

Abstract
Cellular esterases catalyze many essential biological functions by performing hydrolysis reactions on diverse substrates. The promiscuity of esterases complicates assignment of their substrate preferences and biological functions. To identify universal factors controlling esterase substrate recognition, we designed a 32-member structure–activity relationship (SAR) library of fluorogenic ester substrates and used this library to systematically interrogate esterase preference for chain length, branching patterns, and polarity to differentiate common classes of esterase substrates. Two structurally homologous bacterial esterases were screened against this library, refining their previously broad overlapping substrate specificity. Vibrio cholerae esterase ybfF displayed a preference for γ-position thioethers and ethers, whereas Rv0045c from Mycobacterium tuberculosis displayed a preference for branched substrates with and without thioethers. We determined that this substrate differentiation was partially controlled by individual substrate selectivity residues Tyr-119 in ybfF and His-187 in Rv0045c; reciprocal substitution of these residues shifted each esterase's substrate preference. This work demonstrates that the selectivity of esterases is tuned based on transition state stabilization, identifies thioethers as an underutilized functional group for esterase substrates, and provides a rapid method for differentiating structural isozymes. This SAR library could have multifaceted future applications, including in vivo imaging, biocatalyst screening, molecular fingerprinting, and inhibitor design.
Keywords: hydrolase, substrate specificity, fluorescence, chemical biology, carboxylesterase, chemical probes, fluorogenic substrates, structure-activity relationship, serine hydrolase
Introduction
Hydrolases (Enzyme Commission (EC)5 number 3) are ubiquitous cellular enzymes with more than 1200 different members, constituting more than one-third of all enzyme structures in the Protein Data Bank (PDB) and 13 different EC subclasses (1–5). In a classic enzyme mechanism, esterases (EC 3.1), one subclass of serine hydrolases, perform the hydrolysis of an ester using a catalytic triad of serine, histidine, and an acidic residue (6, 7). Esterase catalytic machinery is, however, fairly promiscuous and can also catalyze hydrolysis reactions on varied substrates, including amides, thioesters, phosphoric acid esters, and acid anhydrides (5–9). All of these utilities originate from the same α/β-hydrolase protein fold and employ this catalytic triad, suggesting great plasticity and generality in this common catalytic strategy (9–11). This diverse reactivity and substrate specificity make assigning the biological substrate(s) of different esterases from their sequence alignment difficult (5, 7, 12, 13). A recent algorithm for calculating esterase promiscuity was, however, able to predict promiscuous esterases with 94% accuracy based on calculations of their relevant solvent-accessible surface area (14).
The wide range of substrates and reactivities present among esterases has necessitated the development of diverse substrate analogs and assays for characterizing in vitro and in vivo esterase activity (15–22). Esterase substrate libraries have been used to rapidly fingerprint and classify various bacterial, fungal, and disease states (19, 20, 23–28). High rates of background hydrolysis, however, limit the cellular and high-throughput screening utility of many commonly employed substrates (16, 22, 25, 28). To increase hydrolytic stability and to distance the cleavable moiety from the fluorescent reporter, stable moieties have been inserted between the hydrolytic bond and the fluorophore (16, 18, 24, 30–32). Among these stable moieties, the acyloxymethyl ether class of fluorogenic substrates has found utility in orthogonal cell labeling, substrate specificity screening, and in vivo enzyme characterization (16, 24, 33–36).
Applying these chemically stable substrates and a recently developed synthetic strategy for their production, we have adapted the substrate activity screening (SAS) approach from serine proteases to target esterases (37, 38). In canonical SAS methodology, a broad library of fluorogenic substrate fragments is first screened against an enzyme of interest (37–40). Based on this preliminary screen, the substrate library is then optimized to select for high-activity substrates. We previously developed a small, general library of fluorogenic ester substrates based on acyloxymethyl ether fluorescein (24, 31, 41, 42). We then applied this library to broadly characterize the structural factors controlling the substrate specificity of esterases, to propose biological functions for uncharacterized esterases, and to identify unusual biocatalytic reactions (31, 33, 36, 41–43). This preliminary fluorogenic library provided sensitive detection of even weak binding substrates within a high-throughput and straightforward assay design (35, 41–43).
