

Abstract
Introduction
Epidermal growth factor receptor (EGFR)-targeted therapies with antibodies and small molecular EGFR kinase inhibitors have shown poor efficacy in unselected populations of patients with advanced non-small cell lung carcinomas (NSCLC). In contrast, patients with overexpression of EGFR and activating mutations in EGFR kinase domain demonstrated improved responses to EGFR kinase inhibitors. Therefore, we have developed a novel radiotracer, [18F]F-PEG6-IPQA for PET imaging of EGFR expression-activity in NSCLC, and have described its radiosynthesis and in vitro evaluation in two NSCLC cell lines with wild-type and L858R active mutant EGFR.
Methods
A mesylate precursor was synthesized in multiple steps and radiofluorinated using K18F/Kryptofix. The fluorinated intermediate compound was reduced to an amino derivative then treated with acryloyl isobutyl carbonate, followed by purification by HPLC to obtain the desired product.
Results
Decay-corrected radiochemical yields of [18F]F-PEG6-IPQA were 3.9–17.6%, with an average of 9.0% (n=11). Radiochemical purity was 997% with specific activity of 34 GBq/μmol (mean value, n=10) at the end of synthesis. The accumulation of [18F]F-PEG6-IPQA in H3255 cells was ten-fold higher than in H441 cells, despite a two-fold lower level of activated phospho-EGFR expression in H3255 cells compared with H441 cells. The accumulation of [18F]F-PEG6-IPQA in both cell lines was significantly decreased in the presence of a small molecular EGFR kinase inhibitor, Iressa, at 100 μM concentration in culture medium.
Conclusion
We have synthesized [18F]F-PEG6-IPQA and demonstrated its highly selective accumulation in active mutant L858R EGFR-expressing NSCLC cells in vitro. Further in vivo studies are warranted to assess the ability of PET imaging with [18F]F-PEG6-IPQA to discriminate the active mutant L858R EGFR-expressing NSCLC that are sensitive to therapy with EGFR kinase inhibitors vs NSCLC that express wild-type EGFR.
Keywords: Radiochemistry, [18F]F-PEG6-IPQA, Epidermal growth factor receptor, Non-small cell lung carcinoma, Positron emission tomography
Introduction
According to the American Cancer Society, 219,440 Americans were diagnosed with lung cancer in 2009; among which non-small cell lung carcinomas (NSCLC) represent the majority of lung cancer patients [1]. The outcome of conventional chemotherapy for advanced NSCLCs remains unsatisfactory, with low long-term survival rates. Molecular-targeted therapeutic agents that block pathways important for maintenance and progression of NSCLCs were expected to achieve a significant improvement in disease control and long-term survival. However, only marginal efficacy has been observed with epidermal growth factor receptor (EGFR) inhibitors, anti-EGFR monoclonal antibodies, and angiogenesis inhibitors in unselected populations of patients with advanced NSCLC. In contrast, patients with overexpression of EGFR and activating mutations in EGFR kinase domain demonstrate improved responses when treated with EGFR kinase inhibitors. Therefore, we and others have been developing radiotracers for positron emission tomography (PET) imaging of EGFR expression-activity in tumors and normal tissues at the EGFR kinase level derived from the 4-(anilino)quinazoline pharmacophore [2, 3], including: ML series of 18F-labeled five 4-(anilino)quinazoline derivatives ([18F]ML01) [4], N-{4-[(4,5-dichloro-2-fluorophenyl)-amino]quinazolin-6-yl}-acrylamide labeled with 11C ([11C]-ML03) [5, 6], 4-dimethylamino-but-2-enoic acid [4-(phenylamino)-quinazoline-6-yl]-amides (ML04) labeled with 11C ([11C]ML04) [6], or with 18F ([18F]ML04) [7], [18F]F-PEG4-ML04 [7, 8], as well as [124I]IPQA [9], [18F] gefinitib [10], [11C]PD153035 [11], and [11C]erlotinib [12]. Recent studies by Pantaleo et al. (2010) using different PEG-ylated anilinoquinazoline derivatives labeled with 124I, 18F, and 11C, failed to demonstrate the accumulation of these radiotracers in subcutaneous glioblastoma xenografts in mice [13]. Moreover, no differences in radiotracer accumulation levels were observed between U87MG. wtEGFR tumors overexpressing wild-type EGFR and U138MG tumors lacking EGFR expression [13]. In contrast, clinical studies with [11C]PD153035 have demonstrated some promise for imaging EGFR expression in NSCLC patients [14]. Thus, none of the imaging agents reported to date had demonstrated efficacy and/or selectivity for detection of activated EGFR or active mutant EGFR kinases that confer sensitivity to small molecular EGFR inhibitors currently used in clinical practice [15].
Here, we describe radiosynthesis and initial in vitro evaluation of a novel radiotracer, 4-[(3-iodophenyl)amino]-7-{2-[2-{2-(2-[2-{2-([18F]fluoroethoxy)-ethoxy}-ethoxy]-ethoxy)-ethoxy}-ethoxy]-quinazoline-6-yl-acrylamide ([18F] F-PEG6-IPQA) for PET imaging of EGFR expression-activity. We demonstrate that [18F]F-PEG6-IPQA accumulates in vitro significantly higher in H3255 lung carcinoma cells expressing the L858R active mutant EGFR, compared with H441 lung carcinoma cells overexpressing the wild-type EGFR. This is apparently due to an increased affinity and irreversible binding of [18F]F-PEG6-IPQA to the active mutant L858R EGFR kinase.
Materials and Methods
Reagents and Instrumentation
All reagents and solvents were purchased from Aldrich Chemical Co. (Milwaukee, WI) or Fisher Scientific (Pittsburgh, PA) and used without further purification. Silica gel solid-phase extraction cartridges (Sep-Pak, 900 mg) were purchased from Alltech Associates (Deerfield, IL). Reverse phase C18 Sep-Pak® Plus Environmental cartridges were obtained from Waters (Milford, MA). Fluorine-18 was commercially supplied, as a solution of K[18F/Kryptofix222, by Cyclotope (Houston, TX). Thin layer chromatography (TLC) was performed on silica gel F-254 aluminum-backed plates (Merck, Darmstadt, Germany) with visualization under UV (254 nm) and by staining with potassium permanganate or ceric ammonium molybdate. Flash chromatography was performed using silica gel 60 mesh size 230–400 ASTM (Merck, Darmstadt, Germany) or CombiFlash Companion or SQ16× flash chromatography system (Isco, Lincoln, NE) with RediSep columns (normal phase silica gel; mesh size 230–400 ASTM) and Optima TM grade solvents (Fisher).
