

Abstract
Simultaneous, quantitative determination of intracellular nucleoside triphosphates and other polar metabolites using liquid chromatography with electrospray ionization tandem mass spectrometry (LC-MS/MS) represents a bioanalytic challenge because of charged, highly hydrophilic analytes presented at a large concentration range in a complex matrix. In this study, an ion pair LC-MS/MS method using triethylamine (TEA) – hexafluoroisopropanol (HFIP) ion-pair mobile phase was optimized and validated for simultaneous and unambiguous determination of 8 nucleoside triphosphates (including ATP, CTP, GTP, UTP, dATP, dCTP, dGTP, and dTTP) in cellular samples. Compared to the the less volatile ion-pair reagent, triethylammonium acetate (100 mM, pH 7.0), the combination of HFIP (100 mM) and TEA (8.6 mM) increased the MS signal intensity by about 50-fold, while retaining comparable chromatographic resolution. The isotope-labeled internal standard method was used for the quantitation. Lower limits of quantitation were determined at 0.5 nM for CTP, UTP, dATP, dCTP, and dTTP, at 1 nM for ATP, and at 5 nM for GTP and dGTP. The intra- and inter-day precision and accuracy were within the generally accepted criteria for bioanalytical method validation (< 15%). While the present method was validated for the quantitation of intracellular nucleoside triphosphates, it had a broad application potential for quantitative profiling of nucleoside mono- and bi-phosphates as well as other polar, ionic metabolic intermediates (including carbohydrate derivatives, carboxylic acid derivatives, co-acyl A derivatives, fatty acyls, and others) in biological samples.
Keywords: Ion pair chromatography, Ribonucleoside triphosphate (NTP), Deoxyribonucleoside triphosphate (dNTP), LC-MS/MS, Metabolomics
1. Introduction
Nucleotides are organic molecules that serve as the monomers of nucleic acids such as DNA and RNA. A nucleotide is composed of a nucleobase, a five-carbon sugar (either ribose or 2-deoxyribose), and one, two, or three phosphate groups [1]. Natural ribonucleoside triphosphates (NTPs) include adenosine triphosphate (ATP), guanosine triphosphate (GTP), cytidine triphosphate (CTP), and uridine triphosphate (UTP). Natural deoxyribonucleoside triphophates (dNTPs) include deoxyadenosine triphosphate (dATP), deoxyguanosine triphosphate (dGTP), deoxycytidine triphosphate (dCTP), and thymidine triphosphate (dTTP). In addition to serving as the precursors of RNA and DNA, these nucleotides play a central role in cell metabolism and cellular signaling [1]. In light of their multiple functions, the levels of NTPs and dNTPs in eukaryotic cells maintain a delicate balance under normal physiological conditions. Hence, the development of a sensitive, selective and reliable liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) method that enables quantitative profiling of intracellular nucleotides is important for understanding complex biochemical changes in the nucleotide pool in response to genetic modifications, pathophysiological or environment stimuli.
Because nucleoside triphosphates are composed of three phosphate groups bound to a five-carbon sugar that is linked to a purine or a pyrimidine base, these nucleotide molecules are negatively charged at pH > 2. Due to their polarity, negatively charged nucleotides have a poor retention on conventional reversed-phase liquid chromatography columns because retention on such columns is based on hydrophobic interactions of the analyte with the stationary phase. On the other hand, negatively charged nucleotides can be separated by anion exchange liquid chromatography, where separation is based on ionic interactions of the analytes with the charged stationary phase [2]. However, ion exchange liquid chromatography is incompatible with electrospray ionization mass spectrometry because the elution of charged molecules requires the use of inorganic salts as competing ions in the mobile phase, which are often non-volatile and thus can contaminate the ion source of the mass spectrometer and suppress electrospray ionization. Compared to ion exchange liquid chromatography, ion pair reversed-phase liquid chromatography appears more amenable to electrospray ionization mass spectrometry, where the retention of nucleotides was achieved by the formation of ion pairs between the negatively charged phosphate groups of nucleotides and positively charged ion pair mobile phase that is absorbed on the hydrophobic stationary phase [3].
Several ion pair LC-MS/MS methods have been described in recent years for quantitative analysis of nucleotides in biological samples, where trimethylamine (TEA) – acetic acid or triethylammonium acetate (TEAA) and similar ion-pair reagents (e.g., tributylammonium acetate, tetrabutylammonium acetate, and dimethylhexylamine) were commonly used [4-8]. However, all these methods suffer from a limited sensitivity, with the lower limit of quantitation (LLOQ) ranging from 50 – 5000 nM for individual nucleotides [4-8]. The lack of sensitivity could be attributable to the fact that the ion pair reagents used in these methods are not particularly volatile, thereby resulting in inefficient electrospray ionization of the analytes. It has been reported that the addition of hexafluoroisopropanol (HFIP) into TEA-based ion-pair mobile phase could enhance electrospray ionization signals of oligonucleotides [9]. The proposed mechanism is based on the dynamic adjustment of the pH of the electrospray droplet as a consequence of the preferential evaporation removal of the more volatile anionic counter-ion (i.e., HFIP) from the droplet surface [9]. Specifically, HFIP [boiling point (bp) = 57°C] is more volatile, as compared to TEA (bp = 89°C), acetic acid (bp = 118°C), tributylamine (bp = 214 °C), tetrabutylamine (bp = 100 °C) and dimethylhexylamine (bp = 146 °C). As a buffer system in the mobile phase, the weak acid/weak base system of HFIP/TEA maintains a stable pH at ~ 8.3. However, as the column eluent is electrosprayed and desolvated, the more volatile HFIP is depleted at the droplet surface, thus causing a rise of the pH toward 10 at the surface. At the high pH, the TEA-oligonucleotides ion pairs dissociate, and consequently the oligonucleotides are desorbed into the gas phase and can be ionized efficiently [9].
While TEA-HFIP ion pair mobile phase has been successfully used for the LC-MS/MS analysis of oligonucleotides (with molecular weight > 3000) to achieve reasonable chromatographic resolution and efficient electrospray ionization [9], no studies have been reported on the application of this ion pair mobile phase for the LC-MS/MS analysis of small molecule nucleotides. Here, we investigated if the addition of hexafluoroisopropanol (HFIP) into TEA-based ion-pair mobile phase could enhance electrospray ionization efficiency of nucleoside triphosphates. We optimized and validated an ion pair LC-MS/MS method using TEA – HFIP as the ion-pair mobile phase. Superior to the less volatile ion-pair mobile phase, the TEA – HFIP ion-pair mobile phase produced efficient chromatographic separation as well as high-sensitivity electrospray ionization, which enabled sensitive, selective, and reliable determination of intracellular nucleoside triphosphates. Furthermore, we demonstrated that in addition to the quantitation of nucleoside triphosphates, the developed method had a broad application potential for quantitative analysis of a variety of highly polar, often ionized intracellular metabolites that are unsuitable for analysis with the reversed-phase or hydrophilic interaction liquid chromatography (HILIC) based LC-MS/MS methods.
