

Abstract
Previously, we showed that oral application of the environmental pollutant dibenzo[a,l]pyrene (DB[a,l]P) induces oral tumors in mice. Thus, in the present investigation we examined the effect of alcohol on DB[a,l]P-induced DNA damage and immune regulation; we showed that alcohol (6.4% v/v in the diet, 35% of Calories) significantly enhanced the levels of (−)-anti-trans-DB[a,l]P-dA while decreased the levels of GSH in the mouse oral tissues. Analysis of RNA expression revealed that DB[a,l]P alone upregulates inflammatory genes while alcohol suppresses several markers of immune surveillance. Collectively, these results suggest that alcohol may enhance oral carcinogenesis induced by DB[a,l]P.
Introduction
Head and neck squamous cell carcinoma (HNSCC), the vast majority of which occurs in the oral cavity, is the sixth most common malignancy worldwide, representing a major international health problem (1). HNSCC is the most traumatic form of malignancy due to its visible distortion of the face, and disruption of breathing, speaking and swallowing (2). Quality of life issues and psychosocial dysfunctions are two major challenges that HNSCC patients are facing (3, 4). Early diagnosis for oral cancer has not improved over time.
Epidemiological data strongly support the role of exogenous factors such as tobacco, alcohol and human papilloma virus infection as causative factors; other factors (betel quid chewing, smokeless tobacco, marijuana, and certain dietary components) have also been implicated in the development of HNSCC (1). A recent study found that alcohol consumption combined with tobacco smoking results in a supra-multiplicative synergistic effect which enhances cancer risk up to 14-fold (5). However, avoidance of risk factors has only been partially successful and survival rate remains stagnant at about 50% (6) despite advances in therapeutic approaches. Therefore, new or improved approaches in prevention and/or early detection are critical. To facilitate this, there remains an urgent need to identify the molecular mechanisms responsible for oral carcinogenesis using animal models that can mimic human exposures to relevant risk factors such as alcohol, tobacco and environmental carcinogens
In 2012, it was estimated that 38.1% of the world’s population aged 15 years or older were regular drinkers (7). Alcoholic beverages have been classified as a human carcinogen by The International Agency for Research on Cancer (IARC) (8). The mechanisms by which alcohol consumption enhances oral carcinogenesis remain under investigation; however, numerous lines of research have implicated ethanol-induced oxidative stress/damage as an important component. Other potential mechanisms include possible induction of carcinogen activation pathways and suppression of both innate and adaptive immune systems, providing a window of opportunity for loss of immune surveillance (9, 10).
Chronic alcohol consumption decreased glutathione (GSH) levels and increased the levels of 8-hydroxydeoxyguanosine (8-oxodG) in the blood, liver and gingival tissue of rats (11–13). Acute administration of ethanol led to depleted GSH levels in numerous tissues in both dose and age dependent fashion in mice (13). GSH depletions of 37–48% were observed in the oral cavity after administration of 5 g alcohol/kg b.w. in rats (14–16). The mechanism of GSH depletion has not been elucidated but likely involves direct conjugation with acetaldehyde, a major metabolite derived from alcohol (17–19). GSH, the major intracellular antioxidant (20), reduces oxidative damage to DNA measured as 8-oxodG possibly by inhibiting ROS production (21, 22) and additionally detoxifies many carcinogens including tobacco and environmental polycyclic aromatic hydrocarbons (PAHs) through phase II conjugation (23).
PAHs are formed in tobacco smoke and through other environment combustion sources and are thought to play an important role in cancer development at numerous sites including lung and oral cavity (1). Our previous studies showed that both dibenzo[a,l]pyrene (DB[a,l]P), a representative example of PAHs, and its diol epoxide (DB[a,l]PDE) induced SCC in the oral tissues of mice in a dose-dependent manner. We have also demonstrated the formation of stable covalent DNA adducts derived from DB[a,l]PDE in oral tissues of mice treated with DB[a,l]P (24). Measurement of these adducts provides an experimental approach to monitor carcinogen exposure and provides a risk assessment for susceptibility to carcinogenesis (25). In the current study, experiments were designed to test our hypothesis that chronic alcohol consumption could potentiate DB[a,l]P-induced genotoxicity and suppress immune surveillance in the oral cavity.
