

Abstract
Context: 4-Nerolidylcatechol (4-NRC) has showed antitumor potential through apoptosis. However, its apoptotic mechanisms are still unclear, especially in leukemic cells.
Objectives: To evaluate the cytotoxic potential of 4-NRC and its cell death pathways in p53-null K562 leukemic cells.
Materials and methods: Cytotoxicity of 4-NRC (4.17–534.5 μM) over 24 h of exposure was evaluated by MTT assay. 4-NRC-induced apoptosis in K562 cells was investigated by phosphatidylserine (PS) externalization, cell cycle, sub-G1, mitochondrial evaluation, cytochrome c, cyclin D1 and intracellular reactive oxygen species (ROS) levels, and caspase activity analysis.
Results: IC50 values obtained were 11.40, 27.31, 15.93 and 15.70 μM for lymphocytes, K562, HL-60 and Jurkat cells, respectively. In K562 cells, 4-NRC (27 μM) promoted apoptosis as verified by cellular morphological changes, a significant increase in PS externalization and sub-G1 cells. Moreover, it significantly arrested the cells at the G0/G1 phase due to a reduction in cyclin D1 expression. These effects of 4-NRC also significantly promoted a reduction in mitochondrial activity and membrane depolarization, accumulation of cytosolic cytochrome c and ROS overproduction. Additionally, it triggered an increase in caspases -3/7, -8 and -9 activities. When the cells were pretreated with N-acetyl-l-cysteine ROS scavenger, 4-NRC-induced apoptosis was partially blocked, which suggests that it exerts cytotoxicity though not exclusively through ROS-mediated mechanisms.
Discussion and conclusion: 4-NRC has antileukemic properties, inducing apoptosis mediated by mitochondrial-dependent mechanisms with cyclin D1 inhibition. Given that emerging treatment concepts include novel combinations of well-known agents, 4-NRC could offer a promising alternative for chemotherapeutic combinations to maximize tumour suppression.
Keywords: Cancer, cell cycle arrest, leukaemia, mitochondria, multidrug resistance
Introduction
4-Nerolidylcatechol (4-NRC) (Figure 1) is the main secondary metabolite found in the Brazilian plant Pothomorphe umbellata (L.) Miq. (Piperaceae) (Cunha et al. 2013). Several studies have demonstrated the in vitro and in vivo antioxidant activity of 4-NRC using different experimental models (Desmarchelier et al. 1997; Ropke et al. 2003, 2005, 2006; Barros et al. 2007). In these studies, 4-NRC has showed inhibitory activity against MMP-2 and MMP-9 metalloproteinases, which suggests that this compound has an antioxidant mechanism, which attenuates solar UVB light-induced skin carcinogenesis (Ropke et al. 2006). Moreover, 4-NRC showed a protective effect against cyclophosphamide-induced genotoxicity (Valadares et al. 2007). This compound and/or its semi-synthetic derivatives also presented in vitro antioxidant, antimicrobial, antimalarial and antitumor activities (Brohem et al. 2009; Silva Pinto et al. 2009; Bagatela et al. 2013; Cunha et al. 2013; Cortez et al. 2015). In terms of anticancer properties, it has been shown that apoptosis is the main cell death type triggered by 4-NRC (Brohem et al. 2009, 2012). However, the mechanisms by which it induces apoptosis in cancer cells are still unclear, especially in leukemic cells.
Figure 1.
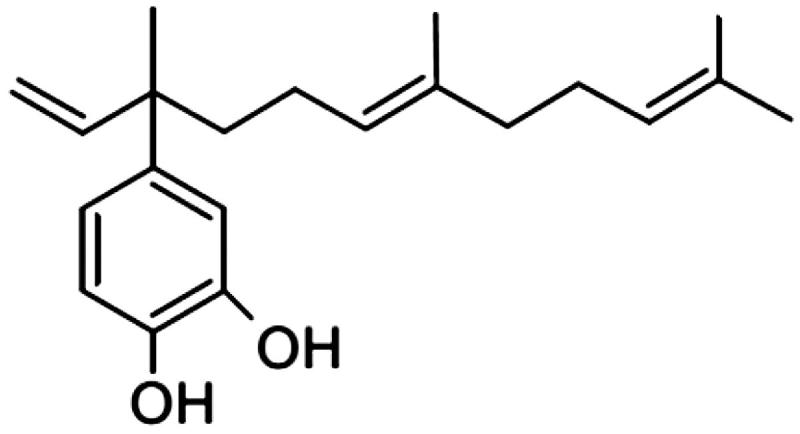
Chemical structure of 4-nerolidylcatechol (4-NRC), the main secondary metabolite found in Brazilian plants such as Pothomorphe umbellata.
In view of this, the K562 human leukemic cell line has been widely used as a model to evaluate the antineoplastic potential of new drugs and to investigate the pathways involved in the cell death. This cell line does not express p53 (Sen et al. 1993), a protein involved in tumour proliferation inhibition (Vousden & Prives 2009). In addition, it is derived from chronic myelogenous leukaemia, characterized by the presence of the Philadelphia (Ph) chromosome, which is formed by translocation between chromosomes 9 and 22 (Miller et al. 2014). This genetic aberration produces the BCR-ABL tyrosinase kinase oncoprotein fusion, active in more than 90% of the patients and responsible for the overproduction of chronic myeloid leukemic (CML) cells (Huang et al. 2011; Mota et al. 2012). In addition, BCR-ABL-expression in K562 cells presents resistance to apoptosis induced by conventional chemotherapeutics and allows for the evaluation of several cellular and molecular events concurrently, which justifies the use of this valuable cellular model in drug discovery (Huang et al. 2011; Mota et al. 2012).
In this respect, mitochondrial alterations have been associated to CML cell drug resistance by mechanisms involving the intrinsic apoptotic pathway (Koptyra et al. 2006; Tokarz & Blasiak 2014). Apoptosis is the type of cell death with different pathways which is most studied. In most of these pathways, mitochondrial membrane permeabilization and/or caspase activation are considered central points of integration of the apoptotic cell death (Savitskaya & Onishchenko 2015). As regards mitochondria, newly accumulated evidence supports the idea that cell cycle regulators, such as cyclin D1, are actively involved in mitochondrial regulation/homeostasis through the phosphorylation of their mitochondrial targets (Pestell 2013; Alexandrou & Li 2014). In this respect, new approaches in mitochondria-oriented anticancer therapy are suggested.
Based on the above, this study evaluated the antileukemic activity and apoptotic pathways involved in 4-NRC-induced cell death in p53-null K562 leukemic cells.