These fluorogenic substrates take advantage of the equilibrium in fluorescein between the highly fluorescent quinoid form and the nonfluorescent lactone form (Fig. 1A) (16, 24, 35, 44). Attachment of the acyloxymethyl ether moieties onto the phenolic oxygens of fluorescein locks it into the nonfluorescent lactone form (16). Selective cleavage of the esters by an esterase leads to the formation of a hemiacetal intermediate, which spontaneously falls apart in water to liberate free fluorescein and a formaldehyde by-product (Fig. 1A) (16, 35). By measuring the increase in fluorescein fluorescence across a range substrate concentrations and ester structures, the substrate specificity of esterases can be mapped and related to their structure (31, 34–36, 41–43).
Figure 1.

Fluorogenic SAR library. A, fluorogenic library of acyloxymethyl ether fluorescein derivatives. Each derivative remains nonfluorescent until activated by an esterase exposing the highly fluorescent fluorescein scaffold (16, 35, 41, 42). Removal of the ester functionalities by an esterase leads to a hemiacetal intermediate, which spontaneously decomposes in water to free fluorescein and formaldehyde (16). Derivatives were designed in 12 series based on changes in chain length, branching, polarity, and sterics. Each series contains carbon, oxygen, and sulfur versions to investigate the importance of hydrogen bonding, polarity, and electron withdrawing character to enzyme activation. Sets of identical compounds 2C/4C and 3C/5C are given different identifiers only for the sake of clearly presenting the C versus O versus S series in this figure. All of the derivatives were synthesized using the recently published synthetic procedure, and full chemical characterization is given in the supporting Methods. Two derivatives (10S and 12C, indicated with asterisks) were not synthesized. B, overall structural alignment of ybfF (green; PDB code 3BF8) (56) and Rv0045c (blue; PDB code 3P2M with modeled dynamic loop) (42, 55). Esterases are shown in cartoon representations with the catalytic serine shown in sticks and colored by atom type. A malonate molecule (gray) from the ybfF structure is included to demarcate the binding pocket (56). Structures were aligned using PyMOL based on total global structural alignment of individual subunits. C, catalytic triad alignment of ybfF and Rv0045c. The catalytic triad is shown in sticks and colored identically to B. D, variation in cap domain of ybfF and Rv0045c. Shown is a surface representation of Rv0045c (blue) with a cartoon representation of ybfF (green). The divergence in cap domain structures is illustrated based on the green helices from ybfF that protrude from the Rv0045c surface.
Among previous esterase targets for substrate specificity mapping were two homologous esterases with high structural similarity but limited sequence similarity (41, 42). These two esterases (ybfF from Vibrio cholerae and Rv0045c from Mycobacterium tuberculosis) showed overlapping substrate specificity profiles against a broad, small library of fluorogenic substrates, which was surprising, given the variation in their binding pocket structures (41, 42). Bacterial esterases like ybfF and Rv0045c have confirmed biological roles in bacterial virulence, survival, and biofilm formation, making them promising therapeutic targets (45–48). The promiscuous and overlapping substrate specificity of bacterial and human esterases, however, limits their utility as therapeutic targets (14, 49). However, simple ester substrates are selectively activated within specific cell types and conditions, suggesting that chemical probes could be designed that selectively target individual bacterial esterases (24, 50).
Herein, we describe the development and application of a refined SAR fluorogenic library for pinpointing the substrate specificity profile of esterases. We then apply this library to differentiating the substrate specificity profiles of two structural isozymes with high structural similarity. Using this optimized substrate library, we dissect molecular differences in esterase substrate specificity based on variations in substrate chain length, branching patterns, and polarity with three parallel series containing carbon, oxygen, and sulfur atoms systematically placed in the alkyl chain (Fig. 1). Combined with protein structural analysis, we identified the unique substrate signatures of these two structural isozymes and partially assigned these signatures to individual substrate differentiation residues, allowing us to uncover previously obfuscated molecular differences in these two homologous esterases. We propose that our library design could have multifaceted future applications, including in vivo imaging, biocatalyst screening, molecular fingerprinting, and inhibitor design.
