

Abstract
The tumor suppressor p53 is often inactivated via its interaction with endogenous inhibitors mouse double minute 4 homolog (MDM4 or MDMX) or mouse double minute 2 homolog (MDM2), which are frequently overexpressed in patients with acute myeloid leukemia (AML) and other cancers. Pharmacological disruption of both of these inter-actions has long been sought after as an attractive strategy to fully restore p53-dependent tumor suppressor activity in cancers with wild-type p53. Selective targeting of this pathway has thus far been limited to MDM2-only small-molecule inhibitors, which lack affinity for MDMX. We demonstrate that dual MDMX/MDM2 inhibition with a stapled a-helical peptide (ALRN-6924), which has recently entered phase I clinical testing, produces marked antileukemic effects. ALRN-6924 robustly activates p53-dependent transcription at the single-cell and single-molecule levels and exhibits biochemical and molecular biological on-target activity in leukemia cells in vitro and in vivo. Dual MDMX/MDM2 inhibition by ALRN-6924 inhibits cellular proliferation by inducing cell cycle arrest and apoptosis in cell lines and primary AML patient cells, including leukemic stem cell-enriched populations, and disrupts functional clonogenic and serial replating capacity. Furthermore, ALRN-6924 markedly improves survival in AML xenograft models. Our study provides mechanistic insight to support further testing of ALRN-6924 as a therapeutic approach in AML and other cancers with wild-type p53.
INTRODUCTION
The p53 protein is the most frequently inactivated tumor suppressor in human cancers (1–4). Its numerous functions include the protection of cells from genomic instability and prevention of progression and dissemination of aberrant cells by responding to cellular stress signals, resulting in a transcriptional response that triggers cell cycle arrest, DNA repair, senescence, and/or apoptosis pathways (5, 6). In the hematopoietic system, p53 protects hematopoietic stem cells (HSCs) against DNA damage by promoting quiescence and inhibiting self-renewal (7–12). Loss of p53 function in stem and myeloid progenitor cells promotes leukemia initiation by enabling aberrant self-renewal (13). In de novo acute myeloid leukemia (AML), TP53 mutations are rare, occurring in less than 10% of patients (14–18). Although in some specific subsets of AML TP53 mutations are more frequent [for example, in up to 80% in patients with complex cytogenetics (19)], p53 inactivation more often results from the overexpression of its endogenous inhibitors MDMX or MDM2, which frequently occurs in p53 wild-type (WT) AML (20–26). It was recently reported that MDMX protein and mRNA are overexpressed in up to 92% of AML cases (27). MDM2 is an E3 ubiquitin ligase that inhibits p53 by targeting it for degradation, whereas MDMX inhibits its transactivation activity and promotes MDM2 activity via direct protein-protein inter-actions (3, 28–35). Seminal work by several groups demonstrated that MDMX and MDM2 play indispensable and nonoverlapping roles in suppressing the normal function of p53 and that dual inhibition of MDMX and MDM2 is essential to fully unleash dormant p53 (36–42).
Given their critical role as negative regulators of p53 tumor suppressor functions, pharmacological disruption of MDMX/MDM2/p53 interactions offers a means to restore p53 activity in p53 WT cancers that might be MDMX-reliant, such as AML. However, efforts to develop small-molecule dual MDMX/MDM2 inhibitors have been largely unsuccessful. The cis-imidazoline compound Nutlin-3 was one of the first antagonists of MDM2-p53 binding shown to restore p53 tumor suppressor function in p53 WT preclinical cancer models; however, due to poor bioavailability, it never advanced to the clinical trial phase (43). Optimized small-molecule inhibitors of MDM2 are currently undergoing clinical testing, with the cis-imidazoline derivatives RG7112 and RG7388 (idasanutlin) being the most advanced, and have generated partially encouraging results (44). However, limitations of these compounds include their lack of affinity for MDMX, which has been reported to antagonize the effects of MDM2 inhibition in tumor cells, as well as dose-limiting toxicities in patients (25, 45–50). More recently, α-helical p53-stapled peptide prototypes have shown promise, demonstrating the feasibility of targeting both MDMX and MDM2 (49,51–54). ALRN-6924 is an optimized α-helical p53-stapled peptide and a dual MDMX/MDM2 inhibitor currently in phase I clinical testing in solid tumors and lymphomas, with thus far excellent tolerability and some promising signs of antitumor activity (55). However, its molecular, cellular, and biochemical mechanisms of action, as well as its therapeutic potential in AML, have yet to be determined.
Here, we demonstrate that MDMX is considerably overexpressed in AML, including in leukemia stem cell-enriched populations, in comparison to age-matched healthy controls. Thus, targeting MDMX with a dual MDMX/MDM2 inhibitor is an attractive therapeutic strategy in AML. We found that ALRN-6924 activates p53-dependent transcription at the single-cell and single-molecule levels and displays robust biochemical and molecular biological on-target activity in leukemia cells. ALRN-6924- mediated dual MDMX/MDM2 inhibition results in genome-wide activation of p53- dependent transcriptional networks. It induces cell cycle arrest and apoptosis pathways in cell lines and primary AML patient cells, including in leukemic stem cell (LSC)-enriched populations. It also disrupts key functional properties, including clonogenic and serial replating capacity of leukemia-initiating cells, with remarkably better activity than RG7388 (idasanutlin), an MDM2-only small-molecule inhibitor, which is currently in phase 3 trials in myelodysplastic syndrome (MDS) and AML. Furthermore, ALRN-6924 inhibits leukemia pathogenesis and increases survival in AML xenograft models in vivo. Our study offers insight into the effects of dual MDMX/MDM2 inhibition using a stapled peptide therapeutic in leukemia and provides a rationale for further development and clinical testing of ALRN- 6924 as a therapeutic approach in cancers with WT p53.
RESULTS
MDMX is highly expressed in AML
MDMX overexpression has been reported in a number of human cancers, including glioma, soft tissue sarcoma, melanoma, retinoblastoma, breast cancers, and hematological malignancies (24,27, 56–61). To compare MDMX expression in patients with different cancer types, we evaluated published gene expression data sets available in The Cancer Genome Atlas (TCGA). Notably, primary cells from patients with AML have very high expression of MDMX compared to most other tumor types (Fig. 1A). Tumor cell lines derived from hematological malignancies also have high expression of MDMX compared to cell lines of other tumor types (fig. S1A). Alternative splicing of MDMX results in the expression of a shorter non-functional isoform, MDMX-S, lacking exon 6, previously reported to attenuate full-length (FL) MDMX protein expression (62). To more carefully examine which isoform of MDMX is most frequently expressed in AML, we calculated the Percent Spliced In (PSI) index, defined as the percentage of FL MDMX mRNA (MDMX-FL) relative to the total of all isoforms, as previously described (61). We found a PSI of 0.4 or higher in 94% of the Acute Myeloid Leukemia data set (LAML) of TCGA patient cohort, indicative of predominant FL MDMX expression (relative to the shorter isoform, MDMX-S) (61) in the vast majority of patients (fig. S1B). Furthermore, we assessed the relative expression of MDMX and MDM2 in highly purified disease-driving stem and progenitor cell compartments in AML. We analyzed gene expression in rigorously defined fluorescence-activated cell sorter (FACS)-fractionated stem (Lin-CD34+ CD38-CD90-) as well as progenitor (Lin-CD34+CD38+CD123+CD45+) cells from patients with AML in comparison to age-matched healthy controls (63). Total MDMX expression was significantly higher in leukemic stem (P < 0.0001) and progenitor cells (P = 0.0013) compared to healthy controls; however, this was not the case for MDM2 (Fig. 1B). These results demonstrate that FL MDMX is overexpressed in AML, including in leukemia stem cells, compared to MDM2, and thus provide a strong rationale for the evaluation of MDMX-targeting compounds in this devastating disease.
Fig. 1. MDMX is targetable in acute myeloid leukemia using an a-helical stapled peptide (ALRN-6924).
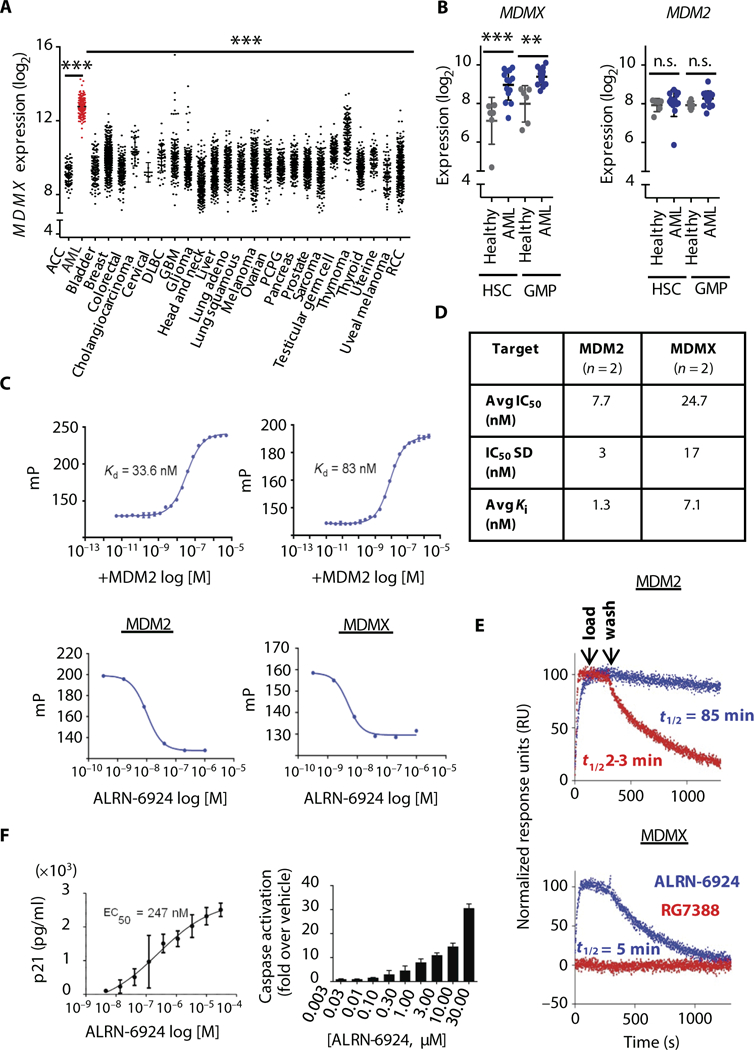
(A) mRNA expression analysis of MDMX in human cancers using The Cancer Genome Atlas (TCGA) data sets and (B) MDMX and MDM2 mRNA expression in hematopoietic stem (HSC) and granulocyte-monocytic progenitor-enriched (GMP) cells from acute myeloid leukemia (AML) patients (n = 14) and healthy controls (n = 6) (data shown as mean ± SD, where **P < 0.01, ***P < 0.001; n.s., not significant). (C) Fluorescence polarization (mP) of a fixed concentration of ALRN-3618 was measured in the presence of varying concentrations (log M) of mouse double minute 2 homolog (MDM2) and MDMX (top panels) (n = 2). Graphs display varying concentrations of ALRN-6924 used to displace ALRN-3618 at its EC80 from each target protein (bottom panels). (D) IC50 and Kl values (n = 2) for each target protein, as determined from a nonlinear four-parameter curve obtained from plotting fluorescence polarization versus drug concentration in (C) (bottom). (E) Sensorgrams for the binding of ALRN-6924 (blue) and RG7388 (red) to surfaces with immobilized MDM2 (top) and MDMX (bottom) (n = 2). (F) Enzyme-linked immunosorbent assay demonstrating the expression of human p21 protein (left) (21 hours after treatment with ALRN-6924) and cellular caspase activity (right) (48 hours after treatment with ALRN-6924) determined using Caspase-Glo 3/7 assay reagent in SJASA-1 cells (n = 2; data shown as mean ± SD). ACC, adrenocortical carcinoma; DLBC, diffuse large B cell lymphoma; PCPG, pheochromocytoma and paraganglioma; RCC, renal cell carcinoma.