Materials and methods
Chemicals
RPMI 1640 medium, fetal bovine serum, streptomycin, penicillin G, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT), propidium iodide (PI), Giemsa dye, RNase type I, dichloro-dihydro-fluorescein diacetate (DCFH-DA), rhodamine 123, 5,5,6,6-tetrachloro-1,1,3,3-tetraethylbenzimidazolylcarbo-cyanine iodide (JC-1), bovine serum albumin (BSA), phytohemagglutinin, ficoll-hypaque, N-acetyl-l-cysteine, protease inhibitor cocktail and bicinchoninic acid protein assay kit were purchased from Sigma-Aldrich (St. Louis, MO). Annexin V Apoptosis Detection Kit FITC was acquired from eBioscience® (San Diego, CA), while a CaspaTagTM Caspase-3/7, -8 and -9 In Situ Assay kit was obtained from MilliporeTM (Temecula, CA). The antibody against cyclin D1 (A-12) (sc-8396), cyclin D1 (H-295) rabbit polyclonal IgG (sc-753) and cytochrome c (6H2) (sc-13561) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA) while BD cell-takTM adhesive and BD Cytofix/Cytoperm™ solution were acquired from BD Biosciences (San Jose, CA). NP-40 lysis buffer was purchased from Amresco (Solon, OH) and antibody against GAPDH and anti-rabbit IgG (Fc), AP conjugate were obtained from Promega (Madison, WI). MitoTracker® Red CMXRos probe and Hoechst 33342 were purchased from Life Technologies (Carlsbad, CA) and Invitrogen (Grand Island, NY), respectively. Acetonitrile, methanol, ethanol, hexane and dichloromethyl were acquired from Merck (Darmstad, Germany), whereas Tween 20 was obtained from Vetec (Rio de Janeiro, RJ, Brazil).
Preparation of P. umbellata root extract
Plant material of P. umbellata was collected from the medicinal herb garden of the University of São Paulo (May–September, 2008), and a sample deposited in the Herbarium of the Institute of Biosciences of the University of São Paulo (Kato-0363). The roots were dried and ground to a powder and finally extracted by percolation, as recommended by method A of the Brazilian Pharmacopoeia, in a 3:1 solution of ethanol and water. The organic solvent was evaporated and the water layer extracted with chloroform. The recovered residue was filtered and quantified for 4-NRC content. The 4-NRC concentration found in the crude extract residue was 21.5% (w/w), as assayed by HPLC-UV detection (Rezende & Barros 2004). Briefly, the crude extract 4-NRC assay was monitored at 282 nm and carried out using a water-acetonitrile-methanol solvent system 18:20:62 as the mobile phase and flow rate was maintained at 1.0 mL/min. HPLC analysis was carried out using a Varian® Prostar HPLC model 210 (Walnut Creek, CA) equipped with a UV/VIS detector (Prostar, model 340), a Reodyne® injector loop (20 μL) and a reverse-phase column Phenomenex® Synergi Fusion 4 μ RP-80 A C18 (150 mm × 4.6 mm) (Torrance, CA), protected by a precolumn cartridge.
Obtaining 4-NRC
4-NRC (molecular weight: 318.4) was isolated from the crude P. umbellata extract, as described elsewhere (Gustafson et al. 1992). Briefly, the ethanol:water extract was submitted to a Sephadex™ LH20 chromatography column (21 × 10 cm) and eluted with hexane: CHCl2:MeOH. The presence of 4-NRC in chromatographic fractions was detected by TLC, by comparing to a previously isolated authentic sample. The structure was confirmed by spectral analysis (1 H, 13 C NMR) in agreement with published data (Gustafson et al. 1992). For the assays, 4-NRC was dissolved in ethanol to a concentration of 5.34 mM and stored at −20 °C.
Cell cultures
The human CML K562, immature T Jurkat and HL-60 cell lines, obtained from the Rio de Janeiro Cell Bank (Federal University of Rio de Janeiro, Rio de Janeiro, Brazil), were cultured in suspensions in RPMI 1640 medium supplemented with 10% foetal bovine serum (FBS), 100 U/mL of penicillin and 100 μg/mL of streptomycin in a humidified atmosphere at 37 °C in 5% CO2. Cells were seeded (1 × 105 cells/mL) in 96-well microtiter plates and incubated with eight concentrations of 4-NRC (4.17–534.5 μM) for 24 h in an incubator (Thermo Scientific, USA) with humidified atmosphere at 37 °C in 5% CO2. Control cells were treated with complete medium only.
Lymphocyte culture
Lymphocytes were obtained according to the standard protocol described by Preston et al. (1987). In brief, a peripheral human blood sample, randomly and anonymously obtained as a residual product from a clinical laboratory, was diluted with an equal volume of RPMI 1640 medium, then layered over Ficoll-Hypaque density gradient separation solution (1.077 g/mL), and centrifuged at 2000 rpm for 20 min at room temperature (Rotina 38 R centrifuge system from Hettich, Baden-Württemberg, Germany). The mononuclear cell layer was removed, washed twice in RPMI 1640 medium and resuspended in RPMI 1640 medium supplemented with 10% FBS and antibiotics. Lymphocytes (1 × 106 cells/mL) were cultured with phytohemagglutinin (10 μg/mL) in 96-well microtiter plates, in the presence and absence of 4-NRC (4.17–534.5 μM), for 24 h in 37 °C in 5% CO2 incubator.
Cytotoxicity assay
After treatment of each cell line and lymphocytes with 4-NRC, cytotoxicity was determined by 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT) reduction assay, adapted from Mosmann (1983). Briefly, the cells were seeded in a 96-well microtiter plates and treated with 4-NRC for 24 h. After that, 10 μL/well MTT (5 mg/mL) were added and the plates were incubated for 3 h at 37 °C. After the plates were centrifuged at 800 rpm for 10 min, the supernatant was removed and 200 μL/well of DMSO were added to solubilize the formazan formed. The absorbance was measured at 560 nm using a spectrophotometer. Each concentration of 4-NRC was tested in three independent experiments in six replicates. The data are expressed as % of the cell proliferation or viability of control cells (100%). For all subsequent assays, IC50 value (concentration that inhibited cell growth in 50% compared to untreated group) obtained in the MTT assay was used.
Morphological changes
K562 cells, treated with 4-NRC (27 μM) for 24 h, were performed on cytospin preparations on microscope slides and stained with Giemsa dye (Fernandes et al. 2015). After drying naturally, the morphological changes in the cells were observed under a light microscopy (AxioZeiss, Zeiss, Germany).