Results and discussion
Structure activity library design
Using a streamlined synthesis for acyloxymethyl ether fluorescein derivatives, we assembled an SAR library of fluorogenic ester substrates (Fig. 1). For library design, we started from the most active substrates in previous broad fluorogenic substrate screens (1C and 1O) and made systematic modifications to optimize these substrates (41, 42). Specifically, we investigated the importance of chain length (series 1–3), ether positioning (series 2 and 3 versus series 4 and 5), branching patterns (series 6–10), and extended modifications (series 11 and 12) on esterase activity. For each of these modifications, we also probed the parallel impact of carbon, oxygen, or sulfur substitution within the alkyl chain, demarcated with superscripts C, O, and S, respectively. Ether substrates were a central point of our current substrate optimization, as ether substrates (1O and 4O) were most active in broad activity screens for multiple esterases (41, 42). Thioethers, which have been only rarely investigated for their impact on esterase activity, were included in the library as a counterpoint to ethers, as thioethers have more constrained angles, lower polarity, and increased ability to interact with aromatic and π-electron donors than ethers (51–53).
Each of these substrates was made using a parallel synthetic procedure (supporting Methods), and in total, 32 unique members were synthesized. Two proposed substrates (10S and 12C) were not synthesized due to preliminary results showing that those series had only minor activity in the substrate specificity screen. Similar SAR libraries have been used to pinpoint the substrate specificity and design inhibitors of various enzyme classes, including serine proteases, kinases, and phosphatases (37–40, 54). The advantages to performing this SAR using fluorogenic substrates over traditional small-molecule screening are that substrate turnover increases the signal of weak substrates while still allowing the catalytic constants to be directly related to the inhibition constants for structurally related inhibitors (37–39). A systematic fluorogenic SAR library was, however, previously unpublished for esterases.
Similar to previous applications, the library was first examined for its ability to deconvolute the substrate specificity of two structural isozymes, ybfF and Rv0045c (41, 42, 55). In prior broad screens, each of these structural isozymes showed nearly perfect overlap in substrate selectivity for substrates 1O and 3O. Structural alignment of these two hydrolases shows the overlap of their α/β-hydrolase domains and catalytic triads (Fig. 1, B and C) (56). This high structural alignment diverges in their cap domains, where cap domains control the differential substrate specificity, enantioselectivity, and conformational dynamics of α/β-hydrolases (Fig. 1D) (4, 21, 43, 57). This differentiation in the structures of the cap domain between ybfF and Rv0045c suggests divergent substrate specificity not observed in previous measurements (Fig. 1) (41, 42). These two structural isozymes provided us with the chance to investigate molecular facets differentiating esterase specificity and to determine the utility of the SAR library within a challenging but well-defined model system.
Multidimensional kinetic analysis
High-throughput kinetic measurements were then completed for both enzymes across the SAR library, and kinetic values were extracted. We chose to perform complete kinetic analysis for each substrate across a wide range of substrate concentrations to provide greater detail about how substrate turnover, binding, and transition state stabilization controlled their substrate specificity (Fig. 2) (38, 39). Specifically, we compared the relative substrate specificity of these two esterases against the entire library using three different kinetic metrics: catalytic specificity (kcat/Km), catalytic effectiveness (kcat/kuncat), and catalytic proficiency ((kcat/Km)/kuncat) (58, 59).
Figure 2.