ALRN-6924 binds with high affinity to both human MDMX and MDM2
pDI is a prototype linear α-helical peptide identified by phage display that shows nanomolar binding affinity to MDMX and MDM2 (64, 65). Unlike ALRN-6924, pDI lacks a chemical linker (“staple”), and it is not cell-permeable. The binding kinetics of ALRN-6924 were measured in a competitive binding assay using a modified pDI peptide (ALRN-3618) synthesized with a fluorescent 5-FAM moiety (5-carboxyfluorescein) appended via a β-alanine spacer to its N terminus (5-FAM-β-alanine-pDI) as a binding probe. ALRN-3618 bound with high affinity to purified MDM2 [dissociation constant (Kd) = 33.6 nM) and MDMX (Kd = 83 nM), demonstrating dual target binding for this linear peptide (Fig. 1C, upper panels). Varying concentrations of ALRN- 6924 were used to displace ALRN-3618 at its EC80 from each target protein. ALRN-6924 displaced ALRN-3618 at its EC80 for the p53-binding site on MDM2, suggesting that they bind to the same site on MDM2 (Fig. 1C, left lower panel). ALRN-6924 displaced ALRN-3618 at its EC80 for the p53-binding site on MDM2 and MDMX, suggesting that they bind to the same site on MDM2/X (Fig. 1C, left and right lower panels, respectively). The results indicate that ALRN-6924 binds with high affinity to both MDMX and MDM2 and are summarized in Fig. 1D. Notably, ALRN-6924 demonstrated superior binding kinetics versus the small- molecule Nutlin derivative RG7388 (Fig. 1E). In binding assays using immobilized MDMX- and MDM2-coated surfaces, ALRN-6924 bind0ing to MDM2 was sustained during wash-off with a t1/2 = 85 min (Fig. 1E, upper panel). In contrast, RG7388 washed off from MDM2- coated surfaces with a shorter half-life (i1/2 = 2 to 3 min). ALRN- 6924 demonstrated specific binding to MDMX with a wash-off i1/2 = 5 min, whereas RG7388 did not bind to MDMX at all (Fig. 1E, lower panel). ALRN-6924 bound to MDMX and MDM2 (Kd = 57 and 10.9 nM, respectively) (table S1) with binding kinetics superior to those of endogenous p53 (Kd = 480 and 770 nM, respectively) (66), an important characteristic of ALRN-6924 that facilitates the disruption of native p53-MDMX/MDM2 complexes.
ALRN-6924 treatment of SJSA-1 cells, which have an intact p53 signaling pathway and were previously reported to be sensitive to dual MDMX/MDM2 inhibition (51), resulted in a concentration- dependent increase in p21 protein and in caspase-3/7 activity indicating that the p53 pathway had been activated (Fig. 1F). To test its efficacy in hematological malignancies, we treated a variety of leukemia cell lines with ALRN-6924 and found that it exhibited potent activity against all five cell lines with WT p53 in the cytotoxicity assay (table S2). Moreover, ALRN-6924 also showed increased cytotoxic activity against one cell line (MOLT16) harboring a heterozygous TP53 mutation (R158H), which has been described as a loss-of-function mutation (http://p53.iarc.fr), but still carries one remaining TP53 WT allele. Thus, ALRN-6924 activated p53-mediated pathways of cell cycle arrest (via p21 up-regulation) and/or apoptosis (via cellular caspase activation) in all tested WT p53 tumor cell lines of hematological origin.
ALRN-6924 rapidly increases transcriptional bursting dynamics from the CDKN1A (p21) gene locus
The majority of mammalian genes are transcribed dynamically over short periods of time, defined as a series of transcriptional bursts (67, 68). Transcription dynamics can be regulated by changes in burst frequency, duration, and intensity. To better understand how ALRN- 6924 affects transcriptional dynamics of p53 target genes, we quantfied the expression of the CDKN1A gene using the MS2 system in live cells (69). Briefly, we used the clustered regularly interspaced short palindromic repeats-CRISPR associated protein 9 (CRISPR-Cas9) system (70) to knock-in 24 MS2 stem-loop (MS2-SL) repeats into the 3’ untranslated region (3’UTR) of the endogenous CDKN1A gene locus in human osteosarcoma (U2OS) cells, which we found to be amenable to CRISPR-Cas9 manipulation, RNA-fluorescence in situ hybridization (RNA-FISH), and live cell imaging (Fig. 2A). This genetically engineered cell line model allowed us to directly measure actual transcriptional bursting dynamics from a canonical p53 target gene locus in response to p53 activation (as opposed to measurements of RNA levels, which are influenced by many factors besides transcription) at the single-molecule (nascent RNAs synthesized at the CDKN1A locus) and single-cell levels. Single-molecule RNA-FISH confirmed that the cell line contained an insertion into a single allele of the p21 gene (fig. S2). This modified cell line also stably expressed a green fluorescent protein (GFP)-tagged MS2 capsid protein (MCP-GFP), which binds RNA harboring the MS2-SL (Fig. 2A) (69). Live cell fluorescence microscopy was used to visualize nascent transcription sites (TSs) containing a single bright MCP-GFP spot bound to MS2-SL-containing RNA on the p21 gene locus (Fig. 2B, top panel). The fluorescence intensity of the MCP-GFP spot is related to the amount of nascent transcripts originating from the p21 gene.
Fig. 2. ALRN-6924 rapidly increases transcription at the p21 locus and affects its bursting dynamics.
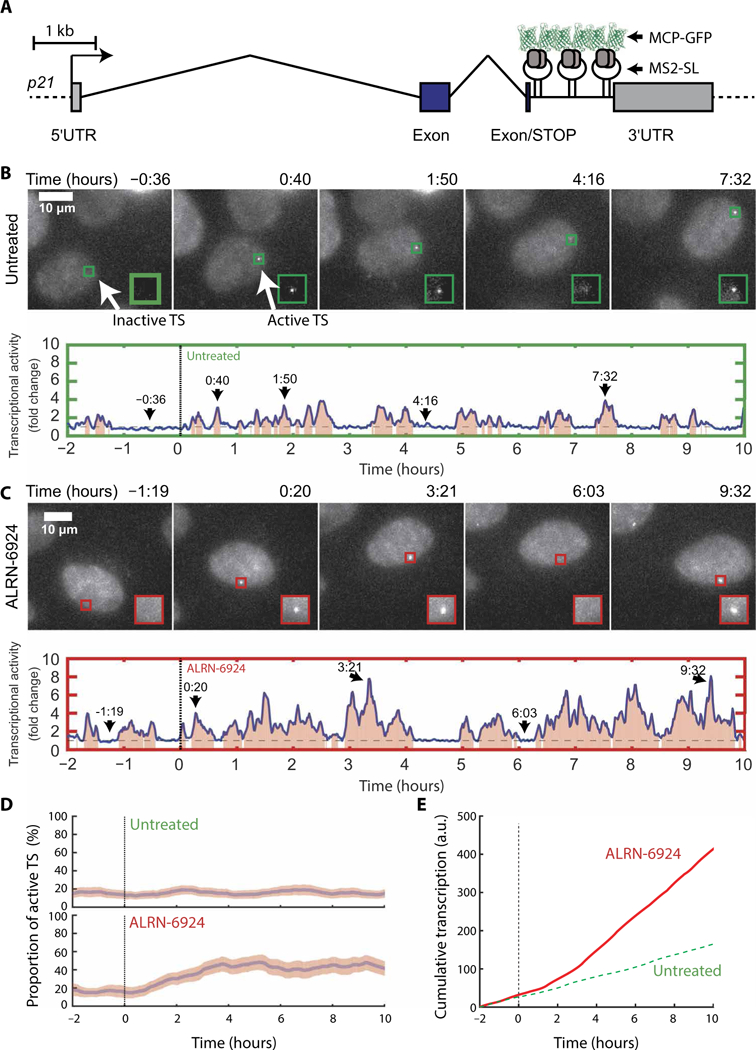
(A) Schematic representation of the engineered p21 gene locus containing MS2 stem-loops (p21-MS2). The transcription start site is shown as an arrow, exons are shown as blue rectangles, introns are shown as angled lines, and 5’ and 3’ untranslated regions (UTRs) are in gray. Scale bar, 1 kb. (B) Live-cell imaging of U2OS p21-MS2 cells in untreated medium. Top panel: Images of cells containing p21-MS2 (maximum intensity projections of z-dimension stacks). Scale bar, 10 μm; the green box outlines the transcription site (TS). Bottom panel: Quantification of the fluorescence intensity at the TS marked in the top panel. (C) Live-cell imaging of U2OS p21-MS2 cells in ALRN-6924-treated medium as described in (B). (D) Proportion of cells containing an active TS (>1.5-fold the background). For each time point and condition, the proportion of active TS in a cell population was calculated using a rolling average of 30 min before and after each time point [85 untreated cells (top) and 54 cells treated with 5 μM ALRN-6924 (bottom)]. The pink area represents the 95% confidence interval. (E) Average cumulative transcription plot of p21-MS2 gene in untreated (green broken lines) versus ALRN-6924-treated cells (red solid lines). MCP-GFP, green fluorescent protein-tagged MS2 capsid protein; a.u., arbitrary units.
At the single-cell level in untreated conditions, transcription of the p21 gene infrequently switches between ON and OFF states over time (Fig. 2B). Quantification showed multiple bursts of transcription during the 10-hour imaging window (Fig. 2B and movie S1), with no major changes before and after the medium replacement. Next, we examined the effect of ALRN-6924 on p21 transcriptional dynamics. Addition of ALRN-6924 resulted in longer and more intense transcriptional bursts compared to untreated cells (Fig. 2, C versus B, and movie S2). To rule out potential effects related to single-cell heterogeneity, we calculated the proportion of cells in a population containing an active TS over time. In an untreated population, 16% of the cells contained an actively transcribing p21 gene at any given time (Fig. 2D, top panel). The proportion of cells harboring an active TS steadily increased to ~44% at 4 hours after the addition of ALRN-6924 (Fig. 2D, top versus bottom panel). Therefore, ALRN-6924 increased the duration and/or frequency of transcriptional bursting at the p21 gene. To further quantitate the effects of ALRN- 6924 on rates of p21 transcription, we plotted the cumulative MS2 signal over time (Fig. 2E). Notably, we observed a considerable difference in the rate of p21 transcription between treated and untreated cells within 2 hours after the addition of ALRN-6924 (compare slopes of ALRN-6924 versus untreated; Fig. 2E). Furthermore, an increased rate of transcription was maintained throughout the 10-hour treatment window, indicating that ALRN-6924 triggered a durable transcriptional response. Collectively, these data suggest that ALRN-6924 rapidly penetrates cellular membranes and triggers on-target downstream p53-dependent transcriptional programs.