FITC annexin V/propidium iodide double staining and analysis
The assay was performed using an Annexin V Apoptosis Detection Kit FITC (eBioscience®, San Diego, CA) in accordance with the manufacturer’s instructions. Briefly, after treatment with 4-NRC (27 μM) for 24 h, K562 cells were harvested, washed twice with cold PBS and resuspended in binding buffer (0.1 M Hepes/NaOH, pH 7.4; 1.4 M NaCl and 25 mM CaCl2). The suspensions were transferred to 5 mL tubes and 5 μL FITC Annexin V and 5 μL of propidium iodide (PI) were added. The cells were incubated at room temperature for 15 min, after which 400 μL binding buffer were added and analysis was performed in a FACSCanto II flow cytometer (Becton Dickinson, CA) using the FACSDiva software. Simultaneously, cell groups were also pretreated with NAC antioxidant agent (2 mM), a ROS scavenger, for 1 h following treatment with 4-NRC to evaluate the involvement of ROS in apoptosis induction. Four cell populations were identified: necrotic cells in the upper-left quadrant (high PI and low FITC signals); early apoptotic cells in the lower-right quadrant (low PI and high FITC signals); late apoptotic cells in the upper-right quadrant; and viable cells in the lower-left quadrant (low PI and FITC signals).
Cell cycle and sub-G1 analysis
After treatment, K562 cells (1 × 106 cells/mL) were harvested, washed with cold PBS and fixed with ice-cold 70% ethanol for at least 24 h. The cells were washed twice with PBS by centrifuging at 1500 rpm for 10 min. Cells were then treated with 4 μg/mL RNase type I for 1 h at 37 °C and resuspended in PBS. Cells were stained with 25 μg/mL of PI and analyzed in a flow cytometer (Ávila et al. 2016).
Cyclin D1 expression analysis
Flow cytometry and western blot analysis were performed for the detection of the expression level of cyclin D1 protein in K562 cells, based on the protocols described by Cortez et al. (2015) and Mota et al. (2016), respectively. After treatment, K562 cells (1 × 106 cells/mL) were harvested by centrifugation, washed with cold PBS and resuspended with BD Cytofix/Cytoperm™ solution for 20 min at 4 °C. Cells were washed twice with cold PBS containing Tween 20 (0.01%) by centrifuging at 1500 rpm for 10 min. After that, 20 μL of antibody anti-cyclin D1 were added and cells were incubated in the dark at room temperature for 30 min. After washing in PBS/1% BSA, cells were resuspended in 400 μL PBS and analyzed in a flow cytometer.
For Western blot analysis, cell lysates were prepared with NP-40 lysis buffer (containing a protease inhibitor cocktail) for 30 min on ice. The cell debris were pelleted by centrifugation at 13,000 rpm and 4 °C for 10 min, and the supernatant was recovered. Protein content of the cell lysates was measured using bicinchoninic acid protein assay kit (Sigma-Aldrich); and 40 μg of protein were separated on a 10% SDS-polyacrylamide gel and electro-transferred to PVDF membranes. After blocking with 5% non-fat skim milk powder in BSA, the membranes were incubated with an antibody against cyclin D1 (1:200). The blots were washed three times with TBST and incubated with the secondary antibody tagged with alkaline phosphatase (1:500) for 1.5 h. After washing with TBST for 30 min, the cyclin D1 protein was detected using the chromogenic substrate BCIP/NBT. Equal protein loading in each lane was confirmed by probing with an anti-GAPDH antibody (1:1000). The bands were visualized and quantified by densitometry using the open-source ImageJ software (https://imagej.nih.gov/ij/). The assays were carried out by two independent experiments.
Evaluation of mitochondrial transmembrane potential (ΔΨm)
The ΔΨm changes were detected using rhodamine 123, a dye sequestered by active mitochondria, and confirmed by JC-1 potentiometric probe that remains in cytoplasma as monomeric form in apoptotic cells, while in viable cells it forms mitochondrial aggregates. Both the assays were conducted in accordance with the manufacturer’s instructions. Similar to that described previously, control and treated cells were incubated with rhodamine 123 (1 μg/mL) in PBS or JC-1 (2.5 μg/mL) in RPMI for 15 min. After that, the cells were washed and analyzed using flow cytometer.
Evaluation of mitochondrial activity
Mitochondrial activity was measured by staining cells with MitoTracker Red, a fluorochrome that measures mitochondrial mass in a ΔΨm-dependent manner, in accordance with the manufacturer’s instructions. After treatment, K562 cells (1 × 106 cells/mL) were washed twice with PBS and incubated with MitoTracker® Red CMXRos (10 nM) in PBS for 15 min at 37 °C in 5% CO2. The cells were washed twice, resuspended in 0.2 mL of PBS and analysis was performed in a flow cytometer.
Cytochrome c release analysis
Cytochrome c release from mitochondria to cytoplasm was evaluated using a fluorescence microscope (DMI 4000 B Leica Microsystems, Bannockburn, IL) coupled to the software LAS-AF. The control and 4-NRC-treated cells (1 × 106 cells/mL) were fixed with methanol and then seeded on BD cell-takTM-coated coverslips. Cells were permeabilized and blocked with PBS solution containing BSA (1%, p/v) and Tween 20 (0.1%, v/v) in humidified chamber for 1 h, at room temperature. Then the cells were incubated with cytochrome c antibody for 1 h and nuclei were stained with Hoechst 33258. The cells were then washed three times with PBS and mounted, using an appropriate mounting medium (PBS with 90% glycerol, v/v).
Measurement of intracellular ROS
Based on the protocol described by Ávila et al. (2016), intracellular ROS production was measured by using a fluorimetric probe, 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA), which can be hydrolyzed to 2,7-dichlorodihydrofluorescein (DCFH) by intracellular esterases when the cells take it up. DCFH is reactive with ROS to give a new highly fluorescent compound, 2,7-dichlorofluorescein (DCF), which can be detected by a flow cytometer. After treatment with 4-NRC, as described above, K562 cells (1 × 106 cells/mL) were incubated with DCFH-DA (10 μM) at 37 °C in 5% CO2 for 1 h and analyzed in a flow cytometer.
Caspase activity
After incubation of K562 cells with 4-NRC (27 μM) for 24 h, the active caspases -3/7, -8 and -9 were assessed by a flow cytometer using the CaspaTagTM Caspase In Situ Assay kit (Millipore, Temecula, CA) in accordance with the manufacturer’s instructions. To evaluate the involvement of ROS in caspase activation, cell groups were also pretreated with NAC (2 mM) for 1 h following treatment with 4-NRC.