Melting points were recorded on a Buchi Melting Point B-545 apparatus and are uncorrected. Proton, 19F, and 13C NMR spectra were recorded on either an 300 or 600 MHz NMR spectrometers (Bruker, Germany) with tetramethylsilane used as an internal reference and hexafluorobenzene as an external reference at The University of Texas MD Anderson Cancer Center. Low resolution mass spectra (ion spray, a variation of electrospray) were acquired on a Perkin-Elmer Sciex API 100 spectrometer or Applied Biosystems Q-trap 2000 LC-MS-MS at The University of Texas MD Anderson Cancer Center. High-resolution mass spectra were obtained on a Bruker BioTOF II mass spectrometer at the University of Minnesota using electrospray ionization technique.
High-performance liquid chromatography (HPLC) was performed with a 1100 series pump (Agilent, Santa Clara, CA), with a built-in UV detector with variable wavelength and a BioScan FlowCount using a PIN Diode for gamma ray detection (Bioscan, Washington DC). Analytical radio-HPLC was conducted on an Agilent system consisting of a 1100 series quaternary pump, vacuum degasser, diode array detector, and a BioScan FlowCount radiodetector equipped with a 1.5×1.5″ NaI(Tl) well-type crystal. Radioactivity was assayed using a Capintec CRC-15R dose calibrator (Ramsey, NJ).
Chemical Syntheses
Compounds 3, 4, and 6 (Scheme 1a) were prepared following literature methods [5, 23].
Scheme 1.

Synthetic schemes for preparation of the non-radioactive compound 4-[(3-iodophenyl)amino]-7-[2-{2-(2-[2-{2-(2-fluoroethoxy)-ethoxy}-ethoxy]-ethoxy)-ethoxy}-ethoxy]-quinazoline-6-yl-acrylamide 1. a Synthesis of 1. b Preparation of 7. c Preparation of precursor 9.
Preparation of 2-[2-{2-(2-[2-{2-(Tert-Butyl-Dimethyl-Silanyloxy)-Ethoxy}-Ethoxy]-Ethoxy)-Ethoxy}-Ethoxy]-Ethanol (7)
A solution of imidazole (1.5 g, 22 mmol) and hexaethylene glycol (10 g, 25 mmol) in dry DMF (25 mL) was cooled to 0°C and stirred for 30 min under argon (Ar). To this solution, tert-butyldimethylsilyl chloride (3.3 g, 22 mmol) in dry dimethylformamide (DMF; 10 mL) was added dropwise and continued stirring at 0°C for another 2 h, then the reaction mixture was allowed to warm up to room temperature. The DMF was removed at 60°C under vacuum, and the resulting mixture was extracted with ethyl acetate (3×100 mL), then the combined organic extracts were washed with brine, dried (Na2SO4), and evaporated under reduced pressure. The crude product was purified by flash chromatography, eluting with 5% MeOH/CH2Cl2 yielding 2-[2-{2-(2-[2-{2-(tert-butyl-dimethyl-silanyloxy)-ethoxy}-ethoxy]-ethoxy)-ethoxy}-ethoxy]-ethanol 7 [7.1 g, 50% yield; TLC: Rf = 0.45, MeOH/CH2Cl2 (1:9)]. 1H NMR (CDCl3) δ—3.64 (m, 24H), 0.87 (s, 9H), 0.04 (s, 6H); 13C NMR δ—77.3, 77.1, 76.9, 72.6, 72.5, 70.7, 70.6, 70.6, 70.6, 70.6, 70.4, 62.7, 61.7, 60.4, 25.9, −5.3; MS (C18H40O7Si)—calculated 396.254, found 397.4 (M + H).
Preparation of 2-(2-[2-{2-(2-[2-{4-(3-Iodophenylamino)-6-Nitro-Quinazoline-7-Yoloxy}-Ethoxy]-Ethoxy)-Ethoxy}-Ethoxy]-Ethoxy)-Ethanol (8)
A solution of 6 (596 mg, 1.6 mmol), 7 (957 mg, 2.4 mmol), and potassium trimethylsilanolate (621 g, 4.74 mmol) in DMSO (35 mL) was stirred together under N2 for 5 h at room temperature. The deep crimson mixture was poured into stirred ice-water (40 mL), and then extracted with ethyl acetate (3 × 50 mL). The combined organic extracts were washed with aqueous 4% NaHCO3 solution (2 × 50 mL) and water (2 × 25 mL), and then concentrated under vacuum. The resulting crude material was purified by flash chromatography eluting with 5% MeOH/CH2Cl2 to obtain 8 [700 g, 71% yield; TLC: Rf = 0.45, MeOH/CH2 Cl2 (1:19)]. 1 H NMR (DMSO-d6) δ—10.08 (s, 1H), 9.25 (s, 1H), 8.69 (s, 1H), 8.29 (s, 1H), 7.94 (dd, J = 1.2, 9.6 Hz, 1H), 7.52 (s, 1H), 7.51 (d, J = 6.0 Hz, 1H), 7.22 (t, J = 8.4 Hz, 1H), 5.77 (s, 1H), 4.59 (t, J = 5.4 Hz, 1H), 4.44 (t, J = 4.2 Hz, 1H), 3.84 (t, J = 4.2 Hz, 2H), 3.62 (m, 2H), 3.54 (m, 2H), 3.49 (m, 14H); 13C NMR δ—157.8, 157.4, 153.7, 153.2, 140.2, 139.0, 132.4, 130.5, 130.2, 121.7, 121.4, 110.5, 108.1, 94.2, 71.3, 68.4, 60.2; MS (C26H33IN4O9)—calculated 672.129, found 673.4 (M+H).