Dual inhibition of MDMX and MDM2 with ALRN-6924 activates the p53 pathway in AML
To directly measure ALRN-6924’s cell permeability, we treated AML cells with increasing concentrations of FAM-tagged ALRN-6924 and then analyzed them by flow cytometry and high-resolution fluorescence microscopy. Intracellular ALRN- 6924, which was detectable both in the cytoplasm and nucleus, as well as an increase in p53 protein, was observed with in 3 hours of treatment (Fig. 3, A and B). We then analyzed its ability to disrupt preformed p53-MDM2 and p53-MDMX- FL complexes [using an MDMX-FL- specific antibody (Millipore 8C6)] by coimmunoprecipitation and immuno- blotting in AML cells treated with ALRN- 6924 or vehicle. Consistent with in vitro biochemical assays, we found that ALRN- 6924 disrupted endogenous p53-MDM2 and p53-MDMX-FL complexes in AML cells (Fig. 3C, left and right panels, respectively, and fig. S3A). Next, we treated patient-derived primary AML cells and AML cell lines expressing WT p53 with increasing concentrations of ALRN-6924 and analyzed cellular lysates for evidence of p53 pathway activation. ALRN-6924 stabilized p53 and up-regulated its downstream targets p21 and MDM2 in p53 WT cells in a dose-dependent manner, but not in p53 null AML cells (Fig. 3D and fig. S3, B and C). MDMX-FL was slightly down-regulated in response to ALRN-6924 (fig. S3D). Furthermore, analysis by RNA sequencing showed that ALRN-6924 triggered differential changes in gene expression compared to vehicle-treated MOLM13 cells within 6 hours after treatment (Fig. 3E). We found 954 differentially expressed genes in response to ALRN- 6924, and gene set enrichment analysis (GSEA) showed a consistent and reproducible enrichment of p53-regulated gene expression signatures (Fig. 3F). Notably, the top up- regulated pathways in ALRN-6924-treated cells included genes involved in p53 signaling, cell cycle arrest, and apoptosis induction (Fig. 3F and fig. S3E). To validate these results, we performed targeted reverse transcription polymerase chain reaction (RT-PCR) of p53 target genes [CDKN1A (p21), MDM2, BAX, PUMA, and GADD45a] previously implicated in cell cycle arrest, apoptosis, and DNA repair. ALRN-6924 up-regulated CDKN1A, MDM2, BAX, PUMA, and GADD45a in a dose-dependent manner after 24 hours of treatment with ALRN-6924 (Fig. 3G and fig. S3, F and G). Together, these results demonstrate that ALRN-6924 is cell-permeable, disrupts both p53-MDMX and p53-MDM2 interactions, and displays on-target p53 pathway activity in AML cells.
Fig. 3. ALRN-6924 blocks MDM2- and MDMX-p53 protein-protein interactions and activates the p53 pathway in AML.
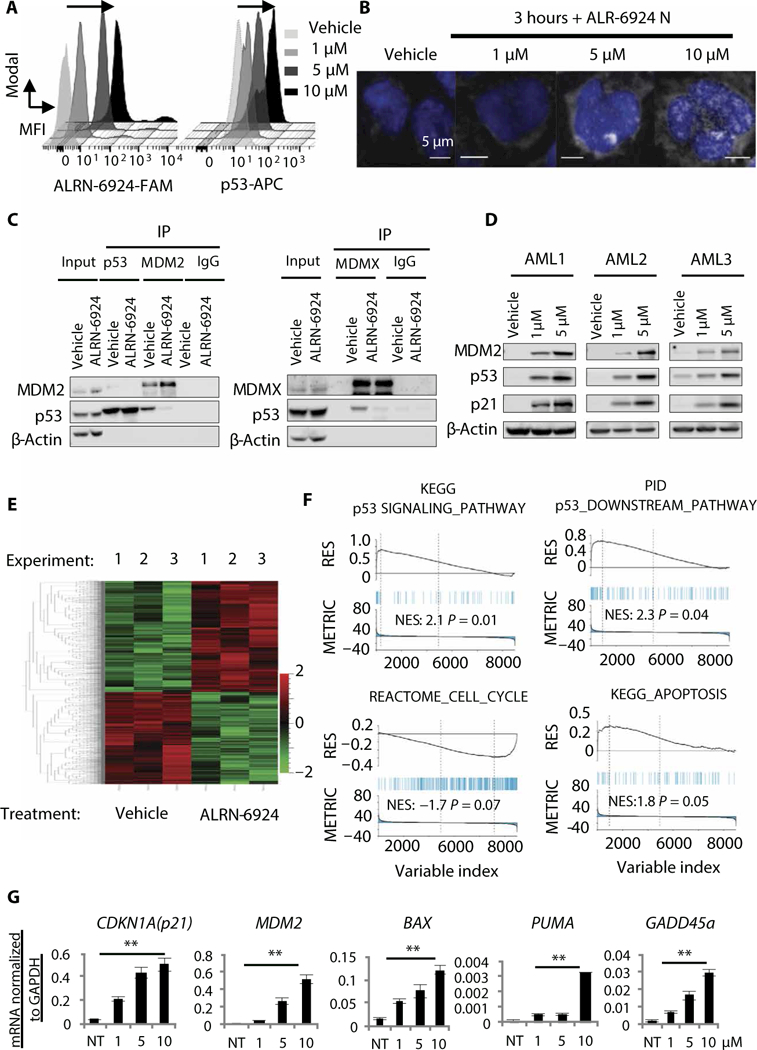
(A) Intracellular staining of FAM-labeled ALRN-6924 and p53-APC 3 hours after treatment with vehicle or ALRN- 6924 at the indicated doses [a representative histogram is shown, black arrow denotes rightward shift (increase) in mean fluorescence intensity (MFI)]. (B) High-resolution fluorescence microscopy of MOLM13 cells treated with FAM- labeled ALRN-6924 3 hours after treatment with the indicated doses (shown is a representative image). (C) Coimmuno- precipitation with anti-MDM2 (left), anti-p53 (left) and anti-MDMX (right) antibodies or immunoglobulin G (IgG) isotype control using MOLM13 cellular extracts followed by Western blot analysis with anti-p53, anti-actin, anti-MDM2, or anti- MDMX antibodies. IP, immunoprecipitation. (D) Western blot analysis showing expression of p53 and p53-target proteins (p21 and MDM2) in primary patient AML cells (representative blots are shown). (E) Hierarchical clustering of genes differentially expressed in MOLM13 cells after exposure to 1 μM ALRN-6924 for 6 hours compared to vehicle-treated cells (n = 3). (F) Gene set enrichment analysis (GSEA) showing the top gene signatures differentially expressed in response to ALRN-6924. (G) Targeted gene expression analysis by quantitative reverse transcription polymerase chain reaction (qRT-PCR) of p53 target genes [CDKN1A (p21), MDM2, BAX, PUMA, and GADD45a] in response to increasing concentrations of ALRN-6924 in MOLM13 cells (n = 3, shown as the mean ± SD; **P < 0.01). KEGG, Kyoto Encyclopedia of Genes and Genomes; PID, Pathway Interaction Database; NES, normalized enrichement score; NT, no treatment.
ALRN-6924 inhibits AML cell growth and clonogenic capacity via induction of cell cycle arrest and apoptosis
To evaluate the relative expression of MDMX-FL, MDMX-S, and MDM2 mRNA in a panel of five p53 WT AML cell lines (MOLM13, MOLM14, ML2, OCI/AML3, and OCI/AML5), we performed RT-PCR analysis. All five AML cell lines expressed higher MDMX-FL compared to MDMX-S, and four of the five AML cell lines had higher MDMX-FL than MDM2 expression (fig. S4, A and B). Moreover, at the protein level, all five cell lines expressed more MDMX than MDM2 (fig. S4C). To investigate the effects of ALRN-6924 on AML cellular proliferation, we treated our panel of p53 WT and two p53-mutant AML cell lines in liquid culture with increasing concentrations of ALRN-6924 (Fig. 4A and fig. S4D). ALRN-6924 inhibited cellular proliferation measured by trypan blue exclusion in a dose- and time-dependent manner (Fig. 4, A and B). Only one p53 WT cell line, OCI/AML3, was less sensitive to treatment with ALRN- 6924 in suspension culture; notably, this cell line had lower MDMX-FL/MDMX-S and MDMX-FL/MDM2 ratios than the other cell lines tested (Fig. 4, A and B, and fig. S4, A and B). Notably, ALRN-6924 had no effect on cell proliferation in HL60 (p53 null) or Kasumi-1 (p53 R248Q mutant) AML cell lines (fig. S4, D and E).
Fig. 4. ALRN-6924 inhibits cellular proliferation and clonogenic capacity, and induces cell cycle arrest and apoptosis in AML cell lines.
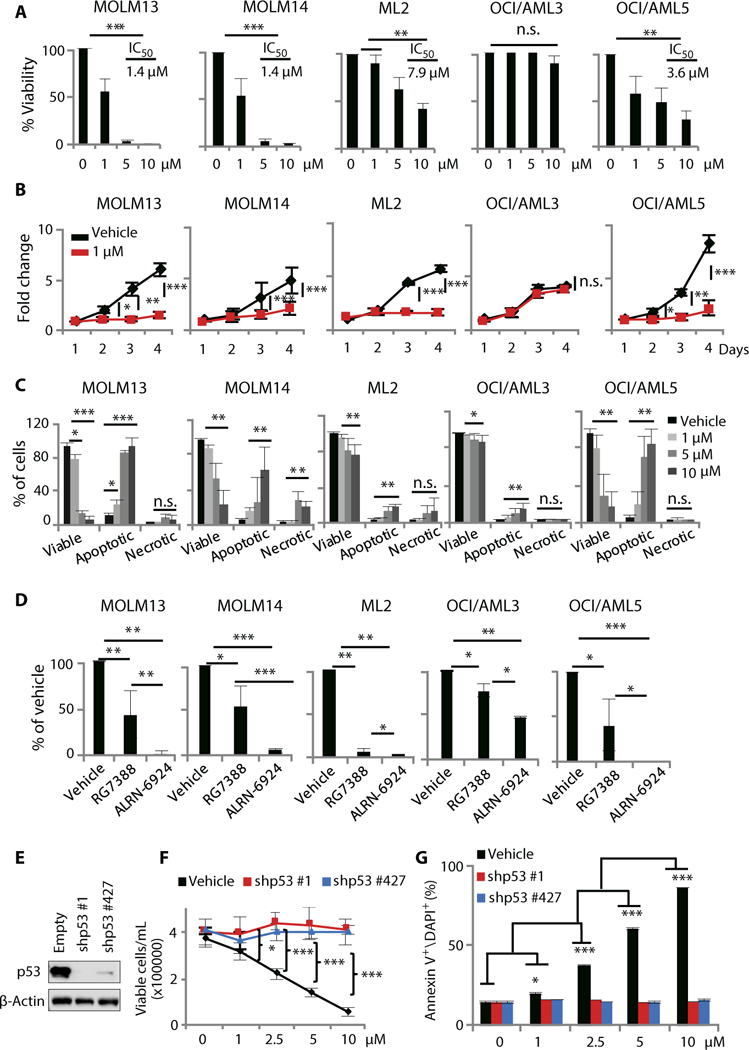
(A) Cellular proliferation measured by trypan blue exclusion in AML cell lines 24 hours after treatment with increasing concentrations of ALRN-6924 (n = 3). (B) Time-dependent changes in proliferation in AML cell lines treated with 1 μM ALRN-6924 (n = 3). (C) Fluorescence-activated cell sorting (FACS) analysis of cellular viability, apoptosis, and necrosis measured by annexin V and 4’,6-diamidino-2-phenylindole (DAPI) staining in AML cell lines 24 hours after treatment with increasing concentration of ALRN-6924 (n = 3). (D) Colony-forming capacity (CFC) assay of AML cell lines that were treated with vehicle, 0.5 pM RG7388, or 0.5 pM ALRN-6924 and grown in cytokine-free methylcellulose semisolid medium for 10 days (n = 3). (E) Western blot analysis of p53 in cell lines expressing an empty vector or one of two different p53 short hairpin RNAs (shRNAs) (a representative blot is shown). (F) Cellular proliferation measured by trypan blue exclusion in isogeneic p53 shRNA AML cell lines treated with ALRN-6924 (n = 3). (G) Apoptosis analysis by annexin V and DAPI staining in isogeneic p53 shRNA AML cell lines treated with ALRN-6924 (n = 3). Data are shown as the mean ± SD, and *P < 0.05, **P < 0.01, ***P < 0.001.