Materials and Methods
Chemicals
DB[a,l]P, (±)-anti-DB[a,l]PDE and DNA adducts were prepared according to published methods by our group (26, 27). These chemicals were characterized on the basis of NMR, mass spectral data and their purity (≥99%) was determined by HPLC.
Animals
Female B6C3F1 mice (Jackson Laboratories, Bar Harbor, ME) were used in our study. Mice were quarantined for 1 week; then they were transferred to the bioassay laboratory. All mice were kept on a 12-h light:12-h dark cycle, maintained at 50% relative humidity and 21 ± 2°C. Water was provided ad libitum. The bioassay was carried out in accordance with the NIH Guide for the Care and Use of Laboratory Animals and was approved by Institutional Animal Care and Use Committee.
Mice were fed Isocaloric Lieber-DeCarli-type liquid diets formulated by BioServ (Frenchtown, N.J.) with or without alcohol (calories increased gradually from 0, 11, 22 to 35%). Diet with alcohol at 35% of total calories contains 6.4% v/v ethanol in the diet. After 3 weeks, mice were treated by topical application into the oral cavity with 2 consecutive daily doses of DB[a,l]P (240 nmol each) in DMSO while mice in the control group were treated with vehicle only (DMSO). Mice were then sacrificed 24 hrs later by CO2 asphyxiation, and tissues (oral tissues and tongues) were harvested and stored at −80°C prior to the analysis.
Analysis of GSH
GSH (including oxidized and protein bound forms) was determined using a previously described method (28). Oral mucosa was homogenized (5% w/v) in ice-cold 5% metaphosphoric acid (MPA) using a Ten-Broeck all-glass homogenizer. After centrifugation at 13,000 × g for 2 min, the resulting supernatants was analyzed in MPA extracts by our HPLC method using dual electrochemical detection (29).
Analysis of DB[a,l]P-induced DNA adducts
We previously reported that DB[a,l]PDE-dA is the major DNA adduct detected in oral tissues of mice treated with DB[a,l]P, and thus this adduct was analyzed in this study (30). The method used for the analysis of DB[a,l]PDE -dA adduct by LC-MS/MS is identical to our previously published procedure (27, 30, 31). Briefly, DNA was isolated using the Qiagen genomic DNA isolation procedure from frozen tissues. A NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE) was used to determine the levels of DNA. Prior to enzymatic digestion, 150 pg of each [15N5]-anti-trans- and [15N5]-anti-cis-DBPDE-dA adducts were added to ~150 μg DNA. DNA was hydrolyzed with 1M MgCl2 (10 μL/mg DNA) and DNase I (0.2 mg/mg DNA) at 37 ºC for 1.5 h, followed with the addition of nuclease P1 (20 μg/mg DNA), snake venom phosphodiesterase (0.08 unit/mg DNA) and alkaline phosphatase (2 units/mg DNA) into the mixture. An Aliquot of the DNA hydrolysate was subjected to dA base analysis by HPLC, and the remaining supernatant was partially purified by an Oasis HLB solid phase extraction column (1 cm3, 30 mg, Waters Ltd.). Then, the sample was analyzed on an API 3200™ LC-MS/MS triple quadrupole mass spectrometer interfaced with an Agilent 1200 series HPLC using an Agilent extend-C18, 5 μm 4.6 × 150 mm column. Adducts were monitored in multiple reaction monitoring (MRM) mode. The MS/MS transitions of m/z 604→ m/z 335, and m/z 609→ m/z 335 were monitored for targeted adducts and internal standards, respectively.
Analysis of Immune-related gene expression.
Total RNA was extracted and purified from oral tissues of DB[a,l]P-treated mice with or without alcohol (n ≥ 3 per group) using the RNeasy kit (Qiagen Inc. Hilden, Germany). The extracted RNA was subjected to the nanoString nCounter® barcoding assay using an nCounter® MAX Analysis System in the Penn State Genomics Core Facility – University Park, PA. RNA expression of immune-related genes was quantified using the Mouse Inflammation v2 panel (256 genes) and data were analyzed using nSolver Ananlysis Software v3.0.