Statistical analysis
The assays were performed as two or three independent experiments. The cytotoxicity results, expressed as the mean ± SD of six replicates, were transformed into percentages of the control, and IC50 values were obtained by non-linear regression. For flow cytometry analysis, 10,000 events were collected per sample of each assay with a FACSCanto II flow cytometer using the FACSDiva software version 6.0. The difference between control and 4-NRC-treated cells was evaluated by unpaired t test or one/two way analysis of variance (ANOVA) and Bonferroni test using GraphPad Prism version 5.01 software for Windows (San Diego, CA). Statistical significance was considered when p < 0.05.
Results
The cytotoxic effects of the eight concentrations of 4-NRC (4.1–534.5 μM) in K562, HL-60 and Jurkat cells by MTT assay are shown in Figure 2(A). The proliferation of these leukemic cells was inhibited by 4-NRC, in a concentration dependent-manner. IC50 values obtained were 27.31 ± 5.6, 15.93 ± 1.9, and 15.70 ± 3.8 μM for K562, HL-60 and Jurkat cells, respectively. In addition, 4-NRC also showed cytotoxicity to normal lymphocytes from human peripheral blood (IC50 = 11.40 ± 0.42 μM) (Figure 2(A)).
Figure 2.

Cytotoxic effects of 4-NRC in lymphocytes and leukemic cells. (A) Lymphocytes and K562, HL-60 and Jurkat cells (1 × 106 cells/mL) were treated with different concentrations of 4-NRC (4.17–534.5 μM) for 24 h and cell viability was determined by MTT assay. Results represent the mean ± SD of three independent experiments in six replicates. (B) Morphological changes promoted by 4-NRC in K562 cells: control cells (2 × 104/mL) stained with Giemsa dye showed normal nucleus while cells treated with 4-NRC (27 μM) for 24 h showed apoptotic nucleus (indicated by arrow). Representative photomicrographs (400 × magnification) of two independent experiments.
Since K562 cells show resistance to apoptosis induced by conventional chemotherapeutics, this cell line was chosen for subsequent assays to evaluate cell death pathways triggered by 4-NRC (IC50 = 27 μM). Figure 2(B) shows the morphological changes in K562 cells after treatment with 4-NRC for 24 h. As can be seen, the treated cells presented apoptotic characteristics when compared to untreated cells. These findings prompted us to investigate the mechanisms of the 4-NRC-induced apoptosis in K562 cells.
Cells undergoing apoptosis suffer changes in their phospholipid bilayer membrane which include phosphatidylserine (PS) externalization to the outer plasma membrane during early and late apoptotic events. Figure 3(A,B) shows the binding of annexin V (A)/propidium iodide (PI) in K562 cells after 4-NRC treatment. As expected, untreated cells showed mainly viable (A-/PI-) (91.0 ± 1.1%). By contrast, cells treated with 4-NRC showed a significant reduction in viability as the following shows: viable (61.2 ± 9.8%, A-/PI-) (p < 0.001), early apoptotic (7.7 ± 1.7%, A+/PI-) (p < 0.01), late apoptotic (21.0 ± 9.0%, A+/PI+) (p < 0.001) and necrotic (10.2 ± 2.0%, A-/PI+) (p < 0.001) cells. When the cells were pretreated with NAC and followed by treatment with 4-NRC, this ROS scavenger partially blocked cell apoptosis, and reached the following values: viable (74.5 ± 15.3%, A-/PI-) (p < 0.001), early apoptotic (4.0 ± 1.2%, A+/PI-), late apoptotic (12.5 ± 9.2%, A+/PI+) and necrotic (9.0 ± 6.1%, A-/PI+) cells. This finding suggests that 4-NRC exerts its cytotoxic effect though not exclusively through ROS-mediated mechanisms.
Figure 3.

Analysis of phosphatidylserine externalization profile in K562 cells after treatment with 4-NRC. The cells (1 × 106 cells/mL) were treated with 4-NRC (27 μM) for 24 h and analyzed using a flow cytometer. Simultaneously, cell groups were also pretreated with N-acetyl-l-cysteine (NAC) antioxidant agent (2 mM) for 1 h following treatment with 4-NRC to evaluate the involvement of ROS in apoptosis induction. Representative distribution (A) and percentage (B) of necrotic cells in the upper-left quadrant (A-/PI+); early apoptotic cells in the lower-right quadrant (A+/PI-); late apoptotic cells in the upper-right quadrant (A+/PI+); and viable cells in the lower-left quadrant (A-/PI-) in control and 4-NRC groups. Each bar presents mean ± SD of three independent experiments (**p < 0.01 and ***p < 0.001 vs. control).
In terms of cell cycle progression, 4-NRC significantly arrested the cells at the G0/G1 phase (59.6 ± 1.5%) (p < 0.001). Consequently, we detected significant reductions of 23.35 ± 0.65 and 8.45 ± 0.05% in both the S and G2/M phases (p < 0.001), respectively. In addition, 4-NRC promoted a significant increase in sub-diploid DNA, a characteristic of apoptotic cells, that accumulated in the sub-G1 position of the cell cycle (p < 0.05), in contrast to untreated cells (Figure 4(A,B)).
Figure 4.

Effects of 4-NRC on the cell cycle and cyclin D1 expression in K562 cells. (A) Representative histograms of the analysis of cell cycle in K562 cells following 24 h treatment with 4-NRC (27 μM). (B) Effects of 4-NRC on cell cycle progression, presented as the percentage of cells in the sub G1, G1, S and G2 phases. Flow cytometry (C) and western blot (D and E) cyclin D1 expression analysis in K562 following treatment with 4-NRC (27 μM) for 24 h. Values represent the means ± SD of two or three independent experiments (*p < 0.05 and **p < 0.001 vs. control).
As 4-NRC promoted changes in the cell cycle of K562 cells, we investigated the effects of this drug on cyclin D1 expression, a regulator of the G0/G1 phase. Flow cytometric analysis of cells treated with 4-NRC showed a cyclin D1 expression of 65.6 ± 3.1%, a significant decrease when compared to the control (p < 0.05) (Figure 4(C)). In addition, western blot analysis also revealed a significant reduction in cyclin D1 (p < 0.001) (Figure 4(D,E)).
