly decreased new RNA production. This implies a critical need for cells to keep their nuclear pA+ RNA concentrations low, which is perhaps instigated because Nab2p levels are limiting and constantly must be recycled from the cytoplasm. Our data support this possibility, as they show a strong correlation between the decay phenotypes elicited by nuclear depletion of Mex67p and Nab2p, despite the weak contribution of Nab2p to pA+ RNA export. Considering that Nab2p binding to nascent pA tails protects the transcripts from degradation (Schmid et al., 2015), we propose that nuclear accumulation of pA+ RNP leads to the out-titration of Nab2p, preventing the protein from protecting newly made RNA and thus eliciting their degradation similarly to conditions of Nab2p nuclear depletion (Figure 5, bottom left). This model is strongly supported by the fact that Nab2p isolated from export-blocked cells binds more RNA than in the WT situation and that effects of depletion of both Mex67p and Nab2p are not additive. Nab2p overexpression can rescue the RNA decay phenotype, indicating that, in accordance with previous in vitro results (Schmid et al., 2015), the protein performs an RNA protective function. This rescue is not strictly proportional to Nab2p levels, perhaps because of detrimental effects of its overexpression or due to an impact of other mRNP protective proteins, which might also become limiting during an export block.
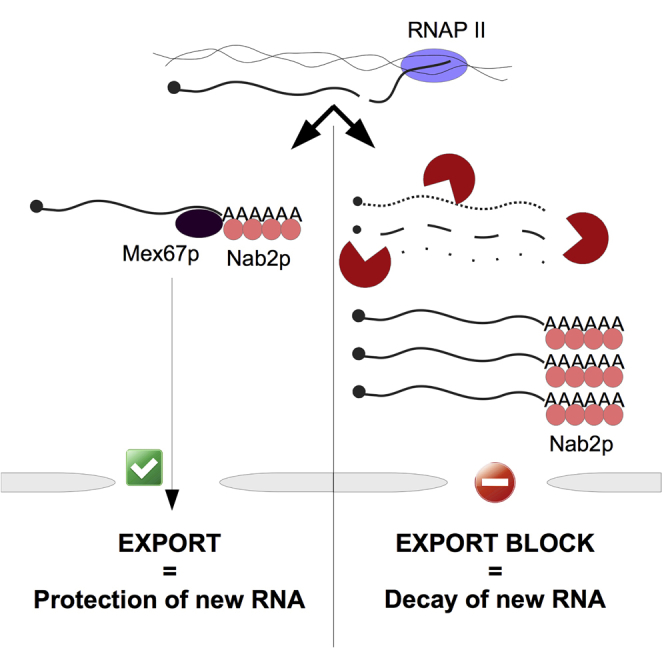
Figure 5.

Nuclear RNA Export Block Sequesters Nab2p on Retained RNAs and Elicits the Decay of Newly Made Transcripts
Model for the decay of newly synthesized pA+ RNA in export compromised cells. In normal conditions (“WT”), Nab2p protects newly made transcripts and stimulates Mex67p-mediated nuclear export (top). When pA+ RNA export is compromised (“EXPORT BLOCK”), Nab2p is inactivated, likely by getting sequestered on retained pA+ RNA (bottom left). Hence, Nab2p is unavailable to protect newly made transcripts, leading to their decay, similarly to conditions of Nab2p nuclear depletion (bottom right).
Some CUTs, the NRD1 mRNA and numerous 3′ extended mRNAs, are upregulated in Mex67p but less strongly in Nab2p nuclear-depleted cells, which indicates impaired NNS and CFI/CPF functions in the absence of Mex67p. Because levels of 3′ extended mRNAs do not correlate with the extent of the decline of their corresponding mRNAs, they cannot be causative for the phenotype. Rather, we suggest that CFI/CPF and NNS complexes are also, at least partially, sequestered on nuclear-retained pA+ RNA and therefore cannot efficiently perform their co-transcriptional functions. More generally, such sequestration of nuclear RNP proteins may be widespread during a nuclear export block, further highlighting the necessity of rapid nuclear RNA clearance, either by export of functional molecules or by decay.
What then is the mechanism of removal of newly produced pA+ RNA in export-deficient cells? It was previously shown that deletion of the nuclear exosome component RRP6 partially restores mRNA levels and rescues the lethality of Nab2p-deficient cells (Schmid et al., 2015). However, we were unable to rescue new mRNA production in Mex67p nuclear-depleted cells by impairing nuclear exosome function or depleting the 5′-3′ exonucleases Rat1p or Xrn1p. Even though it is possible that another undisclosed nuclear nuclease is solely responsible, we find it more likely that nuclear decay systems act redundantly to remove these transcripts. This would also explain why nuclear depletion of Mex67p leads to a stronger phenotype than nuclear loss of Nab2p. In Nab2p-depleted cells, where pA+ RNA export is only mildly affected, newly made transcripts are only exposed to the nuclear degradative environment for a brief time period. In Mex67p-depleted conditions, however, new pA+ RNAs are not only labile, because of the limiting supply of Nab2p, but also trapped in the nucleus. Together with the pronounced nuclear pA+ RNA accumulation in Mex67p-depleted cells, which might also cause shortage of other protective factors, this would create additional decay opportunities, not relevant in the Nab2p-depleted nuclei. Regardless of the exact mechanism of decay, our results demonstrate that maintenance of a low nuclear pA+ RNA concentration is key to proper pA+ RNA production. Under normal conditions, cells prevent nuclear pA+ accumulation by rapid export, whereas RNA decay provides a fail-safe mechanism when export is weakened. Mechanistically, these functions are tightly connected as Nab2p facilitates both the loading of Mex67p on nascent RNA while simultaneously yielding physical protection from decay.