Results
The effects of alcohol on the levels of GSH in mouse oral tissue.
We measured GSH levels in the oral mucosa following administration of alcohol diet for 24 days. GSH levels were decreased 28% (0.89 ± 0.13 μmol/g) significantly in mice fed the alcohol diet at 35% of calories (6.4% v/v alcohol in the diet) compared to those fed the control diet (1.23 ± 0.17 μmol/g), p < 0.05 (Figure 1).
Fig. 1.
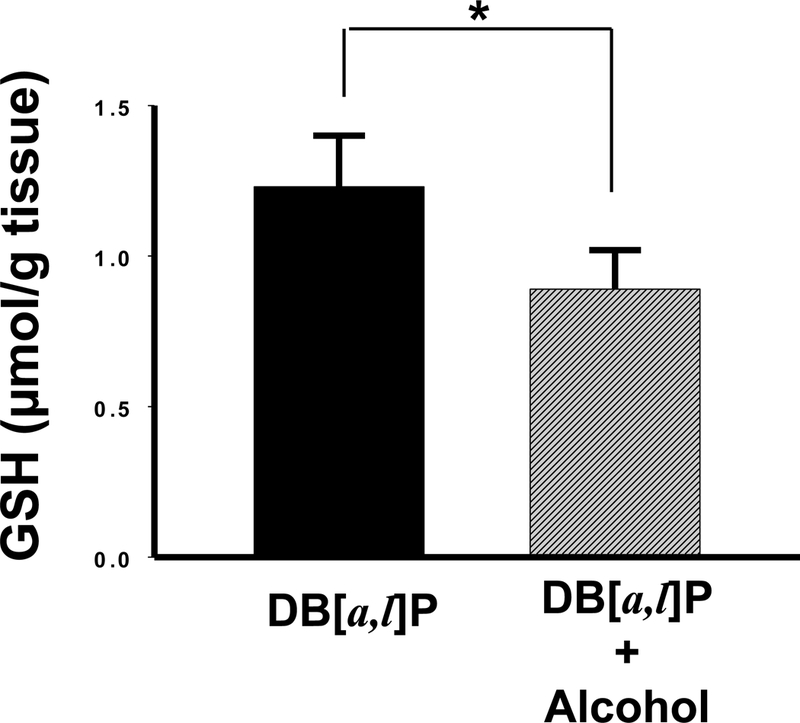
Effect of alcohol on levels of GSH in oral tissues of mice. *, P < 0.05 in a one-tailed t-test.
The effects of alcohol on the levels of DB[a,l]P-induced DNA adducts in mouse oral tissue.
As described above, the decrease in the GSH levels in oral tissues following chronic alcohol consumption suggests a possible reduction in the detoxification of diol epoxides derived from DB[a,l]P in these tissues. Therefore, we subsequently analyzed the levels of DB[a,l]PDE-dA adduct in mouse oral tissues as a measure of DB[a,l]P-induced genotoxicity. We found that alcohol in the diet significantly (p < 0.05) increased the levels of (–)-anti-trans-DB[a,l]P-dA (25%, Figure 2); no liver abnormalities were observed in any animal at termination of the bioassay.
Fig. 2.