To more carefully dissect the mode of ALRN-6924-dependent growth inhibition, we analyzed its effects on cell cycle and apoptosis by propidium iodide and annexin V staining, respectively. ALRN- 6924 induced cell cycle arrest in all p53 WT AML cell lines (fig. S4F). Moreover, ALRN-6924 triggered a dose-dependent increase in apoptosis in all five AML cell lines tested; however, the two cell lines with lower expression of both MDMX and MDM2 (OCI-AML3, ML2) were consistently less responsive (Fig. 4C). We did not observe a significant increase in apoptosis in p53 null (HL60) cells even at the range of doses that were cytotoxic in p53 WT cells, between 5 and 10 μM (fig. S4E). Because MDMX was found to be overexpressed in AML stem and progenitor cells, but not MDM2, we examined the effects of ALRN-6924 or the MDM2-selective inhibitor RG7388 [currently under study in myeloproliferative neoplasms (MPNs) and showing promising results (71)] in long-term clonogenic assays in semisolid medium. P53 reactivation with ALRN- 6924 abrogated the colony-forming capacity (CFC) of AML cells, suggesting that dual MDMX and MDM2 inhibition targets clonogenic potential of immature, leukemia-driving cells (Fig. 4D). Notably, ALRN-6924 exhibited greater inhibition of clonogenicity compared to RG7388 in all five AML cell lines.
To directly test the p53 dependency of ALRN-6924’s effects, we generated two isogenic p53 short hairpin RNA (shRNA) knockdown cell lines derived from MOLM13 cells and assessed the effects of ALRN-6924 treatment on cell proliferation and apoptosis (Fig. 4E). We found that ALRN-6924 inhibited cell growth and induced apoptosis in a completely p53-dependent manner (Fig. 4, F and G).
ALRN-6924 displays robust antileukemic effects in primary patient-derived AML cells and AML xenograft models
To study the antileukemic effects of ALRN- 6924 in p53 WT primary AML cells, patient-derived peripheral blood mononuclear cells (PBMCs) were treated in liquid culture with increasing concentrations of ALRN-6924. Cell viability was inhibited (compared to vehicle-treated cells) in a dose- and time-dependent manner in all samples tested (Fig. 5A). ALRN- 6924 inhibited CFC of primary AML PBMCs (n = 4) compared to healthy controls (n = 3), demonstrating a therapeutic window (Fig. 5B). Next, we treated primary AML patients’ bone marrow (BM) cells (n = 3) and BM cells from healthy donors (n = 3) in suspension culture with a cytotoxic dose of ALRN-6924 and analyzed its effects on cell viability and apoptosis in LSC-enriched compartments (CD34+ CD38-). We observed a significant (P = 9.38 × 10−6) decrease in cell viability accompanied by increased apoptosis specifically in CD34+CD38- LSCs, but not in healthy donor CD34+CD38- cells (Fig. 5C).
Fig. 5. ALRN-6924 shows robust antileukemic activity in primary AML cells and in vivo.
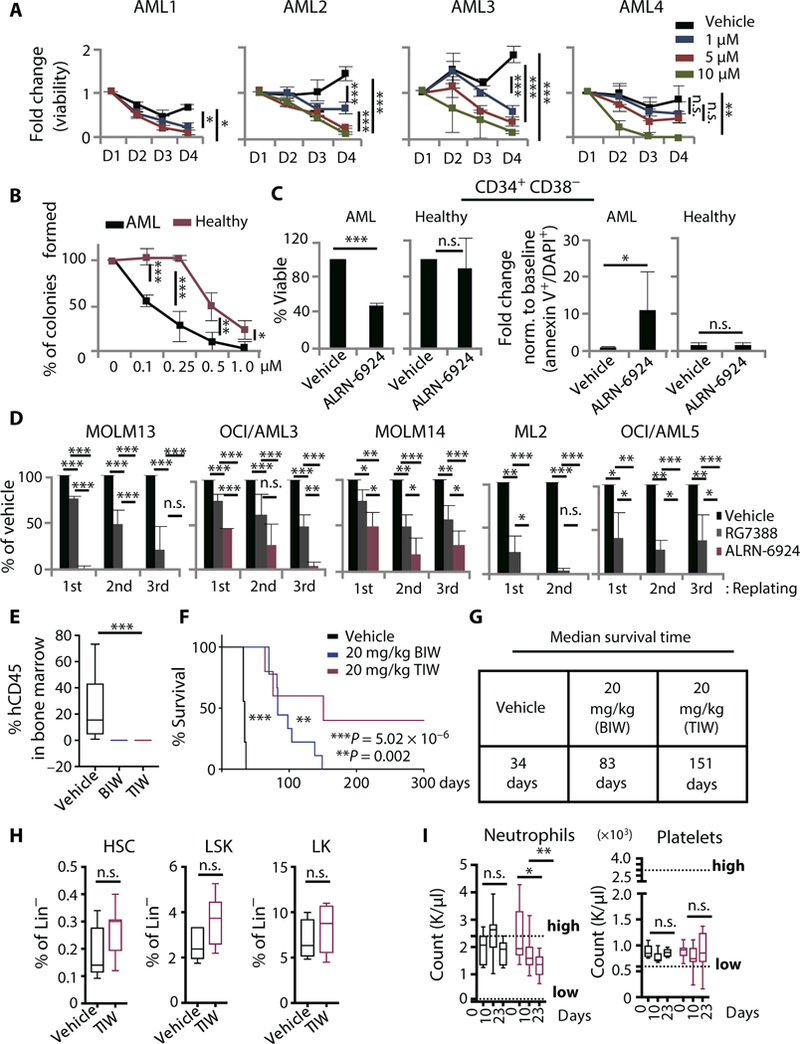
(A) Dose- and time-dependent effects on cellular proliferation measured by trypan blue exclusion in primary AML cells treated with increasing concentrations of ALRN-6924 (data shown as mean ± SD of technical triplicates). (B) Dose-dependent effects of ALRN- 6924 on clonogenic capacity of primary AML cells (n = 4) compared to healthy peripheral blood mononuclear cells (PBMNCs) isolated from cord blood (n = 3) and bone marrow mononuclear cells (BMMNCs) isolated from one AML patient in clinical remission (data shown as mean ± SD; P statistics determined using unpaired two-tailed Student’s t test). (C) FACS analysis of cellular viability (left panel) and apoptosis (right panel, plotted as fold change relative to baseline) measured by annexin V and DAPI staining in leukemia stem cell-enriched (CD34+CD38-) primary bone marrow cells from AML patients (n = 3) and healthy donors (n = 3) 48 hours after treatment with 5 μM ALRN-6924 (data shown as mean ± SD, unpaired one-tailed Student’s t test). (D) Serial replating capacity of AML cell lines treated with either 0.5 μM ALRN-6924 or 0.5 μM RG7388, measured by CFC assay. (E) Human AML cellular engraftment analysis by FACS of mouse bone marrow (BM) from experimental animals 5 weeks after the start of treatment. Human AML cells were identified by costaining mouse BM cells with human CD45 and mouse CD45.1 antibodies [BIW, two times per week (n = 10); TIW, three times per week (n = 5)]. (F) Kaplan-Meier curve showing overall survival of mice xenotransplanted with MOLM13 AML cells and treated with ALRN-6924 or vehicle. (G) Median survival times for the curves shown in (F). (H) FACS analysis of mouse hematopoietic stem and progenitor frequencies after treatment with ALRN-6924 (20 mg/kg) TIW for 4 weeks (HSC = LSK CD150+CD48-, LSK = Lin-Sca+Kit+, and LK = Lin-Kit+ progenitors). (I) Neutrophil and platelet counts assessed by Hemavet analysis of peripheral blood from mice (n = 10) on treatment with ALRN-6924 (20 mg/kg) TIW for 4 weeks. Data shown as the mean ± SD; *P < 0.05, **P < 0.01, ***P < 0.001
Serial replating of CFCs is an important characteristic of the self-renewal properties inherent in LSCs in vitro. There fore, we examined the ability of ALRN- 6924 to inhibit the serial replating capacity of AML cells in comparison to RG7388, several previously characterized p53- stapled peptide prototypes, ATSP-7041 (the previously reported nonclinical MDMX/MDM2 stapled peptide), SAH-p53 (first p53-stapled peptide prototype), a selective stapled peptide targeting MDM2, and a selective nonclinical small-molecule MDMX inhibitor (54, 72). The MDMX inhibitor NSC-207895 downregulated expression of MDMX in AML cells at the mRNA and protein levels (fig. S4, H and I). ALRN-6924 displayed increased potency over RG7388 and all other p53- stapled peptide prototypes tested in CFC and serial replating assays (Fig. 5D and fig. S4G), and in three of the five AML cell lines tested, ALRN-6924 completely abrogated serial replating capacity (Fig. 5D). MDMX inhibition with NSC-207895 demonstrated similar potency to ALRN-6924 in serial replating assays, suggesting that MDMX plays an important functional role for AML cell serial replating capacity.