Regulation of cellular mRNA levels by transcription and cytoplasmic decay has been extensively studied (Castells-Roca et al., 2011, Parker, 2012), whereas contributions from nuclear decay have been reported only recently (Roy and Chanfreau, 2014, Bresson et al., 2017). The strict requirement for Nab2p to confer stability to newly made pA+ RNA raises the possibility of a broader function of this protein in “monitoring” cellular mRNA levels. Nab2p participates in the regulation of its own mRNA (Roth et al., 2009), suggesting that controlling the concentration of this protein is critical. Future efforts will focus on possible roles of Nab2p in affecting cellular pA+ RNA homeostasis, which could be especially important during switches in growth programs, such as during starvation or stress, which require global changes of transcription rates (Castells-Roca et al., 2011, Bresson et al., 2017). Our results positioning Nab2p and nuclear export as a limiting factor for mRNA production therefore invite speculation as to whether changes in the abundance or sub-cellular localization of Nab2p might partake in such responses.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-Rpb3p | Abcam | 1Y26 cat. no.: ab81859; RRID: AB_1658381 |
| anti-Nab2p | Hector et al., 2002 | 3F2 |
| Alexa Fluor 488-labeled goat anti-mouse IgG | Invitrogen | Cat. no.: A32723; RRID: AB_2633275 |
| anti-Pab1 1G1 | Santa Cruz | Cat. no.: sc57953; RRID: AB_672248 |
| anti-Rrp6 | Jensen group | Production bleed AAU1 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Auxin (Indole-3-acetic acid sodium salt) | Sigma Aldrich | Cat. no.: 15148-10G |
| MTSEA-biotin | Biotium | Cat. no.: 90064 |
| ProlongGold with DAPI solution | Life Technologies | Cat. no.: P36935 |
| Rapamycin | Cayman chemicals | Cat. no.: 13346 |
| 4tU | Aldrich | Cat. no.: 440736-1G |
| Critical Commercial Assays | ||
| Turbo DNase free kit | Ambion | Cat. no.: AM1907M |
| SuperScript II | Invitrogen | Cat. no.: 18064-014 |
| Platinum SYBR Green qPCR Super-Mix-UDG kit | Invitrogen | Cat. no.: 11733-046 |
| Escherichia coli poly(A) polymerase kit | ThermoFisher | Cat. no.: AM1350 |
| PureLink micro RNA purification kit | Ambion | Cat. no.: 12183018A |
| Ribo-Zero Gold rRNA Removal kit for yeast | Ilumina | Cat. No.: MRZY1306 |
| RiboLock Rnase Inhibitor | ThermoSceintific | Cat. no.: E00381 |
| Lexogen QuantSeq 3′ mRNA-Seq Library Prep Kit REV | Lexogen | Cat. no.: 016.96 |
| RNase A/T1 | Thermo Scientific | Cat. no. EN0551 |
| PNK enzyme with buffer A | Thermo Scientific | Cat. no.: EK0031 |
| Deposited Data | ||
| 10 min 4tU | This study | GEO: GSE108477 |
| 2 min 4tU | This study | GEO: GSE108550 |
| Raw data for all figures | This study | www.mendeley.com at: https://doi.org/10.17632/jxw2cmh8kp.1 |
| Experimental Models: Organisms/Strains | ||
| S. cerevisiae genome | N/A | UCSC: sacCer3 |
| S. pombe genome | N/A | ENSEMBL: EF2 |
| as W303, tor1-1 fpr1::loxP-LEU2-loxP RPL13-2xFKBP12::loxP-TRP1-loxP | Haruki et al., 2008; Euroscarf | AA WT (HHY212) |
| as W303, tor1-1 fpr1::loxP-LEU2-loxP PMA1-2xFKBP12::loxP-TRP1-loxP Dbp5p::FRB::HIS3 | This study | Dbp5-AA |
| as W303, tor1-1 fpr1::NAT RPL13-2xFKBP12::TRP1 MEX67-FRB::kanMX6 | Haruki et al., 2008; Euroscarf | Mex67-AA (HHY182) |
| as W303, tor1-1 fpr1::NAT RPL13-2xFKBP12::TRP1 MEX67-FRB::kanMX6 NAB2::FRB::HIS3 | This study | Mex67-AA Nab2-AA |
| as W303, tor1-1 fpr1::NAT RPL13-2xFKBP12::TRP1 MEX67-FRB::kanMX6, RRP6::URA3 | This study | Mex67-AA rrp6Δ |
| as W303, tor1-1 fpr1::loxP-LEU-loxP RPL13-2xFKBP12::TRP1 Mtr2p-FRB::HIS3 | This study | Mtr2-AA |
| as W303, tor1-1 fpr1::loxP-LEU-loxP RPL13-2xFKBP12::TRP1 Nab2p-FRB::HIS3 | Schmid et al., 2015 | Nab2-AA |
| as W303, tor1-1 fpr1::loxP-LEU-loxP Pma1::FKBP::TRP1 Npl3p::FRB::GFP::HIS3 | This study | Npl3-AA |
| as W303, tor1-1 fpr1::LEU RPL13-2xFKBP12::loxP-TRP1-loxP, OsTIR::URA, RAT1::AID::KanMX, Mex67::FRB::KanMX | This study | Rat1-AID Mex67-AA |
| as W303, tor1-1 fpr1::LEU RPL13-2xFKBP12::loxP-TRP1-loxP, OsTIR::URA, RAT1::AID::KanMX, XRN1::AID::KanMX, MEX67::FRB::KanMX | This study | Rat1-AID Xrn1-AID Mex67-AA |
| As W303, tor1-1, fpr1::loxP-LEU2-loxP, RPL13-2xFKBP12::loxP-TRP1-loxP, RRP6-FRB::HIS3, DIS3-FRB::KAN | This study | Rrp6-AA Dis3-AA |
| As W303, tor1-1 fpr1::LEU, RPL13-2xFKBP12::loxP-TRP1-loxP, DIS3::FRB::KanMX, RRP6::FRB::HIS, mex67-5i | This study | Rrp6-AA Dis3-AA mex67-5i |
| as W303, tor1-1 fpr1::loxP-LEU-loxP RPL13-2xFKBP12::TRP1 Yra1p-FRB::HIS3 | This study | Yra1-AA |
| leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15 | WT W303 | |
| as W303, MATa, gle1Δ::HIS3 < gle1-37, leu2, cen > | Strahm et al., 1999 | gle1-37 |
| as W303, MATa, mex67-5 | Jimeno et al., 2002 | mex67-5i |
| as W303, mlp1Δ::URA3 | Strambio-de-Castillia et al., 1999 | mlp1Δ |
| as W303, mlp2Δ::HIS33 | Strambio-de-Castillia et al., 1999 | mlp2Δ |
| as BY, MATalpha, his3Δ1, leu2Δ0, lys2Δ0 ura3Δ0W303, MATalpha, nup60Δ::KanMX6 | Lund and Guthrie, 2005 | nup60Δ |
| as W303, MATa, nup116-5::HIS3 | Terry and Wente, 2007 | nup116Δ |
| as W303, MATalpha, nup133::HIS3 | Doye et al., 1994 | nup133Δ |
| as W303, rat7-1, trp1Δ63, ura3-52, leu2Δ1 | Gorsch et al., 1995 | rat1-7 (nup159 mut) |
| Oligonucleotides | ||
| See Table S3 in Supplemental Information | N/A | N/A |
| Recombinant DNA | ||
| 2μ, ori(f1), ori(pMB1), URA3, Ampr, LacZ, MCS, T7 promoter, T3 promoter | ATCC 77107 | pRS426 |
| 2μ, ori(f1), ori(pMB1), URA3, Ampr, LacZ, MCS, T7 promoter, T3 promoter, NAB2 in KpnI-XhoI | PAC717 kind gift from A. Corbett | pRS246-NAB2 |
| 2μ, ori(f1), ori(pMB1), URA3, Ampr, LacZ, MCS, T7 promoter, T3 promoter, FUI1 | Barrass et al., 2015 | p4FUI(URA) |
| 2μ, ori(f1), ori(pUC), URA3, Ampr, TCYC1, MCS, PGAL1, PGAL10, MCS, TADH1 | Stratagene | pESC_URA |
| 2μ, ori(f1), ori(pUC), URA3, Ampr, TCYC1, MCS, PGAL1, PGAL10, NAB2 ORF, TADH1 | This work | pPgal_Nab2 |
| Software and Algorithms | ||
| ImageJ | https://imagej.nih.gov/ij/ | N/A |
| DAPI area selection macro for ImageJ | This study | N/A |
| BBMAP v 35.92 | https://jgi.doe.gov/data-and-tools/bbtools/ | unpublished |
| STAR aligner v GitHub 2016-03-14 | https://github.com/alexdobin/STAR | Dobin et al., 2013 |