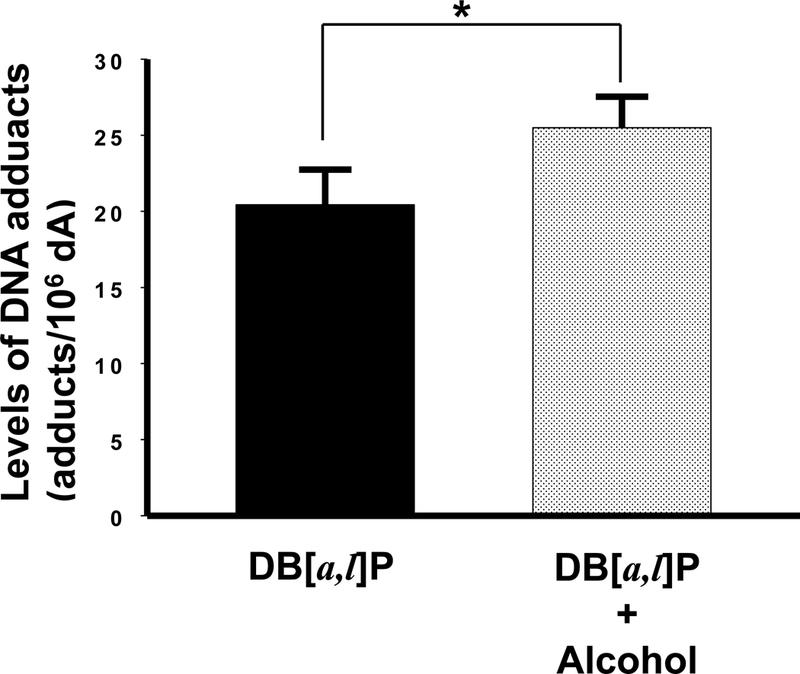
Effect of alcohol on levels of DB[a,l]P-induced DNA adducts in oral tissues of mice. *, P < 0.05 in a one-tailed t-test.
The Role of DB[a,l]P and alcohol on inflammation-related gene expression in oral tissue.
We found that in mice treated with DB[a,l]P a variety of inflammation-related genes were upregulated >2-fold (range 2.1 to 8.9-fold, p<0.05) above the levels expressed in control treated mice while none of the 254 genes evaluated were downregulated at this level of stringency (Figure 3A). Many of these genes are involved in the inflammatory response to carcinogen, including Cox2 (Ptgs2), matrix metallopeptidase 3 (Mmp3), transcripts encoding proteins involved in complement (Masp1) and leukotriene (Alox5) regulation and a number of chemokines important for recruitment of immune cells such as monocytes, neutrophils, NK cells and T cells. Significant increases in the detection of multiple Toll-like receptor transcripts (Tlr3, 5, 8) suggests the presence of infiltrating innate immune cells. Thus, carcinogen alone upregulates the inflammatory microenvironment within the oral tissue.
Fig. 3.
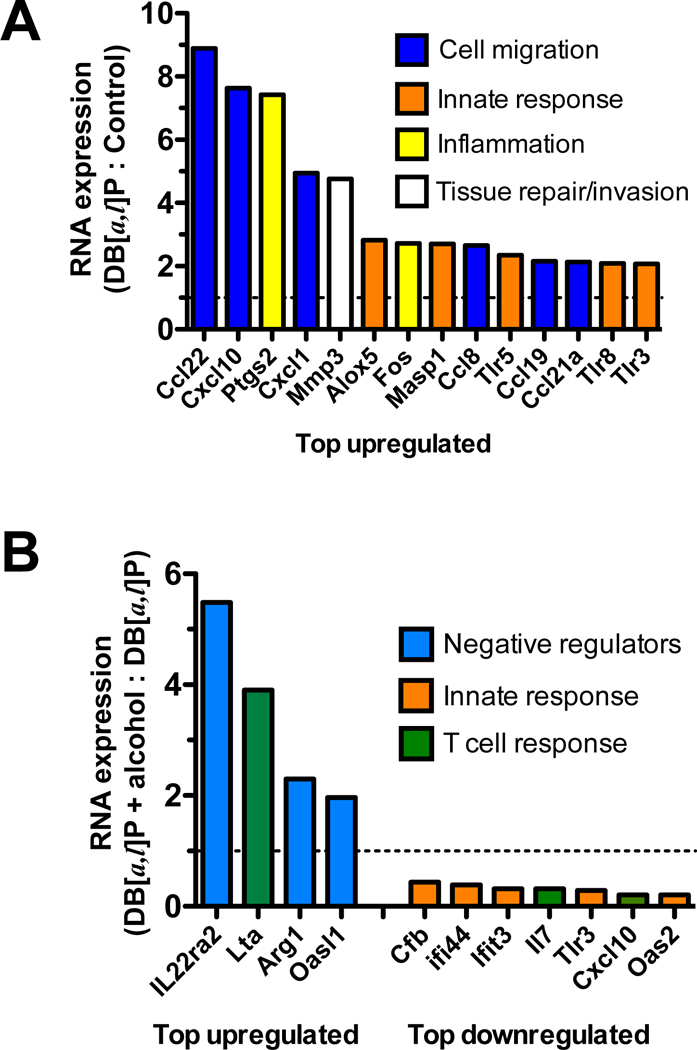
Expression of selected inflammatory genes in mice exposed to DB[a,l]P and alcohol. (A) Fold change in transcripts isolated from the oral tissue of mice pre-exposed to normal diet for three weeks followed by topical application of DB[a,l]P into the oral cavity. (B) Fold change in transcripts isolated from the oral tissue of mice pre-exposed to alcohol diet versus normal diet for three weeks followed by topical application of DB[a,l]P into the oral cavity. N=4 mice per group.
