

Abstract
The accumulation of pathological tau in the brain is associated with neuronal deterioration and cognitive impairments in tauopathies including Alzheimer’s disease. Tau, while primarily localized in axons of healthy neurons, accumulates in the soma and dendrites of neurons under pathogenic conditions. Tau is found in both presynaptic and postsynaptic compartments of neurons in Alzheimer’s disease. New research supports that soluble forms of tau trigger pathophysiology in the brain by altering properties of synaptic and neuronal function at the early stages of disease progression, before neurons die. Here we review current understanding of how tau-mediated synaptic and neuronal dysfunction contributes to cognitive decline. Delineating the mechanisms by which pathogenic tau alters synapses, dendrites and axons will help lay the foundation for new strategies to restore neuronal function in tauopathy.
Introduction
The finely tuned electrochemical communication between neurons in the brain is essential for cognition and behavior. In neurodegenerative tauopathies, the ability of neurons to encode information is compromised leading to cognitive decline and behavioral impairments. Tauopathies, including Alzheimer’s disease (AD), are characterized by increased levels of pathologically aggregated tau in the brain that correlate with the deterioration of synapses and neurons [1,2]. In AD, tau localization is abnormally shifted from axons into the somatodendritic compartment, a process associated with accumulation of amyloid β [3]. The mechanistic links between amyloid β (Aβ) and tau in causing synapse dysfunction have been reviewed by others recently [4,5]. Here, we will focus on tau-mediated neuronal and synapse dysfunction.
Tau is normally enriched in the axons. The current prevailing view is that aberrant forms of mislocalized or secreted tau is toxic to neurons in disease [6]. Importantly, studies on tauopathy mouse models support that toxic tau triggers neuronal dysfunction underlying cognitive impairments without widespread neuron loss [7,8]. While the majority of tauopathy cases are sporadic, there are over fifty familial tau mutations that cause frontotemporal lobar degeneration with tau inclusions (FTLD-tau). A recent review of current strategies for the diagnosis and therapeutic intervention in tauopathies provides insight into the challenges associated with understanding these diseases [9]. Many studies that have shaped the tauopathy field were done on transgenic mice that express human tau carrying familial FTLD-tau mutations that cause frontotemporal dementia (FTD) [7,10–16]. These transgenic mouse lines exhibit different degrees of age-dependent tau pathology, pathophysiology, neuron loss, and behavioral deficits. However, there is a strong link between neuronal dysfunction and cognitive decline among the tauopathy mouse models.
The spreading of toxic tau across neuronal circuits may accelerate pathogenesis in the brain. Growing efforts to uncover the mechanisms that promote tau propagation in neurodegenerative diseases have been reviewed by others recently [17,18]. In this review, we will focus on recent advances to delineate the sequence of events triggered by tau that lead to synapse decline and neuronal dysfunction in tauopathy. We will highlight new findings about the identities of toxic forms of tau how toxic tau disrupts the function of synapses, dendrites and axons during pathogenesis (Figure 1).
Figure 1.
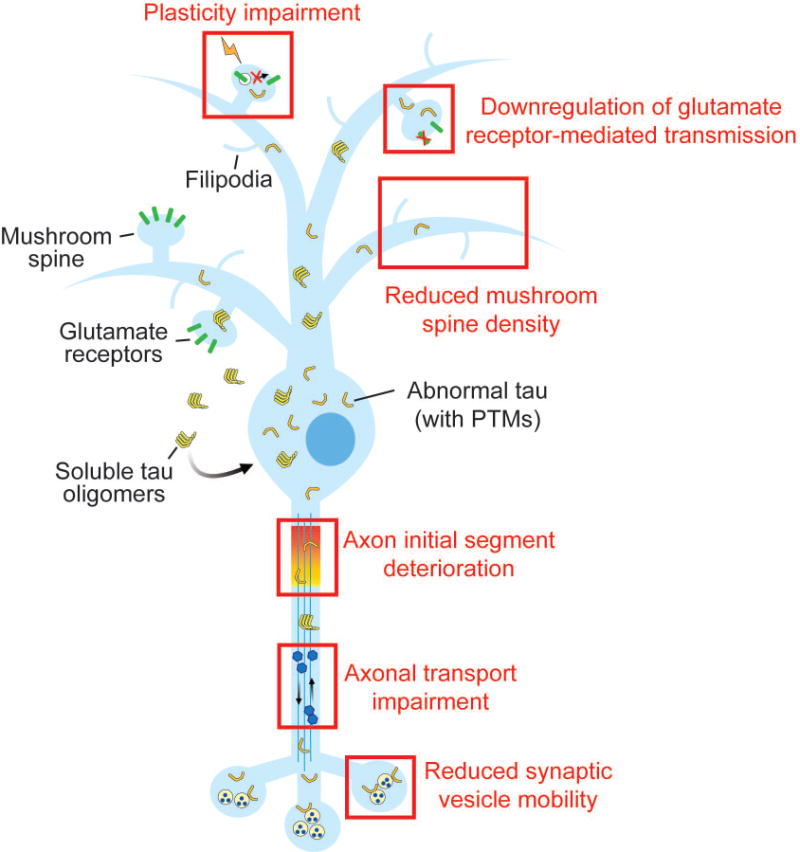