Preparation of Methanesulfonic Acid 2-(2-[2-{2-(2-[2-{4-(3-Iodophenylamino)-6-Nitro-Quinazoline-7-Yoloxy}-Ethoxy]-Ethoxy)-Ethoxy}-Ethoxy]-Ethoxy)-Ethyl Ester (9)
Compound 8 (2 g, 3 mmol) was dissolved in CH2Cl2 (50 mL) and Et3N (2.5 mL, 18 mmol) was added to the solution. Methanesulfonyl chloride (0.7 mL, 9.0 mmol) was added dropwise to the reaction mixture and stirred at room temperature for 3 h under argon. The reaction mixture was extracted with CH2Cl2 (2×100 mL). The combined organic extracts were washed with brine (100 mL) and then dried over anhydrous Na2SO4. After filtration and evaporation of the organic solvent, the residue was purified by flash chromatography, eluting with 3% MeOH/CH2Cl2 to afford 9 (1.5 g, 67% yield). 1H NMR (DMSO-d6) δ—10.07 (s, 1H), 9.23 (s, 1H), 8.68 (s, 1H), 8.28 (s, 1H), 7.93 (dd, J=1.2, 7.8 Hz, 1H), 7.50 (d, J=10.2 Hz, 1H), 7.49 (s, 1H), 7.21 (t, J=7.8 Hz, 1H), 4.43 (t, J=3.6 Hz, 2H), 4.30 (m, 2H), 3.83 (t, J=4.2 Hz, 2H), 3.66 (m, 2H), 3.62 (m, 2H), 3.55 (m, 2H), 3.52 (m, 12H), 3.18 (s, 3H); 13C NMR δ—157.8, 157.4, 153.7, 153.2, 140.1, 139.0, 132.4, 130.5, 130.2, 121.8, 121.4, 110.4, 108.1, 94.1, 70.1, 69.8, 69.7, 69.7, 69.7, 69.7, 69.6, 68.4, 68.2, 36.8; MS (C27H35IN4O11S)—calculated 750.107, found 751.2 (M+H).
Preparation of (3-Iodophenyl)-(7-[2-{2-(2-[2-{2-(2-Fluoroethoxy)-Ethoxy}-Ethoxy]-Ethoxy)-Ethoxy}-Ethoxy]-6-Nitro-Quinazolin-4-yl)-Amine (10)
Compound 8 (350 mg, 0.52 mmol) was dissolved in dichloromethane (10 mL) and cooled to −78°C. A solution of diethylaminosulfur trifluoride (DAST, 0.681 mL, 5.2 mmol) in dichloromethane (5 mL) was added slowly to the cold solution while stirring. The stirred reaction mixture was allowed to warm to room temperature and stirring was continued for 12 h. The reaction mixture was poured into saturated sodium bicarbonate solution (25 mL), diluted with water (25 mL), and extracted with dichloromethane (3 × 50 mL). The combined organic extracts were washed with brine (50 mL), and then dried over anhydrous Na2SO4. After filtration and evaporation of the organic solvent, the residue was purified by flash chromatography, eluting with 3% MeOH/CH2Cl2 to obtain 10 (150 mg, 42% yield). 1H NMR (CDCl3) δ—9.17 (s, 1H), 8.71 (s, 1H), 8.33 (s, 1H), 7.88 (d, J=8.4 Hz, 1H), 7.47 (d, J=7.8 Hz, 1H), 7.20 (s, 1H), 7.10 (t, J=7.8 Hz, 1H), 4.52 (dm, JHF =48 Hz, JHH =4.2 Hz, 2H), 4.25 (t, J=4.2 Hz, 2H), 3.89 (t, J=4.8 Hz, 2H), 3.72 (m, 2H), 3.67 (m, 2H), 3.49 (m, 14H); 13C NMR δ—158.3, 157.8, 154.4, 153.6, 139.6, 139.2, 133.4, 131.2, 130.2, 122.3, 121.8, 110.3, 108.6, 93.8, 83.6, 82.6, 71.1, 70.7, 70.6, 70.6, 70.5, 70.5, 70.4, 70.4, 70.3, 69.6, 69.1; 19F NMR δ, −222.74 (tt, JHF =48 Hz, 28 Hz); MS (C26H32FIN4O8)—calculated 674.125, found 675.2 (M + H).
Preparation of N4-(3-Iodophenyl)-7-[2-{2-(2-[2-{2-(2-Fluoroethoxy)-Ethoxy}-Ethoxy]-Ethoxy)-Ethoxy}-Ethoxy]-Quinazoline-4,6-Diamine (11)
Compound 10 (200 mg, 0.3 mmol) was dissolved in tetrahydrofuran (3 mL). To this solution, stannous chloride (180 mg, 0.95 mmol) was added, and then heated to 60°C for 3 h under argon. The reaction mixture was cooled and water was added (25 mL), then saturated bicarbonate solution (10 mL) was added and extracted with ethyl acetate (3 ×75 mL). The combined organic extracts were concentrated, and the crude product was purified by flash chromatography, eluting with 3% MeOH/CH2Cl2 to yield the amine 11 (150 mg, 43% yield). 1H NMR (CDCl3) δ— 8.54 (s, 1H), 8.16 (s, 1H), 7.74 (d, J=8.4 Hz, 1H), 7.66 (bs, 1H), 7.40 (d, J=7.8 Hz, 1H), 7.14 (s, 1H), 7.07 (m, 2H), 4.53 (dm, JHF =48 Hz, JHH =4.2 Hz, 2H), 4.21 (t, J=4.2 Hz, 2H), 3.88 (t, J= 4.8 Hz, 2H), 3.72 (m, 2H), 3.69 (m, 2H), 3.49 (m, 14H); 13C NMR δ—155.2, 152.4, 151.6, 145.4, 138.4, 132.2, 130.4, 129.7, 120.3, 110.6, 107.3, 101.0, 94.1, 83.8, 82.7, 70.9, 70.8, 70.7, 70.6, 70.6, 70.5, 70.5, 70.4, 70.3, 69.1, 68.1; 19F NMR δ, −221.98 (tt, JHF =51, 28 Hz); MS (C26H34FIN4O6)—calculated 644.151, found 645.3 (M+H).
