

SUMMARY
How transcription affects genome 3D organization is not well understood. We found that during influenza A (IAV) infection, rampant transcription rapidly reorganizes host cell chromatin interactions. These changes occur at the ends of highly transcribed genes, where global inhibition of transcription termination by IAV NS1 protein causes read-through transcription for hundreds of kilobases. In these read-through regions, elongating RNA polymerase II disrupts chromatin interactions by inducing cohesin displacement from CTCF sites, leading to locus decompaction. Read-through transcription into heterochromatin regions switches them from the inert (B) to the permissive (A) chromatin compartment and enables transcription factor binding. Data from non-viral transcription stimuli show that transcription similarly affects cohesin-mediated chromatin contacts within gene bodies. Conversely, inhibition of transcription elongation allows cohesin to accumulate at previously transcribed intragenic CTCF sites and to mediate chromatin looping and compaction. Our data indicate that transcription elongation by RNA polymerase II remodels genome 3D architecture.
Keywords: influenza A virus, transcription, genome 3D structure, cohesin, CTCF, read-through transcription, chromatin compaction
Graphical Abstract
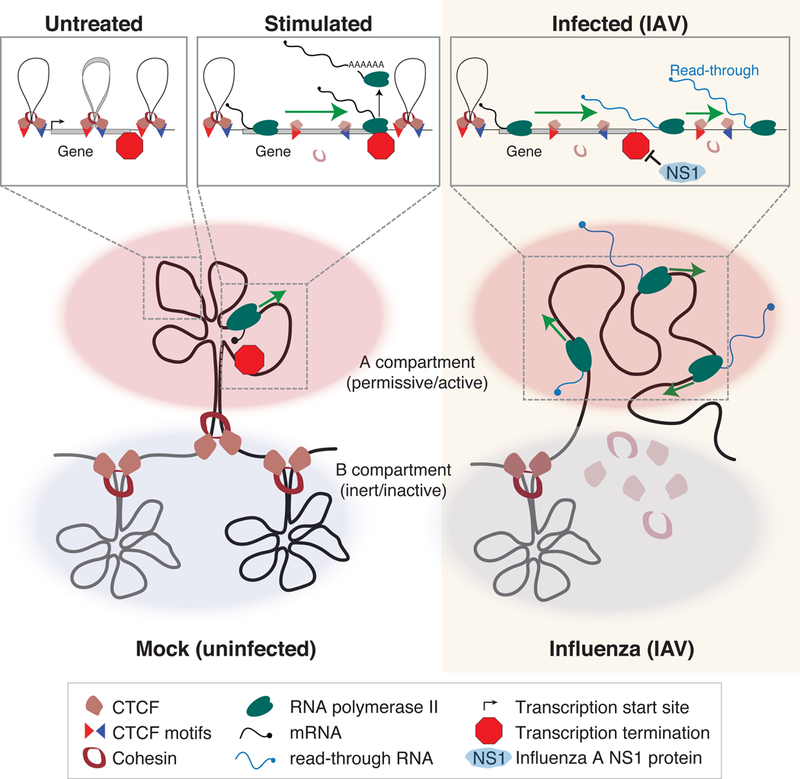
In Brief
Elevated transcription can displace cohesin to remodel genome architecture
INTRODUCTION
In the nucleus of eukaryotic cells, the genome is spatially organized in a non-random fashion. The chromatinized DNA segregates into an active “A” compartment, which contains gene-dense regions that are often transcribed and rich in “active” epigenetic modifications, and an inactive “B” compartment, which encompasses gene-poor, inactive chromatin bearing repressive epigenetic marks (Cremer et al., 2000; Lieberman-Aiden et al., 2009). Compartments represent the long-distance interaction patterns of the enclosed genome segments that preferentially interact with each other, which range in size from ~7 Mb (Lieberman-Aiden et al., 2009) down to 1 kb (Rowley et al., 2017). On a local scale, DNA is folded into loops, loop domains (Rao et al., 2014) and topologically associating domains (TADs) (Dixon et al., 2012; Nora et al., 2012). In mammalian cells, the loop anchors frequently contain convergently oriented motifs for the architectural factor CTCF (Rao et al., 2014). Mounting evidence suggests loop formation is highly dynamic and can be explained by active extrusion of DNA though the ring-shaped cohesin complex. According to the loop extrusion model, cohesin is loaded onto the genome by the Nipped B-like (NIPBL) cohesin loading complex and extrudes DNA, forming a DNA loop (Fudenberg et al., 2016; Gassler et al., 2017; Haarhuis et al., 2017; Sanborn et al., 2015). CTCF bound to convergent CTCF motifs seems to serve as a barrier to extrusion, where cohesin accumulates at CTCF-anchored loops (Haarhuis et al., 2017; Wutz et al., 2017). Depletion of cohesin or its loading factor NIPBL leads to loss of the local interactions underlying loops and loop domains (Rao et al., 2017; Schwarzer et al., 2017). This release of genomic regions from their local environment enables chromatin previously trapped by cohesin to switch compartments and engage in long-range interactions with other chromatin units with similar epigenetic makeup (Haarhuis et al., 2017; Rao et al., 2017; Schwarzer et al., 2017). In the combined absence of CTCF and the cohesin unloading factor WAPL, RNA polymerase II (RNAPII) translocates cohesin from gene bodies to regions downstream, indicating that transcription may play a role in genome 3D organization (Busslinger et al., 2017).
To explore the relationship between transcription and genome structure in a highly dynamic system, we characterized the genome-wide effects of influenza A virus (IAV) infection on transcription, epigenetic status, and chromatin structure. IAV is a negative-stranded RNA orthomyxovirus that replicates its genome in the nucleus of host cells. In addition to eliciting a robust host transcriptional response, IAV has been shown to impact nuclear morphology (Terrier et al., 2014). Viral replication requires transcription of the host genome (Mark et al., 1979), and viral proteins interact with multiple components of the host transcriptional machinery to promote viral replication and limit host defense responses (Marazzi et al., 2012). How the massive viral replicative load and its anti-host defenses affect the spatial configuration of chromatin and function of the host genome in the nucleus is unknown.
We report that IAV infection induces dynamic alterations in host genome 3D organization. These were associated with read-through transcription past the ends of genes, caused by inhibition of transcription termination by IAV non-structural protein 1 (NS1). Read-through transcription resulted in local chromatin decompaction and frequent B-to-A compartment switching downstream of highly transcribed genes. Chromatin loops in read-through regions were disrupted where elongating RNAPII traveled through and cohesin was lost from CTCF sites. We observed a similar relationship between transcription and genome 3D structure in uninfected cells responding to non-viral stimuli within the gene bodies of strongly regulated genes. Our findings suggest that transcribing RNAPII contributes to genome 3D organization dynamics by displacing cohesin from chromatin, leading to loss of local cohesin-mediated interactions and locus decompaction.
RESULTS
Influenza A virus infection affects host cell chromatin organization
Macrophages are among the first responders to IAV infection and play a critical role in disease outcome (Schneider et al., 2014). To examine whether IAV infection affects host genome 3D architecture, we infected post-mitotic human monocyte-derived macrophages (MDM) with IAV and harvested 6, 12, or 18 hours post-infection. For each time point, we captured chromatin interactions by in situ Hi-C (Rao et al., 2014) and sequenced rRNA-depleted RNA (total RNA-seq) of mock- and IAV-infected MDM from two separate donors (Table S1). Exposure to IAV induced rapid and dramatic changes in both host transcriptome and genome 3D structure (Figures S1A-C), including the remodeling of long-range chromatin interactions at anti-viral response gene loci (e.g. IFIT2 locus, Figures 1A and S1C). To characterize locus-specific chromatin interaction changes induced by IAV infection, we introduced two Hi-C metrics that quantify how compact local chromatin is packaged (Figure 1B) and verified them in a system where transcription induces locus decompaction (Jubb et al., 2017) (Figure S1D). We quantify the local compaction level of a region as the ratio of its distal (> 3 Mb distance) to local (< 3 Mb distance) genomic interactions on the same chromosome (distal-to-local ratio, DLR; Figure 1B). To assess a region’s interactions with other chromosomes, we calculate the ratio of inter- chromosomal contacts of a region relative to its total number of interactions (inter-chromosomal fraction, ICF; Figure 1B). Increases in the DLR or ICF imply the locus becomes decompacted, enabling it to make more interactions with distal sites on the same chromosome and with other chromosomes. With these metrics we observed both locus-specific and global changes in 3D structure following IAV infection, and a progressive decline of host cell 3D organization over the infection time course (Figure S1C). Already at 6 hours post-infection (hpi) many specific genomic regions exhibited evidence of switching from the B to the A compartment, decompaction and increased interchromosomal interactions (Figure 1A). To avoid the confounding effects of host shutoff (Bercovich-Kinori et al., 2016) and secondary infection associated with late time points, we focused our subsequent analyses on the 6-hour time point.
Figure 1. Influenza A virus (IAV) infection perturbs host cell genome 3D structure and induces transcription read-through at response gene.
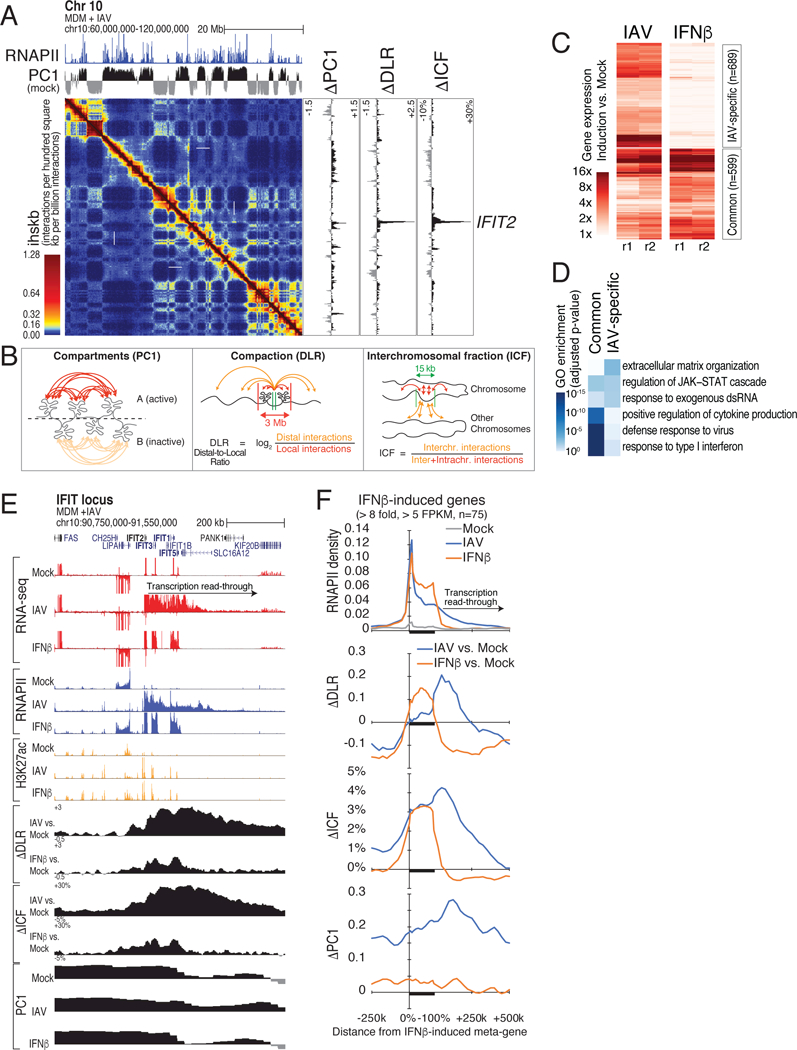
(A) IFIT locus in situ Hi-C contact maps of mock-infected (lower left) and lAV-infected (upper right) primary human monocyte-derived macrophages (MDM) at 6 hours post infection (hpi) (merge of 2 replicates per condition). Top, PC1 values (compartments) and RNAPII ChIP-seq read densities in mock-infected cells. Right, changes in PC1, distal-to-local interaction ratio (DLR) and interchromosomal fraction of interactions (ICF) between IAV and mock conditions. White arrows: IAV-induced changes in chromatin interactions at the IFIT2 locus. (B) Schematic defining chromatin A & B compartments (PC1) and chromatin compaction (DLR, ICF). (C) Hierarchical clustering of genes significantly regulated by IAV and IFNβ (RNA-seq, 2 replicates each, log2FC > 3, FDR<5%). (D) Top-enriched GO terms gene sets induced by both IAV and IFNβ, or IAV alone. (E) Genome browser tracks of total RNA-seq, ChIP-seq (RNAPII/H3K27ac), compartments changes (PC1) and compaction ΔDLR, ΔICF) after IAV infection or IFNβ treatment at the IFIT family locus. (F) Meta-gene profiles of changes in PC1, DLR, ICF and RNAPII after IAV or IFNβ treatment at highly IFNβ-responsive genes (FC > 8, FPKM > 5; 75 genes, see Table S2). See also Figure S1.
IAV infection-induced chromatin changes are associated with transcription downstream of highly active genes
RNA-seq and Hi-C revealed that the most prominent changes in chromatin structure occurred downstream of genes highly induced by IAV infection. Many of these genes are canonical targets of the type I/III interferon pathway (Figures 1C and1D), which dominates the host cell response to IAV infection (Crotta et al., 2013). In order to separate the impact of the virus from the host transcriptional response, we profiled IFNβ-treated cells as a control. While IFNβ stimulation of MDM increased RNA and RNAPII levels at many of the same genes, it only induced chromatin decompaction within gene bodies and did not affect regions downstream (Figures 1E, 1F and S1E). With IAV infection, regions downstream of genes showed increased decompaction and B-to-A compartment switching, and were highly enriched for RNA signal. Based on the RNA-seq data, we identified 533 genes with evidence of extended 3’ RNA signal after IAV infection, continuing up to 850 kb past gene termini (median 170 kb, Table S2). Transcription from these genes gave rise to increased numbers of RNA-seq reads crossing 3’-terminal poly(A) signals (PASs) (Figure S1F) and intergenic and trans-splice events into genomic regions and genes downstream (Figure S1G). ChIP-seq confirmed the presence of RNAPII downstream of genes following IAV infection (Figures 1E, 1F and S1E), which was serine 2-, 5-, and 7-phosphorylated at its C-terminal domain (CTD) (Figure S1H), suggesting that IAV infection causes continued transcription of elongation-competent RNAPII downstream of PAS-terminated genes.
To confirm that the RNA signal downstream of genes was not due to de novo transcription initiation, we performed Start-seq and RNA-seq in IAV-infected THP-1 macrophages. Start-seq detects short transcription start site (TSS)-associated RNAs that are indicative of RNAPII transcription initiation (Scruggs et al., 2015). IAV infection induced robust gene activation and RNA signal downstream of genes in this system, however these regions were devoid of evidence for additional transcription initiation (Figures S1I- K). Taken together, our data established that the IAV infection-induced chromatin interaction changes and transcription downstream of genes were not due to de novo transcription initiation in these regions.
The long-range interaction pattern of a locus often reflects its epigenetic and/or transcriptional state (Rao et al., 2017; Rowley et al., 2017; Schwarzer et al., 2017). To test whether IAV-induced locus decompaction or B-to-A compartment switching were associated with epigenetic changes, we performed ChIP-seq for several eu- and heterochromatin markers and whole-genome bisulfite sequencing in mock- and IAV-infected MDM. IAV infection had little effect on the heterochromatin marks H3K9me3, H3K27me3 and the DNA methylation status (Figures S1E and S1K). In contrast, histone modifications associated with active transcription were elevated in regions switching from the B to the A compartment. While the levels of the promoter mark H3K4me3 and the promoter and enhancer activation mark H3K27ac increased only moderately, the amount of the transcription elongation mark H3K36me3 rose dramatically, along with the increased RNAPII levels in these regions downstream of highly active genes (Figures S1E and S1K). Inspection of the RNA and ChIP-seq data indicated that the observed increases of promoter- and enhancer-associated H3K4me3 and H3K27ac marks were due to activation of canonical gene promoters and enhancers within these regions downstream of highly IAV-activated genes, many of which were also transcriptionally activated by IFNβ (Figures 1E and S1E).
IAV NS1 protein induces read-through transcription and chromatin structure changes
Overall, our data implied that IAV infection was causing “read-through” transcription past the end of genes by globally inhibiting transcription termination. NS1 was previously shown to inhibit PAS-dependent transcription termination of reporter constructs and single genes (Nag et al., 2007; Shimizu et al., 1999), likely as a consequence of inhibiting the PAS-sensing subunit of cleavage and polyadenylation specificity factor, CPSF30 (Nemeroff et al., 1998). Consistent with the proposed role for NS1, non-PAS-terminated histone genes did not exhibit downstream RNA or RNAPII signal in lAV-infected MDM (Figure S2A).
To confirm that NS1 is central to the IAV infection-induced chromatin interactions changes, we infected MDM with a mutant IAV H5N1 strain encoding a C-terminally truncated NS1 of 73 amino acids ΔNS1) that lacks the CPSF interaction domain. This strain is replication-competent, with attenuated virulence (Steel et al., 2009) (Figures S2B-D). Infection with ΔNS1 IAV activated many of the same genes as wild-type (WT) H5N1 IAV and IFΝβ, but did not cause RNAPII or RNA signal downstream of genes, nor did these regions switch chromatin compartments or decompact as observed with WT IAV infection (Figures 2A-C). Notably, this identified many WT IAV-specifically induced genes that were likely upregulated as a result of read-in transcription from upstream genes (Figures 2A and S2A). Together, our results confirmed that NS1 is required for the read-through transcription and genome reconfiguration observed during IAV infection.
Figure 2. IAV-induced read-through and structural changes are NS1-dependent.