Pathogenic forms of tau, including soluble tau oligomers and tau with abnormal post-translational modifications (PTMs), can promote neuronal dysfunction by multiple mechanisms at the early stages of neurodegenerative disease. At postsynaptic sites, pathogenic tau reduces glutamate receptor levels and inhibits receptor trafficking during synaptic plasticity. The tau-mediated loss of mushroom spines is associated with increased filipodia on dendrites. Axonal function is compromised by tau due to the destabilization of the axon initial segment and deficient axonal transport. At the presynaptic terminal, tau decreases the release of neurotransmitter-containing synaptic vesicles by reducing their mobility in the terminal.
Soluble tau oligomers vs. insoluble aggregates in neurons
The accumulation and spread of insoluble tau inclusions in the form of neurofibrillary tangles (NFTs) is considered a pathological hallmark of tauopathies. However, soluble forms of tau oligomers, not NFTs, have emerged a as key toxic species in causing neuronal dysfunction and cognitive decline [10,19]. Tau oligomers accumulate in the human brain in AD [20,21] and progressive supranuclear palsy [22], and increased levels of multimeric tau correlate with memory decline in human tau P301L mutant mice (rTg4510) [23]. Injection of recombinant human tau oligomers, but not human tau monomers or fibrils, into mouse brain impaired memory [24]. Passive immunization targeting soluble tau oligomers exerted protective effects in a mouse model of tauopathy [25].
At the cellular level, exogenously applied human tau oligomers triggered the aggregation and somatodendritic mislocalization of endogenous tau in cultured mouse neurons [26]. At the synapse level, tau oligomers caused internal calcium dysregulation and loss of both synapsin and synaptophysin at presynaptic terminals in human iPSC-derived neurons [27•]. In contrast to the toxicity induced by soluble tau oligomers, there is evidence that insoluble tau oligomers may be non-toxic and even protective for neuron function [28]. Indeed, it is possible that various forms of tau oligomers exist and that their toxicity may depend on oligomer solubility, stoichiometry, structure, or tau post-translational modifications (PTMs).
Familial FTLD tau mutants in synapses
Expression of human tau with FTLD mutations in mice triggers abnormal activity at anatomically distinct synaptic connections. Transgenic mice with human tau carrying P301S or P301L familial FTD-causing mutations have impaired long-term potentiation (LTP) at the Schaffer collateral-CA1 synapses in the hippocampus [7,14]. Mice expressing human tau with the familial FTD V337M mutation have reduced excitatory synaptic transmission in the ventral striatum, associated with decreased PSD-95 and synaptic glutamate receptors levels in both the ventral striatum and the insular cortex [11]. Enhancing NMDA-type glutamate receptor (NMDAR) activation in these mice restored ventral striatal neuron firing and reversed FTD-associated repetitive behaviors. In two independent lines of transgenic mice expressing human tau with the A152T FTD-associated mutation, mossy fiber-CA3 synaptic transmission was found to be elevated without alterations of LTP expression [15,16]. A152T tau or wildtype tau overexpression increased synaptic vesicle release probability in acute slices from aged mice [15], and the A152T mutation increased levels of glutamate and excitotoxicity in hippocampal slice cultures, likely via extra-synaptic mechanisms [16]. The selective vulnerability of particular synapses to tau toxicity may depend on their individual physiological properties and synaptic machinery.