We also found that administration of alcohol in the diet prior to DB[a,l]P increased the expression of several transcripts associated with negative regulation of the immune response (Figure 3B). In particular, transcripts for the IL-22 binding protein (Il22ra2) were dramatically increased, suggesting that tissue repair and survival mechanisms induced by IL-22 could be counteracted in the oral epithelium in the presence of alcohol. Additionally, transcripts for lymphotoxin α (Lta) were increased, indicative of increased inflammation that could drive IL-23-mediated inflammation or apoptosis. However, increased levels of Arginase (Arg1) transcripts, an inhibitor of T cell function, and Oasl1, which can inhibit type I interferon signaling, suggest that multiple levels of immune suppression may be upregulated following administration of alcohol. Consistent with this interpretation is the downregulation of multiple innate immune responsive genes (Cfb, Ifi44, Ifit3, Tlr3) as well as Cxcl10 and IL-7 transcripts, important for T cell chemotaxis and survival. Thus, while the overall microenvironment of the oral cavity appears to be skewed toward inflammation by carcinogen, alcohol may suppress specific immune functions, thereby reducing immune surveillance.
Discussion
Although alcohol has been classified as a human carcinogen, the mechanisms that account for the development of oral carcinogenesis by alcohol consumption remain largely undefined. Acetaldehyde, a principle metabolite of alcohol catalyzed by CYP2E1 and alcohol dehydrogenase (ADH), has been classified as a human carcinogen by the IARC (8) and is known to induce genotoxic effects (32). Reactive oxygen species (ROS), products of alcohol metabolism, can also induce genotoxicity. Furthermore, previous studies showed that alcohol can contribute to epigenetic regulation of oncogenes or tumor suppressor genes (33). Finally, alcohol can suppress and skew aspects of both the innate and adaptive immune system, providing a window of opportunity for loss of immune surveillance (9, 10).
As a nucleophile, GSH could attack electrophiles such as diol epoxides derived from the metabolism of DB[a,l]P; therefore, depleting GSH levels by alcohol and increasing available diol epoxides can account for the enhancement of DB[a,l]P-dA adduct. On the basis of our results we hypothesize that chronic alcohol consumption may increase the carcinogenic potency of DB[a,l]P; further studies will test this hypothesis.