| samtools v 1.3 | http://www.htslib.org/ | Li et al., 2009 |
| HTSeq v 0.6.0 | https://pypi.python.org/pypi/HTSeq | Anders et al., 2015 |
| R package DESeq2 v 1.10.1 | http://bioconductor.org/packages/release/bioc/html/DESeq2.html | Love et al., 2014 |
| deepTools2 software suite v2.2.4 | http://deeptools.readthedocs.io/en/latest/index.html | Ramírez et al., 2016 |
| GitHub | https://github.com/manschmi/MexNab_3seq | N/A |
| Other | ||
| Aria MX Real Time PCR System | Agilent Technologies | Cat. no.: G8830A |
| Axiovert 200M microscope Cy3, DAPI, FITC filters | Zeiss | N/A |
| Covaris S2 Focused Ultrasonicator | Covaris | N/A |
| Bioanalyzer | Agilent | N/A |
| Dynabeads M280 Sheep anti-mouse IgG | Invitrogen | Cat. no.: 11202D |
| Dynabeads MyOne Streptavidin C1 | Invitrogen | Cat. no.: 65002 |
| MG-SR PLUS X-ray films | Konica Minolta | Cat. no.: A3WM |
| NuPAGE 4-12% Bis-Tris Protein Gels | ThermoFisher Scientific | Cat. no.: NP0322BOX |
| NuPage 4x loading dye | Thermo Fisher | Cat. no. NP0007 |
| Typhoon FLA9500 | GeHealthcare | Cat. no.: 28996943 |
| UV Stratalinker 1800 | Stratagene | N/A |
| Zeba Spin Desalting Columns 7KMWCO | Thermo Scientific | Cat. no.: 89890 |
| 3-8% Tris acetate gel | Invitrogen | Cat. no.: EA03755BOX |
| RNAPII ChIP-tiling array | Mayer et al., 2010 | ArrayExpress E-TABM-1033 |
| RNAPII ChIP-seq | Warfield et al., 2017 | sample GSM2551210 from GEO:GSE97081 |
| NETseq | Churchman and Weissman, 2011 | sample GSM617027 from GEO:GSE25107 |
| RNAPII CRAC | Milligan et al., 2016 | sample GSM1706520 from GEO:GSE69676 |
| cDTA | Sun et al., 2012 | ArrayExpress: E-MTAB-760 |
| DTA | Miller et al., 2011 | ArrayExpress: E-MTAB-439 |
| rpb1-1 chase | Presnyak et al., 2015 | GEO: GSE57385 |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Torben Heick Jensen (e-mail: thj@mbg.au.dk).
Experimental Model and Subject Details
Yeast strains, growth conditions and plasmids
AA strains were constructed using materials described in Haruki et al. (2008) and according to standard procedures. For assessment of RNA abundance and localization, cells were grown in YPAD media at 30°C. If HSP104 and SSA4 expression was assessed cells were pre-grown in YPAD media at 25°C and shifted to 38°C for 10-12 min by adding an equal volume of media at 51°C. To test RNA net production by 4tU labeling and perform Rpb3p and Nab2p ChIP analysis, cells were transformed with pESC_URA, pPgal_Nab2, p4FUI(URA3), pRS426(URA3) or pRS426-NAB2(URA3) plasmids and grown in minimal media lacking uracil, to increase 4tU uptake. When needed, rapamycin or auxin were added to the media to a final concentration of 1 μg/ml or 3 mM as specified in (Haruki et al., 2008, Morawska and Ulrich, 2013). If needed cells were grown in 2% galactose media. Cells for RNA extraction were harvested by mixing equal volumes of culture with 96% ethanol pre-cooled on dry-ice. Yeast strains and plasmids used in this study are listed in Key Resource Table sections ‘Experimental Models’: ‘Organisms/Strains’ and ‘Recombinant DNA’, respectively.
Method Details
RNA extraction, reverse-transcription and qPCR
RNAs were extracted using the hot-acid phenol method. Briefly cell pellets were suspended in TES buffer (1% SDS, 5 mM EDTA, 10 mM Tris pH 7.5) and extracted twice with phenol at 65°C with shaking for 30 min and once in chloroform at RT for 5 min. Between washes, samples were centrifuged for 10 min at 16 000 g at 4°C. RNAs were precipitated with ethanol in 20-30 mM LiCl and resuspended in deionized water. Depending on the experiment, 200 ng to 1.5 μg of RNA was selected for DNase treatment using Invitrogen TURBO DNA-free Kit (AM1907M) according to the manufacturer’s recommendation. RNAs were then reverse transcribed using a 100 μM dT18 oligonucleotide with 250 ng/μl random hexamers (Invitrogen, 48190-011) and Invitrogen SuperScript II (18064-014) kit according to the manufacturer’s recommendation. qPCR reactions were prepared using the Invitrogen Platinum SYBR Green qPCR Super-Mix-UDG (11733-046) kit and run on Aria MX Real Time PCR System from Agilent Technologies. Oligonucleotides used are listed in Table S3 in Supplemental Information.
ChIP of Nab2p and Rpb3p
Chromatin from Mex67-AA cells was prepared according to standard procedures. Briefly 50 mL of cells at OD600 = 0.4-0.6 were crosslinked with 1% final paraformaldehyde for 15 min and washed twice with 25 mL of 150 mM NaCl, 10 mM Tris-HCl pH 7.5. After washing, cells were resuspended in 1 mL of FA buffer (50 mL HEPES pH 7.5, 150 mL NaCl, 1 mL EDTA, 1% Triton X-100, 0.1% SDS, 1% Na-deoxycholate) with cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail (04693159001, Roche), supplemented with 0.5 mL of glass beads and vortexed for 20 min at 4°C. Chromatin was fragmented using a Covaris S2 Focused Ultrasonicator (program: 15x freq. Sweeping/duration 30 s, intensity 8.0, cycles/burst 200) and centrifuged for 25 min at 16000 g. 200 μL of the supernatant was taken for IP, which was performed with 30 μL of blocked and pre-washed in FA + 1% BSA buffer Dynabeads M280 Sheep anti-mouse IgG from Invitrogen (11202D) containing immobilized anti-Rpb3p antibody 1Y26 (Abcam, ab202893) or anti-Nab2p 3F2 (Hector et al., 2002). In order to determine the background of the IP for each condition a mock IP was performed in which the sonicated chromatin was incubated with beads not conjugated to the antibodies. After 1h30 min of incubation with the antibody-conjugated beads the resin was washed four times with 0.5 mL of FA buffer, two times with 0.5 mL of FA buffer containing 0.5 M NaCl and one time with TE (10 mM Tris-HCl pH 8.0, 1 mM EDTA pH 8.0). Precipitated chromatin was eluted for 10 min at 65°C with 100 μL of TE with 1% SDS. The elution and 10 μL of the input were treated with 5 μL of 20 mg/ml proteinase K for 30 min at 37°C and DNA fragments were purified using QIAquick PCR purification kit (28106; QIAGEN) according to manufacturer’s instructions, resuspended in 0.2 mL of water and used for real-time PCR. Elutions were normalized to average of input levels. To accurately define the Nab2p/Rpb3p ratio, background was subtracted from raw values.