To test the efficacy of ALRN-6924 to inhibit leukemia initiation and disease progression in vivo, we used xenotransplantation models of AML. First, we transplanted NOD. Cg-PrkdcscidIl2rγtm1Wjl/SzJ NOD-scid IL2rγnuii (NSG) mice with MOLM13 cells and randomly divided them into three groups treated intravenously (IV) with vehicle (group 1), ALRN- 6924 (8 mg/kg, IV) twice per week (BIW) (group 2), or ALRN-6924 (12 mg/kg, IV) BIW (group 3) (fig. S5, A and B). At day 7 after the start of treatment, we observed a dose-dependent decrease in human CD45 leukemic cells in the BM of ALRN- 6924-treated animals (fig. S5, A and B). At this dosing regimen, the median survival was increased by 5.5 days compared to the control (fig. S5C). In the next set of experiments, MOLM13-transplanted animals were randomly divided into three additional groups treated with vehicle (group 1), ALRN-6924 (20 mg/kg, IV) BIW (group 2), or ALRN-6924 (20 mg/kg, intravenously) three times per week (TIW) (group 3) (fig. S5D). The average engraft- ment of human CD45 leukemic cells after 5 weeks of treatment, with 20 mg/kg, was 23.3% in vehicle and 0.00557% in ALRN-6924-treated animals (Fig. 5E). Mice treated with ALRN-6924 showed a remarkable increase in overall survival compared to the vehicle-treated counterparts (Fig. 5F). The median survival for groups 1, 2, and 3 was 34, 83, and 151 days, respectively (Fig. 5G). Notably, 40% of mice in group 3 remained alive 300 days after treatment. In a second xenograft model, with ML2 cells, average median survival for vehicle-treated animals was 28.5 days. In contrast, animals treated with ALRN-6924 (20 mg/kg, IV) BIW and TIW had an average median survival of 38 and 72 days, respectively (fig. S5, E and F).
To assess the effects of ALRN-6924 on healthy hematopoietic cells, we treated non-tumor-bearing C57/BL6 mice with either vehicle or ALRN-6924 (20 mg/kg) TIW for 4 weeks. At the end of treatment, we analyzed the frequency of HSCs and progenitors and found no significant differences between control and ALRN-6924-treated animals (Fig. 5H). ALRN-6924-treated animals appeared healthy throughout the treatment and did not show signs of thrombocytopenia and only a modest reduction in neutrophils [which remained within the normal range for this mouse strain (73)] (Fig. 5I). Together, these data show that ALRN-6924 is a potent and selective inhibitor of primary human AML cells, including immature leukemia stem/progenitor cells, displays strong leukemia-inhibitory activity in vivo, and is well tolerated in mice.
ALRN-6924 selectively activates p53 in leukemia cells of an MDS/AML patient in vivo
We present a case of a 20-year-old female patient with germ-line deletion of one TP53 allele, who was suffering from metastatic breast cancer and developed high-risk MDS with excess of leukemic blasts [refractory anemia with excess blasts (RAEB), 15% blasts, with complex karyotype]. The patient was refractory to conventional chemotherapy; however, cytogenetic analysis revealed that the leukemic blasts had retained a WT copy of TP53. Relative TP53 mRNA expression was fourfold lower in the patient’s MDS/AML cells compared to healthy donor CD34+ cells, and MDMX-FL was the predominant isoform expressed (Fig. 6A). To assess whether the patient could benefit from treatment with ALRN-6924, the patient’s MDS/AML cells were treated in vitro with vehicle, 10 μΜ RG7388, or 10 μΜ ALRN-6924. ALRN-6924 inhibited cell proliferation and clonogenic capacity of the patient’s MDS/AML cells (Fig. 6, B and C) and showed a greater AML-inhibitory effect than RG7388 (Fig. 6B).
Fig. 6. ALRN-6924 selectively activates p53 in leukemia cells of an MDS/AML patient in vivo.
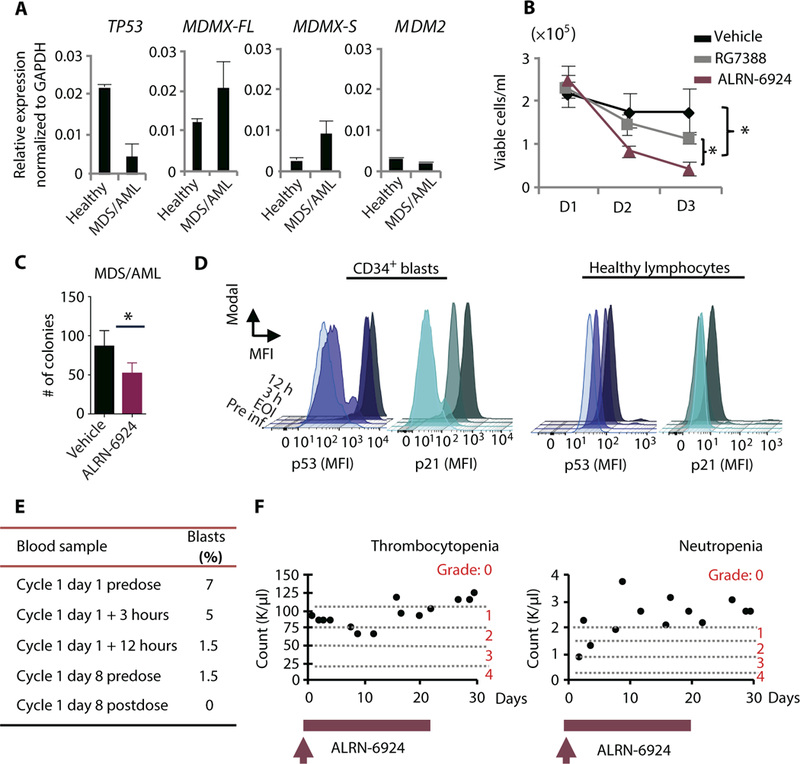
(A) mRNA expression analysis by qRT-PCR of TP53, MDMX-FL, MDMX-S, and MDM2 in leukemic cells isolated from peripheral blood of a patient with myelodysplastic syndrome (MDS)/AML. (B) Cell viability assay using patient-derived leukemic PBMNCs evaluated by trypan blue exclusion. PBMNCs were treated in liquid culture with vehicle, 10 μM ALRN-6924, or 10 μM RG7388 and monitored over time. (C) CFC assay using patient-derived leukemic cells treated with vehicle or 0.5 μM ALRN-6924 as in Fig.5B. Cells were plated in methylcellulose medium containing either vehicle or 1 μM ALRN-6924 for 10 days. (D) Intracellular FACS analysis of PBMNCs isolated from the MDS/AML patient treated with ALRN-6924 at the indicated time points after infusion. Total PBMNCs were isolated and stained with hematopoietic stem cell and progenitor markers CD34 and CD38 and pan-lymphocyte markers followed by intracellular staining of p53 and p21. FACS analysis of p53 activity in AML cells was performed by gating on lymphocyte marker-negative (CD3, CD4, CD8, CD10, and CD19), CD34+CD38- cells. The MFI of p53 and p21 in CD34+CD38- cells compared to the MFI of p53 and p21 in healthy lymphocytes at the indicated times after infusion with ALRN-6924 is shown. (E) Pathological report summarizing the percentages of peripheral blood blasts at the indicated time points after infusion with ALRN-6924. (F) Peripheral blood analysis of the patient’s platelets and neutrophils at the indicated times after infusion with ALRN-6924. Error bars represent the average of technical replicates (n = 3) ± SD; *P < 0.05 unpaired one-tailed Student’s t test.
Because of lack of other therapeutic options, the patient was treated with ALRN-6924 for compassionate use. The patient received treatment with ALRN-6924 at 3.1 mg/kg, IV, once weekly. On the first administration of ALRN-6924, peripheral blood samples were collected at different time intervals [before infusion, end of infusion (EOI), 3 hours after EOI, and 12 hours after EOI] to evaluate p53 and its downstream target, p21, protein in leukemic blasts [defined as having negative staining for lymphocyte markers and CD34+CD38- staining] by intracellular FACS staining (fig. S6A). Notably, p53 protein was rapidly induced in CD34+ leukemic blasts, but not in healthy lymphocytes, suggesting that ALRN- 6924 had a selective effect on the leukemia cells (Fig. 6D). Likewise, p21 was up-regulated in CD34+CD38- p53+ cells, demonstrating activation of the p53 pathway upon ALRN-6924 treatment in vivo. To monitor the percentage of circulating CD34+ leukemic blasts over time, peripheral blood was collected before infusion and 24 hours after EOI in weeks 2 and 3 of the weekly treatment cycles. Consistent with the patient’s pathological report (Fig. 6E), we observed a rapid decrease in circulating CD34+ leukemic blasts from 5 to 7% initially to 1.5 to 2.5% within 12 hours, followed by a further decline to less than 1% in weeks 2 and 3 (Fig. 6E and fig. S6, B and C). At the time of initial treatment, the patient presented with grade 3 neutropenia, which gradually improved while on treatment with ALRN-6924 (Fig. 6F, right panel). Furthermore, no grade 3 or 4 thrombocytopenia was observed while the patient remained on treatment (Fig. 6F, left panel). Despite the observed positive response, the patient could not continue further therapy due to infectious complications from metastatic breast cancer. These data provide proof of principle that a p53-stapled peptide dual MDMX/MDM2 inhibitor (ALRN-6924) engages the p53 pathway and has on-target antileukemic effects in a patient receiving treatment for compassionate use.
DISCUSSION
MDMX and MDM2 are frequently overexpressed in human cancers and exhibit oncogenic activity when overexpressed in experimental animal models (74, 75). Furthermore, they play indispensable and nonoverlapping roles in suppressing the normal function of p53 during embryonic development in genetic mouse models (36–41). Thus, dual inhibition of MDMX and MDM2 can provide a means for reactivating p53 across a broad range of tumors, including AML. ALRN-6924 is an α-helical stapled peptide dual MDMX/MDM2 inhibitor currently being tested in clinical trials (NCT02264613 and NCT02909972). ALRN-6924 represents an example of transforming proof-of-concept stapled peptide prototypes (49, 51, 54) into a bona fide drug candidate. In clinical trials, ALRN-6924 has thus far demonstrated efficacy and safety, with no reported grade 3/4 thrombocytopenias, and grade 3/4 neutropenia reported in less than 5% of patients (55). However, a clear understanding of its molecular and cellular mechanisms of action will provide the rigorous and mechanistic support to compel continued testing of ALRN-6924 in hematologic malignancies and other cancers expressing WT p53.
We report that the MDMX-FL isoform is overexpressed in AML in comparison to other cancers, including at the leukemic stem/progenitor cell level. Thus, MDMX appears to be a particularly relevant target in AML. We demonstrate the molecular and cellular biological effects of pharmacological dual inhibition of MDMX/MDM2 with a p53-stapled peptide in AML. ALRN-6924 rapidly penetrates cellular membranes and stabilizes p53 in AML cells. It binds with high affinity to both MDMX and MDM2, blocking their interaction with p53 in AML cells. ALRN-6924 displays on-target activity at the single-cell level, triggering rapid and sustained transcriptional activation of the p21 gene by increasing the duration and frequency of transcriptional bursting in live cells. Genome-wide, ALRN-6924 activates known p53 transcriptional networks that mediate its tumor suppressor function (76). At the cellular level, ALRN-6924 is a potent inhibitor of leukemia cell proliferation, triggering p53-dependent cell cycle arrest and apoptosis in AML cell lines and primary human leukemia cells. Therefore, we conclude that ALRN-6924 exhibits rapid and sustained on-target intracellular activity in living cells.