2. Materials and Methods
2.1. Chemicals and Reagents
Reference standards for NTPs (including ATP, CTP, GTP, UTP) and dNTPs (including dATP, dCTP, dTTP, and dGTP) were obtained from Sigma Aldrich (St. Louis, MO, USA). Stable isotope-labeled internal standards for each NTP or dNTP, including13C1015N5-dATP, 13C915N3-dCTP, 13C1015N5-dGTP, 13C1015N2-dTTP, 13C1015N5-ATP, 13C915N3-CTP, 13C1015N5-GTP, 13C9-UTP, were purchased in solution from Sigma Aldrich (St. Louis, MO, USA). Ion pairing reagents HFIP and TEA were obtained from Sigma Aldrich (St. Louis, MO, USA) and Alfa Aesar (Ward Hill, MA), respectively. Acetonitrile (LC-MS grade) and methanol (LC-MS grade) was purchased from J.T.Baker (Phillipsburg, NJ). UltraPure™ DNase/RNase- free distilled water was obtained from Invitrogen Life Technologies (Grand Island, NY) and used throughout for aqueous solution preparation.
2.2. Chromatographic and Mass Spectrometric Conditions
2.2.1. Instrumentation
All LC-MS/MS analyses were performed on an AB SCIEX (Foster City, CA) QTRAP 6500 system, which consists of an enhanced high performance hybrid triple quadrupole / linear ion trap mass spectrometer, interfaced with a SHIMADZU (Kyoto, Japan) Nexera high performance liquid chromatography (HPLC) system that is equipped with two X2 LC-30AD pumps, a X2 SIL-30AC autosampler, a CBM-20A communication bus module, a X2 CTO-30A column oven, and DGU-20A degassing units. Analyst® 1.6 software was used for system control and data acquisition and MultiQuant 3.0 software was used for data processing and quantitation.
2.2.2. Liquid chromatography
Chromatographic separation of NTPs and dNTPs was achieved on an Atlantis T3 column (3.0 μm,2.1 mm × 100 mm) (Waters Cor., Milford, MA) at 45°C using a gradient mobile phase containing TEA-HFIP ion pair system. The mobile phase was optimized with different concentration combinations of TEA and HFIP. Based on a technical application note from Waters (http://www.waters.com/webassets/cms/support/docs/715001476.pdf), the ion pair mobile phase containing 8.6 mM TEA and 100 mM HFIP is adequate for oligonucleotide analysis. First, the impact of varying TEAA concentrations on the chromatographic resolution of nucleoside triphosphates was tested. It was found that reasonable chromatographic resolution was achieved as TEAA concentrations ranged from 5 to 100 mM. Therefore, the mobile phase containing 8.6 mM TEA was chosen for further assay development. Next, the impact of HFIP concentrations on the chromatographic resolution and electrospray ionization signal intensity was investigated by adding varying concentrations of HFIP (5, 25, 50 and 100 mM) into TEA (8.6 mM) mobile phase. Furthermore, the mobile phase gradient slope was optimized to achieve a sufficient chromatographic separation. The final selected mobile phase A consisted of 100 mM HFIP and 8.6 mM TEA in water (final pH, 8.3 ± 0.1), and mobile phase B consisted of 10% acetonitrile in mobile phase A. The optimal gradient program was as follows: 0 – 6 min, mobile phase B increasing from 0% to 10%; 6.0 – 6.1 min, mobile phase B decreasing from 10% to 0%; 6.1 – 10 min, mobile phase B maintaining at 0%. Flow rate was 0.5 mL/min. The autosampler temperature was set at 4°C.
2.2.3. Mass spectrometry
The column effluent was monitored using the QTRAP 6500 mass spectrometer, which is a high performance hybrid triple quadrupole/linear ion trap mass spectrometer. Mass spectrometry conditions for individual analytes and internal standards were optimized by direct infusion of the standard solution of individual NTPs, dNTPs and their respective isotope-labeled internal standards into the ion source under both positive and negative ionization mode. It was found that NTPs and dNTPs underwent more efficient ionization in the negative electrospray ionization mode than in the positive ionization mode. Thus, the negative ionization mode, under which the most common fragmentation pathway corresponds to the loss of a phosphate group, was used for monitoring all NTPs, dNTPs and their respective isotope-labeled internal standards (Figure 1 and Supplementary Figure 1). The Turbo ion-spray voltage was set at − 4500 V and the temperature was set at 600°C. Collision gas was optimized at High level, and curtain gas, ion source gas 1 and ion source gas 2 were delivered at 30, 60 and 50 psi, respectively. The optimal mass spectrometric conditions and multiple reaction mass transitions for individual NTPs, dNTPs and isotope-labeled internal standards are shown in Table 1.
Figure 1.

Product ion mass spectra of dATP, dCTP, dGTP, dTTP, ATP, CTP, GTP, and UTP
Table 1.
Optimized mass spectrometric parameters and chromatographic retention time for individual NTPs, dNTPs, and their respective isotope-labeled internal standard
| Analyte | MS Transition (m/z) | Collision energy (V) | Decluster Potential (V) | Retention Time (min) |
|---|---|---|---|---|
| ATP | 506.0 > 272.9 | −40 | −100 | 3.78 |
| CTP | 482.0 > 384.1 | −31 | −100 | 2.77 |
| GTP | 522.0 > 424.2 | −30 | −100 | 2.98 |
| UTP | 483.0 > 385.0 | −27 | −100 | 2.74 |
| dATP | 490.0 > 392.1 | −31 | −100 | 4.96 |
| dCTP | 466.0 > 368.0 | −30 | −100 | 3.27 |
| dGTP | 506.0 > 257.0 | −44 | −100 | 3.91 |
| dTTP | 480.9 > 383.0 | −29 | −100 | 3.97 |
| ATP-13C1015N5 | 521.1 > 278.1 | −45 | −100 | 3.76 |
| CTP-13C915N3 | 494.1 > 396.0 | −28 | −100 | 2.77 |
| GTP-13C1015N5 | 537.0 > 439.0 | −30 | −100 | 2.98 |
| UTP-13C9 | 492.0 > 394.1 | −29 | −100 | 2.75 |
| dATP-13C1015N5 | 505.1 > 407.0 | −33 | −100 | 4.94 |
| dCTP-13C915N3 | 478.1 > 380.1 | −31 | −100 | 3.25 |
| dGTP-13C1015N5 | 521.0 > 262.0 | −45 | −100 | 3.89 |
| dTTP-13C1015N2 | 493.0 > 395.0 | −29 | −100 | 3.95 |
Notably, ATP and dGTP were co-eluted, and they have an identical mass and similar fragmentation pattern. Nevertheless, because of the high ionization efficiency produced by the TEA – HFIP ion-paring mobile phase, two unique, yet sensitive transitions for specifically monitoring ATP (m/z, 506.0 > 272.9) and dGTP (m/z, 506.0 > 256.9) were identified (Figure 1), which enabled unambiguous determination of ATP and dGTP separately.
2.3. Sample Preparation
2.3.1. Stock solutions, calibration standards, and quality control samples
Stock solutions of NTPs, dNTPs and isotope-labeled internal standards were prepared in UltraPure DNAse/RNAse free water at a concentration of 10 mM, and stored at − 20°C. Working solutions of NTPs and dNTPs were prepared freshly on each day of analysis as serial dilutions in UltraPure water. Calibration standards and QC samples [including lower limit of quantitation (LLOQ), low, medium, and high QC] for ATP, CTP, GTP, dATP, dCTP, dGTP and dTTP were prepared by spiking 5 μL of working solution of the nucleotide standard mix (including ATP, CTP, GTP, dATP, dCTP, dGTP and dTTP) into 95 μL of the mobile phase A (containing isotope-labeled internal standards, 50 nM of each) to make the final concentrations of 0.5 – 500 nM. Because CTP (m/z, 482.0 > 384.1) and UTP (m/z, 483.0 > 385.0) exhibited 1 mass difference in their mass transitions and they were not chromatographically separated, the isotope of CTP (with the mass of 484 and abundance of ~11.6%) could influence the quantitation of UTP. To avoid this interference in the assay validation, a separate set of calibration standards and QC samples was prepared for UTP by spiking 5 μL of working solution of UTP standard into 95 μL of the mobile phase A (containing 50 nM of 13C9-UTP) to make the final concentrations of 0.5 – 500 nM. The concentrations of calibration standards and QC samples for individual NTPs and dNTPs are presented in Supplementary Table 1 and Table 2.