Nab2p RNA immunoprecipitation (RIP)
50 mL of Mex67-AA 0.45-0.60 OD cultures were incubated, or not, with rapamycin for 30 min at 30°C. Cells were centrifuged, resuspended in 1 mL of sterile water and spread on a 100 mm diameter Petri dish for cross-linking by a UV Stratalinker (Stratagene). Petri dishes were placed on ice within 5 cm distance from the light source and set to receive 300 or 700 J/cm2. Cells were then washed twice with 1 mL of sterile water and frozen on dry ice. Pellets were resuspended in 1.8 mL of lysis buffer (50 mM Tris-HCl, pH 7.4, 100 mM NaCl, 1% Igepal CA-630, 0.1% SDS, 0.5% sodium deoxycholate) and mixed with 1.5 mL of glass beads. Thereafter, cells were broken in a Precellys®24 tissue homogenizer employing five cycles of 45 s at 6500 rpm with 5 min incubation on ice in between cycles. Broken cell suspensions were recovered and 5 μL of Turbo DNase was added to each sample for 5 min at 37°C. After DNase treatment cells were centrifuged for 3 min at 3000 g to remove unbroken cells and the supernatant was spun again for 5 min at 16 000 g. The supernatant was recovered, and used for IP using sheep anti-mouse IgG magnetic beads M280 (Invitrogen cat no. 11202D), which were pre-incubated in lysis buffer for 90 min with anti-Nab2p antibody 3F2 (Hector et al., 2002) and 1% BSA. Nab2p was IP’ed at 4°C for 1 hour with constant rotation. After IP half of the bead suspension volume was resuspended in 1 mL of RNase A/T1 digestion buffer (10 mM Tris 7.5; 300 mM NaCl, 5 mM EDTA, 2 μl of RNase A/T1 (Thermo Scientific cat. no. EN0551)) and incubated for 7 min at room temperature. After this step both RNase (–) and (+) fractions were washed twice with 1 mL of high salt wash buffer (50 mM Tris-HCl, pH 7.4, 1 M NaCl, 1 mM EDTA, 1% Igepal CA-630, 0.1% SDS, 0.5% sodium deoxycholate). During the second wash, samples were incubated for 5 min with rotation. Beads were washed one more time with 1 mL of PNK labeling buffer (20 mM Tris-HCl, pH 7.4, 10 mM MgCl2, 0.2% Tween-20) and 100 μL of beads in PNK buffer was mixed with 34 ul of NuPage loading dye (Thermo Fisher cat. no. NP0007) and this fraction was used for western blotting analysis (‘elution’ sample). The remaining beads were resuspended in 20 μL of enzymatic PNK suspension prepared with buffer A (Thermo Scientific, cat no. EK0031) and supplemented with gamma-ATP. Samples were incubated for 10 min at 37°C. The reaction mix was removed and beads were resuspended in 1x NuPage elution buffer and incubated for 5 min at 70°C. Samples were loaded on a 3%–8% Tris-acetate gel (Invitrogen cat. no. EA03755BOX). After electrophoresis the gel was washed 5 times with running buffer and imaged with phosphoimager screen on a Typhoon FLA9500 (GE Healthcare) and with MG-SR PLUS X-ray films (Konica Minolta cat. no. A3WM). Signals in the entire lanes of RNaseA/T1 non-treated samples were quantified using ImageJ software. The values displayed on the graph were normalized to the value of the +rapamycin and 700J/cm2 sample and corrected for background.
Isolation of newly synthesized RNAs using 4tU labeling
Isolation of newly synthesized RNA using 4tU labeling was based on (Barrass et. al., 2015) with several modifications. 50 mL of cells at OD600 around 0.4-0.6 were grown in synthetic medium lacking uracil at 30°C. 4tU (Sigma, from a 100mM stock in DMSO) was added to a final concentration of 100 μM for 10 min or 2 min (depending on the experiment) with subsequent cooling of cells by pouring cultures into 50 mL of dry-ice cold ethanol. RNAs were extracted using the hot acid phenol method. All samples were prepared in triplicates and 10 min 4tU labeled S. pombe RNA was added for normalization to all samples (1 μg of S. pombe RNA per 100 μg of S. cerevisiae RNAs; S. pombe spike-in RNA comes from total RNA from cells grown exponentially in YPAD medium at 30°C and labeled with 5 mM 4tU for 10 minutes). Additionally, for both Nab2-AA and Mex67-AA series one negative control from cells that were neither labeled with 4tU nor treated with rapamycin was prepared to determine the background of the 4tU IP procedure. 250-350 μg of total RNA resuspended in 400 μL of 1 mM EDTA, 10 mM HEPES was biotinylated using 10 μL 5mg/ml MTSEA-biotin in DMF (90064; Biotium) for 30 min at 25°C. After biotinylation RNA was desalted using Zeba Spin Desalting Columns 7KMWCO (89890; Thermo Scientific) according to manufacturer’s instructions. After desalting RNA was precipitated with ethanol and resuspended in water. An aliquot of RNA (‘total’ samples) was taken at this step and the remaining RNA was supplemented with salts to the final IP buffer composition: 10 mM Tris-HCl pH 7.0, 0.2 M NaCl, 25 mM MgCl2, 0.1% SDS, 0.1 M NaPi pH 6.8. IP was performed using 30-50 μL (depending on the experiment) of Dynabeads MyOne Streptavidin C1 from Invitrogen (65002) pre-washed in IP buffer and blocked with 200 μL of glycogen. Following 30 min of IP reaction, the beads were washed five times with 0.4 mL of IP buffer and one time with 0.4 mL of TEN1000 (10 mM Tris-HCl pH 7.5, 0.5 mM EDTA, 1 M NaCl). RNA was eluted with 0.1 mL of 0.7 M beta-mercaptoethanol for 10 min at room temperature. Eluted RNA was precipitated with ethanol, 100 μg of glycogen and 20-30 mM final LiCl and resuspended in water. Depending on the application, RNA was subjected to sequencing library preparation or to RT-qPCR analysis (see relevant sections). In the latter case, IP values were normalized to S. pombe spike-in transcripts defined by amplification of cDNA of the SpACT1 mRNA and to input RNA concentrations. Background values, derived from non-4tU-labeled and non-rapamycin treated samples, were then subtracted and final results depicted as ratios between rapamycin treated sample relative to appropriate control.