Preparation of 4-[(3-Iodophenyl)Amino]-7-[2-{2-(2-[2-{2-(2-Fluoroethoxy)-Ethoxy}-Ethoxy]-Ethoxy)-Ethoxy}-Ethoxy]-Quinazoline-6-Yl-Acrylamide (1)
Acryloyl isobutyl carbonate was prepared in situ by adding isobutyl chloroformate (95 μL, 0.72 mmol) dropwise to a stirred solution of acrylic acid (60 μL, 0.86 mmol) and Et3N (200 μL, 1.44 mmol) in tetrahydrofuran (THF; 2.5 mL) at 0°C under N2. Amine 11 (160 mg, 0.25 mmol) was added in one portion to the above solution of acryloyl isobutyl carbonate, and stirring was continued for 10 min. After another 30 min at 0°C, the reaction mixture was applied to a flash chromatography column and was eluted with 5% MeOH/CH2Cl2 to give the desired product 1 (75 mg, 43% yield). 1H NMR (CDCl3) δ—9.12 (s, 1H), 8.96 (s, 1H), 8.67 (s, 1H), 8.22 (s, 1H), 7.76 (m, 2H), 7.47 (d, J=7.8 Hz, 1H), 7.25 (s, 1H), 7.11 (t, J=8.4 Hz, 1H), 6.51 (m, 2H), 5.84 (dd, J=3.6, 8.4 Hz, 1H), 4.55 (dm, JHF =48 Hz, J=4.2 Hz, 2H), 4.38 (t, J=4.2 Hz, 2H), 3.98 (t, J=4.2 Hz, 2H), 3.75 (m, 2H), 3.70 (m, 2H), 3.65 (m, 14H); 13C NMR δ—164.3, 156.7, 154.5, 152.3, 148.3, 139.8, 133.0, 131.3, 130.4, 130.3, 128.3, 128.3, 120.8, 110.2, 109.6, 108.0, 94.1, 83.7, 82.6, 70.8, 70.7, 70.6, 70.5, 70.5, 70.5, 70.5, 70.3, 69.0, 68.55; 19F NMR δ, −222.81 (tt, JHF =48 Hz, 28 Hz); MS (C29H36FIN4O7)—calculated 698.161, found 699.5 (M+H).
Radiosynthesis of 4-[(3-iodophenyl) Amino]-7-{2-[2-{2-(2-[2-{2-([18F] Fluoroethoxy)-Ethoxy}-Ethoxy]-Ethoxy)-Ethoxy}-Ethoxy]-Quinazoline-6-Yl-Acrylamide (14)
Preparation of (3-Iodophenyl)-(7-{2-[2-{2-(2-[2-{2-([18F]Fluoroethoxy)-Ethoxy}-Ethoxy]-Ethoxy)-Ethoxy}-Ethoxy]-6-Nitro-Quinazolin-4-Yl)-Amine 12
The aqueous [18F]fluoride (K18F/Kryptofix 2.2.2) was dried in a two-step evaporation process using the GE TRACERLab FXF-N synthesizer (GE Healthcare). A solution of methanesulfonic acid 2-(2-[2-{2-(2-[2-{4-(3-iodophenylamino)-6-nitro-quinazoline-7-yoloxy}-ethoxy]-ethoxy)-ethoxy}-ethoxy]-ethoxy)-ethyl ester 9 (~10 mg) in anhydrous DMSO (0.4 mL) was added to the dried [18F]-complex. The reaction mixture was heated for 30 min at 120°C. After cooling to room temperature, the reaction mixture was passed through a silica gel cartridge, and the crude product 12 was eluted with 2.5 mL of MeOH/CH2Cl2 (3:7).
Preparation of N4-(3-Iodophenyl)-(7-{2-[2-{2-(2-[2-{2-([18f]Fluoroethoxy)-Ethoxy}-Ethoxy]-Ethoxy)-Ethoxy}-Ethoxy]-Quinazoline-4,6-Diamine 13
The solution of 12 was evaporated to dryness at 80°C under a stream of Ar, then SnCl2 (15 mg) in dry THF (0.4 mL) was added. The mixture was heated for 15 min at 60°C, cooled to room temperature, then the mixture was passed through a silica gel cartridge (previously activated with 2 mL of n-hexane). The cartridge was eluted with dry THF (2×1 mL), and the solvent was evaporated at 60°C under a flow of Ar, down to about 200 μL. The residue, crude N4-(3-iodophenyl)-(7-{2-[2-{2-(2-[2-{2-([18 F]fluoroethoxy)-ethoxy}-ethoxy]-ethoxy)-ethoxy}-ethoxy]-quinazoline-4,6-diamine 13 was cooled to 0°C for the next step.
Preparation of 4-[(3-Iodophenyl)Amino]-7-{2-[2-{2-(2-[2-{2-([18F]Fluoroethoxy)-Ethoxy}-Ethoxy]-Ethoxy)-Ethoxy}-Ethoxy]-Quinazoline-6-Yl-Acrylamide 14
A mixture of acrylic acid (50 μL), isobutyl chloroformate (60 μL), and triethylamine (100 μL) in dry THF (0.2 mL) was prepared under Ar, which then formed a milky white suspension, that was cooled to 0°C. The solution of 13 in dry THF (0.2 mL) was also cooled to 0°C and transferred to the vial containing acryloyl isobutyl carbonate (milky suspension) under Ar. The vial containing 13 was rinsed with 0.1 mL of THF and transferred to the reaction vial containing acryloyl isobutyl carbonate. The reaction mixture was kept at 0°C for 10 min with occasional shaking. The solvent from the reaction mixture was evaporated at 70°C under Ar flow to near dryness and re-dissolved in prep-HPLC solvent (~1 mL) and passed through a 0.45-μm filter (PTFE, 17 mm, Grace, Deerfield, IL). An additional membrane filter (Sartobind Q, 5 cm2, Sartorius AG, Goettingen, Germany) coupled with the PTFE filter was also employed in a few experiments. The filtered solution was injected onto a C8 preparative column (Zorbax CombiHT XDB, 21.2×100 mm) and eluted with acetonitrile/0.1% ammonium formate buffer (45:55) at a flow rate of 6 mL/min. The radioactive peak containing 14 was collected, diluted with 10 mL of water, then passed through a C18 Sep-Pak® (preconditioned with 10 mL of 95% ethanol, then 10 mL of water). The cartridge was washed with 10–15 mL of water and then eluted with 95% ethanol (2.5–3.0 mL) to isolate the product. For in vivo studies, ethanol was concentrated in vacuo to a small volume, and then diluted with saline for injection. The final product 14 was analyzed by radio-HPLC using a Zorbax Eclipse XDB-C8 column (4.6× 150 mm) with a mobile phase consisting of acetonitrile/0.1% ammonium formate buffer (47:53) at 1.0 mL/min flow rate. Radiochemical yield of 14 was in the range of 3.9–17.6% (n= 11, d.c.) from the end of bombardment. The radiochemical purity was 997%, and specific activity was 34 GB/μmol. Total synthesis time was approximately 3 h.