More detailed analyses of FTD-causing tau mutants on presynaptic function was performed at the Drosophila neuromuscular junction. The expression of three human FTLD-tau mutants, R406W, V337M or P301L, increased F-actin enrichment and reduced the mobility of synaptic vesicles in presynaptic boutons [29]. More recently, they showed that tau appears to regulate vesicle release and presynaptic function through a direct interaction with a vesicular protein called Synaptogyrin-3 [30•]. Our understanding of how FTD-causing tau mutations alter presynaptic vesicle release is quickly expanding. Further investigations into the molecular mechanisms by which FTLD mutant tau affects the integrity of synaptic transmission may uncover additional tau-interacting proteins at synapses.
Tau with aberrant post-translational modifications in synapses
Tau undergoes extensive PTMs, including phosphorylation, acetylation, ubiquitination, methylation, glycosylation, and proteolytic cleavages [31]. While some of the PTMs occur under physiological conditions, aberrant PTMs on tau could play critical roles in triggering synapse dysfunction and deterioration. In cultured hippocampal neurons, expression of a tau mutant that mimics the phosphorylation of 14 residues reduced the number of AMPA-type glutamate receptors (AMPAR) at synapses [14]. In a series of studies, we identified aberrantly acetylated lysines on tau that are associated with impairments in hippocampal synaptic plasticity and memory. Mice expressing human tau with mutations to mimic the acetylation of K274 and K281 had deficits in spatial and pattern separation memory associated with obstructed LTP in the dentate gyrus [32•]. Mechanistically, the acetylated tau mimic causes the loss of postsynaptic KIBRA and impaired AMPAR trafficking during LTP. Importantly, treating a tauopathy mouse model with salsalate, a drug that reduces tau acetylation in the brain, protected against memory deficits [33]. New evidence suggests that cleaved forms of tau may also contribute to synaptotoxicity. Blocking the caspase-2 cleavage of human P301L tau by mutating the Asp314 residue on tau restored AMPAR-mediated synaptic transmission in neurons as well as memory deficits in mice [34••]. Moreover, the caspase-3 cleavage of tau at the Asp421 residue may contribute to tau propagation and synaptotoxicity in neurons [35,36]. There are growing efforts to identify key toxic tau PTMs and strategies to reduce their formation or to neutralize their toxicity, which could lead to new therapeutic interventions.
Pathogenic tau impairs dendritic spines
The strength of synaptic transmission is tightly linked to spine structural changes [37]. The size, morphology, or density spines on dendrites can change in response to neuronal activity and experience [37–39]. A recent study provided evidence that in the early stages of AD, spine density is reduced and the retrieval of stored memory, but not the encoding of long-term memory, is impaired. Using mice to model the early stages of AD, they showed that optogenetic induction of LTP in engram-specific dentate granule cells increased dendritic spine density and restored memory deficits [40••]. This works highlights the contribution of spine loss to AD-related cognitive decline.
Consistent with the susceptibility of synaptic transmission to toxic tau, dendritic spines on excitatory neurons are particularly vulnerable in a tauopathy mouse model. Interestingly, in cortical neurons expressing human P301L mutant tau, the number of mushroom spines, considered stable and functional, was reduced. In contrast, the number of filipodia, considered unstable and nonfunctional, was increased [41]. The preservation of filopodia in the presence of toxic tau suggests that it might be possible to restore functional dendritic spines by strengthening synaptic connections on filipodia.
Pathogenic tau impairs axonal functions
Axons support the functional output of neurons. The axon initial segment (AIS) is a specialized region of the axon where action potential firing is initiated. The location and length of the AIS regulates neuronal excitability. In transgenic mice expressing human P301L tau, a shift of the AIS away from the soma was associated with the reduced excitability of CA1 neurons [42]. The AIS also acts as a structural barrier for the axonal compartment. Marked deterioration of the AIS structure was reported in an AD mouse model, which could underlie the missorting of axonal proteins to dendrites [43]. We found that expression of acetyl-mimic tau on K274 and K281 led to destabilization of cytoskeletal-associated proteins in the AIS [44], resulting in missorting of tau to the somatodendritic compartment, where tau is particularly toxic.
Axons maintain neuronal function by the anterograde and retrograde transport of cargo [45]. In Drosophila motor neurons, expression of the human 3 repeat (3R) tau isoform, but not the 4 repeat (4R) isoform, led to the accumulation of vesicles in axons and caused premature lethality [46], most likely by impairing axonal transport. The exact mechanism underlying this impairment is unclear. In transgenic mice expressing Aβ and human tau, a tau-mediated reduction of kinesin-1 light chain was observed in association with impaired anterograde trafficking in axons [47]. Collectively, these studies support that pathogenic tau can disrupt the structural components and molecular machinery in axons, which serve to support the functional output of neurons.