Kinetic plots for fluorogenic SAR substrates against ybfF and Rv0045c. The kinetic activity of ybfF (A; 523 nm) and Rv0045c (B; 423 nm) against series 2 was measured by following the increase in fluorescein fluorescence (λex = 485 nm, λem = 528 nm) over time (7.5 min, collecting data every 50 s) at eight substrate dilutions (100 to 0.78 μm). The saturation enzyme kinetic traces were fitted to the Michaelis–Menten equation, and values for kcat, Km, and kcat/Km were calculated. All measurements were completed in triplicate substrate dilution and are shown ± S.D. (error bars). A, Michaelis–Menten plot of series 2 with ybfF (green). B, Michaelis–Menten plot of series 2 with Rv0045c (blue). Substrates are shown as follows: 2C (black), 2O (red), 2S (yellow).
Using the catalytic specificity metric (kcat/Km) that differentiates the substrate specificity of a single enzyme against multiple substrates (Fig. 3A) (58, 60, 61), kinetic comparisons between the two enzymes recapitulated the basic patterns seen previously for the two structural isozymes, with the highest activity against straight-chain alkyl ether substrates of 4–6 atoms (series 1–3) (41, 42). Rv0045c showed broader substrate specificity based on branching and steric bulk, retaining significant relative activity against series 6–8 with ybfF showing more selectivity toward straight-chain alkyls or with branching significantly beyond the ester carbonyl (series 6) (Fig. 3A). Reinforcing previous results, all of the highest activity substrates were concentrated in the oxygen-containing ether series for both enzymes (36, 41–43). We previously hypothesized that this preference for ether substrates either reflected the natural substrates for each enzyme, probably malonyl- or succinyl-CoA (55, 56), or reflected the activated nature of the ether-containing fluorogenic substrates (43).
Figure 3.

Comprehensive kinetic comparison of two homologous esterases. A, catalytic specificity (kcat/Km) comparison between ybfF (green line with circles) and Rv0045c (blue line with squares). Fluorogenic series are color-coded with carbon substrates in black, oxygen substrates in red, and sulfur substrates in yellow (Fig. 1A). Catalytic specificities were normalized based on the highest specificity for each enzyme. Kinetic constants calculated by fitting the hydrolysis reactions to the Michaelis–Menten equation and solving for values of kcat, Km, and kcat/Km. Complete kinetic values provided in Tables S1–S34. B, uncatalyzed rate of hydrolysis for each substrate. Substrates are colored identically to A. The uncatalyzed rate was calculated by measuring the hydrolysis of each fluorogenic substrate (10 μm) for 6 h in PBS at 25 °C and solving for the rate of hydrolysis by finding the slope of the linear regression. C, catalytic effectiveness (kcat/kuncat) comparison between ybfF and Rv0045c, colored identically to A. D, catalytic proficiency ((kcat/Km)/kuncat) comparison between ybfF and Rv0045c, colored identically to A. E, top 10 catalytic effectiveness substrates for ybfF. Substrates sized based on relative catalytic effectiveness against ybfF. F, top 10 catalytic effectiveness substrates for Rv0045c. Substrates are sized based on relative catalytic effectiveness against Rv0045c.
To differentiate between these hypotheses, kuncat values were measured for each substrate, which, although low, were measurable due to the sensitivity of the fluorogenic substrates (Fig. 3B and Tables S1 and S2). Confirming the activated nature of the ether substrates (43), kuncat measurements were highest for the ether substrates and especially for substrates with the oxygen in the β-position in relation to the ester carbonyl (series 1–3 and 6–7), although additional branching reduced this effect (series 8–10). The kuncat values were then used to calculate the relative catalytic effectiveness, kcat/kuncat (Fig. 3C), and catalytic proficiency, (kcat/Km)/kuncat (Fig. 3D), of each esterase (58, 59, 61–63).
Catalytic effectiveness measures the changing affinity of the enzyme for the substrate in the shift from the ground to transition state and is a more useful measure of enzyme activity for biocatalysis and fermentation applications (58, 61, 63, 64). Transition state stabilization is more important to controlling the catalytic activity of esterases than binding, as esterases are mediocre substrate binders, making catalytic effectiveness a better measure of their catalytic activity (1). Similar to other esterases (1), the catalytic effectiveness of ybfF and Rv0045c fell within a fairly narrow range (1.3 × 106 to 5.8 × 102; Tables S1 and S2), confined by the interplay between their binding energy and transition state stabilization.