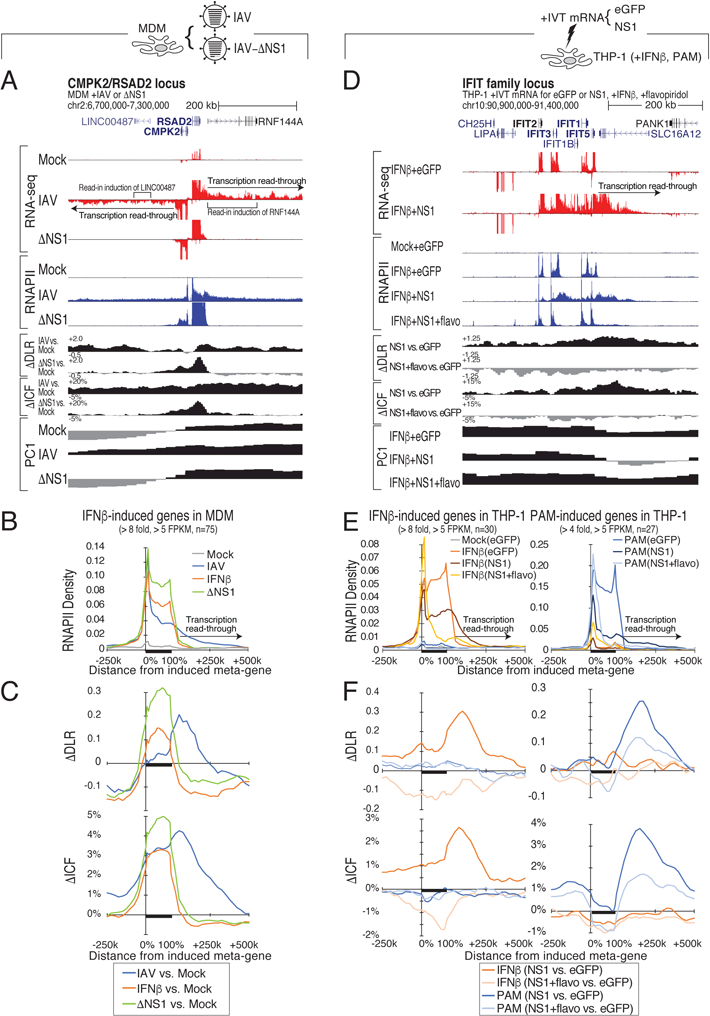
(A) Genome browser tracks of RNA-seq and RNAPII ChIP-seq read densities and chromatin compaction changes at the CMPK2/RSAD2 locus in mock-, wild-type IAV-, and IAV with NS1 truncation ΔNS1)-infected MDM. (B,C) Meta-gene plot of RNAPII ChIP-seq reads (B) and changes in chromatin compaction ΔDLR and ΔICF) (C) after IAV, ΔNS1, or IFNβ treatment of MDM at highly IFNβ-responsive genes defined in Figure 1F. (D) Genome browser tracks of RNA-seq and RNAPII ChIP-seq reads and chromatin compaction changes at the IFIT family locus in THP-1 cells expressing either EGFP or influenza NS1 protein with or without IFNβ and flavopiridol. (E,F) Meta-gene plot of RNAPII ChIP-seq reads (E) or changes in chromatin compaction ΔDLR and ΔICF) (F) in THP-1 cells expressing EGFP or NS1, treated with either IFNβ or PAM (TLR2 agonist) and flavopiridol at genes highly responsive to IFNβ or PAM stimulation in THP-1 (IFNβ: FC > 8, RNAPII FPKM > 5; 30 response genes; PAM: FC > 4, RNAPII FPKM > 5; 27 response genes, see Table S2). See also Figure S2.
Expressing influenza NS1 in THP-1 monocyte cells led to robust RNA and RNAPII signal as well as chromatin decompaction downstream of highly transcribed genes (Figures S2E, S2F and S2I). NS1 also inhibited early RNAPII elongation, leading to accumulation of promoter-proximally paused RNAPII (Figures S2F and S2G). The combined inhibition of transcription termination and elongation by NS1 induced depletion of RNAPII in gene bodies (Figure S2F). Similar accumulation of RNAPII at proximal promoter regions occurred at genes in IAV-infected cells, which may contribute to gene repression by IAV in MDM (Figure S2H). Stimulation with IFNβ established high-level transcription of interferon-inducible genes, which together with NS1 expression induced an RNAPII read-through and regional chromatin decompaction pattern that mimicked that of IAV-infected cells (Figures 2D-F and S2I). In line with NS1 causing read-through transcription, these conditions led to nuclear retention of long transcripts that extend well beyond the normal transcript end sites of PAS-terminated genes (Figures S2J-L).
IAV-induced chromatin interaction changes in read-through regions are caused by transcription elongation
Next, we reasoned that if rampant RNAPII elongation causes the read-through-associated chromatin interaction changes, inhibiting elongation should reverse these changes. We tested this hypothesis in THP-1 cells, where we induced read-through transcription with NS1, followed by treatment with the elongation inhibitor flavopiridol. Using two different stimuli, IFNβ or the TLR2 agonist Pam3CSK4 (PAM), we induced high-level transcription in different gene sets, which directed NSI-dependent RNAPII read-through and decompaction to regions downstream of the respective stimulus-responsive genes (Figures 2D-F and S2I). Subsequent flavopiridol treatment inhibited both elongation and read-through and reversed the locus compaction status and chromatin architecture changes (Figures 2D-F and S2I). These results demonstrated that read-through transcription is causal for the NS1-induced chromatin interaction changes.
Read-through transcription displaces cohesin from CTCF sites
To investigate the mechanism of how read-through transcription affects chromatin structure, we turned our focus to CTCF and cohesin, which are essential for the formation and maintenance of local chromatin interaction domains (< 1 Mb) (Kubo et al., 2017; Nora et al., 2017; Rao et al., 2017; Wutz et al., 2017). Read-through regions in WT IAV-infected MDM were reproducibly associated with widespread loss of chromatin loops and loop domains (Figures 3A and S3A). Out of 9798 loops in MDM, we identified 183 (1.9%) weakened and 118 (1.2%) strengthened loops (p<0.05, log2FC>1.5; Figure S3B), which were associated with large changes in locus compaction levels (Figure S3B). In line with their known functions in loop formation and maintenance, cohesin/RAD21 levels correlated with local compaction levels (Figure S3C) and were increased or decreased at loop anchors of loops strengthened or weakened by IAV infection, respectively (Figures 3A-C). In contrast, RNAPII levels at these loop anchors correlated inversely with cohesin and loop strength and (to a lesser degree) CTCF(Figures 3A-C). The inverse correlation between cohesin and RNAPII levels at loop anchors was most pronounced at promoter-distal CTCF sites devoid of the transcription initiation-associated H3K27ac mark (Figures 3D and S3D). This anticorrelated relationship became stronger with increasing RNAPII occupancy (Figure S3E). Comparison with ΔNS1 IAV-infected and IFNβ-treated MDM showed that WT IAV-specific weakening of chromatin loops occurred mostly in read-through regions while loops weakened in all three conditions were more frequently anchored in gene bodies of interferon-inducible genes (Figures 3E, S3F and Figures 3A S3G and S3H). These data suggested that read-through transcription disrupted cohesin-mediated loops. This was corroborated by the data from NS1-expressing, IFNβ- or PAM-treated THP-1 cells, which exhibited a similar inverse relationship between RNAPII and cohesin at CTCF sites in stimulus-responsive read-through regions and disruption of cohesin-mediated loops (Figure S3I). The latter was reversed by inhibiting elongation with flavopiridol (Figures 3F,S3J and S3K), suggesting that read-through transcription causes chromatin decompaction by disrupting local cohesin-mediated chromatin interactions.
Figure 3. High-level transcription removes cohesin from CTCF sites and eliminates loops.
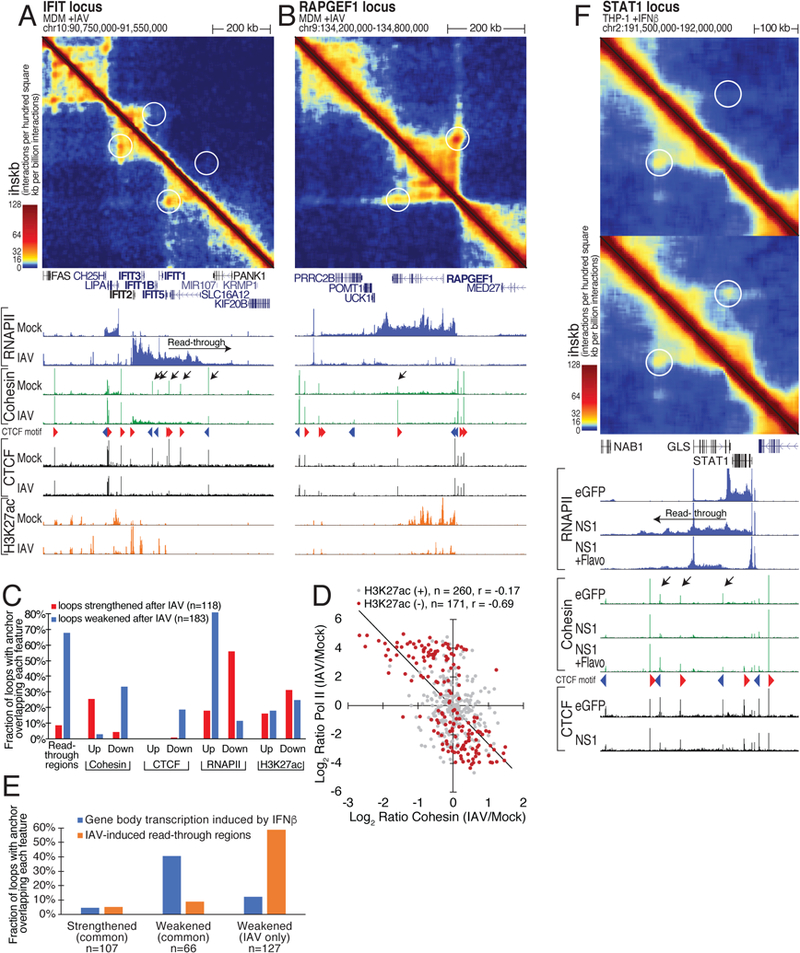
(A) Loss of interactions at the highly induced IFIT family locus following infection. Hi-C contact map in mock- (lower left) and IAV- (upper right) infected MDM (merge of 2 replicates). ChIP-seq for RNAPII, CTCF, RAD21 (cohesin), and H3K27ac below. Red and blue triangles indicate preferred interaction direction of CTCF motifs. Black arrows mark regions with dynamic cohesin changes during infection. (B) Same as (A) for the IAV-repressed RAPGEF1 locus, showing increase of CTCF-colocalizing cohesin (black arrow) and loop strengthening with IAV infection. (C) Weakened loops are preferentially located in read-through regions. Fraction of regulated chromatin loops with at least one anchor overlapping IAV read-through regions or regulated ChIP-seq peaks that either increase or decrease 2-fold after IAV infection (CTCF, cohesin, RNAPII, H3K27ac) (15 kb resolution). (D) Negatively correlation between changes in cohesin and RNAPII levels at CTCF peaks in highly transcribed regions (>7.5 FPKM RNAPII, CTCF, and cohesin signal per CTCF site in either mock or IAV conditions, 431 regions total). Grey points are CTCF peaks by H3K27ac in either condition (FPKM > 2.5), indicating regulatory elements where RNAPII is often paused. (E) Loop weakening with transcription elongation. Percentage of loops regulated in common between IAV, ΔNS1, or IFNβ, or weakened specifically with IAV-infection. At least one loop anchor overlaps IFNβ-regulated gene bodies or IAV read-through regions. Loops connecting an IFNβ-regulated gene and an IAV read-through region were assigned to the gene. (F) Hi-C contact map of the STAT1 locus in EGFP- (lower left) and NS1- (upper right) expressing THP-1 cells stimulated with IFNβ and/or flavopiridol. ChIP-seq for RNAPII, CTCF and cohesin below. Black arrows mark cohesin peaks lost due to read-through transcription and recovered with flavopiridol. See also Figure S3.
To explore how cohesin loading and unloading dynamics may impact transcription read-through-associated changes in genome structure, we knocked down the cohesin loader NIPBL or the cohesin unloader WAPL in THP-1 cells. The Hi-C interaction profile after WAPL depletion showed an increase in local interactions (< ~3 Mb) while NIPBL depletion led to an increase in long-range interactions (> ~3 Mb), confirming previous reports (Figures S4A-C and S4E) (Haarhuis et al., 2017; Schwarzer et al., 2017). Expression of NS1 in PAM-treated cells induced similar levels of read-through transcription regardless of NIPBL/WAPL knockdown (Figures S4D and S4H). Interaction profiles from PAM-NS1 read-through regions phenocopied the depletion of local interactions in favor of long-range interactions seen in IAV-infected MDM read-through regions (Figure S4F), while subsequent treatment with flavopiridol resulted in recovery of local interactions (100 kb - 3 Mb, Figure S4G). WAPL knockdown blunted the NS1-induced loss of local interactions in these regions (Figures S4F and S4H), while NIPBL knockdown resulted in reduced recovery of local interactions following flavopiridol treatment (Figures S4G and S4H), providing additional evidence that transcription and factors controlling cohesin dynamics work together to regulate genome structure.
Genic transcription affects 3D genome structure by eliminating cohesin- mediated loops
Cohesin levels and associated loop interactions also decreased within gene bodies where IAV infection or stimulus responses increased transcription levels (Figures S3F-H). Conversely, cohesin levels and loop interactions rose at CTCF sites in genes where transcription was repressed (Figure 3B). For IFNβ treatment, the anticorrelation of RNAPII and cohesin was strongest at CTCF sites located in gene bodies and weakest at gene promoters, (Figure S5A), suggesting elongating RNAPII affects cohesin and CTCF-anchored loops under normal conditions.
To test the hypothesis that RNAPII-associated locus decompaction and cohesin displacement is a general feature of transcription elongation, we compared the effects of three different non-viral stimuli on transcription and nuclear 3D structure in THP-1 cells. IFNβ and PAM treatment as well as glucocorticoid receptor (GR) activation by dexamethasone (Dex) each activated transcription, local decompaction, and CTCF-anchored loop weakening exclusively at their respective target genes (Figure 4A). We globally assessed the relationship between changes in loop interaction frequencies and transcription at CTCF sites by ranking loops in each experiment by their change in loop strength with treatment and scoring the enrichment of CTCF sites with increased RNAPII levels at their loop anchors by Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2005) (Figure 4B). GSEA across conditions showed significant and selective enrichment of transcribed CTCF sites at the anchors of weakened loops with the corresponding treatment, further confirming the connection between transcription elongation and loss of CTCF-anchored chromatin looping (Figures 4C and S5B).
Figure 4. Intragenic chromatin loops weaken with normal transcriptional responses to cellular signaling.
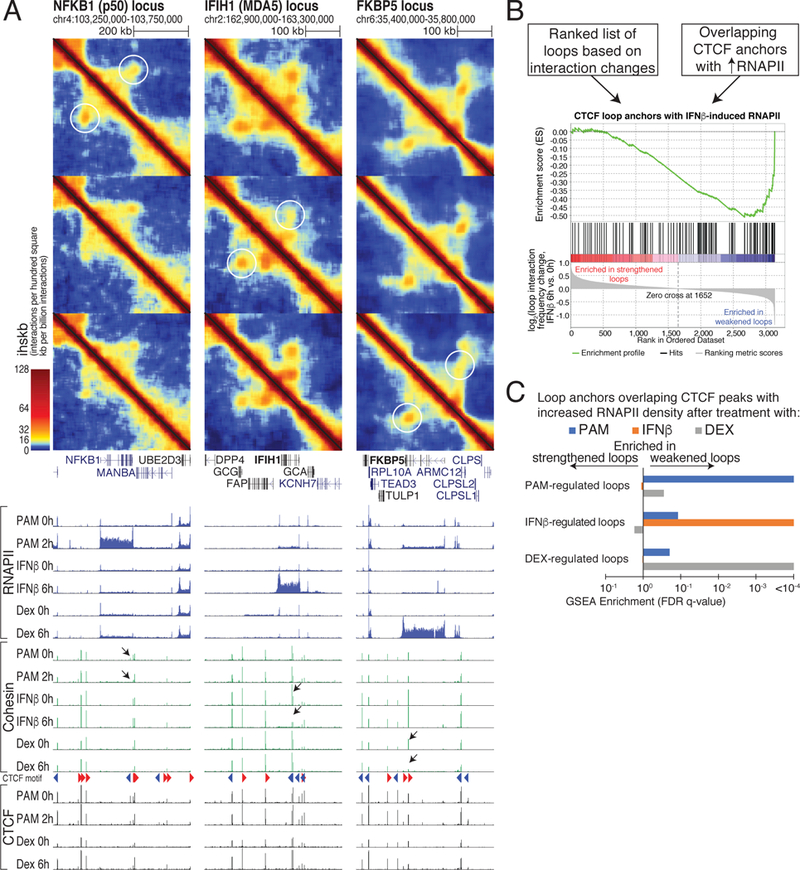
(A) Hi-C contact maps and RNAPII, cohesin, and CTCF ChIP-seq signal at loci that selectively respond to PAM (NFKB1), IFNβ (IFIH1), and dexamethasone (FKBP5) in THP-1 cells. Chromatin loops are weakened only if the corresponding gene is induced. Black arrows mark intragenic CTCF sites where cohesin is lost following transcription induction. (B) Gene Set Enrichment Analysis (GSEA) of ranked lists of chromatin loops for loop anchor overlap at CTCF sites with increased RNAPII following treatment (>1.5 fold). Loops are ranked by their log2 ratio of Hi-C interactions in treated versus control conditions. (C) Association between transcribed CTCF sites and chromatin loop changes is signaling-specific. GSEA enrichment q-values comparing each ranked list of chromatin loop changes with each set of CTCF sites with increased RNAPII density for PAM, IFNβ, and dexamethasone (Dex) treatments. See also Figures S4 and S5.
Cohesin loop formation upon inhibition of transcription elongation
Next, we investigated how steady-state transcription globally affects chromatin loops and locus compaction, using flavopiridol to block transcription elongation in otherwise untreated THP-1 cells. Flavopiridol increased cohesin accumulation and loop strength at CTCF sites located within transcribed regions (Figures 5A-C and S5C). This was most noticeable in long genes (>50 kb), which compacted proportional to their transcription level before inhibiting elongation (Figures 5D, 5E and S5D). Chromatin compartmentalization remained stable, potentially as a result of the continued RNAPII and cohesin occupancy at sites of transcription initiation (Figures S5D and S5E). Collectively, these data suggested that transcription elongation in gene bodies impacts genome 3D organization by weakening cohesin-mediated loops at intragenic CTCF-bound sites, leading to decompaction of transcribed regions.
Figure 5. Loop formation in and compaction of transcribed regions upon blocking transcription elongation with flavopiridol.