Table 2.
Intra- and inter-day precisions and accuracy for quality control (QC) samplesa.
| Compound | Nominal Conc. (nM) | Determined Conc. (nM)c | Accuracy (%)d | Intra-day (%) | Inter-day (%) |
|---|---|---|---|---|---|
| ATP | 1 (LLOQ) | 1.1±0.2 | 114.0 | 18.7 | 9.7 |
| 3 | 2.8±0.2 | 93.3 | 6.7 | 4.6 | |
| 200 | 194.6±14.0 | 97.3 | 6.5 | 3.8 | |
| 400 | 406.6±32.7 | 101.6 | 6.7 | 5.8 | |
| CTP | 0.5 (LLOQ) | 0.52 ± 0.06 | 103.5 | 12.2 | 2.3 |
| 1.5 | 1.5 ± 0.2 | 102.3 | 12.2 | -b | |
| 200 | 194.1 ± 14.2 | 97.0 | 7.4 | -b | |
| 400 | 396.5 ± 18.8 | 99.1 | 3.6 | 4.1 | |
| GTP | 5 (LLOQ) | 4.9 ± 0.8 | 97.8 | 15.7 | 4.9 |
| 15 | 16.2 ±1.9 | 108.1 | 12.3 | -b | |
| 200 | 197.7 ±16.2 | 98.8 | 8.5 | -b | |
| 400 | 413.6 ± 27.5 | 103.4 | 4.6 | -b | |
| UTP | 0.5 (LLOQ) | 0.58 ±0.08 | 115.2 | 14.9 | -b |
| 1.5 | 1.5 ± 0.1 | 99.4 | 7.7 | 3.9 | |
| 200 | 201.2 ±15.3 | 100.6 | 8.0 | -b | |
| 400 | 399.3 ± 30.6 | 99.8 | 7.4 | 2.5 | |
| dATP | 0.5 (LLOQ) | 0.53±0.07 | 105.4 | 16.4 | 7.2 |
| 1.5 | 1.4±0.1 | 95.2 | 10.1 | 2.0 | |
| 200 | 200.4±17.6 | 100.2 | 4.0 | 9.7 | |
| 400 | 410.2±38.7 | 102.6 | 5.8 | 9.7 | |
| dCTP | 0.5 (LLOQ) | 0.52±0.05 | 104.2 | 9.8 | -b |
| 1.5 | 1.4±0.1 | 92.5 | 7.8 | 5.1 | |
| 200 | 189.2±10.8 | 94.6 | 5.9 | -b | |
| 400 | 373.0±18.9 | 93.2 | 4.4 | 3.0 | |
| dGTP | 5 (LLOQ) | 5.6±0.7 | 111.6 | 10.3 | 9.8 |
| 15 | 1 3. 1 ±1 .5 | 87.3 | 10.6 | 4.7 | |
| 200 | 201.3±15.6 | 100.6 | 5.4 | 7.2 | |
| 400 | 442.9±20.9 | 110.7 | 4.6 | 1.4 | |
| dTTP | 0.5 (LLOQ) | 0.54±0.07 | 108.1 | 7.8 | 13.4 |
| 1.5 | 1.4±0.1 | 93.8 | 8.3 | 3.3 | |
| 200 | 199.9±11.5 | 100.0 | 3.0 | 6.4 | |
| 400 | 392.4±14.2 | 98.1 | 2.2 | 3.8 |
Each QC was performed in quintuplicate on three different days.
No additional variation was observed as a result of performing assay in different days.
Data presented as mean ± standard deviation of 15 replicates (quintuplicate on three different days).
Calculated as mean determined concentration / nominal concentration (n = 15).
2.3.2. Cell sample preparation
Frozen cell pellet samples (with 0.1 – 10 million cells per sample) were thawed and kept on ice. One mL of ice-cold methanol/water (60:40, v/v) (containing isotope-labeled internal standards, 4.75 nM of each) was added to each cell sample. The mixture was vortex-mixed, sonicated, and centrifuged (14,000 rpm, 4°C, 5 min). The supernatant was transferred into an Amicon® ultra-centrifugal filter with a molecular weight cutoff of 3000 Da (Merck Millipore Ltd. Ireland). The residual precipitate was further extracted by adding 1 mL of 60% ice-cold methanol, followed by vortex-mixing, sonication and centrifugation (14000 rpm, 4°C, 5 min). The supernatant was combined to the same ultra-centrifugal filter as above. Ultra-filtration was performed by centrifugation at 4°C and 9500 rpm for 4 h. The ultra-filtrate was transferred into a 2 ml tube and dried in a CentriVap Centrifugal Concentrator (Kansas City, MO) under vacuum at 4 °C. The residual was reconstituted in 100 μL of mobile phase A, followed by vortex-mixing and centrifugation (14,000 rpm, 4°C, 5 min). The supernatant was diluted 20-fold with the mobile phase A, and 10 μL was injected into the LC-MS/MS system.
2.4. Method Validation
2.4.1. Calibration curve, precision, and accuracy
Calibration curves were prepared by spiking known concentrations (0.5 – 500 nM) of NTPs and dNTPs in aqueous solution (i.e., mobile phase A) (Supplementary Table 1). Calibration curves were built by fitting the analyte concentrations versus the peak area ratios of the analyte to its respective isotope-labeled internal standard using the least-squares linear regression analysis with a weighting scheme of 1/x2.
Intra- and inter-day precision and accuracy were assessed for the calibration standards (each in duplicate) and QCs (including LLOQ, low, medium, and high QCs, each in quintuplicate) on three days. The accuracy was expressed as the relative percentage of the determined concentration to nominal concentration. The intra- and inter-day precisions were estimated by one-way analysis of variance (ANOVA) using the JMP™ statistical software version 5 (SAS Institute, Cary, NC, USA), as described previously [10].
2.4.2. Matrix effect
Since NTPs and dNTPs are naturally presented in cell samples, their isotope-labeled forms were used for the assessment of the absolute matrix effect of NTPs and dNTPs. Briefly, two sets of calibration standards (at the final concentrations of 10, 25, 50, 100, 250, and 500 nM) were prepared by spiking 5 μL of the working solution of isotople-labeled standard mix (including ATP, CTP, GTP, UTP, dATP, dCTP, dGTP and dTTP) into 95 μL of mobile phase A solution (set 1) and post-extracted cell matrix (prepared from a cell sample with ~ 1 million cells according to the cell sample preparation procedure as described in section 2.3.2) (set 2). Set 1 and 2 calibration standards were directly subjected to the LC-MS/MS analysis. The slope of a calibration curve was obtained by fitting the analyte concentrations versus peak areas using the least-squares linear regression analysis with a weighting scheme of 1/x2. The matrix effect was estimated as the slope ratio of the calibration curve prepared in post-extracted cell matrix (Set 2) to that in mobile phase A solution (Set 1) [11].