For these two structural isozymes, the catalytic effectiveness, unlike catalytic specificity, provided a clear differentiation of their substrate preferences, with ybfF selecting for γ-position thioethers and ethers (series 5) and Rv0045c preferring branched substrates with and without thioethers (series 8–10) (Fig. 4). The strong selectivity based on catalytic effectiveness for substrates containing γ-position heteroatoms for ybfF and for substrates with increased molecular weights and branching for Rv0045c probably reflects their natural substrate preferences. This is supported by the general analysis of factors controlling esterase activity, which showed that features, including increased molecular weight, hydrogen bonding, number of atoms, and rotatable bonds, all generally decreased the transition state stabilization of esterases for their substrates (1). Thus, the selectivity of ybfF for γ-position heteroatoms and branching suggests specific binding and stabilization for these substituents (Fig. 4). Further reinforcing the significance of these substrate differences, highly homologous kinases also differentiated similar small substrate features, and these substrate features provided selectivity among related substrate analogues (40).
Figure 4.

SAR comparisons between the two homologous esterases based on branching and heteroatom positioning. Black, carbon; red, oxygen; yellow, sulfur. A and B, comparison of substrate classes with varying α-position branching. Overall, Rv0045c catalytic effectiveness increases with increased branching, whereas ybfF does not change. C and D, comparison of various substrate classes changing heteroatom from β-position to the γ-position. Overall, catalytic effectiveness is higher in ybfF with the heteroatom in the γ-position than for Rv0045c. Error bars, S.D.
Although catalytic effectiveness provides information about the path to the transition state, catalytic proficiency ((kcat/Km)/kuncat) better reflects an enzyme's ability to lower the activation barrier in a reaction and can be used to compare the relative difficulty of performing chemical transformations with different substrates (58, 59, 61, 65). The catalytic proficiency also provides a direct measure of the binding constant for the transition state, as the inverse version (kuncat·Km/kcat) provides an upper limit for the dissociation constant (KTS) for the transition state in the enzyme (58, 66). For ybfF, catalytic proficiency again showed the relative preference of ybfF for γ-position substrates (series 4 and 5) and indicates its overlapping interaction with these substrates in the ground and transition state, as measures of catalytic effectiveness and catalytic proficiency each showed this preference for γ-position substrates (Fig. 3D). In contrast, the relative catalytic proficiency of Rv0045c shifts toward carbon and thioether substrates with less branching (series 6–8) and with γ-position thioethers (series 4 and 5) (Fig. 3D). Although catalytic proficiency can underestimate the kcat values in enzymes like esterases, which use double displacement enzyme mechanisms and whose transition state in enzymatic and nonenzymatic reactions may differ (58, 67), our fluorogenic substrates simplify these comparisons due to their fluorescence being dependent on only the initial nucleophilic attack step of the classic serine hydrolase mechanism (16). Importantly, the catalytic effectiveness and proficiency measurements overlap for ybfF and Rv0045c (Fig. 3, C and D), illustrating the general utility of the SAR library to pull out essential substrate features that could be adapted for future inhibitor design (37, 39, 58, 67).
Rank comparisons of substrate selectivity
To further dissect these substrate preferences and to use the SAR profile to pinpoint optimal substrates for future applications, we ranked the substrates based on relative enzymatic activity using the three different kinetic metrics toward the two enzymes (Fig. 5). For each enzyme, the highest activity substrate using each kinetic metric was ranked as number 1 with the lowest activity substrate ranked as number 32. The relative ranks of the 32 substrates against ybfF and Rv0045c were then plotted. High-activity substrates against both enzymes will congregate in the lower left quadrant of the plot. Correlations based on substrate preference for the two enzymes are visualized based on a linear relationship, with local aggregations based on substrate series (carbon, oxygen, or sulfur) indicating an overlapping substrate preference by these two esterases.