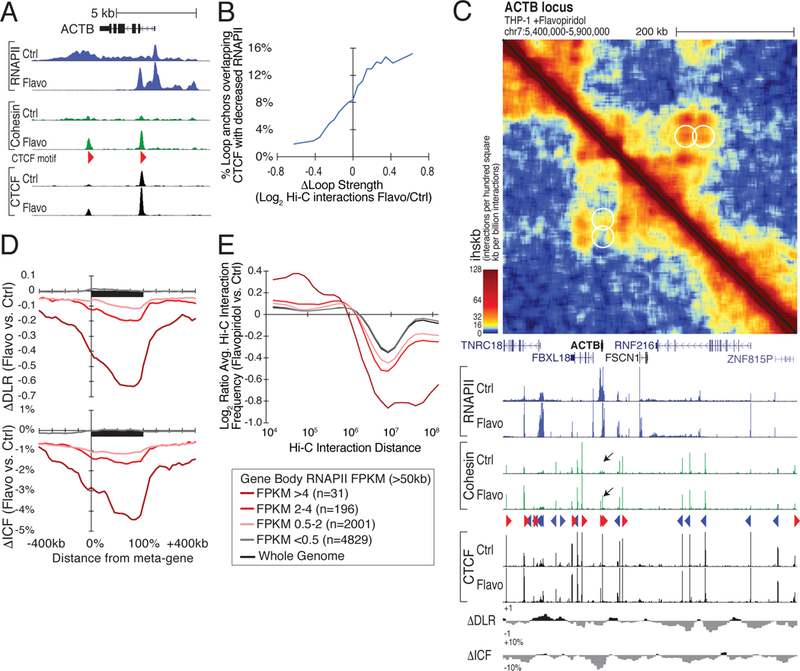
(A) Flavopiridol stalls RNAPII at regulatory elements, enabling cohesin to accumulate at intragenic CTCF sites. Genome browser tracks of RNAPII, cohesin, and CTCF at the ACTB locus in control and flavopiridol-treated THP-1 cells. (B) Loops that strengthen with elongation inhibition overlap CTCF sites that become less transcribed flavopiridol. Moving average showing the fraction of loops with anchors overlapping CTCF sites that lose RNAPII following flavopiridol treatment plotted against the change in loop strength. (C) Gain of loops at CTCF sites located within the ACTB gene body. Hi-C contact map and RNAPII, cohesin, and CTCF ChIP-seq signal at the ACTB locus in control- (lower left) and flavopiridol- (upper right) treated THP-1 cells. Black arrows mark CTCF sites that gain cohesin following flavopiridol treatment. (D) Highly transcribed genes exhibit greater compaction along their gene bodies after flavopiridol treatment. Meta-gene profiles of changes in ΔDLR and ΔICF for genes longer than 50 kb separated by RNAPII levels in control THP-1 cells. (E) Log2 ratio of Hi-C interaction frequencies relative to interaction distance in flavopiridol versus control-treated THP-1 cells at long genes (>50 kb) segregated by their RNAPII levels. See also Figure S5.
Temporal relationship between elongating RNAPII, RAD21 and CTCF
To characterize in detail how transcription elongation affects chromatin looping and cohesin dynamics, we performed a time course of RNAPII, cohesin and CTCF binding during the transcriptional response of THP-1 cells to TLR2 stimulation with PAM and subsequent inhibition of elongation with flavopiridol. RNAPII elongation at PAM-activated genes proceeded at an average ~2 kb/min (Figures 6A and S6A), in agreement with earlier findings (Danko et al., 2013). RNAPII and cohesin were temporally anticorrelated at individual CTCF sites (Figures 6A, 6C and S6B), suggesting that cohesin binding at CTCF sites is directly affected by RNAPII levels. The antagonistic relationship between RNAPII and cohesin was weaker at CTCF sites near initiating RNAPII, and even positively correlated at cohesin peaks near putative promoters and enhancers that lacked CTCF binding (Figure 6C).
Figure 6. Kinetic analysis of RNAPII elongation and cohesin displacement.
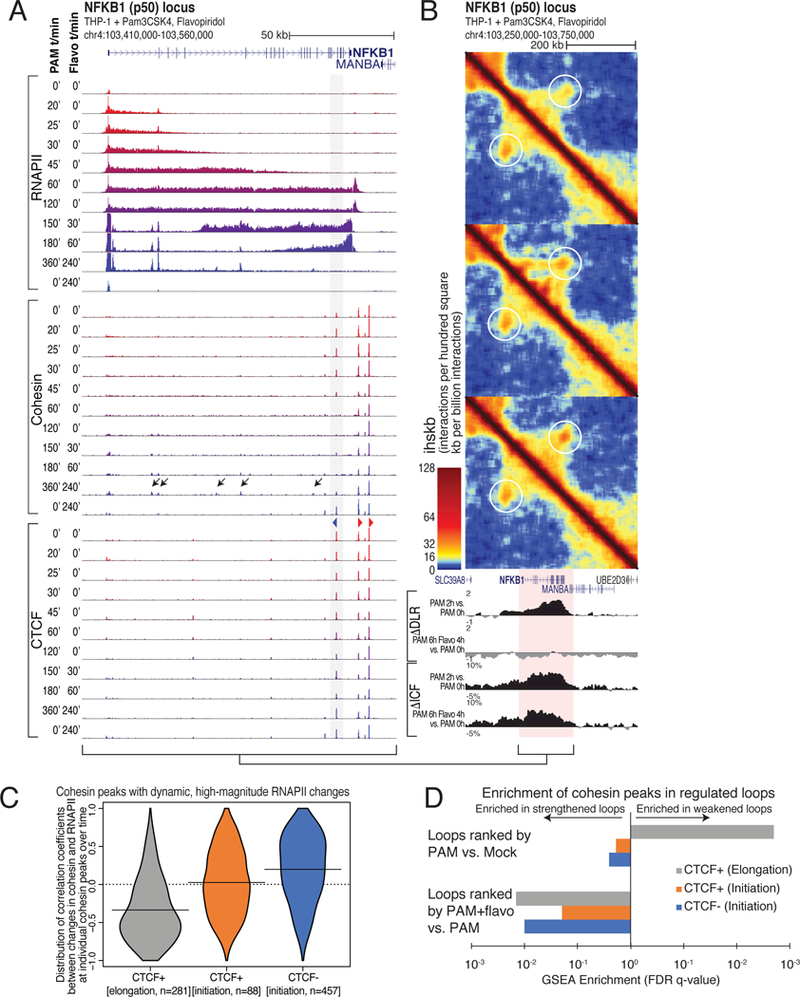
(A) RNAPII, cohesin, and CTCF ChIP-seq at the NFKB1 (p50) locus. THP-1 cells treated with PAM followed by flavopiridol for the indicated times. Flavopiridol treatment for 240 minutes is included as a control. Grey box, CTCF site that loses cohesin when transcribed by RNAPII. Black arrows mark putative PAM-specific enhancers that accumulate RNAPII and cohesin following PAM and flavopiridol treatments. (B) Hi-C contact maps of the NFKB1 locus showing the weakening of the loop formed by the CTCF site highlighted by grey box in (A). Altered interaction patterns are observed near the NFKB1 gene body in PAM+flavopiridol-treated samples. (C) Distribution of correlation coefficients between the temporal levels of RNAPII and RAD21 for each RAD21 peak exhibiting high-magnitude changes in RNAPII density (FC > 2, RNAPII FPKM > 10). Peaks were segregated based on CTCF (>10 FPKM) and whether or not they are found in ‘initiating’ or ‘elongation’ regions based on the increase or decrease in RNAPII density following flavopiridol treatment. (D) GSEA enrichment q-values for changes in loop strength overlapping with the subsets of RAD21 peaks defined in (C). See also Figure S6.
These results are illustrated by the NFKB1 locus, where cohesin disappeared from a 3’-intronic CTCF site when reached by the RNAPII wave front (Figure 6A, grey bar). Elongation inhibition by flavopiridol induced partial recovery of the cohesin signal at this site after the trailing edge of the RNAPII wave had passed (Figure 6A). In situ Hi-C revealed that loop interactions weakened where RNAPII traveled through and cohesin was lost from intragenic CTCF sites, which was accompanied by decompaction of these newly transcribed genomic regions (Figures 6A, 6B, S6B and S6C). Flavopiridol reversed these effects, despite continued TLR2 stimulation (Figures 6A, 6B, S6B and S6C).
Flavopiridol also led to accumulation of cohesin and RNAPII at putative enhancers, many of which were specific to PAM treatment (Figures 6A, S6B and S6E). Analysis of these sites showed strong motif enrichment for NF-kB and AP-1, the main transcription factors activated by TLR2 stimulation, and PU.1 and C/EBP, the main lineage-determining factors in monocytes/macrophages (Heinz et al., 2010) (Figure S6D). Here, new chromatin interactions appeared when elongation was inhibited, as illustrated by multiple intronic sites in the NFKB1 gene and intergenic sites near the CD44 gene (Figures 6A, 6B and S6E), many of which were bound by the predicted transcription factors after treatment with PAM (Figures S6D and S6E). Global interaction analysis further corroborated that interactions at active regulatory elements increased with flavopiridol, along with RNAPII and cohesin accumulation (Figure 6D). Notably, these new interactions occurred both at CTCF-containing and CTCF-less sites.
Taken together, our time course data strongly supports a mechanism where cohesin is displaced from intragenic CTCF sites when reached by elongating RNAPII. Conversely, inhibition of elongation leads to accumulation of cohesin at CTCF and transcription initiation sites, along with an increase in local chromatin interactions and associated chromatin compaction.
DISCUSSION
We have uncovered a previously unknown relationship between RNA polymerase II transcription elongation and CTCF-directed, cohesin-mediated chromatin loops. Our findings provide a mechanism for how chromatin loops, chromatin compartments and regional chromatin compaction status are dynamically regulated by transcription, and how transcription processes interact with the general configuration of chromatin structure in mammalian cells, which appears to be largely governed by dynamic cohesin loop extrusion and CTCF motif orientation (Fudenberg et al., 2016; Gassler et al., 2017; Rao et al., 2017; Sanborn et al., 2015).
Elongating RNAPII disrupts 3D structure by displacing cohesin and CTCF
Our data provide strong evidence that transcription elongation displaces CTCF and cohesin and thereby dissociates chromatin loops as RNA polymerase II transcribes genomic DNA through CTCF loop anchors (Figure 7D). This was most noticeable following IAV/NS1-dependent inhibition of transcription termination, where RNAPII continues to elongate hundreds of kilobases past gene termination sites, eliminating chromatin loops and locally decompacting chromatin. While cohesin binding at CTCF sites diminished along with loop strength, CTCF ChIP-seq signal frequently appeared unperturbed by RNAPII transcription. This discrepancy might reflect the faster binding and recovery kinetics of CTCF relative to cohesin, which in a recent study exhibits 10- and 30-fold shorter residence and search times, respectively, than cohesin in the same cells (Hansen et al., 2017).
Figure 7. Read-through transcription into B compartment chromatin results in enhanced transcription factor binding.
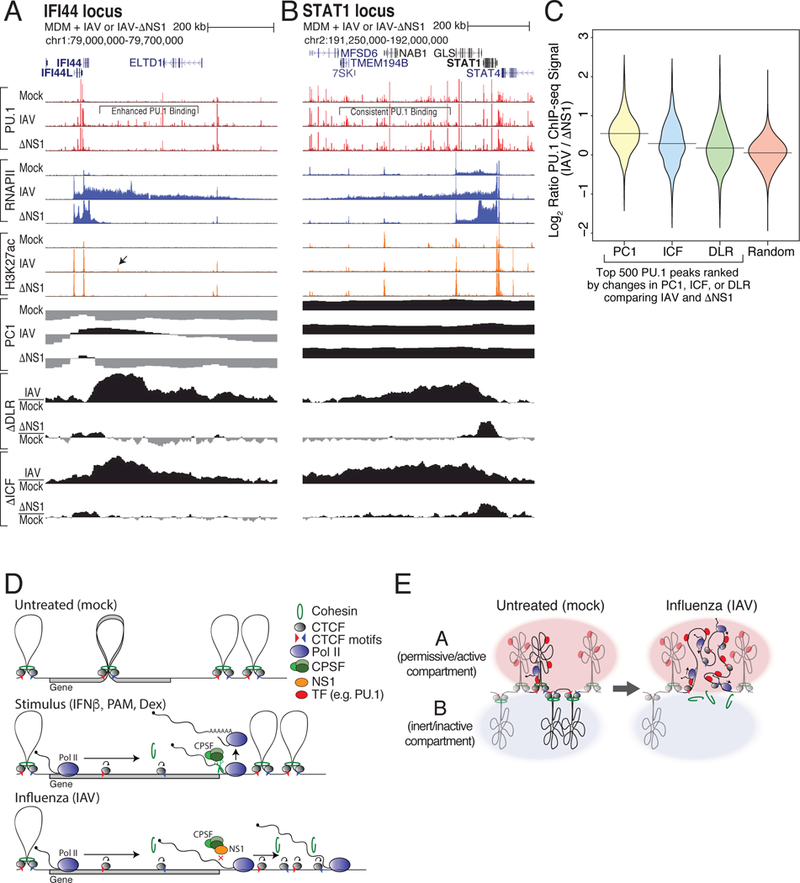
(A,B) Transcription factor binding is increased where B-to-A compartment changes occur. ChIP-seq tracks for PU.1, RNAPII, and H3K27ac at the IFI44 (A) and STAT1 (B) loci in macrophages infected with IAV, IAV lacking NS1 (ΔNS1), or mock. PC1 values and changes in ΔDLR and ΔICF are depicted for each locus. Black arrow highlights new H3K27ac-marked PU.1 site. (C) Distribution of log2 binding ratios between IAV and ΔNS1 conditions for the 500 PU.1 ChIP-seq peaks in regions exhibiting the largest changes in ΔPC1, ΔICF, ΔDLR, or random regions. (D) Model of RNAPII elongation-mediated dissolution of chromatin structure by removal of cohesin at CTCF sites. (E) Model of RNAPII elongation-mediated switching of regions from the B to the A compartment following IAV infection. See also Figure S7.
Transcription has been shown to decompact chromatin and increase mobility of highly transcribed loci (Gu et al., 2018; Jubb et al., 2017). Our results suggest that both are caused by transcription-induced cohesin displacement within transcription units. This is evident at the FKBP5 locus, where Hi-C-derived decompaction after GR activation (Figure S1D) is most pronounced around the cohesin/CTCF binding site in the antepenultimate intron (Figure 4A). Conversely, we see evidence that cohesin-mediated chromatin loops re-form upon down-regulation of highly transcribed genes (Figure 3B), confirming recent results that suggest cohesin loop formation is dynamic, balancing cohesin loading, loop extrusion, RNAPII-mediated redistribution, and unloading (Busslinger et al., 2017; Rao et al., 2017; Schwarzer et al., 2017; Wutz et al., 2017). Although the biochemical mechanisms and cofactors involved in elongating RNAPII-induced cohesin displacement are not yet fully defined, it is likely that WAPL plays a role, based on the diminished regional decompaction following transcriptional activation in WAPL knockdown conditions (Figures S4F and H) and the fact that WAPL knockout leads to cohesin buildup downstream of transcribed genes in mammalian cells (Busslinger et al., 2017).
Given the relative sizes of the cohesin ring and RNAPII, the transcription machinery or associated factors must either clear or move cohesin downstream when melting DNA and transcribing through chromatin. In our experiments, we observed cohesin clearance, as both cohesin and cohesin-mediated interactions were depleted from CTCF sites in regions with high levels of transcription elongation rather than translocated to regions downstream of genes, as observed in yeast (Lengronne et al., 2004). Conversely, initiating or paused RNAPII at regulatory elements correlated positively with cohesin levels. This may be due to cohesin loading by NIPBL at transcription initiation sites (Kagey et al., 2010). Alternatively, paused RNAPII might constitute a localized physical barrier to dynamic cohesin extrusion, which, similar to CTCF, would lead to accumulation of extruding cohesin at RNAPII pause sites. Notably, cohesin accumulated at newly induced intragenic RNAPII pause sites following TLR2 activation and elongation inhibition by flavopiridol. These sites of cohesin accumulation engage in looping interactions with each other (Figures 6A-D and S6B-E) and may form the basis for regulatory enhancer-promoter loop interactions.
Elongating RNAPII causes B to A compartment switching and increases transcription factor accessibility
In addition to causing local decompaction of chromatin, we found that elongating RNAPII can impact chromatin structure by transcribing into B (inert) compartment chromatin and switching its long-range interaction profile to that of the A (permissive) compartment (Figures 1F and S1E). Compartment transitions have been observed on developmental time scales (Dixon et al., 2015; Isoda et al., 2017), but the mechanisms orchestrating compartment changes have remained elusive. The B compartment is thought to consist of mostly compact heterochromatin that is generally refractory to transcription factor binding and transcriptional activation (Lieberman-Aiden et al., 2009; Rao et al., 2014). The formation of heterochromatin domains (i.e. B compartment) has been suggested to be driven by protein interaction-mediated compaction and liquid-liquid phase separation (Larson et al., 2017; Strom et al., 2017). Recent results indicate that transcription itself may be one of the driving forces behind A compartment formation, evidenced by identification of minute gene-sized A compartment “mini-domains” that are similar in size to the transcribed gene loci they encompass (Rowley et al., 2017).
In line with these observations, our data indicate that RNAPII appears to be uninhibited by the presence of B compartment chromatin as it extends A compartment domains during elongation (e.g. Figure S1E). The decompaction and repositioning of B compartment chromatin by RNAPII could be caused by the active translocation of B compartment DNA to a transcription factory or other nuclear sub-structure. Alternatively, the loss of cohesin-mediated 3D structure caused by RNAPII elongation may free the region from local structural constraints that shackle it to neighboring heterochromatin and allow it to interact with other active regions (Rao et al., 2017; Schwarzer et al., 2017). Additionally, RNAPII elongation may clear HP1 or other repressive complexes in a manner similar to how cohesin is cleared as DNA is melted and transcribed.
Transcription-induced compartment switching had functional effects, as evidenced by elevated binding of the macrophage master regulator PU.1 in regions switching from the B to A compartment in IAV-infected cells relative to controls (Figures 7A and7C). This was restricted to B-to-A-switching regions, as PU.1 binding in decompacting regions that were consistently associated with the A compartment remained unchanged (Figures 7B and7C). In some cases, newly bound PU.1 sites acquired H3K27ac, indicative of transcription activation (Figure 7A). We observed similar trends in PU.1 and C/EBPβ binding following NS1 overexpression with PAM treatment in THP-1 cells (Figures S7A and S7B). Together results provide further evidence that transcription elongation into B compartment chromatin produces permissive chromatin capable of increased TF binding and B-to-A compartment transitions, and enables activation of new regulatory elements and transcription programs, similar to a recent study (Isoda et al., 2017).
Virus-induced disruption of genome 3D organization and virulence
Unexpectedly, IAV NS1 protein was sufficient for the transcriptional termination defects and genome structure disruption in IAV-infected cells. Pervasive read-through transcription downstream of genes is not restricted to H5N1 IAV or dependent on the cell type, since analogous results were observed with multiple IAV strains in both macrophages and epithelial cells (Figure S7C). Although not required for viral propagation, NS1 executes a plethora of functions to subvert host antiviral responses and increase virulence. These range from inhibiting intracellular virus sensors and signaling pathways to impairing mRNA processing, export and translation (Krug, 2015). The NS1-mediated inhibition of the CPSF complex plays a major role in host cell shut down, leading to lack of cleavage and polyadenylation of pre-mRNA, thereby substantially suppressing the host cell antiviral response (Noah et al., 2003). The infection-induced nuclear genome 3D structure changes observed in this study may contribute to IAV virulence. However, since NS1-dependent transcription termination defects, read-through and host genome 3D structure alterations are linked, it will be challenging to determine the relative contributions of each phenomenon to NS1-dependent virulence.