Western blotting analysis
Protein extracts were prepared by incubating 4-6 OD units of cells in 8 M urea at 80°C and subsequent disruption of cells using glass beads. Western blotting analysis was performed according to standard procedures. Antibodies used are listed in Resource Table under section Antibodies.
pA+ RNA FISH analysis and protein localization
IF and FISH samples were performed as previously described (Schmid et al., 2015). Briefly, cells were fixed with 4% final paraformaldehyde for 40 min and washed three times with 1.2 M sorbitol, 0.1 M K2HPO4. Cell walls were digested for 25 min at 30°C in 1.2 M sorbitol, 0.1 M K2HPO4, 28mM Beta-mercaptoethanol with zymolyase and spheroblasts were attached to PolyL-lysine coated slides and washed two times with PBS. After these common steps, slides were prepared either for FISH or IF analyses (see below).
For FISH analysis, cells were washed three times with 0.1 M K2HPO4, dehydrated with 80% ethanol at −20°C for at least 30 min, washed twice with 2xSSC for 5 min at 25°C and prehybrydized in 50% formamide, 2xSSC for 10 min 25°C. Hybridization was performed in 50% formamide, 5 mM Na2HPO4, 2xSSC, 200 μg BSA with 1 ng of dT18 LNA Cy3-labeled probe (Thomsen et al., 2005) per sample for 16h at 37°C. After hybridization, slides were washed twice with 50% formamide, 2xSSC for 10 min at 37°C, once with 2xSSC, 0.1% Triton X-100 for 10 min at 25°C, twice with 1xSSC for 10 min at 25°C and twice with PBS for 5 min at 25°C. Slides were dried and mounted with ProlongGold with DAPI solution (Life Technologies, P36935). For IF analysis slides were washed once with PBS, 0.1% Triton X-100 for 5 min at 25°C, then incubated for 30 min at 25°C with PBS, 0.1% Triton X-100, 1% BSA. Nab2p detection was performed using anti-Nab2p 3F2 antibody (Hector et al., 2002) diluted 1:3000 in PBS, 0.1% Triton X-100 for 45 min at 25°C. The secondary antibody Alexa Fluor 488-labeled goat anti-mouse IgG (A32723; Invitrogen) was diluted 1:3000 in PBS, 0.1% Triton X-100 and applied to the slide for 45 min at 25°C. Between and after incubation with both antibodies, slides were washed four times for 5 min at 25°C with PBS, 0.1% Triton X-100. Slides were dried and mounted with ProlongGold with DAPI solution. Detection of Npl3p-GFP-FRB (Npl3-AA) was performed in live cells stained with DAPI.
FISH and IF microscopy pictures were acquired using an Axiovert 200M Zeiss microscope under 63x objective using DAPI, Cy3 or FITC filters. Each FISH picture contained a fixed number of stacks (which varied between experiments but was never smaller than 8) of 0.2 μm combined using maximum intensity projection. In order to quantify the nuclear RNA pA+ signal by FISH, cell nuclei were selected using DAPI images and the following macro in ImageJ: [run(“Subtract Background...,” “rolling=50 sliding”); setOption(“BlackBackground,” false); run(“Make Binary”); run(“Erode”); run(“Dilate”); run(“Analyze Particles...,” “size=80-400 add”); roiManager(“Show All with labels”); roiManager(“Show All”);]. Average nuclear RNA pA+ signal was calculated by dividing the Raw Integrated Density in the nuclear area and subsequently subtracting the value of the minimal pixel (giving a conservative estimate of local background and cytoplasmic signal). Cy3 and FITC channel images were aligned to a common display range (defined as the lowest and highest pixel value for all selected images) to enable comparison between images. Images of DAPI stain were background subtracted in ImageJ (settings: Rolling ball radius 50.0 pixels; sliding paraboloid).
pA+ RNA 3′end sequencing and bioinformatics analysis
RNA quality of total and purified 4tU labeled RNA was tested and quantified using a Bioanalyzer (Agilent). Depending on the sample 200-350 ng of RNA was used to prepare libraries. To generate pA- data, an aliquot of the 2 min 4tU labeled RNAs was in vitro polyadenylated using Escherichia coli poly(A) polymerase (AM1350; ThermoFisher) according to the manufacturer’s instructions. Reactions were then purified using PureLink micro RNA purification kit from Ambion (12183018A) and ribo-depleted using Ribo-Zero Gold rRNA Removal kit for yeast (MRZY1306) from Ilumina according to the manufacturer’s instructions. All libraries were then prepared using Lexogen QuantSeq 3′ mRNA-Seq Library Prep Kit REV (016.96, Lexogen GmbH) for Illumina and sequenced at Vienna Biocenter Core Facilities (Vienna, Austria) multiplexing 32 samples on a HiSeqV4 SR50 (single end 50nt reads) run and using the QuantSeq REV specific primer CSP. An overview of samples, read quality control and mapping statistics are provided in Table S1 and Table S2.
Bioinformatics
Details concerning RNaseq data analysis can be found in (Schmid et al., 2018). All relevant code and analysis scripts are available at GitHub (https://github.com/manschmi/MexNab_3seq).
RNA 3′end sequencing quality control, filtering, mapping and normalization
Lexogen provided barcode splitting and the final data as unmapped bam files. We then applied quality filtering, read trimming and the mapping strategy recommended for QuantSeq REV data by Lexogen and computed genomic coverage of A-addition positions, skipping A-rich positions that allow for internal priming. Raw read coverage was then normalized to the signal obtained for the S. pombe genome. In case of the 4tU-labeled samples, signal from the negative control was subtracted as described in (Schmid et al., 2018). To obtain an estimate pA- signal at each position, we subtracted the S. pombe normalized pA+ signal from the pA+ and pA- signal. For the 2 min 4tU IPs this was done after background subtraction.
Annotations
mRNA and SUT annotations from (Xu et al., 2009) were lifted to UCSC sacCer3. Cryptic Unstable Transcripts (CUTs) and sn/snoRNAs from (Xu et al., 2009) were excluded from analysis since the majority of produced transcripts were not polyadenylated. XUT annotations were taken from (Wery et al., 2016). S. pombe genome and annotations are from ENSEMBL release EF2.
Counting gene body and end signal
Gene body regions were defined from TSSs to regions 200bp upstream of TESs (TSS to TES-200bp). Gene ends were defined as regions from 200bp upstream to 200bp downstream of the annotated TES (TES ± 200bp). Gene body regions shorter than 100bp and parts of gene body regions, which overlapped gene end regions of neighboring genes, were removed from the analysis. For XUTs, end regions overlapping end regions of transcripts from (Xu et al., 2009) were removed from the analysis. Signals within each region were counted by custom scripts using S. pombe normalized, background-subtracted pA+ or pA- bedgraph track files. Distribution of signal was then analyzed using custom scripts in R.