Our data suggest that expression of multiple inflammation-related genes is upregulated in the oral cavity by exposure to DB[a,l]P. While many of these changes were maintained following additional exposure to alcohol, expression of a subset of inflammatory genes was significantly altered such that the tissue microenvironment may be skewed toward an immune suppressive state. Based on these results we propose a working model to further guide investigation of the potential impacts of alcohol on immune surveillance in the oral cavity (Figure 4). In particular, our data suggest that T cell and NK cell responses may be less effective due to the presence of inhibitors such as arginase in the microenvironment that block their effector functions (34). In addition, there may be decreased T cell recruitment due to reduced production of the chemokine Cxcl10. While Increased levels of lymphotoxin indicate the presence of activated T cells or innate lymphoid cells, its presence provides a potential mechanim to upregulate IL-23 production and amplification of the Th17 response, leading to IL-17A and IL-22-induced inflammation of the epithelium (35, 36). We detected small, but not statistically significant increases in the transcripts for these cytokines (data not shown). Thus, further studies will be required to evaluate their expression at the protein level. Our results show increased transcript levels of the IL-22 binding protein following administration of alcohol. A recent study found that conventional dendritic cells can constitutively produce IL-22 binding protein in multiple mouse tissues but that this production can be dramatically upregulated by certain metabolic products, including retinoic acid (37). How alcohol might modulate this effect in the oral cavity remains unknown but could be related to local metabolites produced from alcohol. Since IL-22 can direct both proinflammatory and tissue repair in the epithelium, neutralization of IL-22 could potentially be beneficial or detrimental (35). In addition, we found that transcripts for multiple innate receptors and interferon response genes were reduced following administration of alcohol, suggesting that alcohol may skew the immune cell populations within the oral tissue or may dampen innate sensing. These data support that alcohol skews the immune microenvironment in oral tissues, creating a window of opportunity for escape from immune surveillance.
Fig. 4.
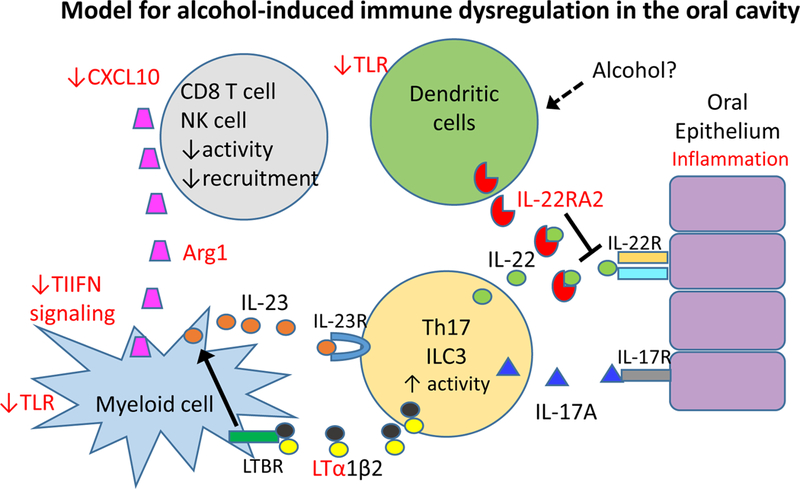
Model of alcohol-induced immune modulation in the oral cavity. Genes in red are modulated by alcohol in the oral cavity. Changes to chemotactic signals and production of Arginase results in reduced T cell and NK cell recruitment activity. Decreased type I interferon (TIIFN) signaling or innate sensing by toll like receptors (TLR) may allow suppressive myeloid cells to produce IL-23 to amplify the Th17 or innate lymphoid cell 3 (ILC3) response, leading to IL-17A and IL-22-induced inflammation of the epithelium; This state may be further amplified by increased lymphotoxin α (LTa) production and increased signaling through the lymphotoxin receptor (LTBR) that upregulates IL-23 production. Alcohol leads to high level production of the IL-22RA2 soluble receptor, perhaps by local or accumulating dendritic cells, which may moderate tissue damage but also block tissue repair.
In summary, we show for the first time that chronic alcohol consumption enhances the genotoxicity induced by DB[a,l]P and promotes gene expression changes that might suppress immune surveillance in the mouse oral cavity. Therefore, our results provided a solid foundation for future studies to test the hypothesis that alcohol can enhance oral cancer induction by the environmental pollutant and tobacco smoke constituent, DB[a,l]P.
Acknowledgement
This work was supported by the National Institutes of Health NCI grant R01-CA173465 and NIEHS R21ES020411.
Footnotes
Statement of publication
This manuscript has not been published elsewhere and it has not been submitted simultaneously for publication elsewhere.