Conclusions
Increasing evidence supports tau playing a key role setting into motion the early pathogenic events that contribute to cognitive decline in neurodegenerative disease. Recent advances reviewed herein shape our current understanding of how pathogenic tau in neurons contributes to synaptic and neuronal dysfunction. Tau-mediated neuronal decline may also involve non-neuronal cells in the brain. Recently, microglia, TREM2 and inflammation have been implicated in tau-mediated pathogenesis and the deterioration of neurons [48,49], but how microglia and inflammation in neurodegenerative disease contribute to synaptic and neuronal dysfunction is not well understood. Given that toxic tau can affect the regulation of postsynaptic strength, dendritic spine density, the AIS, axonal transport, and presynaptic vesicle release dynamics, there are multiple mechanisms by which tau can promote neuronal dysfunction. Moving forward, it will be important to dissect the tau-dependent mechanisms underlying these functional effects and explore new approaches that can restore neuronal function and cognition in tauopathy.
Highlights.
Soluble tau oligomers promote pathophysiology in neurons
Pathogenic tau affects mechanisms that regulate both presynaptic and postsynaptic function
Deterioration of axons in tauopathy involves the dysregulation of the axon initial segment and impaired axonal transport
Loss of dendritic spines underlies memory impairments in tauopathy mouse models
Acknowledgments
We thank Erica Nguyen and Todd Paulson for administrative assistance and Maria Telpoukhovskaia and Grant Kauwe for discussions and comments. This work was supported by the National Institutes of Health (1R01AG054214 and U54NS100717 to L.G), the Tau Consortium (to L.G.), and the BrightFocus Foundation (to T.E.T).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ingelsson M, Fukumoto H, Newell KL, Growdon JH, Hedley-Whyte ET, Frosch MP, Albert MS, Hyman BT, Irizarry MC. Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology. 2004;62:925–931. doi: 10.1212/01.wnl.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- 2.Giannakopoulos P, Herrmann FR, Bussiere T, Bouras C, Kovari E, Perl DP, Morrison JH, Gold G, Hof PR. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology. 2003;60:1495–1500. doi: 10.1212/01.wnl.0000063311.58879.01. [DOI] [PubMed] [Google Scholar]
- 3.Kowall NW, Kosik KS. Axonal disruption and aberrant localization of tau protein characterize the neuropil pathology of Alzheimer’s disease. Ann Neurol. 1987;22:639–643. doi: 10.1002/ana.410220514. [DOI] [PubMed] [Google Scholar]
- 4.Forner S, Baglietto-Vargas D, Martini AC, Trujillo-Estrada L, LaFerla FM. Synaptic Impairment in Alzheimer’s Disease: A Dysregulated Symphony. Trends Neurosci. 2017;40:347–357. doi: 10.1016/j.tins.2017.04.002. [DOI] [PubMed] [Google Scholar]
- 5.Guerrero-Munoz MJ, Gerson J, Castillo-Carranza DL. Tau Oligomers: The Toxic Player at Synapses in Alzheimer’s Disease. Front Cell Neurosci. 2015;9:464. doi: 10.3389/fncel.2015.00464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morris M, Maeda S, Vossel K, Mucke L. The many faces of tau. Neuron. 2011;70:410–426. doi: 10.1016/j.neuron.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 8.Van der Jeugd A, Hochgrafe K, Ahmed T, Decker JM, Sydow A, Hofmann A, Wu D, Messing L, Balschun D, D’Hooge R, et al. Cognitive defects are reversible in inducible mice expressing pro-aggregant full-length human Tau. Acta Neuropathol. 2012;123:787–805. doi: 10.1007/s00401-012-0987-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Orr ME, Sullivan AC, Frost B. A Brief Overview of Tauopathy: Causes, Consequences, and Therapeutic Strategies. Trends Pharmacol Sci. 2017;38:637–648. doi: 10.1016/j.tips.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Warmus BA, Sekar DR, McCutchen E, Schellenberg GD, Roberts RC, McMahon LL, Roberson ED. Tau-mediated NMDA receptor impairment underlies dysfunction of a selectively vulnerable network in a mouse model of frontotemporal dementia. J Neurosci. 