Considering that alterations in mitochondrial function are among the early events in apoptosis, the effects in the Δψm were investigated. The rhodamine 123 staining decreased by 77.2 ± 6.3% (p < 0.001), which suggests the mitochondrial depolarizing ability of 4-NRC (Figure 5(A)). To confirm this result, Δψm changes were also analyzed by using a mitochondria fluorescent JC-1 dye. We verified that 4-NRC significantly promoted a reduction in viable cells with mitochondria-aggregated JC-1 (normal Δψm) to 75.6 ± 2.9% with a consequent increase in apoptotic cells showing monomeric and cytosolic JC-1 to 27 ± 3.3% (p < 0.001), when compared to the control (90 ± 4.7 and 10.6 ± 5.3%, respectively) (Figure 5(B)). The ratios of red (JC-1 aggregates) to green (JC-1 monomers) fluorescence for control and 4-NRC groups were 8.5 and 2.8, respectively, showing that cell apoptosis accompanied the Δψm changes. Moreover, 4-NRC also led to a significant reduction in mitochondrial activity (p < 0.001) due to a decrease of 59.6 ± 6.9% in the MitoTracker Red staining in relation to untreated cells (Figure 5(C)). Since 4-NRC promoted mitochondrial membrane depolarization, we verified a significant accumulation of cytosolic cytochrome c in treated cells, in contrast to the control (Figure 5(D)). These findings demonstrated that a mitochondria-dependent pathway could contribute to 4-NRC-induced apoptosis in K562 cells.
Figure 5.

Effects of 4-NRC on mitochondrial function and ROS production in K562 cells. Cells (1 × 106 cells/mL) were treated with 4-NRC (27 μM) for 24 h and mitochondrial function was analyzed by different assays as follows: (A) 4-NRC-induced Δψm changes were previously evaluated by rhodamine 123 staining; (B) mitochondrial depolarizing ability of 4-NRC was confirmed by JC-1 staining: representative histogram shows viable cells with mitochondria-aggregated JC-1 (normal Δψm) and apoptotic cells with monomeric and cytosolic JC-1 of control and 4-NRC groups; (C) reduction of mitochondrial mass promoted by 4-NRC was evaluated by Mitotracker Red staining; and (D) cytochrome c release by mitochondria of untreated cells and cells treated with 4-NRC was determined by microscopy: the nuclei were visualized by Hoechst staining and merging the cytochrome c and nuclei representative images were carried out (630 × magnification). Dotted circle represents cytochrome c not detectable in control cells. Scale bars =25 μm. (E) Intracellular ROS generation analysis in K562 cells treated with 4-NRC using a fluorimetric probe, 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA). Results represent the mean ± SD. Data are representative of three independent experiments (**p < 0.001 and ***p < 0.0001 vs. control).
In parallel, the ROS generation increased in a time-dependent manner after treatment of K562 cells with 4-NRC (Figure 5(E)). This increase was statistically significant (p < 0.0001), when compared to untreated cells.
Moreover, we observed that caspases were activated during the 4-NRC-induced cell death, since this compound promoted a significant increase of 23.3 ± 6.9 (p < 0.01), 27.5 ± 7.3 (p < 0.01) and 25.2 ± 7.8% (p < 0.001) in caspases -3/7, -8 and -9 activities, respectively. Additionally, when the cells were pretreated with NAC, this substance partially interfered in caspase activation. For this cell group, the values found were 17.7 ± 1.6, 19.0 ± 0.9 and 21.1 ± 2.1% for caspases -3/7, -8 and -9 (p < 0.01), respectively (Figure 6).
Figure 6.

Analysis of the activation of caspases-3/7, -8 and -9 during 4-NRC-induced apoptosis. The cells (1 × 106 cells/mL) were treated with 4-NRC (27 μM) for 24 h and analyzed using a flow cytometer. To evaluate the involvement of ROS in caspase activation, cell groups were also pretreated with N-acetyl-l-cysteine (NAC) (2 mM) for 1 h following treatment with 4-NRC. Each bar presents mean ± SD of three independent experiments (**p < 0.01 and ***p < 0.001 vs. control).
Discussion
Studies have showed cytotoxic effects of 4-NRC against melanoma solid tumour cell lines, which suggests an apoptotic mechanism (Brohem et al. 2009, 2012). In view of that, the present study clarifies that 4-NRC also promotes interesting cytotoxic activity in p53-null K562 leukemic cells. Morphological analysis of K562 cells after treatment with 4-NRC suggested apoptotic characteristics such as cell shrinkage, nuclear condensation and DNA fragmentation. The pattern of apoptotic cell death was also supported by increased sub-G1, cell cycle arrest at the G1/G0 stage and analysis of phosphatidylserine externalization profile.
DNA fragmentation is a major hallmark of apoptosis and promotes an immediate and p53-independent G1 arrest, due to proteolysis of cyclin D1 (Agami & Bernards 2000; Henry et al. 2013). During malignant cell transformation, cyclin D1 can perform multiple functions by enhancing several processes through the regulation of pathways related to abnormal cell growth, angiogenesis, resistance to apoptosis and, in addition, it is actively involved in mitochondrial regulation (Tashiro et al. 2007; Pestell 2013; Alexandrou & Li 2014). Since our data suggested an apoptotic process and cell cycle arrest at the G0/G1 stage triggered by 4-NRC, cyclin D1 expression was evaluated. In fact, reduction in cyclin D1 activity was observed after K562 treatment with 4-NRC which suggests a p53-independent G1 arrest.
The mechanisms of apoptosis are highly complex and sophisticated, involving an energy-dependent cascade of molecular events (Elmore 2007). It has been shown that PS exposure in response to apoptotic stimuli is an evolutionarily conserved event among cells, and is dependent on caspase activity (Henry et al. 2013). From a biochemical standpoint, apoptosis is defined as a caspase-dependent variant of regulated cellular death (Galluzzi et al. 2015). Thus, to further determine the apoptotic mechanism triggered by 4-NRC, cytochrome c release, PS exposure, cell cycle analysis, cyclin D1, Δψm, mitochondrial activity, ROS production and caspase activities were evaluated.
The mitochondria could represent a interesting prospective target for chemotherapy-induced apoptosis (Mow et al. 2001; Park et al. 2008; Lamb et al. 2014, 2015), since the disruption of Δψm can promote ROS overproduction, leading to cell death (Tait & Green 2010; Madamba et al. 2015). It has been suggested that cancer cell mitochondria protect tumour cells from death through the down regulation of mitochondrial depolarization inducers (i.e. ROS), changing of chaperones expression involved in the mitochondrial permeability transition pore opening and activation of kinase signalling pathways (Bonora & Pinton 2014; Rasola & Bernardi 2015). Our data suggest that 4-NRC could interfere with these pathways of resistance to cell death since 4-NRC-induced apoptosis in K562 cells involved a reduction in mitochondrial activity through depolarization and pore formation in the mitochondrial membrane. In addition, this study shows that the pretreatment of K562 cells with NAC followed by treatment with 4-NRC partially blocked cell apoptosis, which suggests that 4-NRC-induced cell death would not seem to be exclusively via ROS-mediated pathways. Similarly, several plant-derived compounds, such as alkaloid berberine (Wang et al. 2012), polyphenols silibinin (Jiang et al. 2015) and curcumin (Zhang et al. 2016), have shown anticancer potential through mitochondrial mechanisms whether associated or otherwise to oxidative stress.