Tumor Cell Lines
Four different NSCLC cell lines were used in this study, expressing different levels of wild-type and active mutant EGFRs and with different sensitivity to EGFR inhibitor Iressa: H441 (wild-type EGFR; resistant [16, 17]) and H3255 (mutant L858R EGFR; sensitive [16-18]). The cells were grown in flasks with D-MEM/F-12 medium supplemented with 10% FBS and antibiotics at 37°C in humidified atmosphere with 5% CO2. Cells were kept in the log phase of proliferative activity.
Assessment of EGFR Expression by Western Blot
The H441 and H3255 NSCLC cell lines expressing wild-type and L858R mutant EGFR, respectively, were grown in 15-cm culture dish until 60–70% confluent. Then the cells were incubated for 30 min in fresh culture medium supplemented with 20% fetal calf serum (FCS) and [18F]F-PEG6-IPQA at 0.37 MBq/mL. Thereafter, the cells were washed with phosphate-buffered saline (PBS), harvested in PBS by scraping, pelleted by centrifugation at 1,500 rpm for 5 min, the supernatant was removed, and the cell pellet was frozen on dry ice. The cell pellet was thawed and lyzed in appropriate volume of buffer (200 uL/100 mg cells) containing 20 mM Tris–HCl pH 7.4, NaCl 150 mM, EDTA 1 mM, EGTA 1 mM, beta-glycerophosphate 1 mM, Na3VO4 1 mM, NaF 1 mM, Triton X-100 1%, and Protease Inhibitor Cocktail 10 uL/mL (all reagents from Sigma-Aldrich, CA) for 1 h at 4°C. Then, the cell lysate was sonicated 3×10 s on ice and cleared by 14,000 rpm centrifugation at 4°C for 10 min. The supernatant was denatured by heating at 70°C with LDS sample buffer (Invitrogen, Carlsbad, CA) and separated by SDS/polyacrylamide gel electrophoresis using precast 4–12% Tris–HCl gel cassettes (Invitrogen, Carlsbad, CA). After transferring proteins onto a nitrocellulose membrane using a semi-dry electroblotting device (Invitrogen, Carlsbad, CA), the membrane was immunostained with a goat polyclonal antibody against phoshpo-tyrosine 845 of EGFR (Santa Cruz, CA) and visualized using the ECL kit (Bio-Rad). For autoradiography, the membrane was exposed for 12 h to HyBlot CL film (Denville Scientific, Metuchen, NJ) to visualize the [18F]-labeled protein bands.
In Vitro Radiotracer Accumulation Assay
Radiotracer uptake studies were performed in monolayer cultures of different NSCLC cell lines as previously described [9, 19]. Briefly, tumor cells were grown in 15-cm culture dishes until 60–70% confluent, at which time the culture medium was depleted of FCS to prevent EGFR activation. Then, the cell cultures were incubated with serum-free medium containing [18F]F-PEG6-IPQA (3.7 MBq/mL) for different time intervals (15, 30, 60, and 120 min). In the first part of the experiment, the cells were harvested by gentle scraping at different time intervals, transferred into 15-mL tubes, and centrifuged at 1,000×g for 2 min. A 100 μL aliquot of supernatant was transferred to a pre-weighed scintillation tube, and the rest of the supernatant was removed by aspiration. The cell pellet was snap-frozen on dry ice and transferred to another pre-weighed scintillation tube. The cell pellet and a sample of radioactive culture medium were weighed assessed for radioactivity concentration using a Packard Quantum gamma counter (Perkin-Elmer, Waltham, MA). The radioactivity concentration was expressed as cpm/g cells and cpm/mL medium, respectively. Cells-to-medium radioactivity concentration ratios were calculated and plotted against time to evaluate the radiotracer accumulation and washout kinetics.
Results and Discussion
Chemistry
The scheme for synthesizing 4-[(3-iodophenyl)amino]-7-{2-[2-{2-(2-[2-{2-(fluoroethoxy)-ethoxy}-ethoxy]-ethoxy)-ethoxy}-ethoxy]-quinazoline-6-yl-acrylamide 1 is shown in Scheme 1a. Compounds 3, 4, and 6 were prepared according to literature methods [20, 22, 23] in 90%, 52%, and 84% yields, respectively. All compounds were fully characterized by spectroscopic methods; the 1H NMR spectra and mass spectra were consistent with those reported previously [20, 22, 23]. Compound 5 was prepared in situ and used without isolation toward the preparation of 6.
Compound 7, 2-[2-{2-(2-[2-{2-(tert-butyl-dimethyl-silanyloxy)-ethoxy}-ethoxy]-ethoxy)-ethoxy}-ethoxy]-ethanol, was prepared according to a literature method [21] (Scheme 1b). Briefly, hexaethylene glycol was allowed to react with tert-butyldimethylsilyl chloride in the presence of imidazole in anhydrous DMF under inert atmosphere. After work-up and chromatographic purification, 7 was isolated in 50% yield. The 1H NMR spectrum was consistent with that reported in the literature [21].
Compound 8 was prepared from 6 by reaction with a mixture of 7 and potassium trimethylsilanolate in DMSO. The key intermediate compound 8 was obtained in 71% yield. The 1H NMR spectrum was consistent with that reported in the literature [22]. Compound 8 was reacted with DAST to get compound 10, which was characterized by 1H and 19F NMR spectroscopy and mass spectrometry. 19F NMR spectrum (coupled) showed a peak at −222.74 ppm as a triplet of triplets with geminal proton–fluorine coupling constant of 48 Hz and a vicinal proton–fluorine coupling constant of 28 Hz. The yield in this fluorination reaction was moderate at 42%. The nitro group of compound 10 was reduced by reaction with SnCl2 to produce the amino-compound 11 in 43% yield. A reduction of such a nitro group was reported previously [22]; however, using that method would result in the iodine being lost, therefore, SnCl2 was used as the reducing agent, which enabled reduction of the nitro group without any loss of iodine. Compound 11 was then reacted with acryloyl isobutyl carbonate, which was freshly prepared by reaction of acrylic acid with isobutyl chloroformate, to produce compound 1. It should be noted that acryloyl isobutyl carbonate is not commercially available due to its high reactivity and instability, therefore, in every synthesis it was prepared and used immediately. Compound 1 was obtained in 43% yield, fully characterized by spectroscopic methods and used as a non-radioactive standard compound for HPLC analysis.