Leukemia initiating cells (LICs) are largely quiescent cells that can escape conventional chemotherapy and can cause disease relapse after a period of latency (77–79). LSCs rely on oncogenic signals that inactivate the p53 pathway, and reactivation of dormant p53 in LICs can be a selective strategy to eradicate LICs (80–83). Our data suggest that targeting of MDMX with a dual MDMX/MDM2 inhibitor is a strategy to target LICs in AML. We found that ALRN-6924, as well as a non- clinical small-molecule MDMX inhibitor, drastically reduced clono- genic and serial replating capacity of AML cells, and with greater activity than the MDM2-only inhibitor RG7388. ALRN-6924 selectively induced apoptosis in immature primary human CD34+CD38- LSC-enriched cells from AML patients, but not in CD34+CD38- BM cells of healthy donors. This is consistent with our finding that specifically MDMX is overexpressed in fractionated stem and progenitor cells of AML patients (in comparison to stem/progenitor cells of age- matched healthy controls), whereas MDM2 is not. In vivo, ALRN- 6924 considerably increased overall survival of experimental animals in AML xenotransplantation mouse models and displayed robust intracellular biological activity in MDS/AML cells derived from a patient on treatment with ALRN-6924 for compassionate use. In this patient, we observed that the p53 pathway was selectively up-regulated in immature CD34+CD38- MDS/AML cells, which resulted in a rapid and sustained reduction in circulating CD34+ leukemic blasts after treatment with ALRN-6924. Our data demonstrate that ALRN-6924 is a biologically active compound exhibiting on-target and on-mechanism activity in AML cells in vitro and in vivo. Our study provides functional and molecular insight into the effects of dual MDMX/MDM2 inhibition using a stapled peptide in leukemia, and provides proof of concept to support continued testing of ALRN-6924 in WT p53 cancers with high expression of MDMX.
MATERIALS AND METHODS
Study design
The purpose of this study was to evaluate the molecular, biochemical, cellular, and in vivo effects of dual targeting of MDMX and MDM2 using an α-helical stapled peptide (ALRN-6924) in AML. In vitro cellular studies were carried out using AML cell lines, primary AML cells, or healthy donor cells treated with the testing compound (ALRN- 6924 or as otherwise specified) at concentrations ranging from 0.003 to 30 μM. End points for viability and apoptosis analyses were selected on the basis of a time course ranging from 0 to 96 hours after addition of the drug to cells in culture. All cellular experiments were conducted in triplicates (technical replicates) and repeated independently at least three times unless otherwise noted. For additional details on experimental procedures, see the Supplementary Materials.
In vivo studies were performed to evaluate the effects of ALRN- 6924 on leukemia initiation and progression, as well as the effects of different dosing concentrations and frequency of treatment. Xenotransplantation studies were carried out using 10 animals per group unless otherwise specified. Animals were transplanted via tail vein injections of 2.0 × 105 cells (unless otherwise specified), using either MOLM13 or ML2 human AML cells and randomly assigned to experimental groups as defined in the figure legends. All groups were dosed with vehicle (phosphate-buffered saline), ALRN-6924 (8 mg/kg), ALRN-6924 (12 mg/kg), or ALRN-6924 (20 mg/kg) at a frequency of twice per week for 10 weeks (unless otherwise noted) or three times per week for 5 weeks. The end point of these in vivo experiments was overall survival. All animals were sacrificed upon visual signs of disease progression. Exclusion criteria included unspecified death and body weight loss of more than 20%. All experiments were carried out as unblinded studies. For additional details on experimental procedures, see the Supplementary Materials.
Statistical analysis
All error bars indicate ± SD, representing at least three independent experiments, unless otherwise specified. Comparisons between two groups were analyzed using two-tailed (unpaired) Student’s t tests unless otherwise specified. Multiple comparisons were analyzed using one-way analysis of variance (ANOVA) with Bonferroni correction in GraphPad Prism. A P value of 0.05 was considered to indicate statistical significance.
Supplementary Material
MDMX is overexpressed in cancers of hematopoietic and lymphoid origin.
MS2-SL repeats were inserted into a single allele of the p21 gene.
ALRN-6924 disrupts the p53-MDM2 and p53-MDMX protein-protein interactions and activates p53-dependent pathways.
MDMX-FL is differentially expressed in p53 WT AML cell lines sensitive to ALRN-6924. Fig. S5. In vivo xenograft models of leukemia development are sensitive to ALRN-6924.
In vivo xenograft models of leukemia development are sensitive to ALRN-6924.
Peripheral blood leukemic blasts from an MDS/AML patient treated with ALRN-6924 were analyzed by flow cytometry.
ALRN-6924 binding kinetics to MDMX and MDM2 compared to RG7388, a small-molecule MDM2 inhibitor.
Sensitivity of leukemia cell lines expressing WT and mutant p53 to ALRN-6924.
Video showing transcriptional bursting of the p21 gene over time in vehicle-treated cells.
Video showing transcriptional bursting of the p21 gene over time in ALRN-6924–treated cells.
Acknowledgments:
We thank D. Sun from the Albert Einstein College of Medicine Stem Cell Isolation and Xenotransplantation Facility and J. Zhang from the Flow Cytometry Core Facility for the expert technical support. We also thank L. Walensky for the helpful discussions and for sharing reagents (SAH-p53).
Funding: This work was supported in part through funding by Aileron Therapeutics to U.S. and L.A.C. and by grants from the NIH (R01CA166429 and R01CA217092 to U.S., U01EB021236 to R.S. and R.C., K01DK105134 to B.W.). C.K. was supported through the Medical Scientist Training Program (T32GM007288), L A.C. through the BETTR (Bronx Einstein Training in Teaching and Research) program (K12GM102779), and J.C.W. through an individual pre-doctoral NIH fellowship (F30GM122308). A.S. was supported by La Fondation pour la Recherche Medicale (SPE20140129393). U.S. is the Diane and Arthur B. Belfer Scholar in Cancer Research of the Albert Einstein College of Medicine and is a Research Scholar of the Leukemia and Lymphoma Society. The Stem Cell Isolation and Xenotransplantation Facility and the Albert Einstein College of Medicine Flow Cytometry Core facility were supported by the New York Stem Cell Science grant (C029154) and NIH grant P30CA013330, respectively.
Footnotes
Competing interests: U.S. and L.A.C. received research funding from Aileron Therapeutics. U.S. received compensation as a consultant to Aileron Therapeutics. L.A.C., V.G., D.A.A., and M.A. are current employees of Aileron Therapeutics. All other authors declare that they have no competing interest.
Data and materials availability: RNA-sequencing data set has been deposited and is available on the gene expression omnibus (GEO) under accession number GSE99255. ALRN-6924 can be provided by Aileron pending scientific review and a completed material transfer agreement. Requests for the compound should be submitted to M. Aivado (maivado@aileronrx.com). All oligonucleotide sequences, plasmids, and genetically engineered cell lines used in this study will be made available upon request.
REFERENCES AND NOTES
- 1.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, Leiserson MDM, Miller CA, Welch JS, Walter MJ, Wendl MC, Ley TJ, Wilson RK, Raphael BJ, Ding L, Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Muller PAJ, Vousden KH, p53 mutations in cancer. Nat. Cell Biol. 15, 2–8 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Shvarts A, Steegenga WT, Riteco N, van Laar T, Dekker P, Bazuine M, van Ham RC, van W der Houven van Oordt, G. Hateboer, A. J. van der Eb, A. G. Jochemsen, MDMX: A novel p53-binding protein with some functional properties of MDM2. EMBO J. 15, 5349–5357 (1996). [PMC free article] [PubMed] [Google Scholar]
- 4.Honda R, Tanaka H, Yasuda H, Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 420, 25–27 (1997). [DOI] [PubMed] [Google Scholar]
- 5.Vousden KH, Lane DP, p53 in health and disease. Nat. Rev. Mol. Cell Biol. 8, 275–283 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Bieging KT, Mello SS, Attardi LD, Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 14, 359–370 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Asai T, Liu Y, Di Giandomenico S, Bae N, Ndiaye-Lobry D, Deblasio A, Menendez S, Antipin Y, Reva B, Wevrick R, Nimer SD, Necdin, a p53 target gene, regulates the quiescence and response to genotoxic stress of hematopoietic stem/progenitor cells. Blood 120, 1601–1612 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abbas HA, Maccio DR, Coskun S, Jackson JG, Hazen AL, Sills TM, You MJ, Hirschi KK, Lozano G, Mdm2 is required for survival of hematopoietic stem cells/ progenitors via dampening of ROS-induced p53 activity. Cell Stem Cell 7, 606–617 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Y, Elf SE, Asai T, Miyata Y, Liu Y, Sashida G, Huang G, Di Giandomenico S, Koff A, Nimer SD, The p53 tumor suppressor protein is a critical regulator of hematopoietic stem cell behavior. Cell Cycle 8, 3120–3124 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jung H, Kim MJ, Kim DO, Kim WS, Yoon SJ, Park YJ, Yoon SR, Kim TD, Suh HW, Yun S, Min JK, Lee HG, Lee YH, Na HJ, Lee DC, Kim HC, Choi I, TXNIP maintains the hematopoietic cell pool by switching the function of p53 under oxidative stress. Cell Metab. 18, 75–85 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Wang YV, Leblanc M, Fox N, Mao JH, Tinkum KL, Krummel K, Engle D, Piwnica-Worms D, Piwnica-Worms H, Balmain A, Kaushansky K, Wahl GM, Fine-tuning p53 activity through C-terminal modification significantly contributes to HSC homeostasis and mouse radiosensitivity. Genes Dev. 25, 1426–1438 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Akala OO, Park IK, Qian D, Pihalja M, Becker MW, Clarke MF, Long-term haematopoietic reconstitution by Trp53−/−p16Ink4a−/−p19Arf−/− multipotent progenitors. Nature 453, 228–232 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Zhao Z, Zuber J, Diaz-Flores E, Lintault L, Kogan SC, Shannon K, Lowe SW, p53 loss promotes acute myeloid leukemia by enabling aberrant self-renewal. Genes Dev. 24, 1389–1402 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kadia TM, Jain P, Ravandi F, Garcia-Manero G, Andreef M, Takahashi K, Borthakur G, E,Jabbour, M. Konopleva, N. G. Daver, C. Dinardo, S. Pierce, R. Kanagal-Shamanna, K. Patel, Z. Estrov, J. Cortes, H. M. Kantarjian, TP53 mutations in newly diagnosed acute myeloid leukemia: Clinicomolecular characteristics, response to therapy, and outcomes. Cancer 122, 3484–3491 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yanada M, Yamamoto Y, Iba S, Okamoto A, Inaguma Y, Tokuda M, Morishima S, Kanie T, Mizuta S, Akatsuka Y, Okamoto M, Emi N, TP53 mutations in older adults with acute myeloid leukemia. Int. J. Hematol. 103, 429–435 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Fenaux P, Preudhomme C, Quiquandon I, Jonveaux P, Lai JL, Vanrumbeke M, Loucheux-Lefebvre MH, Bauters F, Berger R, Kerckaert JP, Mutations of the P53 gene in acute myeloid leukaemia. Br. J. Haematol. 80, 178–183 (1992). [DOI] [PubMed] [Google Scholar]