Figure 5.
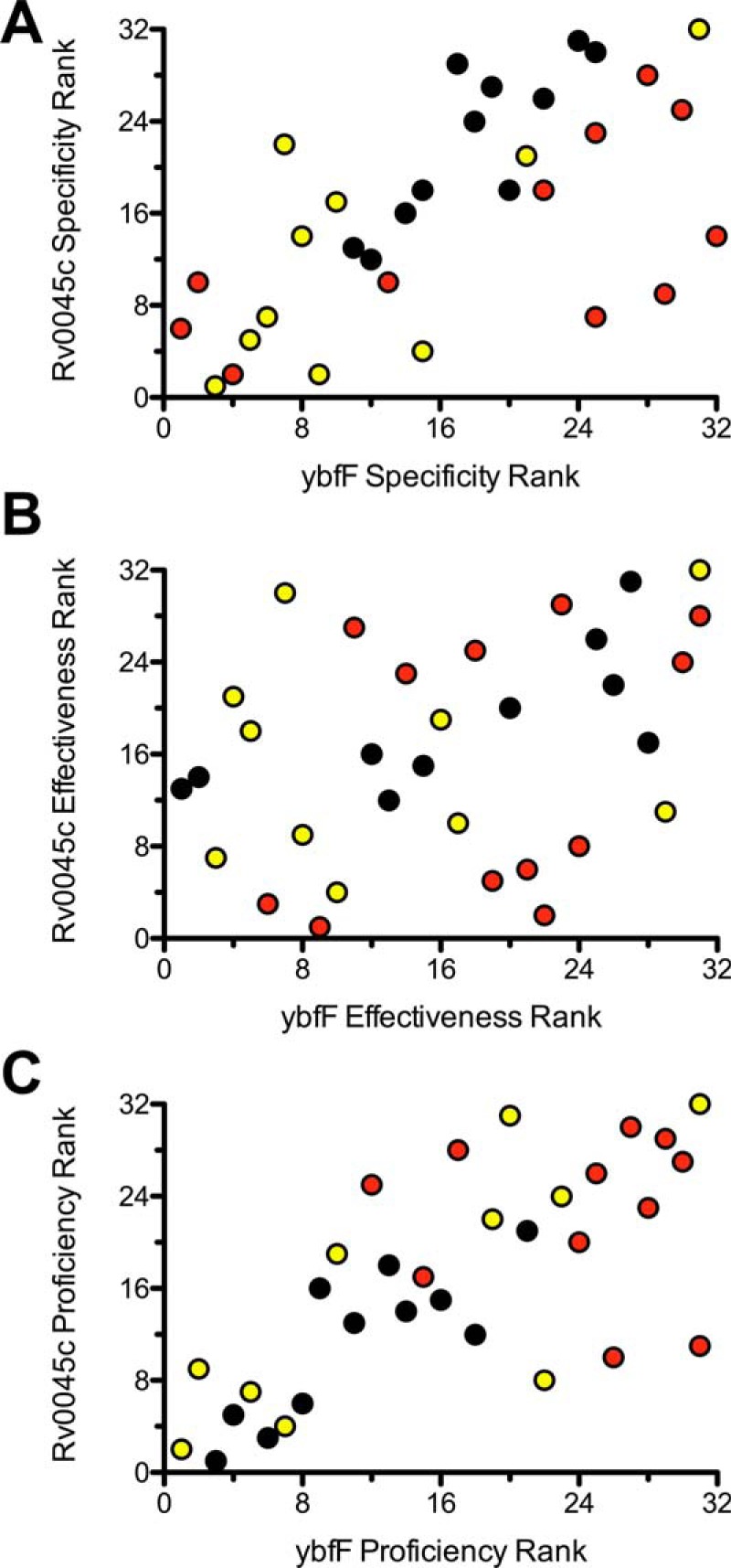
Rank comparison substrate specificity between two homologous esterases. Fluorogenic SAR substrates were ranked 1–32 for ybfF (x axis) and Rv0045c (y axis) based on catalytic specificity (A; kcat/Km), catalytic effectiveness (B; kcat/kuncat), and catalytic proficiency (C; (kcat/Km)/kuncat), and relative rank order is plotted. Substrates are colored based on fluorogenic SAR series (black, carbon; red, oxygen; yellow, sulfur).