Impact of read-through transcription
The read-through transcription reported in this study shares many features of transcription downstream of genes in other systems, including 7SK RNA knockdown (Castelo-Branco et al., 2013), osmotic stress (Vilborg et al., 2015), HSV infection (Rutkowski et al., 2015), depletion of CPSF73 or Xrn2 (Eaton et al., 2018), or mutation of Tyr 1 residues in the RNAPII CTD (Shah et al., 2018). Our findings predict that read-through transcription will have a similar impact on genome structure downstream of highly transcribed genes in these systems. It is also possible that regulation of transcription termination or carefully placed transcription units modulate chromatin structure during development or other cellular responses. Recent studies have shown important regulatory functions for lncRNA in establishing genome structure, and lncRNAs have been shown to be enriched at TAD boundaries where they could play a role in modulating chromatin looping (Amaral et al., 2018; Isoda et al., 2017).
A role for transcription in defining genome 3D structure
This study shows that transcription can directly impact genome 3D structure. Our data indicate that regardless of the epigenetic state or structural configuration of the chromatin it transcribes, the RNAPII elongation complex may only be stopped by mechanisms controlling transcriptional termination. Given that transcription elongation appears refractory to the presence of cohesin, TF binding, or other epigenetic roadblocks, it is tempting to speculate that transcribing RNAPII acts as a temporary eraser of chromatin loops in its path, including enhancer-promoter interactions. How these disruptions of specific chromatin loops ultimately impact transcription regulation is an active debate (Rao et al., 2017), but our results provide a mechanistic explanation for how transcription itself plays an integral role in organizing the genome in the nucleus.
STAR★METHODS
KEY RESOURCES TABLE
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Sven Heinz (sheinz@ucsd.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
IAV-infected human monocyte-derived macrophages
Cellular responses to influenza infection were modeled by infecting human monocyte-derived macrophages with Influenza A/Vietnam/1203/2004 (H5N1) HAIo virus or a mutant H5N1 virus encoding a truncated NS1 protein. Primary blood cells were obtained as de-identified buffy coats from the New York Blood Center from four randomly chosen anonymous healthy blood donors, all of which were male. The New York Blood center obtained informed consent from blood donors for research use of their blood. Experiments were performed with pairs of samples from separate donors to replicate results. No sample size estimation was performed, and due to the anonymous nature of the sample collection, no information is available on a given donor’s drug or procedure history.
THP-1 cells
In addition to primary monocyte-derived macrophages, the THP-1 human monocytic cell line, which originally was isolated from a 1-year old male with acute monocytic leukaemia, was used to model the effects of IAV infection as well as IAV NS1 protein expression and the response to cellular stimulation.
METHOD DETAILS
Cell culture
Monocyte-derived macrophages (MDM) were prepared from buffy coats from anonymous healthy donor blood obtained from the New York Blood Center. Peripheral blood mononuclear cells (PBMC) were isolated by centrifugation on a Ficoll density gradient (Histopaque; Sigma Aldrich), and CD14+ cells were isolated using Miltenyi liquid separation mini columns and magnetic beads labeled with anti-human CD14 antibody (Miltenyi Biotech). For differentiation into macrophages, approximately 3 million CD14+ monocytes were cultured for 10 days in RPMI 1640 medium (CellGro, Corning) containing 10% fetal bovine serum (FBS) (HyClone; Thermo Scientific), 2 mM L-glutamine, 1 mM sodium pyruvate, and 100 U/ml penicillin-100 μg/ml streptomycin (Gibco, Invitrogen), and supplemented with 1000 U/ml human granulocyte-macrophage colony-stimulating factor (hGM-CSF) (Peprotech). THP-1 cells (“THP-1 monocytes”) were propagated in THP-1 medium (RPMI-1640 containing 10% low-endotoxin FBS (Peak Serum Inc.), 1x (100 μM) non-essential amino acids (Life Technologies), 1x MEM Sodium Pyruvate Solution (1 mM) (Life Technologies), 1x GlutaMax I (Life Technologies), 50 μΜ β-mercaptoethanol (Life Technologies)) at 37°C, 5% CO2, 100% humidity.
Unless noted otherwise, agonists were used at the following concentrations: IFΝβ - 1000 U/ml, Pam3CSK4 – 1 μg/ml, Dexamethasone - 1 μΜ, flavopiridol - 2 μΜ. Dexamethasone and, where indicated, IFΝβ treatments were performed on differentiated THP-1 cells (“THP-1 macrophages”) that had been treated with 60 ng/ml phorbol myristate acetate (PMA) for 48 h followed overnight culture without PMA. For IAV infections, THP-1 cells were differentiated in the same manner, except 10 ng/ml PMA were used.
Virus preparation and infection conditions
Influenza A/Vietnam/1203/2004 (H5N1) HAlo virus is a recombinant virus that has been engineered to remove the multibasic cleavage site in the HA protein (Steel et al., 2009). The H5N1 ΔNS1 virus expresses a truncated NS1 protein (residues 1–73) in the H5N1 HAlo background (Steel et al., 2009). Viruses were propagated in ten-day-old embryonated chicken eggs (Charles River Laboratories) and titers were determined by plaque assay in MDCK cells. For infections, MDMs were washed with serum-free RPMI 1640 medium, and infected at a multiplicity of two with H5N1 HAlo virus or H5N1 ΔNS1 virus. Cells were infected to maximize infection rates and generate a synchronized host cell response (MOI 2, 60–80% infected; data not shown). After 1 hour, virus inoculum was removed, cells were washed twice with serum-free RPMI 1640 medium, and then incubated in RPMI 1640 medium containing 10% serum and antibiotics.
THP-1 mRNA electroporation
Since NS1 protein interferes with processing and subsequent translation of mRNAs from DNA expression constructs, including its own, it is advantageous to express NS1 protein from in vitro- transcribed mRNA (IVT mRNA) to induce high levels of NS1 protein expression. IVT mRNA for NS1 of Influenza A/Vietnam/1203/2004 (H5N1) was generated from a T7 promoter-containing synthetic DNA template (T7_NS1(A/Viet Nam/1203/2004(H5N1))), PCR- amplified with primers NS1_T7_f/NS1_T7_r. As a control, EGFP IVT mRNA was transcribed from a T7 promoter-containing template generated by PCR amplification of the EGFP CDS from pEGFP-C1 (Clontech), using primers EGFP_T7f/EGFP_T7r. Sequences for PCR primers and the synthetic T7-NS1 DNA template are listed in the Key Resources Table. PCR-produced IVT templates were phenol:chloroform-purified and mRNA was produced with an mMESSAGE mMACHINE T7 Ultra Kit (Ambion) according to the manufacturer’s instructions, using pseudo-UTP (TriLink) and 5-methyl-CTP (TriLink) instead of UTP and CTP. IVT mRNA was purified with a MEGAClear Transcriptional Clean-up Kit (Ambion), treated with Calf Intestinal Alkaline Phosphatase (NEB), and purified again with phenol:chloroform:isoamyl alcohol (Invitrogen). For electroporations, 3 M THP-1 cells (a gift from Michael Rehli, University Hospital Regensburg, Germany) were pelleted by centrifugation and serum RNases were removed by washing cells twice at room temperature with phenol red-free OptiMEM (Life Technologies). Cells were electroporated in 200 μl phenol red-free OptiMEM in a 4 mm cuvette using a Gene Pulser Xcell (Bio-Rad) with a rectangular pulse of 400 V and 5 ms duration, delivering 9 μg IVT mRNA. Immediately after electroporation, the cell suspension was transferred into a well of a six-well plate filled with 2 ml pre-warmed THP-1 medium (RPMI-1640 containing 10% low-endotoxin FBS (Peak Serum Inc.), 1x (100 μM) non-essential amino acids (Life Technologies), 1x MEM Sodium Pyruvate Solution (1 mM) (Life Technologies), 1x GlutaMax I (Life Technologies), 50 μM β-mercaptoethanol (Life Technologies)) and cultured at 37°C, 5% CO2, 100% humidity for the indicated times. Electroporated cells were 80–90% viable by trypan blue exclusion. Expression of NS1 was verified by Western blot with an antibody against NS1 (Santa Cruz Biotech).
siRNA experiments
All siRNA were manufactured by Dharmacon. THP-1 cells were electroporated with siRNA against WAPL and NIBPL (or control) using the same protocol as described for mRNA electroporation. 3 million cells and 6 μg of siRNA were used per electroporation. After 72 h, electroporated cells were >92% viable by trypan blue exclusion.
In situ Hi-C
In situ Hi-C was performed as described (Rao et al., 2014) with modifications. Cells were pelleted by centrifugation and resuspended and cross-linked in 1% formaldehyde/PBS for 10 minutes at room temperature (RT). After quenching the reaction by adding 1/20th volume of 2.625 M glycine, cells were pelleted by centrifugation and washed twice with ice-cold 0.5% BSA/PBS. Aliquots of 2 M cells each were snap-frozen and stored at −80°C.
After thawing cells on ice, nuclei were isolated by resuspending the cell pellet in 200 μl wash buffer (50 mM Tris/HCl pH 7.5, 10 mM NaCl, 1 mM EDTA, 0.5% SDS, 1x protease inhibitor cocktail (Sigma P8340)). For the initial time course, nuclei were incubated in 0.2 ml tubes at 62°C for 7 minutes in a PCR cycler. Nuclei were spu n down at 2500 g for 5 minutes at room temperature. Most of the supernatant was discarded, leaving the nuclei in 10 μl liquid. Samples were resuspended in reaction buffer (25 μl 10% Triton X-100, 25 μl NEBuffer 2 (initial time course) or Dpn II buffer (later experiments), 195 μl water) and rotated at 37°C for 15 minutes.
Chromatin was digested overnight (ON) with either 4 μΙ (100 U) Mbo I (NEB) and 0.5 μΙ 1 M DTT (initial time course) or 2 μl (100 U) Dpn II (NEB) (later experiments) at 37C, rotating overhead with 8 RPM. The next day, Mbo I digests (not Dpn II digests of later experiments) were inactivated by incubating for 20 minutes at 620, a nd nuclei were collected centrifugation (5 minutes, 1000 g, RT). 225 μl of the supernatant were discarded, leaving the nuclei in 25 μl liquid, and overhangs were filled in with Biotin-14-dCTP (initial time course) or Biotin-14-dATP (later experiments) by adding 75 μl Klenow Master Mix (54.45 μl water, 7.5 μl NEBuffer 2, 0.35 μl 10 mM dATP (or dCTP), 0.35 μl 10 mM dTTP, 0.35 μl 10 mM dGTP, 7.5 μl 0.4 mM Biotin-14-dCTP (or Biotin-14-dATP)(Invitrogen), 2 μl 10% Triton X-100, 2.5 μl (12.5 U) Klenow fragment (Enzymatics) and rotating overhead at RT, 8 RPM for 40 minutes. Reactions were stopped by adding 2.5 μl 0.5 M EDTA and placed on ice. Proximity ligation was performed by transferring the entire reaction into 1.5 ml Eppendorf tubes containing 400 μl ligase mix (320 μl water, 40 μl 10x T4 DNA ligase buffer (Enzymatics), 36 μl 10% Triton X-100, 0.5 μl 10 % BSA, 1 μl (600 U) T4 DNA ligase (HC, Enzymatics) and rotating ON at 160, 8 RPM. Reactions were stopped with 20 μl 0.5 M EDTA, treated with 1 μl 10 mg/ml DNase-free RNAse A for 15 minutes at 420, 33 μl 5 M NaCl and 55 μl 10 % SDS were add ed and crosslinks reversed for 4 h to ON at 650. Proteins were digested with 10 μl 20 mg/ml DNase-free proteinase K (Thermo) for 2 h at 550 while shaking at 800 RPM, then 90 minutes at 65°C. After extraction with 600 μl pH 8-buffered phenol/chloroform/isoamyl alcohol (Ambion) followed by 300 μl chloroform, DNA was precipitated with 1.5 μl (22.5 μg) Glycoblue (Ambion) and 1400 μl 100% ethanol ON at −20°C, pelleted for 20’ at 16000g, 40, washed twice with 80% ethanol, and the pellet air-dried and dissolved in 131 μl TT (0.05% Tween 20/Tris pH 8). DNA (800 ng) was sheared to 300 bp in 130 μl TT on a Covaris M220 or S220, using the manufacturer’s protocol. For the initial time course, fragments were size-selected by first depleting large fragments with 64 μl 20% PEG8000/2.5 M NaCI (6.45% PEG final) and 5 μΙ Speedbeads (3 EDAC, GE Healthcare Life Sciences) for 10’ at RT, collecting beads on a magnet, and isolating the small (<400 bp) fragments from the supernatant by adding an additional 60 μl PEG8000/2.5 M NaCl and 3 μl Speedbeads (9.5% PEG final), incubating at RT for 10 minutes, washing beads twice with 80% ethanol, air-drying, and eluting with 50 μl TT. For all other experiments, size selection was omitted, and DNA was directly bound to streptavidin-coated Dynabeads. Biotinylated DNA was captured on Dynabeads MyOne Streptavidin T1 (Thermo) by combining the sample (50 μl or 130 μl) with 20 μl Dynabeads that had previously been washed once with 1x B&W buffer (2X B&W: 10 mM Tris-HCl pH=7.5, 1 mM EDTA, 2 M NaCl), and had been resuspended in 50 μl or 130 μl 2x B&W containing 0.2% Tween 20. The binding reaction was rotated at RT for 30 minutes, and DNA-bound beads were washed once with 1 ml 1x B&W/0.1% Triton-X 100, once with TET (0.2% Tween 20, 10 mM Tris pH 8, 1 mM EDTA). Beads were resuspended in 100 μl Blunting Mix (74.5 μl water, 0.5 μl 10% Tween 20, 10 μl 10x T4 DNA ligase buffer (Enzymatics), 4 μl 10 mM dNTP, 2 μl (6 U) T4 DNA polymerase (Enzymatics), 0.4 μl (2 U) Klenow fragment (Enzymatics), 2 μl (20 U) T4 polynucleotide kinase (Enzymatics)) and incubated for 30 minutes at 20C. Reactions were stopped by adding 2.5 μl 0. 5 M EDTA, beads collected on a magnet and washed twice with 150 μl 1x B&W/0.1% Triton-X 100, once with 180 μl TET. Beads were resuspended in 50 μl A-tail Mix (44.25 μl water, 0.5 μl 10% Tween 20, 5 μl NEBuffer 2, 0.2 μl 100 mM dATP, 0.3 μl (15 U) Exo--Klenow (Enzymatics) and incubated for 30 minutes at 37C. Reactions were stopped by adding 1.5 μl 0.5 M EDTA, beads collected on a magnet and the SN was discarded. Beads were resuspended on ice in 48 μl ice-cold Ligation mix (25 μl 2x T4 DNA ligation buffer (Enzymatics), 0.5 μl 10% Tween 20, 22.5 μl water), 0.8 μl 25 μM Bioo Nextflex DNA sequencing adapters were added, and the reaction incubated for 20 minutes at RT. Reactions were stopped by adding 5 μl 0.5 M EDTA, and beads were washed twice with 1x B&W/0.1% Triton-X 100, once with TET and resuspended in 30 μΙ 0.05% Tween 20/LoTE (TE dilute 1:4 with water). Libraries were amplified by PCR for 10 or 12 cycles (98°C, 30s; 10/12x[98°C, 10s; 60°C, 25s; 72C, 30s]; 72°C, 5 mi n; 4ºC, ∞), using 10 μΙ of the bead suspension in a 50 μΙ reaction with NEBNext High-Fidelity 2X PCR Master Mix (NEB) and 0.5 μΜ each Solexa 1GA/1GB primers (Solexa 1GA: AATGATACGGCGACCACCGA, Solexa 1GB: CAAGCAGAAGACGGCATACGA). Libraries were precipitated onto magnetic beads by adding 38 μl 20% PEG8000/2.5 M NaCl and 2 μl SpeedBeads (8.6 % PEG final), thorough mixing by vortexing followed by 10 minute incubation at RT. Beads were collected using a magnet and the supernatant discarded. After washing beads twice by adding 180 μl 80% EtOH, moving the tube strip 6x from side to side of the magnet, collecting beads and discarding the supernatant, beads were air-dried, and DNA eluted by adding 20 μl TT. Libraries were sequenced at a minimum of 42 bp paired-end to a depth of approximately 250 million reads per experiment on Illumina HiSeq 4000, HiSeq 2500, or NextSeq 500 instruments. Time-course experiments were performed in biological duplicate with cells from two different donors. Follow-up infections with wild-type H5N1 virus, H5N1-ΔNS1 virus, and IFNβ treatment were performed with cells from a second set of two donors and in THP-1 cells.
RNA-seq Experiments
Cells were lysed in Trizol (Thermo) and RNA was extracted either by adding chloroform, phase separation and isolating RNA from the upper phase using RNeasy columns with on-column DNase treatment (Qiagen) or by using Direct-zol kits (Zymo Research) with on-column DNase treatment, following the manufacturer’s instructions. Strand-specific total RNA-seq libraries from ribosomal RNA-depleted RNA were prepared using the TruSeq kit Stranded Total RNA Library kit (Illumina) according to the manufacturer-supplied protocol. Libraries were sequenced 100 bp paired-end to a depth of approximately 40 million host genome reads on Illumina HiSeq 2500 or NextSeq 500 instruments.
RNA-seq of size-selected large nuclear RNA from NS1- and EGFP-expressing THP-1 cells (“North-seq”)
Twenty-one million THP-1 cells were electroporated in batches of 3 million cells each with IVT mRNA for NS1 or EGFP as described above, and immediately stimulated with 500 U IFNβ (R&D Systems) for 6 hours. Nuclear and cytoplasmic RNA from 15 million live cells was isolated according to (Bhatt et al., 2012). Briefly, cells were washed with ice-cold PBS, resuspended at 75 million cells/ml final in cytoplasmic lysis buffer (0.15% NP-40, 10 mM Tris pH 7.5, 150 mM NaCl) and incubated on ice for 5 minutes. The lysate was layered onto 500 μl ice-cold sucrose buffer (10 mM Tris pH 7.5, 150 mM NaCl, 24% sucrose w/v) and centrifuged in microfuge tubes for 10 minutes at 13,000g, 4°C. The supernatant con taining the cytoplasmic RNA (~ 700 μl) was transferred to a new tube, and the nuclear pellet was resuspended in 500 pl glycerol buffer (20 mM Tris pH 7.9, 75 mM NaCl, 0.5 mM EDTA, 50% glycerol, 0.85 mM DTT). RNA was extracted from the cytoplasmic and nuclear fraction with Trizol LS according to the manufacturer’s instructions, DNase-treated with RQ1 DNase (Promega), extracted once with phenol:chloroform, once with chloroform, and ethanol-precipitated. For size selection of large nuclear RNAs, two micrograms nuclear RNA were denatured with an equal amount of 2x RNA loading dye (NEB, 47.5% formamide final) for 5 minutes at 70°C and se parated on a 1.5% agarose/1x TBE gel for 30 minutes. After staining the RNA with 1x SYBR Gold in 1x TBE for 15 minutes, RNA above the 45S RNA band was excised with a scalpel blade and gel-extracted (Zymoclean Gel RNA Recovery kit, Zymo Research). Stranded RNA-seq libraries were prepared with a TruSeq Stranded Total RNA kit (Illumina), using either the entire amount of size-selected nuclear RNA recovered from the agarose gel without ribosomal RNA depletion, or 200 ng of nuclear or cytoplasmic RNA according to the manufacturer’s instructions. Libraries were sequenced single-end for 75 bp (SE75) on an Illumina NextSeq 500 instrument.