- 17.Peller S, Rotter V, TP53 in hematological cancer: Low incidence of mutations with significant clinical relevance. Hum. Mutat. 21, 277–284 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Zeisig BB, Kulasekararaj AG, Mufti GJ, So CW, SnapShot: Acute myeloid leukemia. Cancer Cell 22, 698–698.e1 (2012). [DOI] [PubMed] [Google Scholar]
- 19.Rucker FG, Schlenk RF, Bullinger L, Kayser S, Teleanu V, Kett H, Habdank M, Kugler CM, Holzmann K, Gaidzik VI, Paschka P, Held G, von Lilienfeld-Toal M, Lubbert M, Frohling S, Zenz T, Krauter J, Schlegelberger B, Ganser A, Lichter P, Dohner K, Dohner H, TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood 119, 2114–2121 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Wade M, Li Y-C, Wahl GM, MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 13, 83–96 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bueso-Ramos CE, Yang Y, deLeon E, McCown P, Stass SA, Albitar M, The human MDM-2 oncogene is overexpressed in leukemias. Blood 82, 2617–2623 (1993). [PubMed] [Google Scholar]
- 22.Seliger B, Papadileris S, Vogel D, Hess G, Brendel C, Storkel S, Ortel J, Kolbe K, Huber C, Huhn D, Neubauer A, Analysis of the p53 and MDM-2 gene in acute myeloid leukemia. Eur. J. Haematol. 57, 230–240 (1996). [DOI] [PubMed] [Google Scholar]
- 23.Faderl S, Kantarjian HM, Estey E, Manshouri T, Chan CY, Rahman Elsaied A, Kornblau SM, Cortes J, Thomas DA, Pierce S, Keating MJ, Estrov Z, Albitar M, The prognostic significance of p16(INK4a)/p14(ARF) locus deletion and MDM-2 protein expression in adult acute myelogenous leukemia. Cancer 89, 1976–1982 (2000). [DOI] [PubMed] [Google Scholar]
- 24.Li L, Tan Y, Chen X, Xu Z, Yang S, Ren F, Guo H, Wang X, Chen Y, Li G, Wang H, MDM4 overexpressed in acute myeloid leukemia patients with complex karyotype and wild-type TP53. PLOS ONE 9, e113088 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tan BX, Khoo KH, Lim TM, Lane DP, High Mdm4 levels suppress p53 activity and enhance its half-life in acute myeloid leukaemia. Oncotarget 5, 933–943 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kojima K, Konopleva M, Samudio IJ, Shikami M, Cabreira-Hansen M, McQueen T, Ruvolo V, Tsao T, Zeng Z, Vassilev LT, Andreeff M, MDM2 antagonists induce p53-dependent apoptosis in AML: Implications for leukemia therapy. Blood 106, 3150–3159 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Han X, Medeiros LJ, Zhang YH, You MJ, Andreeff M, Konopleva M, Bueso-Ramos CE, High expression of human homologue of murine double minute 4 and the short splicing variant, HDM4-S, in bone marrow in patients with acute myeloid leukemia or myelodysplastic syndrome. Clin. Lymphoma Myeloma Leuk. 16, S30–S38 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Haupt Y, Maya R, Kazaz A, Oren M, Mdm2 promotes the rapid degradation of p53. Nature 387, 296–299 (1997). [DOI] [PubMed] [Google Scholar]
- 29.Kubbutat MH, Jones SN, Vousden KH, Regulation of p53 stability by Mdm2. Nature 387, 299–303 (1997). [DOI] [PubMed] [Google Scholar]
- 30.Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, Vogelstein B, Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature 362, 857–860 (1993). [DOI] [PubMed] [Google Scholar]
- 31.Gu J, Kawai H, Nie L, Kitao H, Wiederschain D, Jochemsen AG, Parant J, Lozano G, Yuan Z-M, Mutual dependence of MDM2 and MDMX in their functional inactivation of p53. J. Biol. Chem. 277, 19251–19254 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Stad R, Little NA, Xirodimas DP, Frenk R, van der Eb AJ, Lane DP, Saville MK, Jochemsen AG, Mdmx stabilizes p53 and Mdm2 via two distinct mechanisms. EMBO Rep. 2, 1029–1034 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sharp DA, Kratowicz SA, Sank MJ, George DL, Stabilization of the MDM2 oncoprotein by interaction with the structurally related MDMX protein. J. Biol. Chem. 274, 38189–38196 (1999). [DOI] [PubMed] [Google Scholar]
- 34.Stad R, Ramos YF, Little N, Grivell S, Attema J, van Der Eb AJ, Jochemsen AG, Hdmx stabilizes Mdm2 and p53. J. Biol. Chem. 275, 28039–28044 (2000). [DOI] [PubMed] [Google Scholar]
- 35.Tanimura S, Ohtsuka S, Mitsui K, Shirouzu K, Yoshimura A, Ohtsubo M, MDM2 interacts with MDMX through their RING finger domains. FEBS Lett. 447, 5–9 (1999). [DOI] [PubMed] [Google Scholar]
- 36.Montes R de Oca Luna, D. S. Wagner, G. Lozano, Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 378, 203–206 (1995). [DOI] [PubMed] [Google Scholar]
- 37.Jones SN, Roe AE, Donehower LA, Bradley A, Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 378, 206–208 (1995). [DOI] [PubMed] [Google Scholar]
- 38.Parant J, Chavez-Reyes A, Little NA, Yan W, Reinke V, Jochemsen AG, Lozano G, Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat. Genet. 29, 92–95 (2001). [DOI] [PubMed] [Google Scholar]
- 39.Migliorini D, Lazzerini Denchi E, Danovi D, Jochemsen A, Capillo M, Gobbi A, Helin K, Pelicci PG, Marine JC, Mdm4 (Mdmx) regulates p53-induced growth arrest and neuronal cell death during early embryonic mouse development. Mol. Cell. Biol. 22, 5527–5538 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Francoz S, Froment P, Bogaerts S, De Clercq S, Maetens M, Doumont G, Bellefroid E, Marine JC, Mdm4 and Mdm2 cooperate to inhibit p53 activity in proliferating and quiescent cells in vivo. Proc. Natl. Acad. Sci. U.S.A. 103, 3232–3237 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barboza JA, Iwakuma T, Terzian T, El-Naggar AK, Lozano G, Mdm2 and Mdm4 loss regulates distinct p53 activities. Mol. Cancer Res. 6, 947–954 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xiong S, Van Pelt CS, Elizondo-Fraire AC, Liu G, Lozano G, Synergistic roles of Mdm2 and Mdm4 for p53 inhibition in central nervous system development. Proc. Natl. Acad. Sci. U.S.A. 103, 3226–3231 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA, In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–848 (2004). [DOI] [PubMed] [Google Scholar]
- 44.Zhao Y, Aguilar A, Bernard D, Wang S, Small-molecule inhibitors of the MDM2-p53 protein-protein interaction (MDM2 Inhibitors) in clinical trials for cancer treatment. J. Med. Chem. 58, 1038–1052 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Andreeff M, Kelly KR, Yee K, Assouline S, Strair R, Popplewell L, Bowen D, Martinelli G, Drummond MW, Vyas P, Kirschbaum M, Iyer SP, Ruvolo V, Gonzalez GM, Huang X, Chen G, Graves B, Blotner S, Bridge P, Jukofsky L, Middleton S, Reckner M, Rueger R, Zhi J, Nichols G, Kojima K, Results of the phase I trial of RG7112, a small-molecule MDM2 antagonist in leukemia. Clin. Cancer Res. 22, 868–876 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reis B, Jukofsky L, Chen G, Martinelli G, Zhong H, So WV, Dickinson MJ, Drummond M, Assouline S, Hashemyan M, Theron M, Blotner S, Lee JH, Kasner M, Yoon SS, Rueger R, Seiter K, Middleton SA, Kelly KR, Vey N, Yee K, Nichols G, Chen LC, Pierceall WE, Acute myeloid leukemia patients’ clinical response to idasanutlin (RG7388) is associated with pre-treatment MDM2 protein expression in leukemic blasts. Haematologica 101, e185–e188 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vu B, Wovkulich P, Pizzolato G, Lovey A, Ding Q, Jiang N, Liu J-J, Zhao C, Glenn K, Wen Y, Tovar C, Packman K, Vassilev L, Graves B, Discovery of RG7112: A small-molecule MDM2 inhibitor in clinical development. ACS Med. Chem. Lett. 4, 466–469 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ding Q, Zhang Z, Liu J-J, Jiang N, Zhang J, Ross TM, Chu XJ, Bartkovitz D, Podlaski E, Janson C, Tovar C, Filipovic ZM, Higgins B, Glenn K, Packman K, Vassilev LT, Graves B , Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J. Med. Chem. 56, 5979–5983 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Bernal F, Wade M, Godes M, Davis TN, Whitehead DG, Kung AL, Wahl GM, Walensky LD , A stapled p53 helix overcomes HDMX-mediated suppression of p53. Cancer Cell 18, 411–422 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bo MD, Secchiero P, Degan M, Marconi D, Bomben R, Pozzato G, Gaidano G, Poeta E Del, Forconi F, Zauli G, Gattei V, MDM4 (MDMX) is overexpressed in chronic lymphocytic leukaemia (CLL) and marks a subset of p53wild-type CLL with a poor cytotoxic response to Nutlin-3. Br. J. Haematol. 150, 237–239 (2010). [DOI] [PubMed] [Google Scholar]
- 51.Chang YS, Graves B, Guerlavais V, Tovar C, Packman K, To KH, Olson KA, Kesavan K, Gangurde P, Mukherjee A, Baker T, Darlak K, Elkin C, Filipovic Z, Qureshi FZ, Cai H, Berry P, Feyfant E, Shi XE, Horstick J, Annis DA, Manning AM, Fotouhi N, Nash H, Vassilev LT, Sawyer TK, Stapled a-helical peptide drug development: A potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc. Natl. Acad. Sci. U.S.A. 110, E3445–E3454 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bernal F, Tyler AF, Korsmeyer SJ, Walensky LD, Verdine GL, Reactivation of the p53 tumor suppressor pathway by a stapled p53 peptide. J. Am. Chem. Soc. 129, 2456–2457 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tan BX, Brown CJ, Ferrer FJ, Yuen TY, Quah ST, Chan BH, Jansson AE, Teo HL, Nordlund P, Lane DP, Assessing the efficacy of Mdm2/Mdm4-inhibiting stapled peptides using cellular thermal shift assays. Sci. Rep. 5, 12116 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wachter F, Morgan AM, Godes M, Mourtada R, Bird GH, Walensky LD, Mechanistic validation of a clinical lead stapled peptide that reactivates p53 by dual HDM2 and HDMX targeting. Oncogene 36, 2184–2190 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Funda Meric-Bernstam F, Saleh MS, Infante JR, Goel S, Falchook GS, Shapiro G, Chung KY, Conry RM, Hong DS, Wang JS, Steidl U, Walensky LD, Guerlavais V, Payton M, Annis DA, Aivado M, Patel MR, Phase I trial of a novel stapled peptide ALRN-6924 disrupting MDMX- and MDM2- mediated inhibition of WTp53 in patients with solid tumors and lymphomas. J. Clin. Oncol. 35, 2505–2505 (2017). [Google Scholar]