Differential expression analysis
Comparison of log2 FCs signals between Mex67-AA and Nab2-AA (Figure 1 and S1) was based on 10 min 4tU pA+ samples, where the majority of all genes has signal in the labeled fraction significantly above background (see below). pA+ signal from gene ends (TES ± 200bp) from raw mapped reads (i.e., before normalization to S. pombe reads and without background subtraction) was collected and analyzed using the R package DESeq2 (v 1.16.1). For this, S. pombe based size factors were derived from reads mapping to S. pombe transcripts (TSS to TES+300bp) counted using a custom script based on bedtools and put to the sizeFactors function from DESeq2. pA+ reads from gene ends were counted using the same script and differential expression between 15 min rapamycin treated samples relative to untreated samples analyzed using independent DESeq2 calculations for IP and total RNA samples with otherwise default settings. Differential enrichment of 10 min 4tU IP samples relative to the input and enrichment of IP samples relative to the negative control IP was computed using the same procedure but by combining IP and total RNA data within one DESeq2 run. Plots depicted in Figure 1 show only those RNAs with significant enrichment in the control IP relative to input samples.
Comparison of log2 FCs in gene body and gene end regions of Mex67-AA and Nab2-AA samples in Figure 2 was based on the 2 min 4tU pA+ samples, where the enrichment in the IP relative to negative control was less pronounced. For this analysis, background-subtracted S. pombe-normalized 2 min 4tU IP signals were used to measure pA+ signal from gene ends and pA- signal from gene bodies. Regions where signals were below background were given a pseudocount of 10−6 and plots depicted in Figure 2 show average values from control and 15 min samples on a log-scaled y axis.
Metagene heatmaps
Heatmaps were obtained using computeMatrix, plotHeatmap functions from the deepTools2 software suite v2.2.4 together with custom python scripts (Iasillo et al., 2017). For heatmaps of log2 FCs between two conditions, the minimum positive value within the entire matrix added as pseudocount, and used to replace zero and negative values, mean values from replicates were computed and the log2 FC calculated between depletion and corresponding control libraries using custom python scripts and plotted using plotHeatmap from deepTools2. Sorting according to termination defect was done using the built-in sorting of the plotHeatmap function using a matrix containing log2 FC values for the region 250bp downstream the TES.
Correlation between RNA net production and published transcription and decay-rate data
Transcription and decay-rates were obtained from the relevant publications listed in the ‘Other’ section of the Key Resources Table. Half-life and decay estimates were obtained from the Supplemental Information of the relevant publications. In cases where only half-life information was provided, decay-rates were obtained using equation k = ln(2)/halflife. We also included estimated decay rates derived from the comparison of 4tU- relative to total RNA-levels using the formula: Decay Rate DR = -(1/t) ∗ ln(1 – RNA4tU / RNAtotal), akin to the strategy from (Sun et al., 2012, Miller et al., 2011). RNA4tU and RNAtotal are the S. pombe normalized and, for the 4tU IP background-subtracted, pA+ signal from gene ends (TES ± 200bp) from the 10 min 4tU experiment and labeling time t = 10 min.
Quantification and Statistical Analysis
Quantification of FISH data was performed as described in ‘pA+ RNA FISH analysis and protein localization’ section. 4tU-labeled RNAs that were analyzed by reverse-transcription and qPCR were normalized as described in the ‘Isolation of newly synthesized RNAs using 4tU labeling’. When highlighted, qPCR results were analyzed using a two-tailed paired t test and p value (P) significance is defined as follows: not significant (ns) - p > 0.05; ∗ - p ≤ 0.05; ∗∗ - p ≤ 0.01; ∗∗∗ - p ≤ 0.001 and ∗∗∗∗ - p ≤ 0.0001. Analysis of sequencing data was performed as described in the following sections: ‘Bioinformatics’, ‘RNA 3′end sequencing quality control, filtering, mapping and normalization’, ‘Counting gene body and end signal’, ‘Differential expression analysis’, ‘Metagene heatmaps’ and ‘Correlation between RNA net production and published transcription and decay-rate data’.
Data and Software Availability
The accession number for data from 10 min 4tU labeling experiments reported in this paper is GEO: GSE108477. The accession number for data from 2 min 4tU labeling experiments reported in this paper is GEO: GSE108550. Raw non-sequencing data has been deposited with Mendeley Data at https://doi.org/10.17632/jxw2cmh8kp.1.
Acknowledgments
We thank Marta Lloret Llinares, Toomas Silla, Tommaso Villa, and Domenico Libri for critical comments on the manuscript. Work in the Jensen laboratory was supported by the Danish National Research Council, the Lundbeck Foundation, and the European Research Council (ERC) (grant 339953). J.D. Beggs was supported by a Wellcome Investigator Award (104648) and a Wellcome Centre for Cell Biology core grant (092076). We thank Logan Decker, Rikke Jespersen, Dorthe Caroline Riishøj, Aleksandra Bilska, and Ruta Bernotaite for technical support.
Author Contributions
A.T., M.S., and T.H.J. conceived the project. A.T. designed, conducted, and analyzed the experiments. M.S. contributed to the 4tU-labeling experiments and performed the bioinformatics analysis. M.M. contributed to RNA fluorescence in situ hybridization (FISH) analysis. J.D. Barrass and J.D. Beggs provided advice with 4tU labeling and manuscript corrections. T.H.J. supervised the project. A.T., M.S., and T.H.J. wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: August 28, 2018
Footnotes
Supplemental Information includes four figures and three tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.07.103.