The release of cytochrome c from mitochondria to the cell cytoplasm, due to disruption of Δψm, triggers the activation of the caspase cascade (Stennicke & Salvesen 1998; Chen & Wang 2002; Abondanza et al. 2008), as observed here. In mechanistic terms, this protein causes the activation of apoptosis intrinsic pathway via caspase-9, which further activates caspase-3, thereby establishing the apoptotic process (Galluzzi et al. 2015). In parallel, the extrinsic pathway can be triggered by intrinsic pathway via caspase-8 activation (Billen et al., 2008; Tait & Green 2010). The multi-target action of new drugs has been highlighted as a promising advantage considering cancer cell resistance to conventional therapy (Winter et al. 2014). In this respect, we showed that 4-NRC activated both extrinsic and intrinsic apoptotic pathways.
Furthermore, intensive research into small molecules that target cell cycle regulatory proteins has led to the identification of many candidate inhibitors capable of arresting proliferation and inducing apoptosis in neoplastic cells (Lapenna & Giordano 2009). Brohem et al. (2009) demonstrated that 4-NRC caused G1 growth arrest in SK-Mel-147 and HNF cells, in contrast to other melanoma cell lines studied which did not present any modulation in cell cycle progression. Here, we clearly demonstrated that 4-NRC arrested K562 cells at the G0/G1 phase in the cell cycle progression, parallel to decreased cyclin D1 expression and reduced mitocondrial activity. Each stage of the cell cycle is regulated by cyclin-dependent protein kinases (CDKs) activated at specific points of the cell cycle (Vermeulen et al. 2003; Casimiro et al. 2014). In the case of cyclin D1, it forms a holoenzyme with some subsets of CDKs and exerts influence on the cell cycle progression to provide proliferative signals at the G1 phase (Casimiro et al. 2014). Its overexpression occurs in multiple cancer types, including chronic myelogenous leukaemia (Groot et al. 2000; Casimiro et al. 2014), which shows it is a considerable therapeutic target in oncology.
Inhibitors of protein kinases as anticancer drugs have been invigorated by the successful approval of a number of molecules that target tyrosine kinases, such as imatinib, a BCR-ABL protein kinase inhibitor, used for the treatment of CML (Lapenna & Giordano 2009). Currently, several drugs targeting the cell cycle, such as flavopiridol, indisulam, AZD5438, SNS-032, bryostatin-1, seliciclib, PD 0332991 and SCH 727965 have entered clinical trials. Clinical studies have demonstrated that CDK inhibitors can be administered to patients with advanced cancer (Dickson & Schwartz 2009). The semi-synthetic flavonoid flavopiridol for the treatment of leukaemia has marked antineoplastic properties through inhibition of CDKs (Smolej 2009). Moreover, it was found that flavopiridol synergizes with chemotherapy in a sequence-dependent manner (Robak et al. 2009).
Although there has been great progress in anticancer therapy, most drugs are nonselective in their mechanism of action and can affect all types of proliferating cells (Krishna et al. 2009). 4-NRC also seems to have a nonselective profile since it showed cytotoxicity to normal human lymphocytes. As an alternative, we have proposed liposomes containing 4-NRC as a promising biocompatible and stable drug delivery system (Gaeti et al. 2015).
In conclusion, this study demonstrates that 4-NRC has antileukemic properties, inducing apoptosis via mitochondrial-dependent pathways that involve the inhibition of cyclin D1 and consequent G0/G1 cell cycle arrest (Figure 7). Given that emerging treatment concepts include novel combinations of well-known agents, 4-NRC could be a promising alternative to chemotherapeutic combinations to maximize tumour suppression.
Figure 7.

Multi-target mechanism proposed for 4-NRC-induced apoptosis in multidrug-resistant K562 cells. 4-NRC promotes mitochondrial dysfunction by reduction of mitochondrial activity through loss of Δψm and pore formation in the membrane of the mitochondria. Thus, it culminates with an increase of ROS production and cytochrome c release from mitochondria to cytosol and consequent caspase activation to trigger cell apoptosis. In parallel, 4-NRC induces cell cycle arrest at G1/G0 stage due to reduced activity of cyclin D1.
Funding Statement
This study was supported by Brazilian research funding agencies, such as Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES).
Disclosure statement
The authors report no declarations of interest.
References
- Abondanza TS, Oliveira CR, Barbosa CMV, Pereira FE, Cunha RL, Caires AC, Comasseto JV, Queiroz ML, Valadares MC, Bincoletto C.. 2008. Bcl-2 expression and apoptosis induction in human HL60 leukaemic cells treated with a novel organotellurium (IV) compound RT-04. Food Chem Toxicol. 46:2540–2545. [DOI] [PubMed] [Google Scholar]
- Agami R, Bernards R.. 2000. Distinct initiation and maintenance mechanisms cooperate to induce G1 cell cycle arrest in response to DNA damage. Cell. 102:55–66. [DOI] [PubMed] [Google Scholar]
- Alexandrou AT, Li JJ.. 2014. Cell cycle regulators guide mitochondrial activity in radiation-induced adaptive response. Antioxid Redox Signal. 20:1463–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ávila RI, Alvarenga CBM, Ávila PHM, Moreira RC, Arruda AF, Fernandes TO, Rodrigues BS, Andrade WM, Batista AC, Paula JR, et al. . 2016. Eugenia dysenterica DC. (Myrtaceae) exerts chemopreventive effects against hexavalent chromium-induced damage in vitro and in vivo. Pharm Biol. 54:2652–2663. [DOI] [PubMed] [Google Scholar]
- Bagatela BS, Lopes AP, Fonseca FL, Andreo MA, Nanayakkara DN, Bastos JK, Perazzo FF.. 2013. Evaluation of antimicrobial and antimalarial activities of crude extract, fractions and 4-nerolidylcathecol from the aerial parts of Piper umbellata L. (Piperaceae). Nat Prod Res. 27:2202–2209. [DOI] [PubMed] [Google Scholar]