2.5. Applications
2.5.1. Quantitation of intracellular NTPs and dNTPs
The developed method was applied to determine the intracellular NTP and dNTP levels as a function of the expression of RRM1and RRM2, which are the regulatory and catalytic subunit of ribonucleotide reductase, respectively. One of functions of ribonucleotide reductase is to facilitate the conversion of NTPs to dNTPs, which are required for replication in S phase, and therefore, depletion of RRM1 or RRM2 could induce S-phase arrest in mammalian cells. To investigate the function of the RRM1 and RRM2 genes, intracellular concentrations of NTPs and dNTPs were determined in the non-small-cell lung cancer H23 cells with normal and knocked-down expression of RRM1 and RRM2. In brief, the H23 cells were plated in RPMI 1640 medium containing 10% fetal bovine serum. Twenty-four hours after plating, cells were transfected with siRNA RRM1, RRM2 alone, or both (Dharmacon on-TARGETplus Smartpool siRNA) by using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s protocol. Non-target Pool siRNA was used as the control. At 72 h after the transfection, cells were washed three times with ice-cold PBS and counted. Cell pellets were collected and stored at − 80C until analysis. Intracellular concentrations of NTPs and dNTPs were determined using the validated LC-MS/MS method.
2.5.2. Quantitative profiling of polar cellular metabolites
In an effort to develop a LC-MS based targeted metabolomic profiling platform, we applied the present ion pair LC-MS/MS method, with a minor modification of mobile phase gradient, to determine a number of highly polar, often ionized metabolic intermediates that are unsuitable for analysis with the reversed-phase or hydrophilic interaction liquid chromatography (HILIC) based LC-MS/MS methods. The modified mobile phase gradient was as follows: 0 – 5 min, mobile phase B increasing from 0% to 10%; 5 – 20 min, mobile phase B increasing from 10% to 100%; 20 – 21 min, mobile phase B maintaining at 100%; 21 – 22 min, mobile phase B decreasing from 100% to 0%; 22 – 30 min, mobile phase B maintaining at 0%. Mobile phase A consisted of 100 mM HFIP and 8.6 mM TEA in water (final pH, 8.3 ± 0.1), and mobile phase B consisted of 10% acetonitrile in mobile phase A. Flow rate was 0.5 mL/min. In addition to the quantitation of nucleoside triphosphates, the method could be used for quantitative profiling 74 polar metabolites including nucleoside mono- and bi-phosphates as well as other polar, ionized metabolic intermediates (including carbohydrate derivatives, carboxylic acid derivatives, co-acyl A derivatives, fatty acyls, and others).
The method was applied to profile polar metabolites in human lung cancer cell lines A549 and A549-Snail (with stable overexpression of Snail gene). In brief, A549 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum. DNA constructs for overexpressing Snail gene were generated by cloning the PCR-amplified full-length human Snail cDNA fragment into the doxycycline-inducible lentiviral expression vector pInducer20, as described previously [12]. Lentiviruses were packaged by co-transfection of the resultant pInducer20-Snail construct with 2nd generation packaging plasmids pMD2.G and psPAX2 into 293FT cells [12]. To establish A549-Snail cell lines stably over-expressing Snail in a doxycycline-dependent manner, A549 cells were transduced with the pInducer20-Snail lentivirus, and selected in the media containing 500 ug/ml G418. For inducible overexpression, A549-Snail cells were treated with 2 ug/ml doxycycline for 3 days. Then, cells were rinsed two times with ice-cold water, flashed frozen in liquid nitrogen to quench cellular metabolism, and then scraped in ice-cold 80% methanol. Cell samples were kept at −80°C until analysis.
3. Results and Discussion
3.1. Impact of the ion pair mobile phase on chromatographic resolution and electrospray ionization
Separation of nucleoside triphosphates on an Atlantis T3 column was based on the ion pair, reversed-phase chromatographic principle. The selection of an appropriate ion pairing mobile phase is critical to achieving high-resolution chromatographic separation as well as high-sensitivity electrospray ionization. TEA – acetic acid or TEAA and similar ion pair reagents (e.g., tributylammonium acetate, tetrabutylammonium acetate, and dimethylhexylamine) were commonly used for the ion pair LC-MS/MS analysis of nucleotides [4-8]. Nevertheless, these ion pair reagents are not particularly volatile, thus resulting in inefficient electrospray ionization. Figure 2 shows the extracted chromatograms of a mixture of NTPs and dNTPs (at a concentration of 5 or 1 μM) using the TEAA ion pair mobile phase. While reasonable chromatographic resolution was achieved as TEAA concentrations ranged from 5 to 100 mM (Figure 2A-2C), the electrospray ionization signal intensity was insufficient for accurate quantitation of NTPs/dNTPs at concentrations < 1 μM (Figure 2D). Based on the previous report that the addition of more volatile HFIP into the TEA-based ion-pair mobile phase enhanced electrospray ionization signals of oligonucleotides [9], we investigated if the addition of HFIP into the TEA-based mobile phase could also improve ionization of nucleoside triphosphates. The impact of HFIP concentrations on the chromatographic resolution and MS signal intensity is shown in Figure 3. As increasing concentrations of HFIP (5, 25, 50 and 100 mM) were added into TEA (8.6 mM) mobile phase, the chromatographic resolution was improved and moreover, the MS signal intensity was enhanced (Figure 3A-3D). Notably, compared to the TEAA (100 mM, pH 7.0) mobile phase, the combination of HFIP (100 mM) and TEA (8.6 mM) increased the MS signal intensity by about 50-fold, while retaining comparable chromatographic resolution (Figure 2D and Figure 3D). Also noted was that the mobile phase containing HFIP (100 mM, adjusted pH to 8.3 by NH4OH) alone in the absence of TEA failed to produce chromatographic retention and resolution of NTPs and dNTPs (Figure 3E), suggesting that, although HFIP can enhance electrospray ionization signal, it is alone not an effective ion pair reagent and therefore, it should be used in combination with TEA. Based on these data, an ion pair mobile phase based on TEA (8.6 mM) and HFIP (100 mM) as the buffering acid was selected in the present method.
Figure 2.
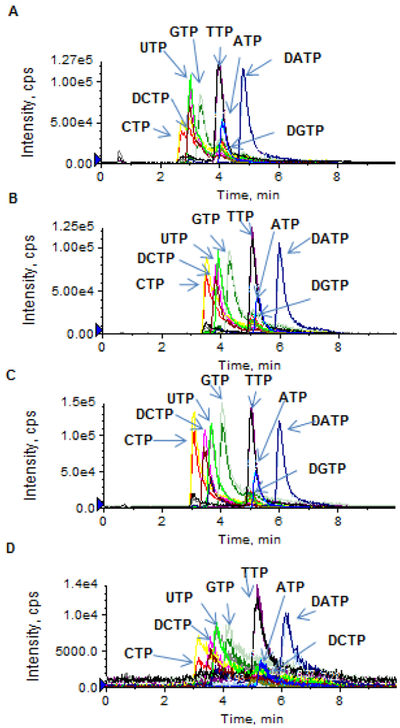
(A – C) Extracted ion chromatograms of a standard mixture of dATP, dCTP, dGTP, dTTP, ATP, CTP, GTP, and UTP (5 μM in aqueous solution), using the traditional TEAA ion-paring mobile phase (pH 7.0) with different TEAA concentrations: 5 mM (A), 25 mM (B), and 100 mM (C). (D) Extracted ion chromatogram of a standard mixture of dATP, dCTP, dGTP, dTTP, ATP, CTP, GTP, and UTP (1 μM in aqueous solution), using the TEAA ion-paring mobile phase (pH 7.0) with the TEAA concentration at 100 mM. Chromatographic separation was performed on an Atlantis T3 (2.1× 100 mm, 3 μm) column, under a gradient mobile phase consisting of mobile phase A (TEAA, pH 7.0) and mobile phase B (10% acetonitrile in mobile phase A), with gradient B% (min) as 0 (0) → 60 (6.0) → 0 (6.1) → 0 (10) at a flow rate of 0.5 mL/min.