For radiosynthesis of [18F]F-PEG6-IPQA, a precursor compound 9 was prepared from 8 (Scheme 1c). The alcohol 8 was treated with methanesulfonyl chloride in CH2Cl2 in the presence of Et3N at room temperature for 3 h. The mesylate precursor 9 was obtained in 67% yield and fully characterized by spectroscopic methods. Fluorination and radiofluorination reactions were performed on 9 with KF/Kryptofix222 or K18F/Kryptofix222 in DMSO to obtain compound 10 and 12, respectively. It was observed that the non-radioactive fluorination reaction gave a better chemical yield of 10 than the DAST reaction from 8. The fluorination reaction afforded a 78% yield in comparison to 42% with the DAST reaction.
Radiochemistry
Starting from the mesylate precursor 9, the radiosynthesis of 14 consists of three steps as shown is Scheme 2. The initial step was accomplished using an automated synthesis module TRACERLab FXF-N (GE Healthcare), and the remaining steps were accomplished by manual operation. The radiofluorination of 9 with K[18F]F/Kryptofix222 in dry DMSO at 120°C (30 min) produced (3-iodophenyl)-(7-{2-[2-{2-(2-[2-{2-([18F] fluoroethoxy)-ethoxy}-ethoxy]-ethoxy)-ethoxy}-ethoxy]-6-nitro-quinazolin-4-yl)-amine 12 in an average radiochemical yield of 52% (d.c.) with a mean radiochemical purity of 72% (n=19). The reduction of the 6-nitro group of 12 was achieved by using stannous chloride in THF at 60°C for 15 min. N4-(3-iodophenyl)-(7-{2-[2-{2-(2-[2-{2-([18F]fluoroethoxy)-ethoxy}-ethoxy]-ethoxy)-ethoxy}-ethoxy]-quinazoline-4,6-diamine 13 was isolated after passing through a silica gel cartridge followed by elution with THF. Conversion of the amine function to an acrylamide group was accomplished by in situ production of acryloyl isobutyl carbonate from the reaction of isobutyl chloroformate, acrylic acid, and triethylamine in THF at 0°C, followed by addition of 13 within 10 min. The crude product, 4-[(3-iodophenyl)amino]-7-{2-[2-{2-(2-[2-{2-([18F]fluoroethoxy)-ethoxy}-ethoxy]-ethoxy)-ethoxy}-ethoxy]-quinazoline-6-yl-acrylamide 14, was then purified by preparative HPLC. The synthesis time was about 3 h from the end of bombardment, including purification and formulation. The intermediate radiofluorinated compounds (12, 13) and the final product (14) were all identified by analytical HPLC at each step of the radiosynthesis and were confirmed by co-elution with their respective non-radioactive standards. The radiochemical yields in labeling step where variable, ranging from 30% to 60%. The two other steps, such as reduction of compound 12 and coupling with acryloyl isobutyl carbonate, also gave variable yields; as a result, the overall yields were more variable than expected. Decay-corrected radiochemical yields ranged from 4% to 17%, with an average yield of 9.0% (n=11). Radiochemical purity was found to be 997%. A representative analytical radio-HPLC chromatogram of 14 is shown in Fig. 1. However, it must be noted that UV detector revealed presence of a double peak indicative of an unidentified impurity (~60%). In contrast, only one peak was detectable by the radiation detector, which corresponded to [18F]F-PEG6-IPQA, based on the elution time of cold F-PEG6-IPQA standard. The average specific activity was determined to be 34 GBq/μmol (n=10), at the end of synthesis, considering only the amount of cold F-PEG6-IPQA without the impurity. Thus, further optimization of radiosynthetic and purification methods will be needed to eliminate or significantly reduce the impurity, which may potentially interfere with binding of [18F]F-PEG6-IPQA to EGFR kinase domain due to possible similarity of structure.
Scheme 2.

Radiosynthesis scheme for the preparation of [18F]F-PEG6-IPQA 14.
Fig. 1.

Analytical radio-HPLC chromatogram of [18F]F-PEG6-IPQA 14 after formulation. The UV trace showed a small amount of unknown impurity.
Initial In Vitro Evaluation
In vitro radiotracer accumulation assay demonstrated a rapid uptake of [18F]F-PEG6-IPQA during the initial phase (first 20 min) in both H441 and H3255 cells. After the first 20 min, the accumulation of [18F]F-PEG6-IPQA has leveled in H441 at a cells/medium ratio of 30–40, while the accumulation in H3255 cell monolayers continued to increase up to 1 h and only thereafter leveled at cells/medium concentration ratio of 400–600 (Fig. 2). The magnitude of [18F]F-PEG6-IPQA accumulation in H3255 cells was more than ten-fold higher than in H441 cells, despite a two-fold lower level of activated phospho-EGFR expression in H3255 cells compared with H441 cells (Fig. 3). Apparently, the presence of impurity in [18F]F-PEG6-IPQA preparation identified by HPLC with a UV detector (Fig. 1) did not interfere with [18F]F-PEG6-IPQA binding of to EGFR kinase and its accumulation in H3255 cells expressing L858R EGFR mutant. The magnitude of [18F]F-PEG6-IPQA accumulation in both cell lines was significantly decreased in presence of a small molecular EGFR kinase inhibitor Iressa at 100 μM in culture medium (Fig. 2). Such significantly higher accumulation of [18F]F-PEG6-IPQA in H3255 tumors can be explained by the presence of L858R activating mutation in the EGFR kinase domain [16-18], and not by the difference in the level of EGFR expression, which was lower in H3255 cells, as compared to H441 cells. As demonstrated by Western blot and autoradiographic analysis of electrophorograms of proteins extracted from H441 and H3255 cells exposed to [18F]F-PEG6-IPQA, the L858R activating mutation, apparently, increases irreversible covalent binding of [18F]F-PEG6-IPQA to the active mutant L858R EGFR kinase domain.
Fig. 2.