For catalytic specificity (kcat/Km), this analysis shows a linear trend, with ether and thioether substrates congregated toward the lower ranks, suggesting a good correlation between substrate selectivity and catalytic specificity between these two homologous esterases (Fig. 5A). Catalytic effectiveness (kcat/kuncat) instead shows a more diffuse correlation with carbon, oxygen, and sulfur substrates distributed fairly evenly. The skewed distribution of the lowest ranked substrates reflects the differential preference for ether substrates by Rv0045c and thioether and branching for ybfF (Fig. 5B). Catalytic proficiency ((kcat/Km)/kuncat) returns to a more linear correlation between the two esterases with a congregation of similar thioether and carbon substrates as the highest proficiency substrates (Fig. 5C). This correlation in catalytic proficiency probably reflects the increased difficulty in catalyzing reactions with carbon and thioether substrates and the relationship between increased hydrophobicity and increased binding free energy favoring hydrolytic reactions and lowered Km values (1, 68). Catalytic proficiency measurements provide a good starting point for the design of esterase inhibitors because the highest-activity substrates best mimic the transition state, and TS mimics are common scaffolds for esterase inhibitor design (3, 58, 67).
The addition of parallel thioether derivatives into the library added a novel dimension to the SAR library, with each esterase showing selectivity toward thioether substrates (Figs. 2–4). Thioethers balanced reduced uncatalyzed hydrolysis rates (Fig. 3B) with increased catalyzed rates (Fig. 3A) to provide high specificity and reactivity. The molecular basis for thioether selectivity could be multifaceted, as thioethers have broad capabilities to interact with electron-poor and electron-rich functional groups (53). This underappreciated functional group makes important stabilizing interactions with aromatic and π-electron donors throughout protein structures in the PDB, suggesting that the thioether-specific interactions with ybfF may be physiologically relevant (51, 52). The more constrained angle of the thioether versus ether and the ability to strengthen its interactions through slow oxidation also make the thioether versatile and potentially tunable (52, 53, 69).
Structural factors controlling substrate selectivity of esterases
To understand the molecular factors controlling the SAR profile of these two structural isozymes, we compared the substrate selectivity contributions of two residues previously identified as selectivity residues between ybfF and Rv0045c (Fig. 6) (41, 42). Previously, conversion of analogous tyrosine (Tyr-119 in ybfF) or histidine (His-187 in Rv0045c) residues to alanine was shown to drastically shift the substrate specificity of each respective enzyme. For instance, conversion of His-187 to tyrosine in Rv0045c endowed this Rv0045c variant with the higher catalytic activity observed in ybfF (41, 42). Using this knowledge and a new reciprocal reversion variant (ybfF Y119H), we measured the changes in the substrate specificity of each variant in relation to their respective WT enzymes (Fig. 6). As seen previously, the Y119A ybfF variant significantly decreased its catalytic activity (Fig. 6D) (41), but interestingly, the Y119H variant endows ybfF with higher activity for extended substrates and those with γ-position thioethers and ethers (series 4–6), similar to WT Rv0045c selectivity (Figs. 3C and 4). Importantly, this Y119H variant also endows ybfF with higher activity for extended substrates and those with γ-position thioethers and ethers (series 4, 5, and 7), as this variant shows higher activity than WT ybfF against these substrates (Fig. 6E). This selectivity for extended substrates is more similar to WT Rv0045c selectivity (Fig. 3A). Rv0045c showed less drastic shifts in specificity between the two variants, with overall improvements in catalytic activity for both variants against the majority of substrates (Fig. 6, F and G). As observed previously for these Rv0045c variants (42), the most extended and branched substrates eventually reach a limit for improvement and impair enzyme activity (series 7–9 and series 11 and 12) (Fig. 6G). Based on these reciprocal reversions, the substrate selectivity between these two homologous esterases can be at least partially attributed to these two analogous residues. Thus, using this SAR library, we were able to identify a major substrate selectivity residue or hot spot (Fig. 6). This hot spot binding and substrate selectivity also supports a future goal to convert these small substrates into specific inhibitors, as substrates or ligands that interact with hot spot or substrate selectivity residues are more likely to maintain affinity upon conversion into full inhibitor structures (70).