Figure 3.
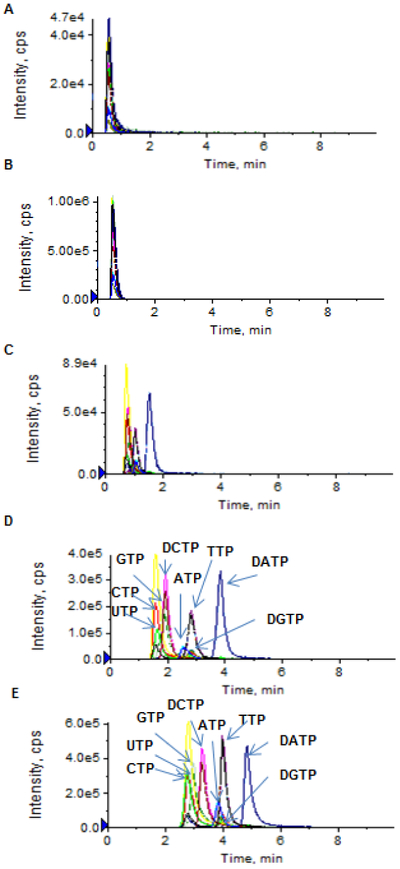
(A – D) Extracted ion chromatograms of a standard mixture of dATP, dCTP, dGTP, dTTP, ATP, CTP, GTP, and UTP (1 μM in aqueous solution), using the TEA-HFIP ion-paring mobile phase with TEA at 8.6 mM and HFIP at different concentrations: 5 mM (A), 20 mM (B), 50 mM (C), and 100 mM (D). (E) Extracted ion chromatograms of dATP, dCTP, dGTP, dTTP, ATP, CTP, GTP, and UTP (at 1 μM in aqueous solution), using a mobile phase containing HFIP only (100 mM, adjusted pH to 8.3 by NH4OH). Chromatographic separation was achieved on an Atlantis T3 (2.1× 100 mm, 3 μm) column, under a gradient mobile phase consisting of mobile phase A (TEA + HFIP or HFIP only) and mobile phase B (10% acetonitrile in mobile phase A), with gradient B% (min) as 0 (0) → 10 (6.0) → 0 (6.1) → 0 (10) at a flow rate of 0.5 mL/min.
In addition to the type of ion pair buffer system, the mobile phase gradient slope has to be well chosen. Since the retention of nucleotides on a reverse-phase HPLC column is based on the formation of ion pairs between the positively charged ion pairing agent and the negatively charged phosphate group of nucleotides, a more aqueous mobile phase appears to retain nucleotides better. As shown in Figure 4, a better retention and separation of NTPs and dNTPs was achieved as the gradient slope was decreased from 0.67% increase of acetonitrile per minute to 0.16% increase of acetonitrile per minute. These data suggest that a shallow gradient slope (i.e., 0.16% increase of acetonitrile per minute) is needed to achieve a good chromatographic resolution. It should be mentioned that the formation of shallow gradients can place performance demands on the HPLC pumps and mixers, and therefore, it is recommended that the mobile phase B contains a premix blend of aqueous and organic solvents (i.e., 10% of acetonitrile in aqueous mobile phase A) to minimize potentially inadequate solvent mixing that can compromise component resolution.
Figure 4.

Extracted ion chromatograms of a standard mixture of dATP, dCTP, dGTP, dTTP, ATP, CTP, GTP, and UTP (200 nM in aqueous solution), using the TEA-HFIP ion-paring mobile phase with different gradient slope of organic solvent. Chromatographic separation was performed on an Atlantis T3 (2.1× 100 mm, 3 μm) column, under a gradient mobile phase consisting of mobile phase A (8.6 mM TEA + 100 mM HELP) and mobile phase B containing 100% (A), 50% (B), or 10% (C) acetonitrile in mobile phase A. The gradient program was set as B% (min): 0(0) → 10 (6.0) → 0 (6.1) → 0 (10).
3.2. Linearity, precision, and accuracy
Several approaches have been used to prepare calibration curves for quantifying endogenous compounds that are naturally presented in biological samples. One approach is the addition calibration method that involves adding known amounts of standard to multiple aliquots of the biological sample [13]. While it can compensate for the matrix effect that could potentially enhance or suppress the analyte signal, this method is time-consuming and difficult to implement for the analysis of a large number of samples containing multiple analytes with concentrations ranging from low nanomolar to high micromolar [13], which is the case of endogenous NTPs and dNTPs in biological samples. A simpler approach is to prepare calibration curve in aqueous solution. However, this method does not take into account potential matrix effects that may modify the MS response. To overcome these limitations, the isotope-labeled internal standard method was applied in the present study, where the calibration curve was prepared by spiking a serial dilution of NTPs/dNTP standards and fixed concentrations of their respective isotope-labeled internal standards in aqueous solution (i.e., mobile phase A). In parallel, the same concentration of individual isotope-labeled internal standard was spiked in processed cell samples. Then, NTP/dNTP concentrations were determined based on the peak area ratio of the analyte to its respective internal standard. As the analyte and its stable isotope-labeled analog share similar chromatographic behavior and MS response, the isotope-labeled internal standard method allows correcting for variations in the matrix effect between the calibrators and biological samples [14].
The LLOQs were determined at 0.5 nM for CTP, UTP, dATP, dCTP and dTTP, at 1 nM for ATP, and at 5 nM for GTP and dGTP, in aquous solution. Linear calibration curves were established over the concentration range of 0.5 – 500 nM for CTP, UTP, dATP, dCTP and dTTP, 1 – 500 nM for ATP, and 5 – 500 nM for GTP and dGTP (Supplementary Table 1). A linear correlation coefficient (R2) of > 0.99 was obtained in all analytical runs. For all calibrator standards and QC samples (including LLOQ, low, medium, and high QCs), the intra- and inter-day precision and accuracy were within the generally accepted criteria for bioanalytical method validation (Table 2 and Supplementary Table 1).
One limitation of the present assay was that the natural isotope of CTP (with the mass of 484.0 and abundance of ~11.6%) may interfere the quantitation of UTP because CTP (m/z, 482.0 > 384.1) and UTP (m/z, 483.0 > 385.0) exhibited 1 mass difference and they were not chromatographically separated. The magnitude of this interference is dependent on the relative abundance of CTP compared to UTP in a sample. In our measured cell samples, CTP concentrations were less than 50% of UTP concentrations in general (Figure 6B), and thus the contribution of CTP isotope (with the mass of 484.0 and abundance of ~11.6%) to determined UTP concentration was insignificant (< 6%). However, caution is needed for samples containing higher CTP concentration than UTP, where UTP concentration could be significantly over-estimated due to the contribution of CTP isotope.