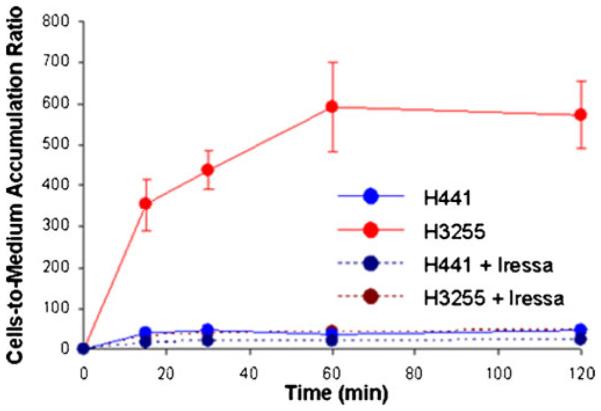
Time-dependent accumulation of [18F]F-PEG6-IPQA 14 in H441 and H3255 tumor cells in vitro, before and after the addition of Iressa (100 μM) into the culture medium.
Fig. 3.
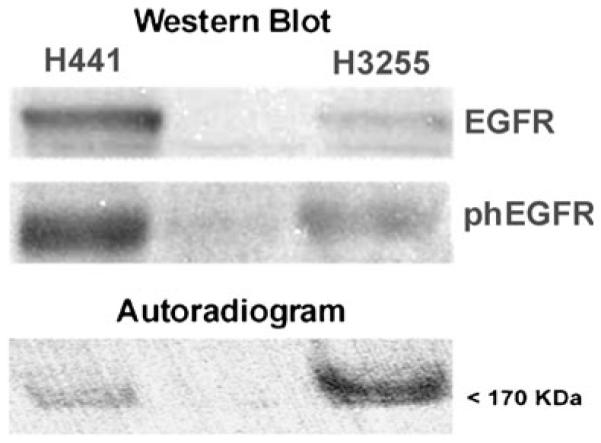
Western blot analysis of total EGFR and phosphotyrosine 845 of EGFR expression in H441 and H3255 tumor cells. The autoradiogram of protein electrophoresis membrane demonstrates preferential irreversible and covalent binding of [18F]F-PEG6-IPQA to active mutant L858R EGFR kinase domain.
Conclusions
We have successfully synthesized [18F]F-PEG6-IPQA with the 18F label attached at the end of the hexaethylene glycol side chain and demonstrated highly selective accumulation in active mutant L858R EGFR-expressing NSCLC cells in vitro. Further, in vivo studies are warranted to assess the ability of PET imaging with [18F]F-PEG6-IPQA to discriminate the active mutant L858R EGFR-expressing NSCLC that are sensitive to therapy with EGFR kinase inhibitors vs NSCLC that express wild-type EGFR.
Significance.
Non-small cell lung carcinoma (NSCLC) represents the majority of lung cancers. The response rate to EGFR inhibitors in patients with NSCLC exhibiting activating mutations of EGFR is approximately 85–90%, suggesting that these mutations, at least in part, may have caused malignant transformation and contribute in large to the tumor maintenance pathway. Therefore, we have been developing small molecular radiolabeled agents with preferential irreversible binding to active mutant EGFR kinases (i.e., L858R). Positron emission tomography imaging using such as selective radiolabeled agent could potentially allow for visualization of primary and metastatic tumor lesions expressing active mutant EGFR kinase, and for selection of patients who may benefit from therapy with EGFR kinase inhibitors.
Acknowledgments
This work was supported by the following grants: W81XWH-05-2-0027 (J.G.—Project Leader; Imaging Core Leader) and U24CA126577 (J.G. co-PI). We thank Karen Yoas, M.S., for the excellent coordination of studies.
Footnotes
Conflict of interest. None of the authors of this manuscript has any conflict of interest in relation to studies and results presented herein.
References
- 1.ACS Cancer Reference Information. 2009 http://wwwcancerorg/docroot/CRI/content/CRI_2_4_1x_What_Are_the_Key_Statistics_About_ Lung_Cancer_15asp?sitearea=
- 2.Mishani E, Abourbeh G. Cancer molecular imaging: radio-nuclide-based biomarkers of the epidermal growth factor receptor (EGFR) Curr Top Med Chem. 2007;7:1755–1772. doi: 10.2174/156802607782507457. [DOI] [PubMed] [Google Scholar]
- 3.Mishani E, Hagooly A. Strategies for molecular imaging of epidermal growth factor receptor tyrosine kinase in cancer. J Nucl Med. 2009;50:1199–1202. doi: 10.2967/jnumed.109.062117. [DOI] [PubMed] [Google Scholar]
- 4.Bonasera TA, Ortu G, Rozen Y, Krais R, Freedman NM, Chisin R, Gazit A, Levitzki A, Mishani E. Potential (18)F-labeled biomarkers for epidermal growth factor receptor tyrosine kinase. Nucl Med Biol. 2001;28:359–374. doi: 10.1016/s0969-8051(01)00200-1. [DOI] [PubMed] [Google Scholar]
- 5.Ortu G, Ben-David I, Rozen Y, Freedman NM, Chisin R, Levitzki A, Mishani E. Labeled EGFr-TK irreversible inhibitor (ML03): in vitro and in vivo properties, potential as PET biomarker for cancer and feasibility as anticancer drug. Int J Cancer. 2002;101:360–370. doi: 10.1002/ijc.10619. [DOI] [PubMed] [Google Scholar]
- 6.Mishani E, Abourbeh G, Rozen Y, Jacobson O, Laky D, Ben David I, Levitzki A, Shaul M. Novel carbon-11 labeled 4-dimethylamino-but-2-enoic acid [4-(phenylamino)-quinazoline-6-yl]-amides: potential PET bioprobes for molecular imaging of EGFR-positive tumors. Nucl Med Biol. 2004;31:469–476. doi: 10.1016/j.nucmedbio.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 7.Abourbeh G, Dissoki S, Jacobson O, Litchi A, Ben Daniel R, Laki D, Levitzki A, Mishani E. Evaluation of radiolabeled ML04, a putative irreversible inhibitor of epidermal growth factor receptor, as a bioprobe for PET imaging of EGFR-overexpressing tumors. Nucl Med Biol. 2007;34:55–70. doi: 10.1016/j.nucmedbio.2006.10.012. [DOI] [PubMed] [Google Scholar]
- 8.Dissoki S, Aviv Y, Laky D, Abourbeh G, Levitzki A, Mishani E. The effect of the [18F]-PEG group on tracer qualification of [4-(phenylamino)-quinazoline-6-YL]-amide moiety–an EGFR putative irreversible inhibitor. Appl Radiat Isot. 2007;65:1140–1151. doi: 10.1016/j.apradiso.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 9.Pal A, Glekas A, Doubrovin M, Balatoni J, Namavari M, Beresten T, Maxwell D, Soghomonyan S, Shavrin A, Ageyeva L, Finn R, Larson SM, Bornmann W, Gelovani JG. Molecular imaging of EGFR kinase activity in tumors with 124I-labeled small molecular tracer and positron emission tomography. Mol Imaging Biol. 2006;8:262–277. doi: 10.1007/s11307-006-0049-0. [DOI] [PubMed] [Google Scholar]