Supplemental Information
References
- Aitchison J.D., Blobel G., Rout M.P. Kap104p: a karyopherin involved in the nuclear transport of messenger RNA binding proteins. Science. 1996;274:624–627. doi: 10.1126/science.274.5287.624. [DOI] [PubMed] [Google Scholar]
- Anders S., Pyl P.T., Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assenholt J., Mouaikel J., Andersen K.R., Brodersen D.E., Libri D., Jensen T.H. Exonucleolysis is required for nuclear mRNA quality control in yeast THO mutants. RNA. 2008;14:2305–2313. doi: 10.1261/rna.1108008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrass J.D., Reid J.E., Huang Y., Hector R.D., Sanguinetti G., Beggs J.D., Granneman S. Transcriptome-wide RNA processing kinetics revealed using extremely short 4tU labeling. Genome Biol. 2015;16:282. doi: 10.1186/s13059-015-0848-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batisse J., Batisse C., Budd A., Böttcher B., Hurt E. Purification of nuclear poly(A)-binding protein Nab2 reveals association with the yeast transcriptome and a messenger ribonucleoprotein core structure. J. Biol. Chem. 2009;284:34911–34917. doi: 10.1074/jbc.M109.062034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossie M.A., DeHoratius C., Barcelo G., Silver P. A mutant nuclear protein with similarity to RNA binding proteins interferes with nuclear import in yeast. Mol. Biol. Cell. 1992;3:875–893. doi: 10.1091/mbc.3.8.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bresson S., Tuck A., Staneva D., Tollervey D. Nuclear RNA decay pathways aid rapid remodeling of gene expression in yeast. Mol. Cell. 2017;65:787–800.e5. doi: 10.1016/j.molcel.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castells-Roca L., García-Martínez J., Moreno J., Herrero E., Bellí G., Pérez-Ortín J.E. Heat shock response in yeast involves changes in both transcription rates and mRNA stabilities. PLoS ONE. 2011;6:e17272. doi: 10.1371/journal.pone.0017272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchman L.S., Weissman J.S. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature. 2011;469:368–373. doi: 10.1038/nature09652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doye V., Wepf R., Hurt E.C. A novel nuclear pore protein Nup133p with distinct roles in poly(A)+ RNA transport and nuclear pore distribution. EMBO J. 1994;13:6062–6075. doi: 10.1002/j.1460-2075.1994.tb06953.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert W., Guthrie C. The Glc7p nuclear phosphatase promotes mRNA export by facilitating association of Mex67p with mRNA. Mol. Cell. 2004;13:201–212. doi: 10.1016/s1097-2765(04)00030-9. [DOI] [PubMed] [Google Scholar]
- Gorsch L.C., Dockendorff T.C., Cole C.N. A conditional allele of the novel repeat-containing yeast nucleoporin RAT7/NUP159 causes both rapid cessation of mRNA export and reversible clustering of nuclear pore complexes. J. Cell Biol. 1995;129:939–955. doi: 10.1083/jcb.129.4.939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant R.P., Marshall N.J., Yang J.C., Fasken M.B., Kelly S.M., Harreman M.T., Neuhaus D., Corbett A.H., Stewart M. Structure of the N-terminal Mlp1-binding domain of the Saccharomyces cerevisiae mRNA-binding protein, Nab2. J. Mol. Biol. 2008;376:1048–1059. doi: 10.1016/j.jmb.2007.11.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green D.M., Marfatia K.A., Crafton E.B., Zhang X., Cheng X., Corbett A.H. Nab2p is required for poly(A) RNA export in Saccharomyces cerevisiae and is regulated by arginine methylation via Hmt1p. J. Biol. Chem. 2002;277:7752–7760. doi: 10.1074/jbc.M110053200. [DOI] [PubMed] [Google Scholar]
- Hammell C.M., Gross S., Zenklusen D., Heath C.V., Stutz F., Moore C., Cole C.N. Coupling of termination, 3′ processing, and mRNA export. Mol. Cell. Biol. 2002;22:6441–6457. doi: 10.1128/MCB.22.18.6441-6457.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haruki H., Nishikawa J., Laemmli U.K. The anchor-away technique: rapid, conditional establishment of yeast mutant phenotypes. Mol. Cell. 2008;31:925–932. doi: 10.1016/j.molcel.2008.07.020. [DOI] [PubMed] [Google Scholar]
- Hautbergue G.M., Hung M.L., Golovanov A.P., Lian L.Y., Wilson S.A. Mutually exclusive interactions drive handover of mRNA from export adaptors to TAP. Proc. Natl. Acad. Sci. U S A. 2008;105:5154–5159. doi: 10.1073/pnas.0709167105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hector R.E., Nykamp K.R., Dheur S., Anderson J.T., Non P.J., Urbinati C.R., Wilson S.M., Minvielle-Sebastia L., Swanson M.S. Dual requirement for yeast hnRNP Nab2p in mRNA poly(A) tail length control and nuclear export. EMBO J. 2002;21:1800–1810. doi: 10.1093/emboj/21.7.1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge C.A., Colot H.V., Stafford P., Cole C.N. Rat8p/Dbp5p is a shuttling transport factor that interacts with Rat7p/Nup159p and Gle1p and suppresses the mRNA export defect of xpo1-1 cells. EMBO J. 1999;18:5778–5788. doi: 10.1093/emboj/18.20.5778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iasillo C., Schmid M., Yahia Y., Maqbool M.A., Descostes N., Karadoulama E., Bertrand E., Andrau J.C., Jensen T.H. ARS2 is a general suppressor of pervasive transcription. Nucleic Acids Res. 2017;45:10229–10241. doi: 10.1093/nar/gkx647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias N., Tutucci E., Gwizdek C., Vinciguerra P., Von Dach E., Corbett A.H., Dargemont C., Stutz F. Ubiquitin-mediated mRNP dynamics and surveillance prior to budding yeast mRNA export. Genes Dev. 2010;24:1927–1938. doi: 10.1101/gad.583310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimeno S., Rondón A.G., Luna R., Aguilera A. The yeast THO complex and mRNA export factors link RNA metabolism with transcription and genome instability. EMBO J. 2002;21:3526–3535. doi: 10.1093/emboj/cdf335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly S.M., Corbett A.H. Messenger RNA export from the nucleus: a series of molecular wardrobe changes. Traffic. 2009;10:1199–1208. doi: 10.1111/j.1600-0854.2009.00944.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly S.M., Pabit S.A., Kitchen C.M., Guo P., Marfatia K.A., Murphy T.J., Corbett A.H., Berland K.M. Recognition of polyadenosine RNA by zinc finger proteins. Proc. Natl. Acad. Sci. U S A. 2007;104:12306–12311. doi: 10.1073/pnas.0701244104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libri D., Dower K., Boulay J., Thomsen R., Rosbash M., Jensen T.H. Interactions between mRNA export commitment, 3′-end quality control, and nuclear degradation. Mol. Cell. Biol. 2002;22:8254–8266. doi: 10.1128/MCB.22.23.8254-8266.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund M.K., Guthrie C. The DEAD-box protein Dbp5p is required to dissociate Mex67p from exported mRNPs at the nuclear rim. Mol. Cell. 2005;20:645–651. doi: 10.1016/j.molcel.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Marfatia K.A., Crafton E.B., Green D.M., Corbett A.H. Domain analysis of the Saccharomyces cerevisiae heterogeneous nuclear ribonucleoprotein, Nab2p. Dissecting the requirements for Nab2p-facilitated poly(A) RNA export. J. Biol. Chem. 2003;278:6731–6740. doi: 10.1074/jbc.M207571200. [DOI] [PubMed] [Google Scholar]
- Mayer A., Lidschreiber M., Siebert M., Leike K., Söding J., Cramer P. Uniform transitions of the general RNA polymerase II transcription complex. Nat. Struct. Mol. Biol. 2010;17:1272–1278. doi: 10.1038/nsmb.1903. [DOI] [PubMed] [Google Scholar]