ChIP-seq
ChIP-seq was essentially performed as described (Heinz et al., 2013). Briefly, cells were fixed in either in 2 mM DSG/PBS for 30 minutes at room temperature followed by an additional 15 minutes with 37% formaldehyde to 1% formaldehyde final (if an experiment contained dimeric transcription factors), or in 1% formaldehyde/PBS for 10 minutes at room temperature (for all other experiments), the reactions quenched by adding 2.625 M glycine to 125 mM final and cells pelleted by centrifugation for 5 minutes at 1,000 g, 4ºC. Cells were washed twice with ice-cold PBS and cell pellets were snap-frozen in dry ice/methanol and stored at −80ºC. Fixed cells were thawed on ice, resuspended in either ice-cold L2 (0.5% Empigen BB, 1% SDS, 50 mM Tris/HCl pH 7.5, 1 mM EDTA, 1 ͯ protease inhibitor cocktail) or ice-cold LB3 (10 mM Tris/HCl pH 7.5, 100 mM NaCl, 0.5mM EGTA, 0.1% deoxcycholate, 0.5% sarkosyl, 1 ͯ protease inhibitor cocktail) and chromatin was sheared to an average DNA size of 300–500 bp by administering 7 pulses of 10 seconds duration at 13 W power output with 30 seconds pause on wet ice using a Misonix 3000 sonicator. The lysate was diluted either 2.5-fold (L2) with ice-cold L2 dilution buffer (20 mM Tris/HCl pH 7.4@20ºC, 100 mM NaCl, 0.5% Triton X-100, 2 mM EDTA, 1 ͯ protease inhibitor cocktail) or 1.1-fold (LB3) with ice-cold 10% Triton X-100. One percent of the lysate was kept as ChIP input. For each immunoprecipitation, aliquots of diluted lysate equivalent to 150,000 to 1 million cells, 20 μl of Dynabeads Protein A (for rabbit polyclonal antibodies) or Dynabeads protein G (for murine monoclonal antibodies) and 2 μg antibody were combined and rotated overnight at 8 RPM and 4ºC. The next day, b eads were collected on a magnet and washed three times each with wash buffer I (10 mM Tris/HCl pH 7.5, 150 mM NaCI, 1% Triton X-100, 0.1% SDS, 2 mM EDTA), wash buffer III (10 mM Tris/HCl pH 7.5, 250 mM LiCl, 1% IGEPAL CA-630, 0.7% Deoxycholate, 1 mM EDTA) and twice with ice-cold TET (10 mM Tris/HCl pH7.5, 1 mM EDTA, 0.2% Tween-20). Libraries were prepared with NEBNext Ultra II DNA library prep kit reagents according to the manufacturer’s protocol on the beads suspended in 25 μl TT (10 mM Tris/HCl pH7.5, 0.05% Tween-20), with reagent volumes reduced by half. DNA was eluted and crosslinks reversed by adding 4 μl 10% SDS, 4.5 μl 5 M NaCl, 3 μl EDTA, 1 μl proteinase K (20 mg/ml), 20 μl water, incubating for 1 h at 55ºC, then 30 min to overnight at 65ºC. DNA was cleaned up by adding 2 μl SpeedBeads 3 EDAC in 61 pl of 20% PEG 8000/1.5 M NaCl, mixing and incubating for 10 minutes at room temperature. SpeedBeads were collected on a magnet, washed twice by adding 150 μl 80 % EtOH for 30 seconds each, collecting beads and aspirating the supernatant. After air-drying the SpeedBeads, DNA was eluted in 25 μl TT and the DNA contained in the eluate was amplified for 12 cycles in 50 μl PCR reactions using NEBNext High-Fidelity 2X PCR Master Mix or NEBNext Ultra II PCR master mix and 0.5 μM each of primers Solexa 1GA and Solexa 1GB. Libraries were cleaned up as above by adding 36.5 μl 20% PEG 8000/2.5 M NaCl and 2 pl Speedbeads, two washes with 150 μl 80 % EtOH for 30 seconds each, air-drying beads and eluting the DNA into 20 μl TT. ChIP library size distributions were estimated following 2% agarose/TBE gel electrophoresis of 2 μl library, and library DNA amounts measured using a Qubit HS dsDNA kit on a Qubit fluorometer. ChIP input material (1 percent of sheared DNA) was treated with RNAse for 15 min at 37°C in EB buffer (10 mM Tris pH 8, 0.5% SDS, 5 mM EDTA, 280 mM NaCl), then digested with Proteinase K for 1 h at 55°C and crosslinks reversed at 65°C for 30 min to overnight. DNA was cleaned up with 2 μl SpeedBeads 3 EDAC in 61 μl of 20% PEG 8000/1.5 M NaCl and washed with 80% ethanol as described above. DNA was eluted from the magnetic beads with 25 μl of TT and library prep and amplification were performed as described for ChIP samples. Antibodies were from Genetex (RNA polymerase II (RPB2): GTX102535, RAD21: GTX106012), Active Motif (CTCF: 61311, H3K27ac: 39133), Diagenode (RNAPII S2P: C15200005, S5P: C15200007), EMD Millipore (RNAPII S7P: 04–1570, H3K27me3: 07–0449), Abcam (H3K4me3: ab8580, H3K9me3: ab8898, H3K36me3: ab9050), Santa Cruz Biotech (PU.1: sc-352, NF-kB p65: sc-372, C/EBPβ: sc-150, c-FOS: sc-7202). Libraries were sequenced single-end for a minimum of 75 bp (SE75) or paired-end for 42 bp (PE42) to a depth of 10–20 million reads on an Illumina NextSeq 500 instrument.
MethylC-seq
MethylC-seq libraries were prepared from ChIP input DNA that had been isolated as from formaldehyde-fixed samples as previously described (Heinz et al., 2013). Briefly, chromatin was sheared to an average size of 300 bp in ChIP lysis buffer, crosslinks reversed in 1% SDS/TE/300 mM NaCl ON at 65ºC, treated for 30 minut es with 5 μg RNase A, digested for 2 h with 20 μg proteinase K and DNA was isolated with Speedbeads/PEG as described above. DNA libraries were made from 100 ng input DNA by adding methylated DNA sequencing adapters (Bioo) with NEB Ultra II DNA library prep kit reagents and cleaned up using Speedbeads/PEG/NaCl as above. Fifty nanograms of adapter-ligated DNA were bisulfite-treated using a Zymo EZ DNA methylation kit (Zymo Research) according to the manufacturer’s instructions, and PCR-amplified using KAPA HiFi U+ ReadyMix and Solexa 1GA/1GB primers, followed by SpeedBead cleanup of the PCR product as described above. Libraries were sequenced paired-end for 2×150 bp (PE150) to a depth of approximately 100 million reads on an Illumina NextSeq 500 instrument.
Start-seq
Start-seq was essentially performed as described (Scruggs et al., 2015), from total RNA with minor modifications. These comprised replacing phenol/chloroform extractions with Trizol LS (Invitrogen), using RppH (NEB) instead of tobacco acid pyrophosphatase and generating sequencing libraries with a NEBNext Small RNA Library Prep kit (NEB), according to the manufacturer’s instructions. Briefly, 10 μg Trizol-isolated total RNA were separated on a 15% Urea-TBE PAGE gel, 25–65 nt RNA extracted from the gel in a buffer containing 300 mM sodium acetate pH 5.5, 10 mM Tris/HCl pH 7.5, 1 mM EDTA, 0.05% Tween 20. RNA was precipitated with 3 M sodium acetate pH 5.5 (added to 300 mM final) and 2.5 volumes of 100% ethanol overnight at −20°C. Non-capped phosphorylat ed RNA was degraded with 2 U Terminator nuclease (Lucigen), followed by RNA dephosphorylation with 10 U Calf Intestinal Phosphatase (CIP, NEB) and RNA cleanup by Trizol LS according to modified manufacturer’s instructions (1/10th volume 3 M sodium acetate/pH 5.5 was added before RNA precipitation with an equal volume isopropanol). RNA was dephosphorylated a second time with 10 U CIP and Trizol LS-purified as before. After decapping RNA with 5 U RppH, sequencing libraries were prepared using a small RNA library prep kit (NEB) according to the manufacturer’s instructions, and sequenced for 75 cycles on an Illumina NextSeq 500 instrument. To control for non-capped RNA signal in the input RNA, the same overall protocol was followed but omitting the RppH decapping step.
QUANTIFICATION AND STATISTICAL ANALYSIS
Hi-C Analysis
Hi-C reads were trimmed at Mbo I/Dpn II recognition sites (GATC) and aligned to the human genome (GRCh37/hg19) using bowtie2 (Langmead and Salzberg, 2012), keeping only read pairs that both map to unique genomic locations for further analysis (MAPQ > 10). All PCR duplicates were also removed. Subsequent Hi-C analysis was performed using HOMER (Lin et al., 2012), including Hi-C normalization and structural feature identification. All software tools and a description of how to use them are freely available at http://homer.ucsd.edu/homer/interactions2/. The monocyte-derived macrophage Hi-C samples generated by this study can be divided into two separate sets of experiments. The first set of macrophage Hi-C experiments was an initial pilot study performed as a time course of mock- and IAV-infected samples (6, 12, 18 h.p.i.) in replicate from two separate donors. The second set of macrophage Hi-C experiments was performed with mock, IAV-, IAV-ΔNS1-infected (6 hpi) and IFNβ-treated samples in replicate from an additional two donors. To ensure the fair comparison across experimental conditions with equitable samples, only the second set of experiments were used in the bulk of analysis presented throughout the main study. The initial time-course samples were only used to perform an initial evaluation of IAV’s temporal impact on chromatin structure (Figure S1C) and to demonstrate the reproducibility of IAV-mediated changes in local chromatin structure as demonstrated at the IFIT locus (donors 3 & 4, Figure S3A).
Hi-C Normalization and Visualization
To construct Hi-C contact maps, Hi-C interaction counts were initially normalized by the total Hi-C read coverage at each of the two interacting regions (e.g. window size of 15 kb), which reflects the number of restriction sites, read mapping rate, and many of the other technical biases (such as open chromatin) that lead some regions to accumulate more reads than others. Given the total coverage of a region i (ni) and the total number of reads N, the expected interaction counts between any two regions is then given by ni*nj/N. Interaction totals at chromosomal or sub-chromosomal scales were further normalized using iterative interaction read balancing to ensure interaction totals were consistent across all 15 kb regions (Imakaev et al., 2012). With the exception of the PCA analysis of chromatin compartments described below, all of the analysis in this study was performed using a window resolution size of 15 kb unless explicitly stated otherwise. To reduce edge-effects introduced by arbitrary binning the genome at specific intervals, 15 kb Hi-C contact maps were resampled with a step size of 5 kb resolution to provide a smoothed contact map. For visualization, Hi-C contact map pixels were normalized to interactions per hundred square kilobases per billion interactions (ihskb) to normalize for the size of bins used and the depth of sequencing such that the matrices are readily comparable across experiments. Normalized Hi-C matrices generated using HOMER were then visualized using TreeView3 (Keil et al., 2016). THP-1 HiC experiments were visualized using contact maps with a window size of 20 kb to account for their reduced sequencing depth (~50% that of the MDM samples). Hi-C matrices displayed in each of the figures were generated by pooling reads across replicate experiments unless otherwise specified, and the same color scheme is used throughout the figure panels.
Hi-C Compartment Analysis:
PCA analysis of Hi-C experiments used to define chromatin compartments were performed with HOMER (Lin et al., 2012). For each chromosome, a balanced and distance normalized contact matrix was generated using window size of 50 kb sampled every 25 kb, reporting the log2 ratios of observed to expected contact frequencies for any two regions. This resolution represented the smallest size that yielded consistent PC1 results between replicates. The correlation coefficient of the interaction profiles for any two regions across the entire chromosome were then calculated to generate a correlation matrix. This matrix was then analyzed using Principal Component Analysis (PCA) from the prcomp function in R (www.r-project.org), and the eigenvector loadings for each 25 kb region along the first principal component were assigned to each region (PC1 values). The PC1 values from each chromosome were scaled by their standard deviation to make them more comparable across chromosomes and analysis parameters. For each chromosome, PC1 values are multiplied by −1 if negative PC1 regions are more strongly enriched for active chromatin regions defined by H3K27ac peaks to ensure the positive PC1 values align with the A/permissive compartment (as opposed to the B/inert compartment). Sex chromosomes were excluded from the PCA analysis. Due to the abnormal karyotype of THP-1 cells, the PCA calculation of THP-1 cells was performed on individual chromosome arms separately. Furthermore, chromosome arms with major duplications, deletions, and/or rearrangements were excluded from further analysis (chr11q, chr12p, chr21p) as these regions occasionally produced results that were inconsistent with A-B compartment structure. PCA calculations were performed on individual replicates as well as combined experiments derived from pooling replicates, the later of which were used in figures and other summary analysis.
In order to identify confidently switching compartments during IAV infection, PCA analysis was first independently carried out on mock- and IAV- infected replicate experiments. Replicate PC1 values across the genome were then analyzed using the R package limma (lmFit/eBayes) to identify regions with significantly changing PC1 values (FDR < 5% and greater than 0.5 absolute change in PC1 value). Significantly changing regions were then stitched together into larger domains if they were located adjacent to one another or separated by no more than 50 kb and the intervening regions had a change in PC1 greater than 0.5. Regions at least 100 kb or larger were reported in Table S3 (B-to-A: 23 regions, A-to-B: 9 regions).
Hi-C compaction analysis (DLR/ICF):
The compaction status of each 15 kb region in the genome was considered by calculating the distal-to-local log2 ratio (DLR) and the interchromosomal fraction (ICF) of Hi-C interactions at that locus. To calculate the DLR, coverage-normalized Hi-C interaction frequencies were used to calculate the total number of interactions interacting with the 15 kb region of interest to regions located either greater than 3 Mb (i.e. distal) versus those less than 3 Mb away (i.e. local). A distal interaction threshold of 3 Mb segregates interactions that are predominantly mediated by cohesin (i.e. < 3 Mb) from those mediated by other mechanisms. This distance threshold was defined based on the interaction distance with the maximum difference in Hi-C interaction frequency observed between NIPBL and WAPL knockdown (Figure S4E).
Interactions less than 15 kb were excluded from the analysis (i.e. potential re-ligation products). When computing the ICF, the total number of coverage normalized Hi-C interactions interacting between the 15 kb region of interest and other chromosomes were compared to the total number of normalized interactions at that locus. The DLR and ICF calculations were performed at 3 kb intervals (i.e. on overlapping 15 kb regions) along the genome. Since the specific chromatin looping structure of a region or its placement along the chromosome can have a significant impact on the absolute values of the DLR and ICF (i.e. regions near telomeres usually have higher ICF), each value is normally reported as the change in DLR or ICF between conditions. Furthermore, to account for any global impact that technical variation can have on systemic DLR and ICF values, ΔDLR and ΔICF vectors are centered on zero. For example, replicate Hi-C experiments can differ in their global fraction of interchromosomal interactions by 1–2%, and removing this global shift helps limit changes in ICF to specific loci with strong changes in compaction.
Hi-C Loop Analysis
Pairs of genomic regions exhibiting elevated numbers of Hi-C interactions, or loops, were found using HOMER. First, balanced Hi-C interaction matrices were generated for each chromosome with a window size of 15 kb sampled every 3 kb, considering only interactions between regions within 2 Mb of one another. Putative loops were identified by identifying local maxima in the contact map satisfying the following criteria: (1) The putative loop interaction count total (at 15 kb resolution) must be 1.5-fold greater than the local average interaction count from the surrounding 75 kb region (i.e. 5x window size), (2) the putative loop interaction count total must must be 2-fold greater than the global average interaction count for regions with the same interaction distance along the chromosome. Both thresholds were further controlled by modeling the average local (1) and global (2) expected loop interaction counts using a Poisson distribution and requiring the observed loop interaction total to exceed p-values of 0.01 and 10−5, respectively, to avoid calling putative loops based on low interaction counts. To help standardize the analysis across experiments, only putative loops greater than or equal to 45 kb in length (i.e. 3x window size) and those with at least 20 normalized Hi-C interactions were considered for further analysis. Loops were further filtered if the read coverage at their anchor points was lower than half of the average coverage of similarly sized regions in the Hi-C experiment or if they overlapped regions annotated as segmental duplications defined in the UCSC Genome Browser (Bailey et al., 2002). In addition, putative loops on the sex chromosomes were excluded from THP-1 datasets when considering further analysis.
In order to identify loops that weaken or strengthen between conditions, loops are first found separately in each condition using experiments that pooled replicates to maximize sensitivity for loop identification. Then, lists of loops between conditions were merged, combining the loops where both sets of loop anchors were located within 15 kb of one another, replacing the merged loops with a single loop specified by the average loop anchor positions of the initial loops. The resulting merged loop list (including unmerged, experiment-specific loops) was then used to reanalyze Hi-C matrices from each individual replicate in each condition to calculate a matrix of normalized Hi-C interaction counts for each loop across each experiment. Loop interaction counts were then rounded to the nearest integer and the resulting matrix was analyzed using edgeR (Robinson et al., 2010) to identify differentially regulated loops between conditions from the interaction count data. The edgeR design matrix was modified to reflect that each replicate sample in Mock-, IAV-, IAV-ΔNS1-, and IFNβ-treated conditions were paired across each donor. Differentially regulated loops were defined as loops with changes of normalized interaction counts exceeding 1.5 fold and with an edgeR-derived p-value less than 0.05. Adjusted p-values reflecting the estimated false discovery rate (FDR) were also calculated and reported in Table S3.
Hi-C GSEA Analysis
Gene Set Enrichment Analysis (GSEA, (Subramanian et al., 2005)) was used to assess the significance of overlap between specific genomic features of interest (e.g. RNAPII transcribed CTCF sites) and changes in loop strength between conditions (Figures 4B, 4C, 6D and S5B).