- 10.Su H, Seimbille Y, Ferl GZ, Bodenstein C, Fueger B, Kim KJ, Hsu YT, Dubinett SM, Phelps ME, Czernin J, Weber WA. Evaluation of [(18)F]gefitinib as a molecular imaging probe for the assessment of the epidermal growth factor receptor status in malignant tumors. Eur J Nucl Med Mol Imaging. 2008;35:1089–1099. doi: 10.1007/s00259-007-0636-6. [DOI] [PubMed] [Google Scholar]
- 11.Wang H, Yu JM, Yang GR, Song XR, Sun XR, Zhao SQ, Wang XW, Zhao W. Further characterization of the epidermal growth factor receptor ligand 11C-PD153035. Chin Med J (Engl) 2007;120:960–964. [PubMed] [Google Scholar]
- 12.Memon AA, Jakobsen S, Dagnaes-Hansen F, Sorensen BS, Keiding S, Nexo E. Positron emission tomography (PET) imaging with [11C]-labeled erlotinib: a micro-PET study on mice with lung tumor xenografts. Cancer Res. 2009;69:873–878. doi: 10.1158/0008-5472.CAN-08-3118. [DOI] [PubMed] [Google Scholar]
- 13.Pantaleo MA, Mishani E, Nanni C, Landuzzi L, Boschi S, Nicoletti G, Dissoki S, Paterini P, Piccaluga PP, Lodi F, Lollini PL, Fanti S, Biasco G. Evaluation of modified PEG-anilinoquinazoline derivatives as potential agents for EGFR imaging in cancer by small animal PET. Mol Imaging Biol. 2010 doi: 10.1007/s11307-010-0315-z. doi:10.1007/s11307-010-0315-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu N, Li M, Li X, Meng X, Yang G, Zhao S, Yang Y, Ma L, Fu Z, Yu J. PET-based biodistribution and radiation dosimetry of epidermal growth factor receptor-selective tracer 11C-PD153035 in humans. J Nucl Med. 2009;50:303–308. doi: 10.2967/jnumed.108.056556. [DOI] [PubMed] [Google Scholar]
- 15.Gelovani JG. Molecular imaging of epidermal growth factor receptor expression-activity at the kinase level in tumors with positron emission tomography. Cancer Metastasis Rev. 2008;27:645–653. doi: 10.1007/s10555-008-9156-5. [DOI] [PubMed] [Google Scholar]
- 16.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science (New York, NY) 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 17.Mukohara T, Engelman JA, Hanna NH, Yeap BY, Kobayashi S, Lindeman N, Halmos B, Pearlberg J, Tsuchihashi Z, Cantley LC, Tenen DG, Johnson BE, Janne PA. Differential effects of gefitinib and cetuximab on non-small-cell lung cancers bearing epidermal growth factor receptor mutations. J Natl Cancer Inst. 2005;97:1185–1194. doi: 10.1093/jnci/dji238. [DOI] [PubMed] [Google Scholar]
- 18.Tracy S, Mukohara T, Hansen M, Meyerson M, Johnson BE, Janne PA. Gefitinib induces apoptosis in the EGFRL858R non-small-cell lung cancer cell line H3255. Cancer Res. 2004;64:7241–7244. doi: 10.1158/0008-5472.CAN-04-1905. [DOI] [PubMed] [Google Scholar]
- 19.Najjar AM, Nishii R, Maxwell DS, Volgin A, Mukhopadhyay U, Bornmann WG, Tong W, Alauddin M, Gelovani JG. Molecular-genetic PET imaging using an HSV1-tk mutant reporter gene with enhanced specificity to acycloguanosine nucleoside analogs. J Nucl Med. 2009;50:409–416. doi: 10.2967/jnumed.108.058735. [DOI] [PubMed] [Google Scholar]
- 20.Wissner A, Fraser HL, Ingalls CL, Dushin RG, Floyd MB, Cheung K, Nittoli T, Ravi MR, Tan X, Loganzo F. Dual irreversible kinase inhibitors: quinazoline-based inhibitors incorporating two independent reactive centers with each targeting different cysteine residues in the kinase domains of EGFR and VEGFR-2. Bioorg Med Chem. 2007;15:3635–3648. doi: 10.1016/j.bmc.2007.03.055. [DOI] [PubMed] [Google Scholar]
- 21.MacMahon S, Fong R, 2nd, Baran PS, Safonov I, Wilson SR, Schuster DI. Synthetic approaches to a variety of covalently linked porphyrin–fullerene hybrids. J Org Chem. 2001;66:5449–5455. doi: 10.1021/jo010317x. [DOI] [PubMed] [Google Scholar]
- 22.Dissoki S, Eshet R, Billauer H, Mishani E. Modified PEG-anilinoquinazoline derivatives as potential EGFR PET agents. J Label Comp Radiopharm. 2009;52:41–52. [Google Scholar]
- 23.Iino T, Sasaki Y, Bamba M, Mitsuya M, Ohno A, Kamata K, Hosaka H, Maruki H, Futamura M, Yoshimoto R, Ohyama S, Sasaki K, Chiba M, Ohtake N, Nagata Y, Eiki J, Nishimura T. Discovery and structure-activity relationships of a novel class of quinazoline glucokinase activators. Bioorganic & Medicinal Chemistry Letters. 2009;19(19):5531–5538. doi: 10.1016/j.bmcl.2009.08.064. [DOI] [PubMed] [Google Scholar]