2014;34:16482–16495. doi: 10.1523/JNEUROSCI.3418-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tatebayashi Y, Miyasaka T, Chui DH, Akagi T, Mishima K, Iwasaki K, Fujiwara M, Tanemura K, Murayama M, Ishiguro K, et al. Tau filament formation and associative memory deficit in aged mice expressing mutant (R406W) human tau. Proc Natl Acad Sci U S A. 2002;99:13896–13901. doi: 10.1073/pnas.202205599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang B, Higuchi M, Yoshiyama Y, Ishihara T, Forman MS, Martinez D, Joyce S, Trojanowski JQ, Lee VM. Retarded axonal transport of R406W mutant tau in transgenic mice with a neurodegenerative tauopathy. J Neurosci. 2004;24:4657–4667. doi: 10.1523/JNEUROSCI.0797-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, Lanier LM, Yuan LL, et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68:1067–1081. doi: 10.1016/j.neuron.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maeda S, Djukic B, Taneja P, Yu GQ, Lo I, Davis A, Craft R, Guo W, Wang X, Kim D, et al. Expression of A152T human tau causes age-dependent neuronal dysfunction and loss in transgenic mice. EMBO Rep. 2016;17:530–551. doi: 10.15252/embr.201541438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Decker JM, Kruger L, Sydow A, Dennissen FJ, Siskova Z, Mandelkow E, Mandelkow EM. The Tau/A152T mutation, a risk factor for frontotemporal-spectrum disorders, leads to NR2B receptor-mediated excitotoxicity. EMBO Rep. 2016;17:552–569. doi: 10.15252/embr.201541439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goedert M, Eisenberg DS, Crowther RA. Propagation of Tau Aggregates and Neurodegeneration. Annu Rev Neurosci. 2017;40:189–210. doi: 10.1146/annurev-neuro-072116-031153. [DOI] [PubMed] [Google Scholar]
- 18.Kruger L, Mandelkow EM. Tau neurotoxicity and rescue in animal models of human Tauopathies. Curr Opin Neurobiol. 2016;36:52–58. doi: 10.1016/j.conb.2015.09.004. [DOI] [PubMed] [Google Scholar]
- 19.Sydow A, Van der Jeugd A, Zheng F, Ahmed T, Balschun D, Petrova O, Drexler D, Zhou L, Rune G, Mandelkow E, et al. Tau-induced defects in synaptic plasticity, learning, and memory are reversible in transgenic mice after switching off the toxic Tau mutant. J Neurosci. 2011;31:2511–2525. doi: 10.1523/JNEUROSCI.5245-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patterson KR, Remmers C, Fu Y, Brooker S, Kanaan NM, Vana L, Ward S, Reyes JF, Philibert K, Glucksman MJ, et al. Characterization of prefibrillar Tau oligomers in vitro and in Alzheimer disease. J Biol Chem. 2011;286:23063–23076. doi: 10.1074/jbc.M111.237974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Sarmiento J, Troncoso J, Jackson GR, Kayed R. Identification of oligomers at early stages of tau aggregation in Alzheimer’s disease. Faseb j. 2012;26:1946–1959. doi: 10.1096/fj.11-199851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gerson JE, Sengupta U, Lasagna-Reeves CA, Guerrero-Munoz MJ, Troncoso J, Kayed R. Characterization of tau oligomeric seeds in progressive supranuclear palsy. Acta Neuropathol Commun. 2014;2:73. doi: 10.1186/2051-5960-2-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berger Z, Roder H, Hanna A, Carlson A, Rangachari V, Yue M, Wszolek Z, Ashe K, Knight J, Dickson D, et al. Accumulation of pathological tau species and memory loss in a conditional model of tauopathy. J Neurosci. 2007;27:3650–3662. doi: 10.1523/JNEUROSCI.0587-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Clos AL, Jackson GR, Kayed R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol Neurodegener. 2011;6:39. doi: 10.1186/1750-1326-6-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Castillo-Carranza DL, Gerson JE, Sengupta U, Guerrero-Munoz MJ, Lasagna-Reeves CA, Kayed R. Specific targeting of tau oligomers in Htau mice prevents cognitive impairment and tau toxicity following injection with brain-derived tau oligomeric seeds. J Alzheimers Dis. 2014;40(Suppl 1):S97–s111. doi: 10.3233/JAD-132477. [DOI] [PubMed] [Google Scholar]