- Barros LFM, Barros PSM, Röpke CD, Silva VV, Sawada TC, Barros SB, Belfort R. Jr.. 2007. Dose-dependent in vitro inhibition of rabbit corneal matrix metalloproteinases by an extract of Pothomorphe umbellata after alkali injury. Braz J Med Biol Res. 40:1129–1132. [DOI] [PubMed] [Google Scholar]
- Billen LP, Shamas-Din A, Andrews DW.. 2008. Bid: A Bax-like BH3 protein. Oncogene. 27:S93–S104. [DOI] [PubMed] [Google Scholar]
- Bonora M, Pinton P.. 2014. The mitochondrial permeability transition pore and cancer: Molecular mechanisms involved in cell death. Front Oncol. 4:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brohem CA, Sawada TCH, Massaro RR, Almeida RL, Rivelli DP, Ropke CD, da Silva VV, de Lima TM, Curi R, Barros SB, et al. . 2009. Apoptosis induction by 4-nerolidylcatechol in melanoma cell lines. Toxicol in Vitro. 23:111–119. [DOI] [PubMed] [Google Scholar]
- Brohem CA, Massaro RR, Tiago M, Marinho CE, Jasiulionis MG, de Almeida RL, Rivelli DP, Albuquerque RC, de Oliveira TF, de Melo Loureiro AP, et al. . 2012. Proteasome inhibition and ROS generation by 4-nerolidylcatechol induces melanoma cell death. Pigment Cell Melanoma Res. 25:354–369. [DOI] [PubMed] [Google Scholar]
- Casimiro MC, Velasco-Velázquez M, Aguirre-Alvarado C, Pestell RG.. 2014. Overview of cyclins D1 function in cancer and the CDK inhibitor landscape: Past and present. Expert Opin Investig Drugs. 23:295–304. [DOI] [PubMed] [Google Scholar]
- Chen M, Wang J.. 2002. Initiator caspases in apoptosis signaling pathways. Apoptosis. 7:313–319. [DOI] [PubMed] [Google Scholar]
- Cortez AP, de Ávila RI, da Cunha CR, Santos AP, Menegatti R, Rezende KR, Valadares MC.. 2015. 4-Nerolidylcatechol analogues as promising anticancer agents. Eur J Pharmacol. 765:517–524. [DOI] [PubMed] [Google Scholar]
- Cunha CRM, Mendanha Neto SA, Silva CC, Cortez AP, Gomed MN, Martins FI, Alonso A, Rezende KR, Menegatti R, de Magalhães MT, et al. . 2013. 4-Nerolidylcatechol and its synthetic analogues: Antioxidant activity and toxicity evaluation. Eur J Med Chem. 62:371–378. [DOI] [PubMed] [Google Scholar]
- Desmarchelier C, Barros S, Repetto M, Latorre LR, Kato M, Coussio J, Ciccia G.. 1997. 4-Nerolidylcatechol from Pothomorphe spp. scavenges peroxyl radicals and inhibits Fe(II)-dependent DNA damage. Planta Med. 63:561–563. [DOI] [PubMed] [Google Scholar]
- Dickson MA, Schwartz GK.. 2009. Development of cell-cycle inhibitors for cancer therapy. Curr Oncol. 16:36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore S.2007. Apoptosis: a review of programmed cell death. Toxicol Pathol. 35:495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes TO, Ávila RI, Moura SS, Ribeiro GA, Naves MMV, Valadares MC.. 2015. Campomanesia adamantium (Myrtaceae) fruits protect HepG2 cells against carbon tetrachloride-induced toxicity. Toxicol Rep. 2:184–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaeti MPN, Benfica PL, Mendes LP, Vieira MS, Anjos JL, Alonso A, Rezende KR, Valadares MC, Lima EM.. 2015. Liposomal entrapment of 4-nerolidylcatechol: Impact on phospholipid dynamics, drug stability and bioactivity. J Nanosci Nanotechnol. 15:838–847. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Bravo-San Pedro JM, Vitale I, Aaronson SA, Abrams JM, Adam D, Alnemri ES, Altucci L, Andrews D, Annicchiarico-Petruzzelli M, et al. . 2015. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ. 22:58–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groot RP, Raaijmakers JA, Lammers JW, Koenderman L.. 2000. STAT5-dependent cyclinD1 and Bcl-xL expression in Bcr-Abl-transformed cells. Mol Cell Biol Res Commun. 3:299–305. [DOI] [PubMed] [Google Scholar]
- Gustafson KR, Cardellinall JH, McMahon JB, Pannell LK, Cragg GM, Boyd MR.. 1992. HIV inhibitory natural products. 6. The peltatols, novel HIV-inhibitory catechol derivatives from Pothomorphe peltata. J Org Chem. 57:2809–2811. [Google Scholar]
- Henry CM, Hollville E, Martin SJ.. 2013. Measuring apoptosis by microscopy and flow cytometry. Methods. 61:90–97. [DOI] [PubMed] [Google Scholar]
- Huang H-L, Chen Y-C, Huang Y-C, Yang KC, Pan H, Shih SP, Chen YJ.. 2011. Lapatinib induces autophagy, apoptosis and megakaryocytic differentiation in chronic myelogenous leukemia K562 cells. PLoS One. 6:e29014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang K, Wang W, Jin X, Wang Z, Ji Z, Meng G.. 2015. Silibinin, a natural flavonoid, induces autophagy via ROS-dependent mitochondrial dysfunction and loss of ATP involving BNIP3 in human MCF7 breast cancer cells. Oncol Rep. 33:2711–2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koptyra M, Falinski R, Nowicki MO, Stoklosa T, Majsterek I, Nieborowska-Skorska M, Blasiak J, Skorski T.. 2006. BCR/ABL kinase induces self-mutagenesis via reactive oxygen species to encode imatinib resistance. Blood. 108:319–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna IV, Vanaja GR, Kumar NS, Suman G.. 2009. Cytotoxic studies of anti-neoplastic drugs on human lymphocytes-in vitro studies. Cancer Biomark. 5:261–272. [DOI] [PubMed] [Google Scholar]
- Lamb R, Harrison H, Hulit J, Smith DL, Lisanti MP, Sotgia F.. 2014. Mitochondria as new therapeutic targets for eradicating cancer stem cells: quantitative proteomics and functional validation via MCT1/2 inhibition. Oncotarget. 5:11029–11037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb R, Ozsvari B, Lisanti CL, Tanowitz HB, Howell A, Martinez-Outschoorn Sotgia F, Lisanti MP.. 2015. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: treating cancer like an infectious disease. Oncotarget. 6:4569–4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapenna S, Giordano A.. 2009. Cell cycle kinases as therapeutic targets for cancer. Nat Rev Drug Discov. 8:547–566. [DOI] [PubMed] [Google Scholar]