References
- 1.El-Bayoumy K, et al. , Carcinogenesis of the Oral Cavity: Environmental Causes and Potential Prevention by Black Raspberry. Chem Res Toxicol, 2017; 30: 126–144. [DOI] [PubMed] [Google Scholar]
- 2.Koster ME and Bergsma J, Problems and coping behaviour of facial cancer patients. Soc Sci Med, 1990; 30: 569–78. [DOI] [PubMed] [Google Scholar]
- 3.Talmi YP, Quality of life issues in cancer of the oral cavity. J Laryngol Otol, 2002; 116: 785–90. [DOI] [PubMed] [Google Scholar]
- 4.Hammerlid E, et al. , Quality-of-life effects of psychosocial intervention in patients with head and neck cancer. Otolaryngol Head Neck Surg, 1999; 120: 507–16. [DOI] [PubMed] [Google Scholar]
- 5.Scoccianti C, et al. , European Code against Cancer 4th Edition: Alcohol drinking and cancer. Cancer Epidemiol, 2016; 45: 181–188. [DOI] [PubMed] [Google Scholar]
- 6.Siegel RL, Miller KD and Jemal A, Cancer statistics, 2016. CA Cancer J Clin, 2016; 66: 7–30. [DOI] [PubMed] [Google Scholar]
- 7.Praud D, et al. , Cancer incidence and mortality attributable to alcohol consumption. Int J Cancer, 2016; 138: 1380–7. [DOI] [PubMed] [Google Scholar]
- 8.IARC IARC monographs on the evaluation of carcinogenic risk to humans; Alcohol drinking , V. 44 1988, Lyon, France: 1–378. [PMC free article] [PubMed] [Google Scholar]
- 9.Guttenplan JB, et al. , Mutagenesis and carcinogenesis induced by dibenzo[a,l]pyrene in the mouse oral cavity: a potential new model for oral cancer. Int J Cancer, 2012; 130: 2783–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen KM, et al. , Mechanisms of oral carcinogenesis induced by dibenzo[a,l]pyrene: an environmental pollutant and a tobacco smoke constituent. Int J Cancer, 2013; 133: 1300–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Irie K, et al. , Effects of ethanol consumption on periodontal inflammation in rats. J Dent Res, 2008; 87: 456–60. [DOI] [PubMed] [Google Scholar]
- 12.Das SK and Vasudevan DM, Alcohol-induced oxidative stress. Life Sci, 2007; 81: 177–87. [DOI] [PubMed] [Google Scholar]
- 13.Vogt BL and Richie JP Jr., Glutathione depletion and recovery after acute ethanol administration in the aging mouse. Biochem Pharmacol, 2007; 73: 1613–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mallikarjuna K, Nishanth K and Reddy KS, Hepatic glutathione mediated antioxidant system in ethanol treated rats: Decline with age. Pathophysiology, 2007; 14: 17–21. [DOI] [PubMed] [Google Scholar]
- 15.Videla L and Guerri C, Glutathion and alcohol. 1990: Boca Raton: CRC Press. [Google Scholar]
- 16.Videla LA, Fernandez V and Valenzuela A, Age-dependent changes in rat liver lipid peroxidation and glutathione content induced by acute ethanol ingestion. Cell Biochem Funct, 1987; 5: 273–80. [DOI] [PubMed] [Google Scholar]
- 17.Kallama S and Hemminki K, Urinary excretion products after the administration of 14C-acetaldehyde to rats. J Appl Toxicol, 1983; 3: 313–6. [DOI] [PubMed] [Google Scholar]
- 18.Hemminki K, Urinary sulfur containing metabolites after administration of ethanol, acetaldehyde and formaldehyde to rats. Toxicol Lett, 1982; 11: 1–6. [DOI] [PubMed] [Google Scholar]
- 19.Kera Y, Kiriyama T and Komura S, Conjugation of acetaldehyde with cysteinylglycine, the first metabolite in glutathione breakdown by gamma-glutamyltranspeptidase. Agents Actions, 1985; 17: 48–52. [DOI] [PubMed] [Google Scholar]
- 20.Locigno R and Castronovo V, Reduced glutathione system: role in cancer development, prevention and treatment (review). Int J Oncol, 2001; 19: 221–36. [DOI] [PubMed] [Google Scholar]