- 26.Swanson E, Breckenridge L, McMahon L, Som S, McConnell I, Bloom GS. Extracellular Tau Oligomers Induce Invasion of Endogenous Tau into the Somatodendritic Compartment and Axonal Transport Dysfunction. J Alzheimers Dis. 2017;58:803–820. doi: 10.3233/JAD-170168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27•.Usenovic M, Niroomand S, Drolet RE, Yao L, Gaspar RC, Hatcher NG, Schachter J, Renger JJ, Parmentier-Batteur S. Internalized Tau Oligomers Cause Neurodegeneration by Inducing Accumulation of Pathogenic Tau in Human Neurons Derived from Induced Pluripotent Stem Cells. J Neurosci. 2015;35:14234–14250. doi: 10.1523/JNEUROSCI.1523-15.2015. This study demonstrates that extracellular tau oligomers, but not monomers, promote the accumulation of intracellular hyperphosphorylated tau associated with synapse deterioration in human iPSC-derived neurons. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cowan CM, Quraishe S, Hands S, Sealey M, Mahajan S, Allan DW, Mudher A. Rescue from tau-induced neuronal dysfunction produces insoluble tau oligomers. Sci Rep. 2015;5:17191. doi: 10.1038/srep17191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou L, McInnes J, Wierda K, Holt M, Herrmann AG, Jackson RJ, Wang YC, Swerts J, Beyens J, Miskiewicz K, et al. Tau association with synaptic vesicles causes presynaptic dysfunction. Nat Commun. 2017;8:15295. doi: 10.1038/ncomms15295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30•.McInnes J, Wierda K, Snellinx A, Bounti L, Wang YC, Stancu IC, Apostolo N, Gevaert K, Dewachter I, Spires-Jones TL, et al. Synaptogyrin-3 Mediates Presynaptic Dysfunction Induced by Tau. Neuron. 2018 doi: 10.1016/j.neuron.2018.01.022. This study identifies a novel interaction of tau with Synaptogyrin-3, a transmembrane synaptic vesicle protein. Reducing Synaptogyrin-3 expression restored vesicle mobility and release in neurons that express pathogenic tau. [DOI] [PubMed] [Google Scholar]
- 31.Morris M, Knudsen GM, Maeda S, Trinidad JC, Ioanoviciu A, Burlingame AL, Mucke L. Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat Neurosci. 2015;18:1183–1189. doi: 10.1038/nn.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32•.Tracy TE, Sohn PD, Minami SS, Wang C, Min SW, Li Y, Zhou Y, Le D, Lo I, Ponnusamy R, et al. Acetylated Tau Obstructs KIBRA-Mediated Signaling in Synaptic Plasticity and Promotes Tauopathy-Related Memory Loss. Neuron. 2016;90:245–260. doi: 10.1016/j.neuron.2016.03.005. This paper shows that LTP and hippocampal-associated memory in mice is disrupted by the expression of tau with mutations to mimic acetylation on two lysine residues. The tau-induced plasticity deficit was due to the loss of postsynaptic KIBRA and impaired AMPA receptor trafficking. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Min SW, Chen X, Tracy TE, Li Y, Zhou Y, Wang C, Shirakawa K, Minami SS, Defensor E, Mok SA, et al. Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat Med. 2015;21:1154–1162. doi: 10.1038/nm.3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34••.Zhao X, Kotilinek LA, Smith B, Hlynialuk C, Zahs K, Ramsden M, Cleary J, Ashe KH. Caspase-2 cleavage of tau reversibly impairs memory. Nat Med. 2016;22:1268–1276. doi: 10.1038/nm.4199. This study demonstrates that blocking the caspase-2 mediated cleavage of tau at Asp 314 restores synapse function and prevents the mislocalization of tau into dendritic spines. The reduction of caspase-2 levels ameliorated memory deficits in a tauopathy mouse model. [DOI] [PubMed] [Google Scholar]
- 35.Kim Y, Choi H, Lee W, Park H, Kam TI, Hong SH, Nah J, Jung S, Shin B, Lee H, et al. Caspase-cleaved tau exhibits rapid memory impairment associated with tau oligomers in a transgenic mouse model. Neurobiol Dis. 2016;87:19–28. doi: 10.1016/j.nbd.2015.12.006. [DOI] [PubMed] [Google Scholar]