- 56.Burgess A, Chia KM, Haupt S, Thomas D, Haupt Y, Lim E, Clinical overview of MDM2/X-targeted therapies. Front. Oncol. 6, 7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gembarska A, Luciani F, Fedele C, Russell EA, Dewaele M, Villar S, Zwolinska A, Haupt S, de Lange J, Yip D, Goydos J, Haigh JJ, Haupt Y, Larue L, Jochemsen A, Shi H, Moriceau G, Lo RS, Ghanem G, Shackleton M, Bernal F, Marine JC, MDM4 is a key therapeutic target in cutaneous melanoma. Nat. Med. 18, 1239–1247 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Touqan N, Diggle CP, Verghese ET, Perry S, Horgan K, Merchant W, Anwar R, Markham AF, Carr IM, Achuthan R, An observational study on the expression levels of MDM2 and MDMX proteins, and associated effects on P53 in a series of human liposarcomas. BMC Clin. Pathol. 13, 32 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Furgason JM, Koncar RF, Michelhaugh SK, Sarkar FH, Mittal S, Sloan AE, Barnholtz-Sloan JS, Bahassi el M, Whole genome sequence analysis links chromothripsis to EGFR, MDM2, MDM4, and CDK4 amplification in glioblastoma. Oncoscience 2, 618–628 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Haupt S, Buckley D, Pang JM, Panimaya J, Paul PJ, Gamell C, Takano EA, Lee YY, Hiddingh S, Rogers TM, Teunisse AF, Herold MJ, Marine JC, Fox SB, Jochemsen A, Haupt Y, Targeting Mdmx to treat breast cancers with wild-type p53. Cell Death Dis. 6, e1821 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dewaele M, Tabaglio T, Willekens K, Bezzi M, Teo SX, Low DH, Koh CM, Rambow F, Fiers M, Rogiers A, Radaelli E, Al-Haddawi M, Tan SY, Hermans E, Amant F, Yan H, Lakshmanan M, Koumar RC, Lim ST, Derheimer FA, Campbell RM, Bonday Z, Tergaonkar V, Shackleton M, Blattner C, Marine JC, Guccione E, Antisense oligonucleotide-mediated MDM4 exon 6 skipping impairs tumor growth. J. Clin. Invest. 126, 68–84 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lenos K, Grawenda AM, Lodder K, Kuijjer ML, Teunisse AF, Repapi E, Grochola LF, Bartel F, Hogendoorn PC, Wuerl P, Taubert H, Cleton-Jansen AM, Bond GL, Jochemsen AG, Alternate splicing of the p53 inhibitor HDMX offers a superior prognostic biomarker than p53 mutation in human cancer. Cancer Res. 72, 4074–4084 (2012). [DOI] [PubMed] [Google Scholar]
- 63.Barreyro L, Will B, Bartholdy B, Zhou L, Todorova TI, Stanley RF, Ben-Neriah S, Montagna C, Parekh S, Pellagatti A, Boultwood J, Paietta E, Ketterling RP, Cripe L, Fernandez GF, Greenberg PL, Tallman MS, Steidl C, Mitsiades CS, Verma A, Steidl U, Overexpression of IL-1 receptor accessory protein in stem and progenitor cells and outcome correlation in AML and MDS. Blood 120, 1290–1298 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hu B, Gilkes DM, Chen J, Efficient p53 activation and apoptosis by simultaneous disruption of binding to MDM2 and MDMX. Cancer Res. 67, 8810–8817 (2007). [DOI] [PubMed] [Google Scholar]
- 65.Phan J, Li Z, Kasprzak A, Li B, Sebti S, Guida W, Schonbrunn E, Chen J, Structure-based design of high affinity peptides inhibiting the interaction of p53 with MDM2 and MDMX. J. Biol. Chem. 285, 2174–2183 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Popowicz GM, Czarna A, Rothweiler U, Szwagierczak A, Krajewski M, Weber L, Holak TA, Molecular basis for the inhibition of p53 by Mdmx. Cell Cycle 6, 2386–2392 (2007). [DOI] [PubMed] [Google Scholar]
- 67.Suter DM, Molina N, Gatfield D, Schneider K, Schibler U, Naef F, Mammalian genes are transcribed with widely different bursting kinetics. Science 332, 472–474 (2011). [DOI] [PubMed] [Google Scholar]
- 68.Vera M, Biswas J, Senecal A, Singer RH, Park HY, Single-cell and single-molecule analysis of gene expression regulation. Annu. Rev. Genet. 50, 267–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bertrand E, Chartrand P, Schaefer M, Shenoy SM, Singer RH, Long RM, Localization of ASH1 mRNA particles in living yeast. Mol. Cell 2, 437–445 (1998). [DOI] [PubMed] [Google Scholar]
- 70.Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, Zhang F, Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154, 1380–1389 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mascarenhas J, Virtgaym E, Kosiorek H, Stal M, Sandy L, Orellana A, Xia L, Kremyanskaya M, Petersen B, Dueck A, Hoffman R, 254 open label phase I study of single agent oral RG7388 (idasanutlin) in patients with polycythemia vera and essential thrombocythemia. Blood 130, 254 (2017). [Google Scholar]
- 72.Wang H, Ma X, Ren S, Buolamwini JK, Yan C, A small-molecule inhibitor of MDMX activates p53 and induces apoptosis. Mol. Cancer Ther. 10, 69–79 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Serfilippi LM, Pallman DR, Russell B, Serum clinical chemistry and hematology reference values in outbred stocks of albino mice from three commonly used vendors and two inbred strains of albino mice. Contemp. Top. Lab. Anim. Sci. 42, 46–52 (2003). [PubMed] [Google Scholar]
- 74.Jones SN, Hancock AR, Vogel H, Donehower LA, Bradley A, Overexpression of Mdm2 in mice reveals a p53-independent role for Mdm2 in tumorigenesis. Proc. Natl. Acad. Sci. U.S.A. 95, 15608–15612 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xiong S, Pant V, Suh YA, Van Pelt CS, Wang Y, Valentin-Vega YA, Post SM, Lozano F, Spontaneous tumorigenesis in mice overexpressing the p53-negative regulator Mdm4. Cancer Res. 70, 7148–7154 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vousden KH, Prives C, Blinded by the light: The growing complexity of p53. Cell 137, 413–431 (2009). [DOI] [PubMed] [Google Scholar]
- 77.Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson A, Caligiuri MA, Dick JE, A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 367, 645–648 (1994). [DOI] [PubMed] [Google Scholar]
- 78.Passegue E, Jamieson CH, Ailles LE, Weissman IL, Normal and leukemic hematopoiesis: Are leukemias a stem cell disorder or a reacquisition of stem cell characteristics? Proc. Natl. Acad. Sci. U.S.A. 100, (Suppl. 1) 11842–11849 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, Tanaka S, Nakamura R, Tanaka T, Tomiyama H, Saito N, Fukata M, Miyamoto T, Lyons B, Ohshima K, Uchida N, Taniguchi S, Ohara O, Akashi K, Harada M, Shultz LD, Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat. Biotechnol. 25, 1315–1321 (2007). [DOI] [PubMed] [Google Scholar]
- 80.Qi J, Singh S, Hua WK, Cai Q, Chao SW, Li L, Liu H, Ho Y, McDonald T, Lin A, Marcucci G, Bhatia R, Huang WJ, Chang CI, Kuo YH, HDAC8 inhibition specifically targets Inv(16) acute myeloid leukemic stem cells by restoring p53 acetylation. Cell Stem Cell 17, 597–610 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Abraham SA, Hopcroft LE, Carrick E, Drotar ME, Dunn K, Williamson AJ, Korfi K, Baquero P, Park LE, Scott MT, Pellicano F, Pierce A, Copland M, Nourse C, Grimmond SM, Vetrie D, Whetton AD, Holyoake TL, Dual targeting of p53 and c-MYC selectively eliminates leukaemic stem cells. Nature 534, 341–346 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li L, Wang L, Li L, Wang Z, Ho Y, McDonald T, Holyoake TL, Chen W, Bhatia R, Activation of p53 by SIRT1 inhibition enhances elimination of CML leukemia stem cells in combination with imatinib. Cancer Cell 21,266–281 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Takeishi S, Matsumoto A, Onoyama I, Naka K, Hirao A, Nakayama KI, Ablation of Fbxw7 eliminates leukemia-initiating cells by preventing quiescence. Cancer Cell 23, 347–361 (2013). [DOI] [PubMed] [Google Scholar]
- 84.Yu C, Liu Y, Ma T, Liu K, Xu S, Zhang Y, Liu H, La Russa M, Xie M, Ding S, Qi LS, Small molecules enhance CRISPR genome editing in pluripotent stem cells. Cell Stem Cell 16, 142–147 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Beard C, Hochedlinger K, Plath K, Wutz A, Jaenisch R, Efficient method to generate single-copy transgenic mice by site-specific integration in embryonic stem cells. Genesis 44, 23–28 (2006). [DOI] [PubMed] [Google Scholar]
- 86.Wu B, Chao JA, Singer RH, Fluorescence fluctuation spectroscopy enables quantitative imaging of single mRNAs in living cells. Biophys. J. 102, 2936–2944 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D, In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272, 263–267 (1996). [DOI] [PubMed] [Google Scholar]
- 88.Pandolfi A, Stanley RF, Yu Y, Bartholdy B, Pendurti G, Gritsman K, Boultwood J, Chernoff J, Verma A, Steidl U, PAK1 is a therapeutic target in acute myeloid leukemia and myelodysplastic syndrome. Blood 126, 1118–1127 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Carvajal LA, Hamard PJ, Tonnessen C, Manfredi JJ, E2F7, a novel target, is up-regulated by p53 and mediates DNA damage-dependent transcriptional repression. Genes Dev. 26, 1533–1545 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hamard P-J, Lukin DJ, Manfredi JJ, p53 basic C terminus regulates p53 functions through DNA binding modulation of subset of target genes. J. Biol. Chem. 287, 22397–22407 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
MDMX is overexpressed in cancers of hematopoietic and lymphoid origin.
MS2-SL repeats were inserted into a single allele of the p21 gene.
ALRN-6924 disrupts the p53-MDM2 and p53-MDMX protein-protein interactions and activates p53-dependent pathways.
MDMX-FL is differentially expressed in p53 WT AML cell lines sensitive to ALRN-6924. Fig. S5. In vivo xenograft models of leukemia development are sensitive to ALRN-6924.
In vivo xenograft models of leukemia development are sensitive to ALRN-6924.
Peripheral blood leukemic blasts from an MDS/AML patient treated with ALRN-6924 were analyzed by flow cytometry.
ALRN-6924 binding kinetics to MDMX and MDM2 compared to RG7388, a small-molecule MDM2 inhibitor.
Sensitivity of leukemia cell lines expressing WT and mutant p53 to ALRN-6924.
Video showing transcriptional bursting of the p21 gene over time in vehicle-treated cells.
Video showing transcriptional bursting of the p21 gene over time in ALRN-6924–treated cells.