- Miller C., Schwalb B., Maier K., Schulz D., Dümcke S., Zacher B., Mayer A., Sydow J., Marcinowski L., Dölken L. Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Mol. Syst. Biol. 2011;7:458. doi: 10.1038/msb.2010.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan L., Huynh-Thu V.A., Delan-Forino C., Tuck A., Petfalski E., Lombraña R., Sanguinetti G., Kudla G., Tollervey D. Strand-specific, high-resolution mapping of modified RNA polymerase II. Mol. Syst. Biol. 2016;12:874. doi: 10.15252/msb.20166869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minvielle-Sebastia L., Winsor B., Bonneaud N., Lacroute F. Mutations in the yeast RNA14 and RNA15 genes result in an abnormal mRNA decay rate; sequence analysis reveals an RNA-binding domain in the RNA15 protein. Mol. Cell. Biol. 1991;11:3075–3087. doi: 10.1128/mcb.11.6.3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morawska M., Ulrich H.D. An expanded tool kit for the auxin-inducible degron system in budding yeast. Yeast. 2013;30:341–351. doi: 10.1002/yea.2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker R. RNA degradation in Saccharomyces cerevisae. Genetics. 2012;191:671–702. doi: 10.1534/genetics.111.137265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porrua O., Libri D. Transcription termination and the control of the transcriptome: why, where and how to stop. Nat. Rev. Mol. Cell Biol. 2015;16:190–202. doi: 10.1038/nrm3943. [DOI] [PubMed] [Google Scholar]
- Portman D.S., O’Connor J.P., Dreyfuss G. YRA1, an essential Saccharomyces cerevisiae gene, encodes a novel nuclear protein with RNA annealing activity. RNA. 1997;3:527–537. [PMC free article] [PubMed] [Google Scholar]
- Presnyak V., Alhusaini N., Chen Y.H., Martin S., Morris N., Kline N., Olson S., Weinberg D., Baker K.E., Graveley B.R., Coller J. Codon optimality is a major determinant of mRNA stability. Cell. 2015;160:1111–1124. doi: 10.1016/j.cell.2015.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F., Ryan D.P., Grüning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dündar F., Manke T. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44(W1):W160–W165. doi: 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth K.M., Byam J., Fang F., Butler J.S. Regulation of NAB2 mRNA 3′-end formation requires the core exosome and the Trf4p component of the TRAMP complex. RNA. 2009;15:1045–1058. doi: 10.1261/rna.709609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rougemaille M., Gudipati R.K., Olesen J.R., Thomsen R., Seraphin B., Libri D., Jensen T.H. Dissecting mechanisms of nuclear mRNA surveillance in THO/sub2 complex mutants. EMBO J. 2007;26:2317–2326. doi: 10.1038/sj.emboj.7601669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy K., Chanfreau G. Stress-induced nuclear RNA degradation pathways regulate yeast bromodomain factor 2 to promote cell survival. PLoS Genet. 2014;10:e1004661. doi: 10.1371/journal.pgen.1004661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Rosa H., Moreno H., Simos G., Segref A., Fahrenkrog B., Panté N., Hurt E. Nuclear mRNA export requires complex formation between Mex67p and Mtr2p at the nuclear pores. Mol. Cell. Biol. 1998;18:6826–6838. doi: 10.1128/mcb.18.11.6826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid M., Olszewski P., Pelechano V., Gupta I., Steinmetz L.M., Jensen T.H. The nuclear polyA-binding protein Nab2p is essential for mRNA production. Cell Rep. 2015;12:128–139. doi: 10.1016/j.celrep.2015.06.008. [DOI] [PubMed] [Google Scholar]
- Schmid M., Tudek A., Jensen T.H. Simultaneous Measurement of Transcriptional and Post-transcriptional Parameters by 3'end RNA-seq. Cell Rep. 2018;24:2468–2478. doi: 10.1016/j.celrep.2018.07.104. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segref A., Sharma K., Doye V., Hellwig A., Huber J., Lührmann R., Hurt E. Mex67p, a novel factor for nuclear mRNA export, binds to both poly(A)+ RNA and nuclear pores. EMBO J. 1997;16:3256–3271. doi: 10.1093/emboj/16.11.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahm Y., Fahrenkrog B., Zenklusen D., Rychner E., Kantor J., Rosbach M., Stutz F. The RNA export factor Gle1p is located on the cytoplasmic fibrils of the NPC and physically interacts with the FG-nucleoporin Rip1p, the DEAD-box protein Rat8p/Dbp5p and a new protein Ymr 255p. EMBO J. 1999;18:5761–5777. doi: 10.1093/emboj/18.20.5761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strambio-de-Castillia C., Blobel G., Rout M.P. Proteins connecting the nuclear pore complex with the nuclear interior. J. Cell Biol. 1999;144:839–855. doi: 10.1083/jcb.144.5.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M., Schwalb B., Schulz D., Pirkl N., Etzold S., Larivière L., Maier K.C., Seizl M., Tresch A., Cramer P. Comparative dynamic transcriptome analysis (cDTA) reveals mutual feedback between mRNA synthesis and degradation. Genome Res. 2012;22:1350–1359. doi: 10.1101/gr.130161.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry L.J., Wente S.R. Nuclear mRNA export requires specific FG nucleoporins for translocation through the nuclear pore complex. J. Cell Biol. 2007;178:1121–1132. doi: 10.1083/jcb.200704174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen R., Nielsen P.S., Jensen T.H. Dramatically improved RNA in situ hybridization signals using LNA-modified probes. RNA. 2005;11:1745–1748. doi: 10.1261/rna.2139705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran E.J., Zhou Y., Corbett A.H., Wente S.R. The DEAD-box protein Dbp5 controls mRNA export by triggering specific RNA:protein remodeling events. Mol. Cell. 2007;28:850–859. doi: 10.1016/j.molcel.2007.09.019. [DOI] [PubMed] [Google Scholar]
- van Dijk E.L., Chen C.L., d’Aubenton-Carafa Y., Gourvennec S., Kwapisz M., Roche V., Bertrand C., Silvain M., Legoix-Né P., Loeillet S. XUTs are a class of Xrn1-sensitive antisense regulatory non-coding RNA in yeast. Nature. 2011;475:114–117. doi: 10.1038/nature10118. [DOI] [PubMed] [Google Scholar]
- Vasiljeva L., Buratowski S. Nrd1 interacts with the nuclear exosome for 3′ processing of RNA polymerase II transcripts. Mol. Cell. 2006;21:239–248. doi: 10.1016/j.molcel.2005.11.028. [DOI] [PubMed] [Google Scholar]
- Warfield L., Ramachandran S., Baptista T., Devys D., Tora L., Hahn S. Transcription of nearly all yeast RNA polymerase II-transcribed genes is dependent on transcription factor TFIID. Mol. Cell. 2017;68:118–129.e5. doi: 10.1016/j.molcel.2017.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wery M., Descrimes M., Vogt N., Dallongeville A.S., Gautheret D., Morillon A. Nonsense-mediated decay restricts LncRNA levels in yeast unless blocked by double-stranded RNA structure. Mol. Cell. 2016;61:379–392. doi: 10.1016/j.molcel.2015.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyers F., Rougemaille M., Badis G., Rousselle J.C., Dufour M.E., Boulay J., Régnault B., Devaux F., Namane A., Séraphin B. Cryptic pol II transcripts are degraded by a nuclear quality control pathway involving a new poly(A) polymerase. Cell. 2005;121:725–737. doi: 10.1016/j.cell.2005.04.030. [DOI] [PubMed] [Google Scholar]
- Xu Z., Wei W., Gagneur J., Perocchi F., Clauder-Münster S., Camblong J., Guffanti E., Stutz F., Huber W., Steinmetz L.M. Bidirectional promoters generate pervasive transcription in yeast. Nature. 2009;457:1033–1037. doi: 10.1038/nature07728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao W., Roser D., Köhler A., Bradatsch B., Bassler J., Hurt E. Nuclear export of ribosomal 60S subunits by the general mRNA export receptor Mex67-Mtr2. Mol. Cell. 2007;26:51–62. doi: 10.1016/j.molcel.2007.02.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.