- 36.Nicholls SB, DeVos SL, Commins C, Nobuhara C, Bennett RE, Corjuc DL, Maury E, Eftekharzadeh B, Akingbade O, Fan Z, et al. Characterization of TauC3 antibody and demonstration of its potential to block tau propagation. PLoS One. 2017;12:e0177914. doi: 10.1371/journal.pone.0177914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Engert F, Bonhoeffer T. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature. 1999;399:66–70. doi: 10.1038/19978. [DOI] [PubMed] [Google Scholar]
- 39.Trachtenberg JT, Chen BE, Knott GW, Feng G, Sanes JR, Welker E, Svoboda K. Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature. 2002;420:788–794. doi: 10.1038/nature01273. [DOI] [PubMed] [Google Scholar]
- 40••.Roy DS, Arons A, Mitchell TI, Pignatelli M, Ryan TJ, Tonegawa S. Memory retrieval by activating engram cells in mouse models of early Alzheimer’s disease. Nature. 2016;531:508–512. doi: 10.1038/nature17172. This study provides evidence that a mouse model of AD retains the ability to store memory, but the mice have an impairment in memory retrieval. They further show that optogenetic stimulation of memory engram neurons in vivo can restore dendritic spines and memory in the AD mouse model. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crimins JL, Rocher AB, Luebke JI. Electrophysiological changes precede morphological changes to frontal cortical pyramidal neurons in the rTg4510 mouse model of progressive tauopathy. Acta Neuropathol. 2012;124:777–795. doi: 10.1007/s00401-012-1038-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hatch RJ, Wei Y, Xia D, Gotz J. Hyperphosphorylated tau causes reduced hippocampal CA1 excitability by relocating the axon initial segment. Acta Neuropathol. 2017;133:717–730. doi: 10.1007/s00401-017-1674-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marin MA, Ziburkus J, Jankowsky J, Rasband MN. Amyloid-beta plaques disrupt axon initial segments. Exp Neurol. 2016;281:93–98. doi: 10.1016/j.expneurol.2016.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44•.Sohn PD, Tracy TE, Son HI, Zhou Y, Leite RE, Miller BL, Seeley WW, Grinberg LT, Gan L. Acetylated tau destabilizes the cytoskeleton in the axon initial segment and is mislocalized to the somatodendritic compartment. Mol Neurodegener. 2016;11:47. doi: 10.1186/s13024-016-0109-0. This study demonstrates that abnormally acetylated tau reduces the cytoskeletal-associated proteins in the AIS. The compromised AIS enables the mislocalization of tau into the somatodendritic compartment of neurons. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maday S, Twelvetrees AE, Moughamian AJ, Holzbaur EL. Axonal transport: cargo-specific mechanisms of motility and regulation. Neuron. 2014;84:292–309. doi: 10.1016/j.neuron.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sealey MA, Vourkou E, Cowan CM, Bossing T, Quraishe S, Grammenoudi S, Skoulakis EMC, Mudher A. Distinct phenotypes of three-repeat and four-repeat human tau in a transgenic model of tauopathy. Neurobiol Dis. 2017;105:74–83. doi: 10.1016/j.nbd.2017.05.003. [DOI] [PubMed] [Google Scholar]
- 47.Sherman MA, LaCroix M, Amar F, Larson ME, Forster C, Aguzzi A, Bennett DA, Ramsden M, Lesne SE. Soluble Conformers of Abeta and Tau Alter Selective Proteins Governing Axonal Transport. J Neurosci. 2016;36:9647–9658. doi: 10.1523/JNEUROSCI.1899-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leyns CEG, Ulrich JD, Finn MB, Stewart FR, Koscal LJ, Remolina Serrano J, Robinson GO, Anderson E, Colonna M, Holtzman DM. TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc Natl Acad Sci U S A. 2017;114:11524–11529. doi: 10.1073/pnas.1710311114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bemiller SM, McCray TJ, Allan K, Formica SV, Xu G, Wilson G, Kokiko-Cochran ON, Crish SD, Lasagna-Reeves CA, Ransohoff RM, et al. TREM2 deficiency exacerbates tau pathology through dysregulated kinase signaling in a mouse model of tauopathy. Mol Neurodegener. 2017;12:74. doi: 10.1186/s13024-017-0216-6. [DOI] [PMC free article] [PubMed] [Google Scholar]