First, a pre-ranked list of Hi-C loops was generated by calculating the Hi-C loop interaction log2 fold changes by dividing the normalized Hi-C interactions between treatment and control conditions while adding a pseudocount of 5 normalized interactions to both conditions to help stabilize the variance of loops with lower interaction counts. GSEA “gene sets” were generated by assigning loops to the list if they were associated with specific genomic features where at least one of their two loop anchors overlapped a genomic feature (resolution 15 kb). GSEA v3.0 was run in pre-ranked list mode with 10k randomizations across each gene set to score enrichment and the results were summarized with the FDR q-values.
RNA-seq Analysis
Sequencing reads were mapped to the human and influenza genomes using STAR (Dobin et al., 2013). The human hg38 genome index incorporated RefSeq gene annotation to aid in spliced alignment. The influenza index was built from the influenza A/Viet Nam/2013/2004 sequence alone. Raw uniquely mapped reads counts per human gene were calculated using FeatureCounts (Liao et al., 2014). In experiments where technical replicates using the same biological donors were available, technical replicates were merged. Differential expression analysis was carried out using DESeq2 (Love et al., 2014), with comparisons carried out against time-matched mock-infected controls. Gene expression heatmaps were generated based on the regularized logarithm transformation (rlog) from DESeq2, then hierarchically clustered and visualized with the pheatmap package in R. Statistically enriched Gene Ontology biological process terms were identified and refined using clusterProfiler (Yu et al., 2012), then visualized with pheatmap. Genes highly induced by IFΝβ relative to mock were defined as genes with greater than 8-fold induction and a maximum FPKM greater than 5 (n = 75 genes, Figure 1F; Table S2). Poly(A) site cleavage efficiency for each gene was defined as the ratio of RNA-seq reads directly spanning the RefSeq-defined transcription end site (normalized to FPKM) to the gene FPKM levels (Figure S1F). The annotation of splicing frequencies were calculated using the splice event tables produced during the STAR alignment. Unannotated splicing events were classified as intra- or intergenic using RefSeq gene annotations and then normalized per million mapped reads (Figure S1G).
Read-through regions defined by RNA-seq were identified by finding contiguous intervals of RNA-Seq read coverage. Briefly, stranded read coverage and magnitude was calculated for all 1 kb intervals of the hg38 genome. Using bedtools (Quinlan and Hall, 2010), intervals with at least 0.05 reads per million mapped reads were merged with a maximum distance of 2 kb. Merged intervals of at least 25 kb were re-merged with a maximum distance of 10 kb, then annotated with the gene closest to the upstream end of the interval and overlapping gene with highest expression. Intervals primarily overlapping genes were filtered out. Potential read-through regions were then scored based on normalized read count and coverage (Table S2).
ChIP-seq Analysis
Reads were aligned to the human genome (GRCh37/hg19) using bowtie2 (Langmead and Salzberg, 2012), keeping only reads that map to a single unique genomic location for further analysis (MAPQ > 10). ChIP-seq peaks were called using HOMER’s findPeaks program in either “histone” (histone marks, RNAPII) or “factor” (CTCF, RAD21, PU.1, C/EBPβ, Fos, NF-kB p65) mode using default parameters to identify broad or focal peaks, respectively. HOMER was also used to generate sequencing-depth normalized bedGraph/bigWig files for visualization in the UCSC Genome Browser (http://genome.ucsc.edu). De novo motif discovery was performed using HOMER using the 100 bp around ChIP-seq peak centers. CTCF motif direction was calculated using HOMER by finding the best match to the known CTCF motif within 50 bp of the peak summit and assigning the peak direction based on the DNA strand the CTCF motif matched best. Meta-gene (e.g. Figure 1F), meta-loop (Figure S3B), and ChIP-seq heatmap (Figure S6A) plots were generated using the HOMER annotatePeaks.pl and makeMetaGeneProfile.pl programs. Overlapping peak locations were identified using the HOMER mergePeaks program. ChIP-seq peaks were defined as overlapping loop anchor regions if they were found within the 7.5 kb of the center of the loop anchor (loop analysis was carried out with window resolution of 15 kb). Whenever ChIP-seq log2 fold changes are reported or used (e.g. Figures 3D, 7C, S4D, S6A and S7A), normalized read densities were calculated +/− 200 bp from the peak center and a pseudocount of 0.5 FPKM was added to both the numerator and denominator to dampen low intensity noise and avoid divide-by-zero errors. Differential ChIP-seq peaks were calculated using the HOMER tool getDifferentialPeaks program to identify regions with greater than 2-fold normalized read coverage and p-value less than 1e-7 (Poisson, Figure 3C).
The anticorrelation analysis of changes in RNAPII and RAD21 levels (Figures 3D, S3D, S3E, S3I and S5A) was performed at CTCF peaks merged between control and treated conditions which met the following criterion: (1) The maximum level of CTCF, RAD21, and RNAPII in either control or treated conditions must exceed 7.5 FPKM (for MDM) or 10 FPKM (for THP-1 cells) and be less than 1.5 FPKM in ChIP-input samples to define peaks with high magnitude ChIP signal for all three proteins. (2) CTCF sites were classified as H3K27ac positive if they had greater than 2.5 FPKM (for MDM) or 1.5 FPKM (THP-1) H3K27ac ChIP-seq signal in either condition. The different thresholds for MDM versus THP-1 cells reflects the differing ChIP-efficiency for the CTCF and H3K27ac ChIP-seq data generated in each cell type. The Pearson correlation was calculated using the log2 ratios for RNAPII and RAD21 between treated and control conditions at each CTCF peak that passed the above criteria.
To analyze the temporal correlation between RNAPII and RAD21/cohesin levels at individual peaks in the PAM- and flavopiridol-treated THP-1 time course (Figure 6C), RAD21 peaks were first merged across all time points into a single set of RAD21 peaks. Next, RNAPII and RAD21 levels were calculated at each peak over the time course and their levels quantile normalized among RNAPII or RAD21 samples to standardize their signal distribution across the dataset. Only peaks where RNAPII is induced by PAM at least 2-fold relative to control cells with a maximum RNAPII level greater than 10 FPKM in any of the PAM-treated conditions were considered further to focus on sites experiencing induced, high magnitude transcription regulation. To determine if each peak was found in a region more strongly associated with RNAPII initiation or RNAPII elongation, the ratio of RNAPII in flavopiridol and non-flavopiridol treated samples was computed for both PAM treated and untreated conditions. RAD21 peaks in regions where RNAPII is likely initiating where defined as those with >1.5 increases in RNAPII after flavopiridol treatment. Conversely, RAD21 peaks in regions where RNAPII is likely elongating where defined as those with a <−1.5-fold decrease in RNAPII density after flavopiridol treatment in either condition. Sites were further divided based on the presence of CTCF (>10 FPKM). Pearson correlation was calculated for each peak by comparing the log2 ratios of RNAPII and RAD21 relative to their controls (i.e. untreated cells) across each time point.
To identify CTCF peaks with changes in RNAPII transcription following cellular stimulus (Figure 4C), CTCF peaks were first merged between treated and untreated conditions into a unified set of CTCF peaks. From this set, only the top 30,000 peaks (~ >10 FPKM) were considered per treatment to avoid inclusion of weaker peaks where CTCF often associates with regulatory elements [presumably] in an indirect manner. RNAPII ChIP-seq reads were then counted at each CTCF peak in both treated and untreated conditions, and edgeR was used to identify CTCF peaks with induced levels of RNAPII following stimulus by setting the common dispersion to 10−5 and using thresholds of FDR < 10% and log 2 ratio > 1.5.
RNAPII ChIP-seq was also used to approximate gene expression levels in THP-1 cells by using HOMER annotatePeaks.pl program to calculate the RNAPII FPKM levels at GENCODE defined protein coding genes. Genes less than 250 bp in length were excluded from this analysis.
Genes highly induced by IFNβ or PAM were defined as genes with a fold-change greater than 8 (IFNβ) or 4 (PAM) and a maximum RNAPII-defined expression greater than 5 FPKM. Proximal promoter RNAPII pausing ratios were calculated as the log2 ratio of proximal promoter RNAPII FPKM (−50 to +200 nt relative to the TSS) to the gene body (+200 nt to +5 kb relative to the TSS).
RNAPII ChIP-seq data was also used to define read-through transcription in IFNβ and PAM-treated THP-1 cells (Figures S3J, S4F and S4G). To identify regions with significant read-through transcription, RNAPII peaks were found using NS1+IFNβ or NS1+PAM with the HOMER findPeaks program with parameters “-style histone -minDist 5000”, which merges adjacent peaks found within 5 kb to assemble broad regions of continuous enrichment. Considering only peaks that are greater 10 kb in length (i.e. likely transcripts), RNAPII ChIP-seq reads were counted across both eGFP and NS1 conditions, keeping only peaks that contained greater than 2-fold more reads with NS1.
Start-seq Analysis
Start-seq reads were trimmed for 3’ adapter sequences and then aligned to the human genome (GRCh37/hg19) using bowtie2 (Langmead and Salzberg, 2012), keeping only reads that map to a single unique genomic location for further analysis (MAPQ > 10). Transcription start sites were defined using HOMER by running findPeaks in ‘tss’ mode, which looks for strand-specific clusters of Start-seq reads within 150 bp regions in a similar manner to the discovery of ChIP- seq peaks. Visualization and quantification of Start-seq reads was carried out as described for ChIP-seq.
MethylC-seq Analysis
MethylC-seq reads were aligned with Bismark/Bowtie2 (Krueger and Andrews, 2011) and reported as the number of observed methylated and unmethylated cytosines in the CpG context. HOMER was used process the methylation data into average methylation rates for B-to-A switched regions (Figure S1K) and create bedGraph genome browser tracks reporting the average %mCpG rates in 1 kb bins across the genome (Figure S1E).
DATA AND SOFTWARE AVAILABILITY
Time course Hi-C data from IAV-infected macrophages are available in the NCBI GEO database under accession number GSE113703. All other sequencing data analyzed in this study (Hi-C, ChIP-seq, RNA-seq, North-seq, Start-seq, and methylC-seq) are available under accession number GSE103477. Time course RNA-seq data from IAV-infected macrophages and IAV-infected epithelial cells are available under accession number GSE97672 and GSE89008, respectively. Analysis software developed for this study is included in HOMER and is available at http://homer.ucsd.edu/homer/.
Supplementary Material
(A) Total number of up- and down-regulated genes at 3, 6, 12, and 18 hours post infection (hpi) between IAV and time-matched mock controls (2 replicates per IAV and mock infection time point). (B) Ratio of viral RNA to host RNA in rRNA-depleted total RNA-seq experiments at each time point. (C) IAV infection leads to gradual loss of genome structure over time. In situ Hi-C contact maps for 60 Mb of Chr 10 in mock- and influenza A virus (IAV)-infected primary human monocyte-derived macrophages (MDM) at 6, 12, and 18 hpi. RNA polymerase II (RNAPII) ChIP-seq read densities from mock-infected cells and PC1 values (compartments) for each time point are depicted above each contact map, while changes in PC1, DLR (distal-to-local ratio), and ICF (interchromosomal interaction frequency) between time-matched IAV and mock conditions are depicted to the right (Hi-C data based on 2 replicates per IAV- and mock-infected time point). Asterisk denotes that the global changes in DLR and ICF were not normalized to zero across the genome for this analysis (see methods). (D) Changes in locus-specific compaction as measured by changes in DLR and ICF confirm that the FKBP5 locus decompacts in THP-1 cells treated with dexamethasone (Dex). (E) RNA-seq and ChIP-seq read density and chromatin structure tracks at the IFIH1 locus, which displays B-to-A compartment switching and IAV-induced transcriptional read-through both upstream and downstream of the locus. Tracks for mock- and IAV-infected, and IFNβ-treated MDMs are shown for RNA-seq, RNAPII (transcription), H3K36me3 (transcriptional elongation), H3K27ac (promoter/enhancer activation), H3K4me3 (promoter activation), H3K9me3 (repression), H3K27me3 (repression), 5-methylcytosine percentage per kb (CpG context only, repression). Changes in DLR and ICF between mock- and either IAV-infected or IFNβ-treated cells are reported at 15 kb resolution. PC1 eigenvector loadings are reported at 50 kb resolution. (F) Ratio of reads spanning the poly(A) cleavage site as a function of time post infection in IAV- and mock-infected cells. (G) Distribution of splice sites identified from RNA-seq reads in mock and IAV infection conditions as a function of time. Annotated genic sites correspond to annotated splice junctions. Unannotated splice sites are segregated based on whether they were found in intragenic or intergenic regions. (H) Meta-gene profiles for RNAPII CTD phosphorylation status (Ser5/initiating, Ser7/initiating, Ser2/elongating) in MDM after IAV infection or IFΝβ treatment at highly IFNβ-responsive genes. (I) Meta-gene profiles for RNAPII, H3K27ac, H3K4me3, and Start-seq (transcription initiation) in mock- and IAV-infected MDM and THP-1 macrophages at at highly IFNβ-responsive genes. (J) Transcription initiation occurs only at responsive gene promoters and not read-through regions. Genome browser screenshots of RNA-seq and Start-seq tracks at the CMPK2/RSAD2 locus in mock- and IAV-infected THP-1 macrophages. (K) Distribution of RNA-seq and ChIP-seq read densities in mock-, IAV-, and IFΝβ-treated cells measured by fragments per kb per million mapped reads (FPKM) in regions that switch from the B to A compartment (n = 23 regions, see Table S3). The distribution of average mCpG% for each region is shown for Mock- and IAV-infected cells.
(A) RNAPII ChIP-seq density at different classes of response genes in MDM after mock-, IAV-, dNS1-, or IFNβ-treatment. (B) Ratio of viral RNA to host RNA in rRNA-depleted total RNA-seq experiments for wild-type IAV (H5N1) and NS1 truncation mutant H5N1 virus (ΔNS1) at 6 hpi. (C) Total number of up- and down-regulated genes after IAV infection, ΔNS1 infection, or IFNβ treatment 6 hpi relative to mock infected controls (two replicates per condition, >3 fold, FDR < 5%). (D) Hierarchical gene expression clustering of significantly regulated genes in IAV, ΔΝS1, and ΙFΝβ-treated conditions. Top enriched GO terms for major clusters are shown to the left. (E) Western blot showing the protein levels of NS1 after electroporation of in vitro transcribed mRNA for influenza NS1 in THP-1 cells. (F) Meta-gene plot of RNAPII ChIP-seq density in THP-1 cells expressing EGFP (control) or NS1 at all genes with RNAPII expression > 1 FPKM. NS1 expression results in read-through transcription downstream of gene 3’ ends and an increase in RNAPII promoter-proximal pausing at gene 5’ ends. (G) Distribution of RNAPII pause ratios (ratio of promoter to gene body density) in EGFP and NS1 expressing THP-1 cells. (H) Meta-gene plot of RNAPII ChIP-seq density in IAV-, ΔNS1-, or Mock-infected MDM at genes down-regulated by IAV in RNA-seq (< 1/3 FC, < 5% FDR, n = 587 genes, two replicates per condition). IAV infection results in increased RNAPII promoter-proximal pausing relative to ΔNS1 in down-regulated genes. (I) RNAPII ChIP-seq read densities and changes in DLR and ICF in THP-1 cells expressing EGFP (control) or NS1 and treated with PAM, IFNβ, and/or flavopiridol. Transcriptional and structural effects are specific for target genes and reversible following flavopiridol treatment. (J) Northern blot of nuclear RNA showing the regions of the gel extracted for the sequencing of long RNA species (North-seq). (K) Meta-gene plot of RNA-seq read density of cytoplasmic, nuclear, and long (>13kb) nuclear RNA isolated from THP-1 cells treated with IFNβ overexpressing either EGFP or NS1. RNA from read-through regions was highly enriched for nuclear and long nuclear fractions. (L) Browser tracks showing RNA-seq/North-seq read densities at the STAT1 locus for THP-1 cells over expressing EGFP or NS1.
(A) Normalized Hi-C 2D contact maps for each mock- and lAV-infected replicate Hi-C experiments at the IFIT family locus showing reproducible changes in chromatin interactions after infection. (B) Meta-loop plot showing the average ΔDLR and ΔICF at loops strengthened or weakened during IAV infection. 0% and 100% represent the aggregate positions of loop anchors. Randomly chosen loops (200) identified in MDM were included as a control. (C) Distribution of DLR values relative to RAD21/cohesin peaks are shown for RAD21 peaks separated into quantiles based on the number of reads per peak. The percentage of RAD21 peaks overlapping CTCF and RNAPII peaks, respectively, are displayed for each quantile. CTCF-associated and RNAP-associated RAD21 peaks lead to increased and decreased compaction, respectively. (D) Correlation coefficient between changes in RNAPII and changes in RAD21/cohesin at CTCF sites after IAV infection as a function of distance relative to the TSS. Only CTCF sites with high levels of RNAPII, RAD21, and CTCF in either mock- or IAV-infected cells were considered (>7.5 FPKM). (E) Negative correlation between cohesin and RNAPII is dependent on high levels of transcription. Correlation between the changes in RAD21/cohesin and RNAPII levels at CTCF sites lacking H3K27ac is shown at different levels of maximal RNAPII loading at each site. (F) Comparison of changes in loop normalized Hi-C interactions in IAV and ΔNS1 infections and IFNβ treatments compared to mock infection in macrophages for all loops identified as differentially regulated following treatment (6 hpi, log2 vs. mock). Interaction ratios were clustered using hierarchical clustering and grouped by loops that strengthen across all three treatment conditions, weaken across all three conditions, or are selectively weakened with IAV infection. (G) Normalized Hi-C interaction matrix at the highly induced IFIT family locus in mock- (lower left) and ΔNS1- or IFNβ- (upper right) treated macrophages (compare to IAV-infection in Figure 3A). ChIP-seq read densities for RNAPII, CTCF, RAD21 (cohesin), and H3K27ac are shown below. Red and blue triangles indicate preferred interaction direction of highest matching CTCF motif found at each CTCF peak. Black arrows above RAD21 peaks indicate regions with dynamic cohesin changes during infection/treatment. (H) Same as (G) depicting changes at the MX1/MX2 locus, which shows similar changes across IAV, ΔNS1, and IFNβ treatment. Cohesin and loop loss at the CTCF site at the 3’ end of the MX1 gene is observed across all treatments (black arrow). (I) Negative correlation between cohesin and RNAPII is dependent on high levels of transcription in both PAM- and IFΝβ-treated THP-1 cells overexpressing NS1. Correlation between the changes in RAD21 and RNAPII levels at CTCF sites is shown at different levels of maximal RNAPII loading at each site. (J) RNAPII and RAD21/cohesin changes at CTCF sites in NS1-induced read-through regions in THP-1 cells stimulated with IFNβ ± flavopiridol. Changes in RNAPII and Rad21/cohesin observed with IFNβ are reverted after flavopiridol treatment. Flavopiridol significantly changed the log2 ratios of both RNAPII and RAD21 (t-test). (K) Normalized Hi-C interaction matrix at the highly induced TNFAIP3 locus in EGFP- (lower left) and NS1- (upper right) expressing THP-1 cells stimulated with PAM. ChIP-seq read densities for RNAPII, CTCF and RAD21 (cohesin) are shown below. Black arrows above RAD21 peaks indicate peaks lost due to NSI-induced read-through transcription and partially recovered following flavopiridol treatment.