- 21.Hamilos DL and Wedner HJ, The role of glutathione in lymphocyte activation. I. Comparison of inhibitory effects of buthionine sulfoximine and 2-cyclohexene-1-one by nuclear size transformation. J Immunol, 1985; 135: 2740–7. [PubMed] [Google Scholar]
- 22.Wu G, et al. , Glutathione metabolism and its implications for health. J Nutr, 2004; 134: 489–92. [DOI] [PubMed] [Google Scholar]
- 23.Coles B and Ketterer B, The role of glutathione and glutathione transferases in chemical carcinogenesis. Crit Rev Biochem Mol Biol, 1990; 25: 47–70. [DOI] [PubMed] [Google Scholar]
- 24.Zhang SM, et al. , Simultaneous detection of deoxyadenosine and deoxyguanosine adducts in the tongue and other oral tissues of mice treated with Dibenzo[a,l]pyrene. Chem Res Toxicol, 2014; 27: 1199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Penning TM, Polycyclic Aromatic Hydrocarbons: Multiple Metabolic Pathways and the DNA Lesions Formed The Chemical Biology of DNA Damage. 2010: Wiley-VCH Verlag GmbH & Co. KGaA; 131–155. [Google Scholar]
- 26.Krzeminski J, et al. , Synthesis of Fjord region diol epoxides as potential ultimate carcinogens of dibenzo[a,l]pyrene. Chem Res Toxicol, 1994; 7: 125–9. [DOI] [PubMed] [Google Scholar]
- 27.Zhang SM, et al. , Identification and quantification of DNA adducts in the oral tissues of mice treated with the environmental carcinogen dibenzo[a,l]pyrene by HPLC-MS/MS. Chem Res Toxicol, 2011; 24: 1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang Z, et al. , Enhanced levels of glutathione and protein glutathiolation in rat tongue epithelium during 4-NQO-induced carcinogenesis. Int J Cancer, 2007; 120: 1396–401. [DOI] [PubMed] [Google Scholar]
- 29.Kleinman WA and Richie JP, Jr., Determination of thiols and disulfides using high-performance liquid chromatography with electrochemical detection. J Chromatogr B Biomed Appl, 1995; 672: 73–80. [DOI] [PubMed] [Google Scholar]
- 30.Zhang SM, et al. , Simultaneous detection of deoxyadenosine and deoxyguanosine adducts in the tongue and other oral tissues of mice treated with dibenzo[a,l]pyrene. Chem Res Toxicol, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen KM, et al. , Induction of Ovarian Cancer and DNA Adducts by Dibenzo[a,l]pyrene in the Mouse. Chem Res Toxicol, 2012; 25: 374–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu HS, et al. , Formation of acetaldehyde-derived DNA adducts due to alcohol exposure. Chem Biol Interact, 2010; 188: 367–75. [DOI] [PubMed] [Google Scholar]
- 33.Mathers JC, Strathdee G and Relton CL, Induction of epigenetic alterations by dietary and other environmental factors. Adv Genet, 2010; 71: 3–39. [DOI] [PubMed] [Google Scholar]
- 34.Bronte V, et al. , L-arginine metabolism in myeloid cells controls T-lymphocyte functions. Trends Immunol, 2003; 24: 302–6. [DOI] [PubMed] [Google Scholar]
- 35.Sonnenberg GF, Fouser LA and Artis D, Functional biology of the IL-22-IL-22R pathway in regulating immunity and inflammation at barrier surfaces. Adv Immunol, 2010; 107: 1–29. [DOI] [PubMed] [Google Scholar]
- 36.Tumanov AV, et al. , Lymphotoxin controls the IL-22 protection pathway in gut innate lymphoid cells during mucosal pathogen challenge. Cell Host Microbe, 2011; 10: 44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martin JC, et al. , Interleukin-22 binding protein (IL-22BP) is constitutively expressed by a subset of conventional dendritic cells and is strongly induced by retinoic acid. Mucosal Immunol, 2014; 7: 101–13. [DOI] [PMC free article] [PubMed] [Google Scholar]