Figure 6.
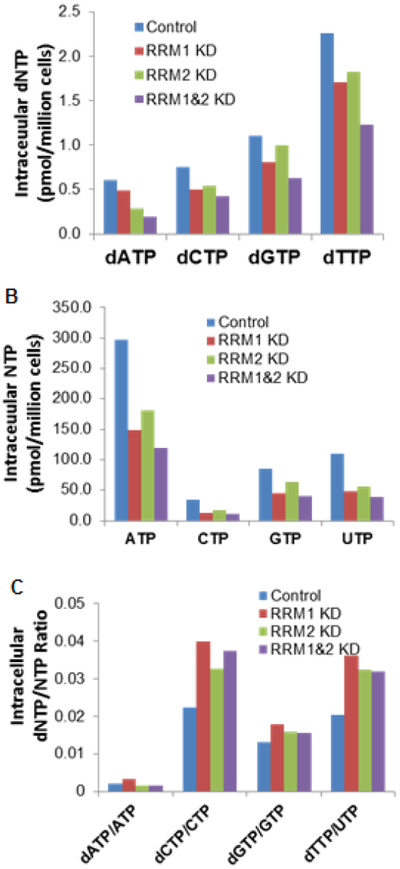
(A and B) Intracellular concentrations of dNTPs and NTPs, and (C) intracellular dNTP/NTP ratios, in H23 non-small cell lung cancer cells with normal and knocked-down expression of RRM1/RRM2.
3.3. Matrix effect
Given the virtually identical chemical structure of an analyte and its stable isotope-labeled analog, the matrix effect of isotope-labeled NTP and dNTP analogs determined from cell matrix were used to represent the absolute matrix effect of their respective NTPs and dNTPs. No apparent matrix effect influencing ionization of NTPs and dNTPs (with matrix effect of 72% – 95%) was observed for all isotople-labeled NTPs and dNTPs in the cell extract (prepared from ~ 1 million cells) (Table 3), suggesting the sample preparation procedure (involving protein precipitation and ultrafiltration) effectively reduced matrix effect from cellular samples. However, it was observed that the matrix effect became significant in cellular samples containing > 10 million cells, which could impact the chromatographic resolution and ionization. As shown in Figure 5, the retention time and peak shape of individual NTP/dNTP in the 20-fold diluted cell extract were comparable to those of the standards in aqueous solution; whereas, in the undiluted cell extract (containing ~10 million cells), the retention time significantly shifted and the peak shape was distorted and moreover, the ionization efficiency was reduced. These observations suggest that cell matrix components such as other competing ions, if presented at a high concentration, could not only greatly disrupt the normal nucleotide-TEA ion-paring equilibrium, but also suppress the ionization signal. Dilution of cell extracts with the mobile phase A (containing 100 mM HFIP and 8.6 mM TEA in water, pH 8.3) can effectively reduce, if not completely abolish, the matrix effect. Based on these data and given the high sensitivity of the method, cell extracts were diluted 20-fold with the mobile phase A for all cell sample analyses.
Table 3.
Matrix effect of isotope-labeled NTPs and dNTPs from cell pellets (with ~1 million cells)
| Setl curvea | Set2 curvea | Matrix factorb | |
|---|---|---|---|
| ATP | A=2822C – 2821, R2= 0.998 | A=2394C – 1622, R2= 0.999 | 0.85 |
| CTP | A=14308C – 15077, R2= 0.999 | A=11335C – 24583, R2=0.999 | 0.79 |
| GTP | A=11233C – 11401, R2 = 0.998 | A=10441C – 20729, R2=0.999 | 0.93 |
| UTP | A=6977C – 3371, R2=0.999 | A=5015C – 7256, R2=0.999 | 0.72 |
| dATP | A=10957C – 1951, R2=0.999 | A=9587C – 14755, R2=0.999 | 0.88 |
| dCTP | A=7535C – 3241, R2=0.998 | A=5761C – 5377, R2=0.999 | 0.76 |
| dGTP | A=1528C – 3667, R2=0.998 | A=1448C – 2030, R2=0.999 | 0.95 |
| dTTP | A=7196C ± 2428, R2=0.998 | A=5741C – 8465, R2=0.999 | 0.80 |
Two sets of calibration standards (at the final concentrations of 10, 25, 50, 100, 250, and 500 nM) were prepared by spiking 10 μL of the working solution of isotople-labeled standard mix into 90 μL of mobile phase A solution (set 1), or 90 μL of post-extracted cell matrix that was prepared from a cell sample with ~ 1 million cells (set 2). Set 1 and 2 calibration standards were directly subjected to the LC-MS/MS analysis.
The matrix factor was calculated as the slope ratio of the calibration curve prepared in post-extracted cell matrix (Set 2) to that in mobile phase A solution (Set 1). Matrix factor within 0.85 – 1.15 indicates no apparent matrix effect; matrix factor < 0.85 indicates ion suppression; and matrix factor > 1.15 indicates ion enhancement.
Figure 5.

Multiple-reaction monitoring (MRM) chromatograms of dATP (490.0 > 392.1), dCTP (466.0 > 367.9), dGTP (506.0 >256.9), dTTP (480.9 > 383.1), ATP (506.0 > 272.9), CTP (482.0 > 384.1), GTP (522.0 > 424.0), and UTP (483.0 > 384.9) for: (A1-A8) a standard mixture (containing 200 nM of ATP, CTP, GTP, UTP, dATP, dCTP, dGTP and dTTP) in aqueous solution; (B1-B8) a cellular extract sample diluted 20-fold with the mobile phase A (8.6 mM TEA + 100 mM HFIP); and (C) the undiluted cellular extract sample (containing ~10 million cells). Chromatographic separation was achieved on an Atlantis T3 (2.1× 100 mm, 3 μm) column, under gradient mobile phase consisting of mobile phase A (8.6 mM TEA + 100 mM HFIP) and mobile phase B (10% acetonitrile in mobile phase A), with gradient B% (min) as 0 (0) → 10 (6.0) → 0 (6.1) → 0 (10) at a flow rate of 0.5 mL/min.
3.4. Application
As an application example, the developed method was applied to determine intracellular concentrations of NTPs and dNTPs in the H23 cells with normal and silent RRM1 and RRM2 expression (Figure 6). Notably, the intracellular levels of dNTPs were ~ 2 orders of magnitude lower than those of NTPs. The knockdown of either the RRM1 or RRM2 gene reduced the intracellular levels of all dNTPs (including dATP, dCTP, dGTP, and dTTP), and the knockdown of both genes resulted in an additive effect on the reduction of dNTP levels (Figure 6A). These data are in line with the notion that the RRM1 and RRM2 play an important role in facilitating the formation of dNTPs. Interestingly, the knockdown of RRM1, RRM2, or both genes resulted in the decrease of intracellular NTP levels to a greater extent than the decrease of dNTP levels (Figure 6B). Consequently, the dNTP/NTP ratios were increased in the RRM1/RRM2 knock-down cells, as compared to those in the control cells (Figure 6C). Studies are currently ongoing to elucidate the exact molecular mechanism underlying these observations.