- Madamba SM, Damri KN, Dejean LM, Peixoto PM.. 2015. Mitochondrial ion channels in cancer transformation. Front Oncol. 5:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller GD, Bruno BJ, Lim CS.. 2014. Resistant mutations in CML and Ph(+)ALL-role of ponatinib. Biologics. 8:243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosmann T.1983. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J Immunol Methods. 65:55–63. [DOI] [PubMed] [Google Scholar]
- Mota MF, Benfica PL, Valadares MC.. 2012. Synadenium umbellatum Pax. promotes cell cycle arrest and induces apoptosis in K-562 leukemia cells. Braz J Pharm Sci. 48:497–505. [Google Scholar]
- Mota MF, Cortez AP, Benfica PL, Rodrigues BS, Castro TF, Macedo LM, Castro CH, Lião LM, Carvalho FS, Romeiro LAS, et al. . 2016. Induction of apoptosis in Ehrlich ascites tumour cells via p53 activation by a novel small-molecule MDM2 inhibitor – LQFM030. J Pharm Pharmacol. 68:1143–1159. [DOI] [PubMed] [Google Scholar]
- Mow BM, Blajeski AL, Chandra J, Kaufmann SH.. 2001. Apoptosis and the response to anticancer therapy. Curr Opin Oncol. 13:453–462. [DOI] [PubMed] [Google Scholar]
- Park JH, Jin C-Y, Lee BK, Kim GY, Choi YH, Jeong YK.. 2008. Naringenin induces apoptosis through downregulation of Akt and caspase-3 activation in human leukemia THP-1 cells. Food Chem Toxicol. 46:3684–3690. [DOI] [PubMed] [Google Scholar]
- Pestell RG.2013. New roles of cyclin D1. Am J Pathol. 183:3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston RJ, San Sebastian JR, McFee AF.. 1987. The in vitro human lymphocyte assay for assessing the clastogenicity of chemical agents. Mutat Res. 189:175–183. [DOI] [PubMed] [Google Scholar]
- Rasola A, Bernardi P.. 2015. Reprint of 'The mitochondrial permeability transition pore and its adaptive responses in tumor cells'. Cell Calcium. 58:18–26. [DOI] [PubMed] [Google Scholar]
- Rezende KR, Barros SBDM.. 2004. Quantification of 4-nerolidylcatechol from Pothomorphe umbellata (Piperaceae) in rat plasma samples by HPLC-UV. Rev Bras Cienc Farm. 40:373–380. [Google Scholar]
- Robak T, Jamroziak K, Robak P.. 2009. Current and emerging treatments for chronic lymphocytic leukaemia. Drugs. 69:2415–2449. [DOI] [PubMed] [Google Scholar]
- Ropke CD, Meirelles RR, da Silva VV, Sawada TC, Barros SB.. 2003. Pothomorphe umbellata extract prevents alpha-tocopherol depletion after UV-irradiation. Photochem Photobiol. 78:436–439. [DOI] [PubMed] [Google Scholar]
- Ropke CD, Sawada TCH, da Silva VV, Michalany NS, de Moraes Barros SB.. 2005. Photoprotective effect of Pothomorphe umbellata root extract against ultraviolet radiation induced chronic skin damage in the hairless mouse. Clin Exp Dermatol. 30:272–276. [DOI] [PubMed] [Google Scholar]
- Ropke CD, Silva VV, Kera CZ, Miranda DV, de Almeida RL, Sawada TC, Barros SB.. 2006. In vitro and in vivo inhibition of skin matrix metalloproteinases by Pothomorphe umbellata root extract. Photochem Photobiol. 82:439–442. [DOI] [PubMed] [Google Scholar]
- Savitskaya MA, Onishchenko MA.. 2015. Mechanisms of apoptosis. Biochemistry. 80:1693–1405. [DOI] [PubMed] [Google Scholar]
- Sen S, Takahashi R, Rani S, Freireich EJ, Stass AS.. 1993. Expression of differentially phosphorylated Rb and mutant p53 proteins in myeloid leukemia cell lines. Leuk Res. 17:639–647. [DOI] [PubMed] [Google Scholar]
- Silva Pinto AC, Silva LFR, Cavalcanti BC, Melo MR, Chaves FC, Lotufo LV, de Moraes MO, de Andrade-Neto VF, Tadei WP, Pessoa CO, et al. . 2009. New antimalarial and cytotoxic 4-nerolidylcatechol derivatives. Eur J Med Chem. 44:2731–2735. [DOI] [PubMed] [Google Scholar]
- Smolej L.2009. Modern concepts in the treatment of chronic lymphocytic leukemia. Hematology. 14:249–254. [DOI] [PubMed] [Google Scholar]
- Stennicke HR, Salvesen GS.. 1998. Properties of the caspases. Biochim Biophys Acta. 1387:17–31. [DOI] [PubMed] [Google Scholar]
- Tait SW, Green DR.. 2010. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 11:621–632. [DOI] [PubMed] [Google Scholar]
- Tashiro E, Tsuchiya A, Imoto M.. 2007. Functions of cyclin D1 as an oncogene and regulation of cyclin D1 expression. Cancer Sci. 98:629–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokarz P, Blasiak J.. 2014. Role of mitochondria in carcinogenesis. Acta Biochim Pol. 61:671–678. [PubMed] [Google Scholar]
- Valadares MC, Rezende KR, Pereira ER, Sousa MC, Gonçalves B, de Assis JC, Kato MJ.. 2007. Protective effects of 4-nerolidylcatechol against genotoxicity induced by cyclophosphamide. Food Chem Toxicol. 45:1975–1978. [DOI] [PubMed] [Google Scholar]
- Vermeulen K, Van Bockstaele DR, Berneman ZN.. 2003. The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 36:131–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vousden KH, Prives C.. 2009. Blinded by the light: the growing complexity of p53. Cell. 137:413–431. [DOI] [PubMed] [Google Scholar]
- Wang L, Liu L, Shi Y, Cao H, Chaturvedi R, Calcutt MW, Hu T, Ren X, Wilson KT, Polk DB, et al. . 2012. Berberine induces caspase-independent cell death in colon tumor cells through activation of apoptosis-inducing factor. PLoS One. 7:e36418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter E, Chiaradia LD, Silva AH, Nunes RJ, Yunes RA, Creczynski-Pasa TB.. 2014. Involvement of extrinsic and intrinsic apoptotic pathways together with endoplasmic reticulum stress in cell death induced by naphthylchalcones in a leukemic cell line: advantages of multi-target action. Toxicol in Vitro. 28:769–777. [DOI] [PubMed] [Google Scholar]
- Zhang L, Cheng X, Gao Y, Bao J, Guan H, Lu R, Yu H, Xu Q, Sun Y.. 2016. Induction of ROS-independent DNA damage by curcumin leads to G2/M cell cycle arrest and apoptosis in human papillary thyroid carcinoma BCPAP cells. Food Funct. 7:315–325. [DOI] [PubMed] [Google Scholar]