(A) Western blot depicting NIPBL and WAPL protein levels following siRNA-mediated knockdown for 48 hours. Samples with siRNAs labeled in black (WAPL-3, control, NIPBL-pool) were used in subsequent experiments. (B,C) Normalized Hi-C interaction matrix for siNIPBL and siWAPL THP-1 cells at the PDE4B locus (B) and 60 Mb of chr10 (C). WAPL knockdown results in additional loops at the local scale (B, e.g. white circle) while NIPBL knockdown yields an increase in long range interactions (C, white arrows). (D) Log2 ratios of gene body RNAPII density in PAM-treated vs untreated THP-1 cells for all RNAi conditions for both eGFP and NS1 expressing cells. siRNA against NIPBL and WAPL have a relatively minor impact on RNAPII levels in PAM response genes. (E) Effect of NIPBL or WAPL depletion on chromosome-wide Hi-C interaction frequencies relative to Hi-C interaction distance in PAM-treated THP-1 cells. (F) WAPL depletion inhibits transcription-associated decompaction of highly transcribed read-through regions. Log2 differences in interaction frequencies in NS1- versus EGFP-expressing THP-1 cells as a function of interaction distance in Ctrl, NIPBL or WAPL knockdown conditions at PAM+NS1-induced read-through regions. (G) NIPBL depletion inhibits flavopiridol-induced recompaction of PAM+NS1-induced read-through regions. Distribution of Hi-C interaction frequencies relative to interaction distance in Ctrl, NIPBL or WAPL knockdown in PAM+NS1-induced read-through regions in THP-1 before or after flavopiridol inhibition of elongation. (H) Normalized Hi-C interaction matrices at the PAM-activated SOD2 locus, comparing EGFP, NS1, and NS1+flavopiridol THP-1 cells treated with PAM. ChIP-seq read densities for RNAPII, RAD21 (cohesin), and CTCF are shown below for Ctrl, NIPBL, and WAPL knockdown conditions.
(A) Without read-through, the anticorrelation of RNAPII and cohesin/RAD21 at CTCF sites is strongest within gene bodies of highly transcription genes. Pearson correlation of the ratios of RNAPII and RAD21 ChIP-seq signal at CTCF peaks in the respective gene annotation regions IFNβ vs non-treated THP-1 cells. (B) CTCF sites in gene bodies of genes that are highly transcriptionally activated by a stimulus are enriched for loops that are specifically lost with the respective stimulus. GSEA results from cross-comparing ranked lists of loops and transcribed CTCF sites in PAM-, IFNβ-, and Dex-treated THP-1 cells. (C) Elongation inhibition increases RAD21, CTCF, and cohesin-mediated loop interaction frequencies in the gene body of the constitutively transcribed HNRNPC and CHD8 genes in THP-1 cells, strengthening the loop connecting the two genes. (D) Elongation inhibition increases cohesin/RAD21 levels at intragenic CTCF sites and increases chromatin compaction ΔDLR/ΔICF) in the LYST gene, but the region remains in the A compartment (no change in PC1). (E) RNAPII and cohesin/RAD21 accumulate at TSS following inhibition of transcription elongation with flavopiridol.
(A) Heat map of RNAPII log2 ratios across PAM responsive genes in THP-1 cells. (B) RNAPII, RAD21/cohesin, and CTCF normalized ChIP-seq density at the SOD2 locus. THP-1 cells treated with PAM for 0, 20, 25, 30, 45, 60, or 120 minutes followed by flavopiridol treatment for an additional 30, 60, or 240 minutes are shown. Flavopiridol treatment alone for 240 minutes is included as a control. Grey box corresponds to CTCF site that loses RAD21/cohesin as RNAPII levels increase during transcription activation. (C) Normalized Hi-C contact maps showing the weakening of the loops formed by the CTCF site highlighted in (B). (D) Top 4 de novo motifs identified in Rad21 peaks lacking CTCF from Figure 6C. (E) Normalized Hi-C contact maps and ChIP-seq densities at the CD44 locus following PAM and flavopiridol treatment. White boxes highlight loops that are specifically strengthened following PAM and flavopiridol treatment (middle upper-right Hi-C contact map). Regulatory elements activated by PAM accumulate RNAPII and cohesin following treatment with flavopiridol (grey boxes).
(A) Average of log2 binding ratios for PU.1 and C/ΕΒΡβ comparing NS1- and EGFP-expressing THP-1 cells treated with PAM. Average changes in ChIP-seq ratios are reported as a function of the change in PC1 values. (B) ChIP-seq tracks showing PU.1, C/EBPβ, RNAPII, and PC1 levels at the SOD2 locus in PAM-treated THP-1 cells overexpressing EGFP or NS1. The read-through region downstream of SOD2 exhibits B-to-A compartment changes and enhanced transcription factor binding following NS1-induced read-through transcription. (C) Read-through transcription is a general feature of most strains of influenza viruses and can be seen in multiple cell types. RNA-seq read densities are shown at the IFIT family locus for macrophages (MDM) and epithelial cells (HTBE) that are mock- or IAV-infected with either H1N1, H3N2, or H5N1 strains of IAV.
Excel file containing the list of Hi-C, RNA-seq, ChIP-seq, methylC-seq, Start-seq, and North-seq experiments performed in primary macrophages and THP-1 cells. The total number of uniquely aligned reads used in the analysis are reported for each experiment. RNA-seq experiment performed in infection conditions separate the read totals based on human and influenza genome alignments. ChIP-seq experiments also report the total number of peaks identified in each sample.
Excel file containing two worksheets specifying IAV read-through regions and lists of highly induced genes by IFΝβ or PAM. The first worksheet contains 533 regions with elevated intergenic RNA-seq levels in IAV- vs. mock-infected macrophages at 6 hpi. The “score” column is calculated as the product of the median fractional coverage in the interval and the sum of normalized read counts over the interval. The “primary gene FPKM” column provides the expression level of the highest expressed gene across the interval (likely corresponding to the primary source of the read-through expression). The “gene overlap fraction” column describes the fraction of the region overlapping annotated genes on the same DNA strand. Genomic coordinates are relative to the GRCh37/hg19 version of the human genome. The second worksheet contains the lists of highly induced genes regulated by IFΝβ in MDM, IFΝβ in THP-1, and PAM in THP-1 as measured by RNAPII ChIP-seq.
Differentially regulated chromatin loops and regions changing chromatin compartments during IAV infection. Related to Figures 3 and S3. Excel file containing two worksheets specifying regulated loops and regions changing compartments. The first worksheet contains the chromatin loops that are regulated during IAV infection in primary macrophages (>1.5 FC, < 0.05 p-value). The second worksheet contains regions that confidently change chromatin compartments during IAV infection (>0.5 ΔPC1, < 5% FDR). Genomic coordinates are relative to the GRCh37/hg19 version of the human genome.
Juicer quality control and summary statistics for all Hi-C experiments. Related to Figures 1–7 and S1-S7. Excel file containing Hi-C metrics for all experiments.
Excel file containing DNA and RNA oligonucleotide sequences for the PCR primers and siRNAs used in this study.
Highlights.
Influenza A virus NS1 protein causes global read-through transcription past gene ends
Read-through transcription remodels genome 3D organization downstream of genes
Transcription elongation disrupts cohesin-mediated CTCF-anchored chromatin loops
Transcription into heterochromatin can cause B-to-A compartment transition
ACKNOWLEDGEMENTS
We thank Georg and Meinrad Busslinger for early access to results, discussion and the suggestion to analyze cohesin, Julia Minderjahn and Michael Rehli for advice on mRNA electroporation, Sascha Duttke for Start-seq advice, and Bing Ren, Christopher Glass and Kees Murre for comments on the manuscript. This study was funded by NIH-NIAID award U19 AI106754 (Fluomics) to AGS, CB, MLS, IM and RAA and a Pilot Project Grant sub-award to SH. LT was supported by an SNSF Advanced Postdoc Mobility fellowship. This work was supported by the Salk Institute’s NGS Core Facility with funding from NIH-NCI CCSG: P30 014195, the Chapman Foundation and the Helmsley Charitable Trust. Additional RNA-seq library prep and sequencing was conducted at UCSD’s IGM Genomics Center.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Amaral PP, Leonardi T, Han N, Viré E, Gascoigne DK, Arias-Carrasco R, Büscher M, Pandolfini L, Zhang A, Pluchino S, et al. (2018). Genomic positional conservation identifies topological anchor point RNAs linked to developmental loci. Genome Biol. 19, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, Adams MD, Myers EW, Li PW, and Eichler EE (2002). Recent segmental duplications in the human genome. Science 297, 1003–1007. [DOI] [PubMed] [Google Scholar]
- Bercovich-Kinori A, Tai J, Gelbart IA, Shitrit A, Ben-Moshe S, Drori Y, Itzkovitz S, Mandelboim M, and Stern-Ginossar N (2016). A systematic view on influenza induced host shutoff. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt DM, Pandya-Jones A, Tong A-J, Barozzi I, Lissner MM, Natoli G, Black DL, and Smale ST (2012). Transcript dynamics of proinflammatory genes revealed by sequence analysis of subcellular RNA fractions. Cell 150, 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busslinger GA, Stocsits RR, van der Lelij P, Axelsson E, Tedeschi A, Galjart N, and Peters J-M (2017). Cohesin is positioned in mammalian genomes by transcription, CTCF and Wapl. Nature 544, 503–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelo-Branco G, Amaral PP, Engstrom PG, Robson SC, Marques SC, Bertone P, and Kouzarides T (2013). The non-coding snRNA 7SK controls transcriptional termination, poising, and bidirectionality in embryonic stem cells. Genome Biol. 14, R98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremer T, Kreth G, Koester H, Fink RH, Heintzmann R, Cremer M, Solovei I, Zink D, and Cremer C (2000). Chromosome territories, interchromatin domain compartment, and nuclear matrix: an integrated view of the functional nuclear architecture. Crit. Rev. Eukaryot. Gene Expr. 10, 179–212. [PubMed] [Google Scholar]
- Crotta S, Davidson S, Mahlakoiv T, Desmet CJ, Buckwalter MR, Albert ML, Staeheli P, and Wack A (2013). Type I and type III interferons drive redundant amplification loops to induce a transcriptional signature in influenza-infected airway epithelia. PLoS Pathog. 9, e1003773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danko CG, Hah N, Luo X, Martins AL, Core L, Lis JT, Siepel A, and Kraus WL (2013). Signaling pathways differentially affect RNA polymerase II initiation, pausing, and elongation rate in cells. Mol. Cell 50, 212–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, and Ren B (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget JE, Lee AY, Ye Z, Kim A, Rajagopal N, Xie W, et al. (2015). Chromatin architecture reorganization during stem cell differentiation. Nature 518, 331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton JD, Davidson L, Bauer DLV, Natsume T, Kanemaki MT, and West S (2018). Xrn2 accelerates termination by RNA polymerase II, which is underpinned by CPSF73 activity. Genes Dev. 32, 127–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fudenberg G, Imakaev M, Lu C, Goloborodko A, Abdennur N, and Mirny LA (2016). Formation of Chromosomal Domains by Loop Extrusion. Cell Rep. 15, 2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gassler J, Brandão HB, Imakaev M, Flyamer IM, Ladstätter S, Bickmore WA, Peters JM, Mirny LA, and Tachibana K (2017). A mechanism of cohesin-dependent loop extrusion organizes zygotic genome architecture. EMBO J. 36, 3600–3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu B, Swigut T, Spencley A, Bauer MR, Chung M, Meyer T, and Wysocka J (2018). Transcription-coupled changes in nuclear mobility of mammalian cis-regulatory elements. Science 359, 1050–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haarhuis JHI, Elbatsh AMO, van den Broek B, Camps D, Erkan H, Jalink K, Medema RH, and Rowland BD (2013). WAPL-mediated removal of cohesin protects against segregation errors and aneuploidy. Curr. Biol. 23, 2071–2077. [DOI] [PubMed] [Google Scholar]
- Haarhuis JHI, van der Weide RH, Blomen VA, Yáñez-Cuna JO, Amendola M, van Ruiten MS, Krijger PHL, Teunissen H, Medema RH, van Steensel B, et al. (2017). The Cohesin Release Factor WAPL Restricts Chromatin Loop Extension. Cell 169, 693–707.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahne F, and Ivanek R (2016). Visualizing Genomic Data Using Gviz and Bioconductor In Statistical Genomics, (Humana Press, New York, NY: ), pp. 335–351. [DOI] [PubMed] [Google Scholar]
- Hansen AS, Pustova I, Cattoglio C, Tjian R, and Darzacq X (2017). CTCF and cohesin regulate chromatin loop stability with distinct dynamics. eLife Sciences 6, e25776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Romanoski CE, Benner C, Allison KA, Kaikkonen MU, Orozco LD, and Glass CK (2013). Effect of natural genetic variation on enhancer selection and function. Nature 503, 487–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imakaev M, Fudenberg G, McCord RP, Naumova N, Goloborodko A, Lajoie BR, Dekker J, and Mirny LA (2012). Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat. Methods 9, 999–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isoda T, Moore AJ, He Z, Chandra V, Aida M, Denholtz M, Piet van Hamburg J, Fisch KM, Chang AN, Fahl SP, et al. (2017). Non-coding Transcription Instructs Chromatin Folding and Compartmentalization to Dictate Enhancer-Promoter Communication and T Cell Fate. Cell 171, 103–119.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jubb AW, Boyle S, Hume DA, and Bickmore WA (2017). Glucocorticoid Receptor Binding Induces Rapid and Prolonged Large-Scale Chromatin Decompaction at Multiple Target Loci. Cell Rep. 21, 3022–3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, van Berkum NL, Ebmeier CC, Goossens J, Rahl PB, Levine SS, et al. (2010). Mediator and cohesin connect gene expression and chromatin architecture. Nature 467, 430–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keil C, Leach RW, Faizaan SM, Bezawada S, Parsons L, and Baryshnikova A (2016). Treeview 3.0 (alpha 3) - Visualization and analysis of large data matrices (Zenodo). [Google Scholar]
- Krueger F, and Andrews SR (2011). Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27, 1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug RM (2015). Functions of the influenza A virus NS1 protein in antiviral defense. Curr. Opin. Virol. 12, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo N, Ishii H, Gorkin D, Meitinger F, Xiong X, Fang R, Liu T, Ye Z, Li B, Dixon J, et al. (2017). Preservation of Chromatin Organization after Acute Loss of CTCF in Mouse Embryonic Stem Cells. bioRxiv 118737. [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson AG, Elnatan D, Keenen MM, Trnka MJ, Johnston JB, Burlingame AL, Agard DA, Redding S, and Narlikar GJ (2017). Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin. Nature 547, 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengronne A, Katou Y, Mori S, Yokobayashi S, Kelly GP, Itoh T, Watanabe Y, Shirahige K, and Uhlmann F (2004). Cohesin relocation from sites of chromosomal loading to places of convergent transcription. Nature 430, 573–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YC, Benner C, Mansson R, Heinz S, Miyazaki K, Miyazaki M, Chandra V, Bossen C, Glass CK, and Murre C (2012). Global changes in the nuclear positioning of genes and intra- and interdomain genomic interactions that orchestrate B cell fate. Nat. Immunol. 13, 1196–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marazzi I, Ho JSY, Kim J, Manicassamy B, Dewell S, Albrecht RA, Seibert CW, Schaefer U, Jeffrey KL, Prinjha RK, et al. (2012). Suppression of the antiviral response by an influenza histone mimic. Nature 483, 428–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark GE, Taylor JM, Broni B, and Krug RM (1979). Nuclear Accumulation of Influenza Viral RNA Transcripts and the Effects of Cycloheximide, Actinomycin D, and α-Amanitin. J. Virol. 29, 744–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nag A, Narsinh K, and Martinson HG (2007). The poly(A)-dependent transcriptional pause is mediated by CPSF acting on the body of the polymerase. Nat. Struct. Mol. Biol. 14, 662–669. [DOI] [PubMed] [Google Scholar]
- Nemeroff ME, Barabino SM, Li Y, Keller W, and Krug RM (1998). Influenza virus NS1 protein interacts with the cellular 30 kDa subunit of CPSF and inhibits 3’end formation of cellular pre-mRNAs. Mol. Cell 1, 991–1000. [DOI] [PubMed] [Google Scholar]
- Noah DL, Twu KY, and Krug RM (2003). Cellular antiviral responses against influenza A virus are countered at the posttranscriptional level by the viral NS1A protein via its binding to a cellular protein required for the 3’ end processing of cellular pre-mRNAS. Virology 307, 386–395. [DOI] [PubMed] [Google Scholar]
- Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, et al. (2012). Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485, 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora EP, Goloborodko A, Valton A-L, Gibcus JH, Uebersohn A, Abdennur N, Dekker J, Mirny LA, and Bruneau BG (2017). Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell 169, 930–944.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP, Huang S-C, Glenn St Hilaire B, Engreitz JM, Perez EM, Kieffer-Kwon K-R, Sanborn AL, Johnstone SE, Bascom GD, Bochkov ID, et al. (2017). Cohesin Loss Eliminates All Loop Domains. Cell 171, 305–320.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley MJ, Nichols MH, Lyu X, Ando-Kuri M, Rivera ISM, Hermetz K, Wang P, Ruan Y, and Corces VG (2017). Evolutionarily Conserved Principles Predict 3D Chromatin Organization. Mol. Cell 67, 837–852.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowski AJ, Erhard F, L’Hernault A, Bonfert T, Schilhabel M, Crump C, Rosenstiel P, Efstathiou S, Zimmer R, Friedel CC, et al. (2015). Widespread disruption of host transcription termination in HSV-1 infection. Nat. Commun. 6, 7126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanborn AL, Rao SSP, Huang S-C, Durand NC, Huntley MH, Jewett AI, Bochkov ID, Chinnappan D, Cutkosky A, Li J, et al. (2015). Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc. Natl. Acad. Sci. U. S. A. 112, E6456–E6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C, Nobs SP, Heer AK, Kurrer M, Klinke G, van Rooijen N, Vogel J, and Kopf M (2014). Alveolar macrophages are essential for protection from respiratory failure and associated morbidity following influenza virus infection. PLoS Pathog. 10, e1004053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzer W, Abdennur N, Goloborodko A, Pekowska A, Fudenberg G, Loe-Mie Y, Fonseca NA, Huber W, H Haering C, Mirny L, et al. (2017). Two independent modes of chromatin organization revealed by cohesin removal. Nature 551, 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scruggs BS, Gilchrist DA, Nechaev S, Muse GW, Burkholder A, Fargo DC, and Adelman K (2015). Bidirectional Transcription Arises from Two Distinct Hubs of Transcription Factor Binding and Active Chromatin. Mol. Cell 58, 1101–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah N, Maqbool MA, Yahia Y, El Aabidine AZ, Esnault C, Forné I, Decker T-M, Martin D, Schüller R, Krebs S, et al. (2018). Tyrosine-1 of RNA Polymerase II CTD Controls Global Termination of Gene Transcription in Mammals. Mol. Cell 69, 48–61.e6. [DOI] [PubMed] [Google Scholar]