In addition to the determination of intracellular NTPs and dNTPs, the developed ion pair LC-MS/MS method, with a minor modification, had a broad application for quantitative profiling of 74 polar, ionized intracellular metabolites including nucleoside mono- and bi-phosphates, carbohydrate derivatives, carboxylic acid derivatives, co-acyl A derivatives, fatty acyls, and others (Table 4). The retention time and calibration curve range for these metabolites are summarized in Table 4. The quantitative reliability of the method was demonstrated by the negligible shift of retention times (i.e., < 5% for all analytes during 2 month period), correlation coefficient (R) > 0.99 for linear calibration curves, and intra- and inter-day variation < 15% for repetitive quality control sample analysis. The method was successfully applied to quantitative profiling of these highly polar metabolic intermediates in lung cancer cell lines, A549 and A549-Snail (with overexpression of Snail). The partial least square discriminant analysis (PLS-DA) indicated a clear separation between A549 and A549-Snail cell lines, suggesting the overexpression of Snail gene significantly influenced the metabolomic profile of the lung cancer cell line A549 (Fig. 7). The underlying molecular mechanism is currently under investigation.
Table 4.
The list of metabolites that can be quantitatively determined by the developed ion pair LC-MS/MS method.
| Metabolite Name | MS/MS transition |
Retention Time (min)a |
Curve Range (μM)b |
Rc |
|---|---|---|---|---|
| Carbohydrate derivatives | ||||
| 2-Phosphoglyceric acid | 185.1 > 78.9 | 1.46±0.02 | 0.01-10 | 0.997 |
| ADP-glucose | 590.0 > 428.1 | 1.02±0.05 | 0.05-1 | 0.989 |
| Deoxyribose 5-phosphate | 213.0 > 78.9 | 0.96±0.01 | 0.01-1 | 0.994 |
| D-Erythrose 4-phosphate | 199.0 > 96.9 | 0.86±0.01 | 0.01-1 | 0.993 |
| D-Glyceraldehyde 3-phosphate | 169.0 > 151.0 | 0.91±0.01 | 0.05-2 | 0.981 |
| D-Ribose 5-phosphate | 213.0 > 78.9 | 0.87±0.01 | 0.01-1 | 0.994 |
| D-Sedoheptulose 7-phosphate | 289.0 > 97.0 | 0.84±0.01 | 0.01-1 | 0.994 |
| Fructose 1,6-bisphosphate | 339.0 > 97.0 | 2.67±0.06 | 0.02-0.5 | 0.993 |
| Fructose 6-phosphate | 258.9 > 96.9 | 0.85±0.01 | 0.01-1 | 0.994 |
| Gluconic acid | 195.1 >129.0 | 0.67±0.01 | 0.01-1 | 0.995 |
| Glucose 1-phosphate | 259.0 > 241.0 | 0.85±0.01 | 0.01-1 | 0.995 |
| Glucose 6-phosphate | 259.0 > 96.7 | 0.84±0.01 | 0.01-1 | 0.995 |
| Glucuronic acid | 193.0 > 113.0 | 0.66±0.01 | 0.01-2 | 0.989 |
| N-Acetyl-glucosamine 1-phosphate | 300.0 > 79.0 | 0.91±0.01 | 0.01-1 | 0.995 |
| Sucrose | 341.1 > 178.9 | 0.60±0.00 | 0.01-0.5 | 0.990 |
| UDP-glucose | 565.1 > 323.0 | 0.84±0.01 | 0.01-2 | 0.998 |
| Carboxylic acids and derivatives | ||||
| 1-Methylhistidine | 170.1 > 124.1 | 1.50±0.01 | 0.05-5 | 0.985 |
| Argininosuccinic acid | 291.1 > 70.1 | 0.71±0.01 | 0.05-0.5 | 0.951 |
| Asymmetric dimethylarginine | 203.2 > 70.2 | 1.09±0.01 | 0.01-2 | 0.995 |
| N-Acetylputrescine | 131.2 > 114.1 | 0.97±0.05 | 0.2-2 | 0.985 |
| CoA derivatives | ||||
| 3-Hydroxy-3-methylglutaryl-CoA | 912.1 > 405.1 | 10.02±0.06 | 0.01-10 | 0.992 |
| 3-Hydroxybutyryl-CoA | 854.3 > 347.2 | 10.48±0.07 | 0.01-5 | 0.992 |
| Acetyl-CoA | 810.1 > 303.2 | 11.08±0.07 | 0.02-10 | 0.970 |
| Coenzyme A | 768.3 > 261.2 | 8.71±0.10 | 0.1-1 | 0.988 |
| Dephospho-CoA | 686.5 > 339.1 | 4.72±0.09 | 0.1-10 | 0.996 |
| Propionyl-CoA | 824.3 > 317.2 | 13.17±0.09 | 0.05-5 | 0.994 |
| Succinyl-CoA | 866.2 > 408.2 | 9.52±0.06 | 0.01-10 | 0.998 |
| Fatty acyls | ||||
| 2-keto-D-gluconic acid | 193.0 > 103.0 | 0.68±0.00 | 0.01-1 | 0.996 |
| Carnitine | 162.3 > 103.0 | 0.81±0.01 | 0.01-1 | 0.987 |
| Hydroxyisocaproic acid | 131.2 > 85.0 | 2.15±0.02 | 0.01-10 | 0.996 |
| L-2-Hydroxyglutaric acid | 147.0 > 128.9 | 0.93±0.01 | 0.01-1 | 0.995 |
| L-Acetylcarnitine | 204.1 > 85.0 | 1.71±0.02 | 0.01-10 | 0.994 |
| L-Malic acid | 132.9 > 114.9 | 0.91±0.01 | 0.01-2 | 0.991 |
| Miscellaneous Metabolites | ||||
| 1-Methylnicotinamide | 137.1 > 94.0 | 0.76±0.01 | 0.01-1 | 0.994 |
| 7-Methylguanosine | 298.1 > 166.1 | 0.82±0.01 | 0.01-0.5 | 0.989 |
| Adenosine | 268.2 >136.1 | 0.93±0.11 | 0.2-10 | 0.991 |
| Choline | 104.1 > 60.1 | 0.87±0.06 | 0.2-1 | 0.991 |
| Oxoglutaric acid | 144.9 > 101.0 | 1.01±0.08 | 0.1-10 | 0.991 |
| Phosphorylcholine | 184.2 > 125.0 | 0.97±0.01 | 0.02-2 | 0.980 |
| S-Adenosylmethionine | 399.2 > 250.0 | 0.87±0.02 | 0.02-2 | 0.986 |
| Shikimic acid | 173.0 > 93.0 | 0.69±0.01 | 0.05-2 | 0.991 |
| Thiamine | 265.2 > 122.1 | 3.18±0.04 | 0.01-2 | 0.992 |
| Thiamine pyrophosphate | 425.2 > 122.1 | 4.39±0.07 | 0.05-1 | 0.991 |
| Nucleotides | ||||
| Adenosine diphosphate ribose | 558.1 > 346.1 | 1.03±0.02 | 0.01-10 | 0.997 |
| Adenosine phosphosulfate | 426.0 > 346.0 | 1.13±0.02 | 0.01-2 | 0.997 |
| ADP | 428.2 > 136.0 | 1.92±0.54 | 0.1-5 | 0.989 |
| AMP | 348.1 > 136.1 | 1.20±0.02 | 0.02-1 | 0.990 |
| ATP | 506.0 > 272.9 | 3.69±0.07 | 0.05-1 | 0.993 |
| CDP | 402.0 > 158.8 | 1.53±0.03 | 0.01-5 | 0.996 |
| CTP | 482.0 > 384.1 | 2.79±0.06 | 0.02-0.5 | 0.992 |
| dAMP | 332.2 > 136.0 | 1.87±0.02 | 0.02-2 | 0.993 |
| dATP | 490.0 > 392.1 | 4.68±0.08 | 0.01-1 | 0.993 |
| dCDP | 386.1 > 159.0 | 1.902±0.04 | 0.01-10 | 0.996 |
| dCTP | 466.0 > 368.0 | 3.24±0.06 | 0.01-2 | 0.995 |
| dGDP | 428.2 > 152.1 | 2.46±0.11 | 0.05-10 | 0.987 |
| dGMP | 348.2 > 152.1 | 1.37±0.03 | 0.1-2 | 0.981 |
| dGTP | 506.0 > 257.0 | 3.80±0.07 | 0.05-0.5 | 0.990 |
| dTDP | 401.0 > 158.8 | 2.60±0.05 | 0.01-10 | 0.997 |