- Shimizu K, Iguchi A, Gomyou R, and Ono Y (1999). Influenza virus inhibits cleavage of the HSP70 pre-mRNAs at the polyadenylation site. Virology 254, 213–219. [DOI] [PubMed] [Google Scholar]
- Steel J, Lowen AC, Pena L, Angel M, Solórzano A, Albrecht R, Perez DR, Garcίa-Sastre A, and Palese P (2009). Live attenuated influenza viruses containing NS1 truncations as vaccine candidates against H5N1 highly pathogenic avian influenza. J. Virol. 83, 1742–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom AR, Emelyanov AV, Mir M, Fyodorov DV, Darzacq X, and Karpen GH (2017). Phase separation drives heterochromatin domain formation. Nature 547, 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrier O, Carron C, Cartet G, Traversier A, Julien T, Valette M, Lina B, Moules V, and Rosa-Calatrava M (2014). Ultrastructural fingerprints of avian influenza A (H7N9) virus in infected human lung cells. Virology 456, 39–42. [DOI] [PubMed] [Google Scholar]
- Vilborg A, Passarelli MC, Yario TA, Tycowski KT, and Steitz JA (2015). Widespread Inducible Transcription Downstream of Human Genes. Mol. Cell 59, 449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wutz G, Várnai C, Nagasaka K, Cisneros DA, Stocsits RR, Tang W, Schoenfelder S, Jessberger G, Muhar M, Hossain MJ, et al. (2017). Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. EMBO J. 36, 3573–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang L-G, Han Y, and He Q-Y (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuin J, Franke V, van Ijcken WFJ, van der Sloot A, Krantz ID, van der Reijden MIJA, Nakato R, Lenhard B, and Wendt KS (2014). A cohesin-independent role for NIPBL at promoters provides insights in CdLS. PLoS Genet. 10, e1004153. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Total number of up- and down-regulated genes at 3, 6, 12, and 18 hours post infection (hpi) between IAV and time-matched mock controls (2 replicates per IAV and mock infection time point). (B) Ratio of viral RNA to host RNA in rRNA-depleted total RNA-seq experiments at each time point. (C) IAV infection leads to gradual loss of genome structure over time. In situ Hi-C contact maps for 60 Mb of Chr 10 in mock- and influenza A virus (IAV)-infected primary human monocyte-derived macrophages (MDM) at 6, 12, and 18 hpi. RNA polymerase II (RNAPII) ChIP-seq read densities from mock-infected cells and PC1 values (compartments) for each time point are depicted above each contact map, while changes in PC1, DLR (distal-to-local ratio), and ICF (interchromosomal interaction frequency) between time-matched IAV and mock conditions are depicted to the right (Hi-C data based on 2 replicates per IAV- and mock-infected time point). Asterisk denotes that the global changes in DLR and ICF were not normalized to zero across the genome for this analysis (see methods). (D) Changes in locus-specific compaction as measured by changes in DLR and ICF confirm that the FKBP5 locus decompacts in THP-1 cells treated with dexamethasone (Dex). (E) RNA-seq and ChIP-seq read density and chromatin structure tracks at the IFIH1 locus, which displays B-to-A compartment switching and IAV-induced transcriptional read-through both upstream and downstream of the locus. Tracks for mock- and IAV-infected, and IFNβ-treated MDMs are shown for RNA-seq, RNAPII (transcription), H3K36me3 (transcriptional elongation), H3K27ac (promoter/enhancer activation), H3K4me3 (promoter activation), H3K9me3 (repression), H3K27me3 (repression), 5-methylcytosine percentage per kb (CpG context only, repression). Changes in DLR and ICF between mock- and either IAV-infected or IFNβ-treated cells are reported at 15 kb resolution. PC1 eigenvector loadings are reported at 50 kb resolution. (F) Ratio of reads spanning the poly(A) cleavage site as a function of time post infection in IAV- and mock-infected cells. (G) Distribution of splice sites identified from RNA-seq reads in mock and IAV infection conditions as a function of time. Annotated genic sites correspond to annotated splice junctions. Unannotated splice sites are segregated based on whether they were found in intragenic or intergenic regions. (H) Meta-gene profiles for RNAPII CTD phosphorylation status (Ser5/initiating, Ser7/initiating, Ser2/elongating) in MDM after IAV infection or IFΝβ treatment at highly IFNβ-responsive genes. (I) Meta-gene profiles for RNAPII, H3K27ac, H3K4me3, and Start-seq (transcription initiation) in mock- and IAV-infected MDM and THP-1 macrophages at at highly IFNβ-responsive genes. (J) Transcription initiation occurs only at responsive gene promoters and not read-through regions. Genome browser screenshots of RNA-seq and Start-seq tracks at the CMPK2/RSAD2 locus in mock- and IAV-infected THP-1 macrophages. (K) Distribution of RNA-seq and ChIP-seq read densities in mock-, IAV-, and IFΝβ-treated cells measured by fragments per kb per million mapped reads (FPKM) in regions that switch from the B to A compartment (n = 23 regions, see Table S3). The distribution of average mCpG% for each region is shown for Mock- and IAV-infected cells.
(A) RNAPII ChIP-seq density at different classes of response genes in MDM after mock-, IAV-, dNS1-, or IFNβ-treatment. (B) Ratio of viral RNA to host RNA in rRNA-depleted total RNA-seq experiments for wild-type IAV (H5N1) and NS1 truncation mutant H5N1 virus (ΔNS1) at 6 hpi. (C) Total number of up- and down-regulated genes after IAV infection, ΔNS1 infection, or IFNβ treatment 6 hpi relative to mock infected controls (two replicates per condition, >3 fold, FDR < 5%). (D) Hierarchical gene expression clustering of significantly regulated genes in IAV, ΔΝS1, and ΙFΝβ-treated conditions. Top enriched GO terms for major clusters are shown to the left. (E) Western blot showing the protein levels of NS1 after electroporation of in vitro transcribed mRNA for influenza NS1 in THP-1 cells. (F) Meta-gene plot of RNAPII ChIP-seq density in THP-1 cells expressing EGFP (control) or NS1 at all genes with RNAPII expression > 1 FPKM. NS1 expression results in read-through transcription downstream of gene 3’ ends and an increase in RNAPII promoter-proximal pausing at gene 5’ ends. (G) Distribution of RNAPII pause ratios (ratio of promoter to gene body density) in EGFP and NS1 expressing THP-1 cells. (H) Meta-gene plot of RNAPII ChIP-seq density in IAV-, ΔNS1-, or Mock-infected MDM at genes down-regulated by IAV in RNA-seq (< 1/3 FC, < 5% FDR, n = 587 genes, two replicates per condition). IAV infection results in increased RNAPII promoter-proximal pausing relative to ΔNS1 in down-regulated genes. (I) RNAPII ChIP-seq read densities and changes in DLR and ICF in THP-1 cells expressing EGFP (control) or NS1 and treated with PAM, IFNβ, and/or flavopiridol. Transcriptional and structural effects are specific for target genes and reversible following flavopiridol treatment. (J) Northern blot of nuclear RNA showing the regions of the gel extracted for the sequencing of long RNA species (North-seq). (K) Meta-gene plot of RNA-seq read density of cytoplasmic, nuclear, and long (>13kb) nuclear RNA isolated from THP-1 cells treated with IFNβ overexpressing either EGFP or NS1. RNA from read-through regions was highly enriched for nuclear and long nuclear fractions. (L) Browser tracks showing RNA-seq/North-seq read densities at the STAT1 locus for THP-1 cells over expressing EGFP or NS1.
(A) Normalized Hi-C 2D contact maps for each mock- and lAV-infected replicate Hi-C experiments at the IFIT family locus showing reproducible changes in chromatin interactions after infection. (B) Meta-loop plot showing the average ΔDLR and ΔICF at loops strengthened or weakened during IAV infection. 0% and 100% represent the aggregate positions of loop anchors. Randomly chosen loops (200) identified in MDM were included as a control. (C) Distribution of DLR values relative to RAD21/cohesin peaks are shown for RAD21 peaks separated into quantiles based on the number of reads per peak. The percentage of RAD21 peaks overlapping CTCF and RNAPII peaks, respectively, are displayed for each quantile. CTCF-associated and RNAP-associated RAD21 peaks lead to increased and decreased compaction, respectively. (D) Correlation coefficient between changes in RNAPII and changes in RAD21/cohesin at CTCF sites after IAV infection as a function of distance relative to the TSS. Only CTCF sites with high levels of RNAPII, RAD21, and CTCF in either mock- or IAV-infected cells were considered (>7.5 FPKM). (E) Negative correlation between cohesin and RNAPII is dependent on high levels of transcription. Correlation between the changes in RAD21/cohesin and RNAPII levels at CTCF sites lacking H3K27ac is shown at different levels of maximal RNAPII loading at each site. (F) Comparison of changes in loop normalized Hi-C interactions in IAV and ΔNS1 infections and IFNβ treatments compared to mock infection in macrophages for all loops identified as differentially regulated following treatment (6 hpi, log2 vs. mock). Interaction ratios were clustered using hierarchical clustering and grouped by loops that strengthen across all three treatment conditions, weaken across all three conditions, or are selectively weakened with IAV infection. (G) Normalized Hi-C interaction matrix at the highly induced IFIT family locus in mock- (lower left) and ΔNS1- or IFNβ- (upper right) treated macrophages (compare to IAV-infection in Figure 3A). ChIP-seq read densities for RNAPII, CTCF, RAD21 (cohesin), and H3K27ac are shown below. Red and blue triangles indicate preferred interaction direction of highest matching CTCF motif found at each CTCF peak. Black arrows above RAD21 peaks indicate regions with dynamic cohesin changes during infection/treatment. (H) Same as (G) depicting changes at the MX1/MX2 locus, which shows similar changes across IAV, ΔNS1, and IFNβ treatment. Cohesin and loop loss at the CTCF site at the 3’ end of the MX1 gene is observed across all treatments (black arrow). (I) Negative correlation between cohesin and RNAPII is dependent on high levels of transcription in both PAM- and IFΝβ-treated THP-1 cells overexpressing NS1. Correlation between the changes in RAD21 and RNAPII levels at CTCF sites is shown at different levels of maximal RNAPII loading at each site. (J) RNAPII and RAD21/cohesin changes at CTCF sites in NS1-induced read-through regions in THP-1 cells stimulated with IFNβ ± flavopiridol. Changes in RNAPII and Rad21/cohesin observed with IFNβ are reverted after flavopiridol treatment. Flavopiridol significantly changed the log2 ratios of both RNAPII and RAD21 (t-test). (K) Normalized Hi-C interaction matrix at the highly induced TNFAIP3 locus in EGFP- (lower left) and NS1- (upper right) expressing THP-1 cells stimulated with PAM. ChIP-seq read densities for RNAPII, CTCF and RAD21 (cohesin) are shown below. Black arrows above RAD21 peaks indicate peaks lost due to NSI-induced read-through transcription and partially recovered following flavopiridol treatment.
(A) Western blot depicting NIPBL and WAPL protein levels following siRNA-mediated knockdown for 48 hours. Samples with siRNAs labeled in black (WAPL-3, control, NIPBL-pool) were used in subsequent experiments. (B,C) Normalized Hi-C interaction matrix for siNIPBL and siWAPL THP-1 cells at the PDE4B locus (B) and 60 Mb of chr10 (C). WAPL knockdown results in additional loops at the local scale (B, e.g. white circle) while NIPBL knockdown yields an increase in long range interactions (C, white arrows). (D) Log2 ratios of gene body RNAPII density in PAM-treated vs untreated THP-1 cells for all RNAi conditions for both eGFP and NS1 expressing cells. siRNA against NIPBL and WAPL have a relatively minor impact on RNAPII levels in PAM response genes. (E) Effect of NIPBL or WAPL depletion on chromosome-wide Hi-C interaction frequencies relative to Hi-C interaction distance in PAM-treated THP-1 cells. (F) WAPL depletion inhibits transcription-associated decompaction of highly transcribed read-through regions. Log2 differences in interaction frequencies in NS1- versus EGFP-expressing THP-1 cells as a function of interaction distance in Ctrl, NIPBL or WAPL knockdown conditions at PAM+NS1-induced read-through regions. (G) NIPBL depletion inhibits flavopiridol-induced recompaction of PAM+NS1-induced read-through regions. Distribution of Hi-C interaction frequencies relative to interaction distance in Ctrl, NIPBL or WAPL knockdown in PAM+NS1-induced read-through regions in THP-1 before or after flavopiridol inhibition of elongation. (H) Normalized Hi-C interaction matrices at the PAM-activated SOD2 locus, comparing EGFP, NS1, and NS1+flavopiridol THP-1 cells treated with PAM. ChIP-seq read densities for RNAPII, RAD21 (cohesin), and CTCF are shown below for Ctrl, NIPBL, and WAPL knockdown conditions.
(A) Without read-through, the anticorrelation of RNAPII and cohesin/RAD21 at CTCF sites is strongest within gene bodies of highly transcription genes. Pearson correlation of the ratios of RNAPII and RAD21 ChIP-seq signal at CTCF peaks in the respective gene annotation regions IFNβ vs non-treated THP-1 cells. (B) CTCF sites in gene bodies of genes that are highly transcriptionally activated by a stimulus are enriched for loops that are specifically lost with the respective stimulus. GSEA results from cross-comparing ranked lists of loops and transcribed CTCF sites in PAM-, IFNβ-, and Dex-treated THP-1 cells. (C) Elongation inhibition increases RAD21, CTCF, and cohesin-mediated loop interaction frequencies in the gene body of the constitutively transcribed HNRNPC and CHD8 genes in THP-1 cells, strengthening the loop connecting the two genes. (D) Elongation inhibition increases cohesin/RAD21 levels at intragenic CTCF sites and increases chromatin compaction ΔDLR/ΔICF) in the LYST gene, but the region remains in the A compartment (no change in PC1). (E) RNAPII and cohesin/RAD21 accumulate at TSS following inhibition of transcription elongation with flavopiridol.
(A) Heat map of RNAPII log2 ratios across PAM responsive genes in THP-1 cells. (B) RNAPII, RAD21/cohesin, and CTCF normalized ChIP-seq density at the SOD2 locus. THP-1 cells treated with PAM for 0, 20, 25, 30, 45, 60, or 120 minutes followed by flavopiridol treatment for an additional 30, 60, or 240 minutes are shown. Flavopiridol treatment alone for 240 minutes is included as a control. Grey box corresponds to CTCF site that loses RAD21/cohesin as RNAPII levels increase during transcription activation. (C) Normalized Hi-C contact maps showing the weakening of the loops formed by the CTCF site highlighted in (B). (D) Top 4 de novo motifs identified in Rad21 peaks lacking CTCF from Figure 6C. (E) Normalized Hi-C contact maps and ChIP-seq densities at the CD44 locus following PAM and flavopiridol treatment. White boxes highlight loops that are specifically strengthened following PAM and flavopiridol treatment (middle upper-right Hi-C contact map). Regulatory elements activated by PAM accumulate RNAPII and cohesin following treatment with flavopiridol (grey boxes).
(A) Average of log2 binding ratios for PU.1 and C/ΕΒΡβ comparing NS1- and EGFP-expressing THP-1 cells treated with PAM. Average changes in ChIP-seq ratios are reported as a function of the change in PC1 values. (B) ChIP-seq tracks showing PU.1, C/EBPβ, RNAPII, and PC1 levels at the SOD2 locus in PAM-treated THP-1 cells overexpressing EGFP or NS1. The read-through region downstream of SOD2 exhibits B-to-A compartment changes and enhanced transcription factor binding following NS1-induced read-through transcription. (C) Read-through transcription is a general feature of most strains of influenza viruses and can be seen in multiple cell types. RNA-seq read densities are shown at the IFIT family locus for macrophages (MDM) and epithelial cells (HTBE) that are mock- or IAV-infected with either H1N1, H3N2, or H5N1 strains of IAV.
Excel file containing the list of Hi-C, RNA-seq, ChIP-seq, methylC-seq, Start-seq, and North-seq experiments performed in primary macrophages and THP-1 cells. The total number of uniquely aligned reads used in the analysis are reported for each experiment. RNA-seq experiment performed in infection conditions separate the read totals based on human and influenza genome alignments. ChIP-seq experiments also report the total number of peaks identified in each sample.
Excel file containing two worksheets specifying IAV read-through regions and lists of highly induced genes by IFΝβ or PAM. The first worksheet contains 533 regions with elevated intergenic RNA-seq levels in IAV- vs. mock-infected macrophages at 6 hpi. The “score” column is calculated as the product of the median fractional coverage in the interval and the sum of normalized read counts over the interval. The “primary gene FPKM” column provides the expression level of the highest expressed gene across the interval (likely corresponding to the primary source of the read-through expression). The “gene overlap fraction” column describes the fraction of the region overlapping annotated genes on the same DNA strand. Genomic coordinates are relative to the GRCh37/hg19 version of the human genome. The second worksheet contains the lists of highly induced genes regulated by IFΝβ in MDM, IFΝβ in THP-1, and PAM in THP-1 as measured by RNAPII ChIP-seq.
Differentially regulated chromatin loops and regions changing chromatin compartments during IAV infection. Related to Figures 3 and S3. Excel file containing two worksheets specifying regulated loops and regions changing compartments. The first worksheet contains the chromatin loops that are regulated during IAV infection in primary macrophages (>1.5 FC, < 0.05 p-value). The second worksheet contains regions that confidently change chromatin compartments during IAV infection (>0.5 ΔPC1, < 5% FDR). Genomic coordinates are relative to the GRCh37/hg19 version of the human genome.
Juicer quality control and summary statistics for all Hi-C experiments. Related to Figures 1–7 and S1-S7. Excel file containing Hi-C metrics for all experiments.
Excel file containing DNA and RNA oligonucleotide sequences for the PCR primers and siRNAs used in this study.