| dTMP | 321.1 > 195.0 | 1.60±0.03 | 0.01-2 | 0.996 |
| dTTP | 480.9 > 383.0 | 3.867±0.07 | 0.01-2 | 0.994 |
| dUMP | 307.0 > 195.0 | 1.15±0.02 | 0.01-2 | 0.997 |
| FAD | 784.1 > 437.0 | 8.69±0.08 | 0.01-10 | 0.995 |
| Flavin Mononucleotide | 457.2 > 439.1 | 10.38±0.14 | 0.2-10 | 0.996 |
| GDP | 442.0 > 344.0 | 1.67±0.04 | 0.02-10 | 0.997 |
| GMP | 364.2 > 152.0 | 1.02±0.02 | 0.02-0.2 | 0.988 |
| GTP | 522.0 > 424.2 | 2.99±0.07 | 0.1-5 | 0.990 |
| NAD± | 664.2 > 136.1 | 0.85±0.01 | 0.01-0.5 | 0.991 |
| NADP± | 744.0 > 604.1 | 2.24±0.05 | 0.02-10 | 0.992 |
| UDP | 403.1 > 158.8 | 1.51±0.03 | 0.01-10 | 0.997 |
| Uridine diphosphate-N- acetylglucosamine |
606.1 > 385.0 | 0.88±0.01 | 0.01-10 | 0.997 |
| UTP | 483.0 > 385.0 | 2.77±0.07 | 0.02-1 | 0.994 |
Shown as the mean ± standard deviation from 4 independent calibration curves.
Calibration curves were prepared by spiking the standards into Mobile phase A (consisting of 100 mM HFIP and 8.6 mM TEA in water, pH = 8.3).
Shown as the mean from 4 independent calibration curves.
Figure 7.

Partial least square discriminant analysis (PLS-DA) score plots indicating discrimination between the parental A549 cells and A549-Snail cells (with overexpression of Snail). Symbols (▲ or +) represent three biological replicates. Ellipses represent 95% confidence intervals.
4. Conclusion
An ion pair LC-MS/MS method using TEA – HFIP ion-pair mobile phase was optimized and validated for simultaneous and unambiguous determination of 8 nucleoside triphosphates (including ATP, CTP, GTP, UTP, dATP, dCTP, dGTP, and dTTP) in cellular samples. Compared to the less volatile ion-pair reagent (e.g., TEAA), the TEA – HFIP ion-pair reagent produced efficient chromatographic separation as well as high-sensitivity electrospray ionization. A sample preparation procedure involving protein precipitation and ultrafiltration effectively reduced the matrix effect. The isotope-labeled internal standard method was used for the quantitation. Lower limits of quantitation were determined at 0.5 nM for CTP, UTP, dATP, dCTP, and dTTP, at 1 nM for ATP, and at 5 nM for GTP and dGTP. The intra- and inter-day precision and accuracy were within the generally accepted criteria for bioanalytical method validation. While the present method was validated for the quantitation of intracellular nucleoside triphosphates, it had a broad application potential for quantitative profiling of a variety of polar intracellular metabolites including nucleoside mono- and bi-phosphates, carbohydrate derivatives, carboxylic acid derivatives, co-acyl A derivatives, fatty acyls, and others in biological samples.
Supplementary Material
Highlights:
An ion pair LC-MS/MS method using trimethylamine – hexafluoroisopropanol
Validated for the quantitation of intracellular nucleoside triphosphates
Broad application potential for profiling of a variety of highly polar metabolites
Acknowledgments
This study was supported by the United States Public Health Service Cancer Center Support Grant P30 CA022453 and the National Institute of Health Grant R01 CA129343.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Stryer L, Biochemistry (3rd ed.), W. H. Freeman, New York, 1988. [Google Scholar]
- [2].Walton HF, Analytical chemistry, 40 (1968) 51R–62R. [DOI] [PubMed] [Google Scholar]
- [3].Witters E, Van Dongen W, Esmans EL, Van Onckelen HA, Journal of chromatography. B, Biomedical sciences and applications, 694 (1997) 55–63. [DOI] [PubMed] [Google Scholar]
- [4].Cohen S, Megherbi M, Jordheim LP, Lefebvre I, Perigaud C, Dumontet C, Guitton J, Journal of chromatography. B, Analytical technologies in the biomedical and life sciences, 877 (2009) 3831–3840. [DOI] [PubMed] [Google Scholar]
- [5].Claire RL 3rd, Rapid communications in mass spectrometry : RCM, 14 (2000) 1625–1634. [DOI] [PubMed] [Google Scholar]
- [6].Chen P, Liu Z, Liu S, Xie Z, Aimiuwu J, Pang J, Klisovic R, Blum W, Grever MR, Marcucci G, Chan KK, Pharmaceutical research, 26 (2009) 1504–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Qian T, Cai Z, Yang MS, Analytical biochemistry, 325 (2004) 77–84. [DOI] [PubMed] [Google Scholar]
- [8].Michopoulos F, Whalley N, Theodoridis G, Wilson ID, Dunkley TP, Critchlow SE, Journal of chromatography. A, 1349 (2014) 60–68. [DOI] [PubMed] [Google Scholar]
- [9].Apffel A, Chakel JA, Fischer S, Lichtenwalter K, Hancock WS, Analytical chemistry, 69 (1997) 1320–1325. [DOI] [PubMed] [Google Scholar]
- [10].Wiegand R, Wu J, Sha X, LoRusso P, Li J, Journal of chromatography. B, Analytical technologies in the biomedical and life sciences, 878 (2010) 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Matuszewski BK, Journal of chromatography. B, Analytical technologies in the biomedical and life sciences, 830 (2006) 293–300. [DOI] [PubMed] [Google Scholar]
- [12].Meerbrey KL, Hu G, Kessler JD, Roarty K, Li MZ, Fang JE, Herschkowitz JI, Burrows AE, Ciccia A, Sun T, Schmitt EM, Bernardi RJ, Fu X, Bland CS, Cooper TA, Schiff R, Rosen JM, Westbrook TF, Elledge SJ, Proceedings of the National Academy of Sciences of the United States of America, 108 (2011) 3665–3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Harris DC, Quantitative Chemical Analysis, W.H. Freeman, New York, 2003. [Google Scholar]
- [14].Wu J, Wiegand R, LoRusso P, Li J, Journal of chromatography. B, Analytical technologies in the biomedical and life sciences, 941 (2013) 100–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.