Figure 6.

Reciprocal substrate discrimination residues. A and B, comparison of the binding pocket structures between ybfF (A; green) and Rv0045c (B; blue) showing the catalytic serine and substrate differentiation residues in sticks. For ybfF, the bound malonate is also shown in sticks. C, aligned binding pocket structure between ybfF (green) and Rv0045c (blue). The binding pocket surface for ybfF is shown to illustrate the relative positioning of the two differentiation residues within the pocket. Coloration and representation are identical to those in A and B. D–G, relative shifts in catalytic specificity (kcat/Km) for ybfF and Rv0045c upon substitution of substrate discrimination residues to alanine (D and F) and to reciprocal residues (E and G). Catalytic specificity ratios of variants with higher activity than the WT enzyme were calculated by (kcat/Km)variant/(kcat/Km)WT. Relative ratios for variants with higher activity than the WT enzyme were assigned positive values to showcase the increased activity of that variant from the WT enzyme. Catalytic specificity ratios of variants with lower activity than the WT enzyme were calculated by dividing the (kcat/Km)WT/(kcat/Km)variant. Relative ratios for variants with lower activity than the WT enzyme were assigned negative values to showcase the decreased activity of that variant from the WT enzyme. Detailed kinetic values are given in Tables S3–S34. Error bars, S.D.
Conclusions
Fluorogenic enzyme probes allow measurement of dynamic spatiotemporal regulation of enzymatic activity (27, 44) and have confirmed therapeutic and diagnostic applications, including surgical cancer labeling and point of care bacterial pathogen identification (71, 72). These applications, however, require molecular differentiation between highly similar enzyme substrates and identification of unique enzyme signatures that demarcate disease conditions (27, 71, 72). Esterases are a large class of enzymes with potential diagnostic applications in labeling infectious bacteria and therapeutic applications combating cancer, inflammation, and pathogenic infections (3, 45, 73). The substrate promiscuity of esterases has, however, hindered the construction of selective esterase probes for these applications (24, 74).
To design selective esterase probes, we systematically investigated the substrate specificity of two structurally and functionally overlapping esterases using a fluorogenic SAR library. Our fluorogenic SAR library provides an intricate picture of the general substrate specificity determinants in esterases (Fig. 1). Starting with a model system of two structural isozymes and using three different catalytic measurements (Figs. 2–4), the previously broad overlapping substrate specificity of these esterases was narrowly defined to preferences for γ-position thioethers and ethers for one isozyme (ybfF) and branched substrates with and without thioethers for the other isozyme (Rv0045c) (Figs. 3 and 4). These selectivities highlight the general substrate recognition principles of esterases, including increased substrate hydrophobicity and binding free energy favoring hydrolysis, but decreased transition state stabilization with increasing substrate molecular weight and heteroatoms (Fig. 5) (1) The biological significance of these assignments was confirmed based on the ability to modify these substrate preferences with single-residue reversions (Fig. 6). Moving forward, our measurements with model esterases are being transitioned into bacterial systems like M. tuberculosis to determine whether disease-relevant esterase activity can be isolated and probed using this SAR library (49). The fluorogenic SAR library could also be applied to more complex systems, including screening for orthogonal esterase activity and novel biocatalytic reactivity (19, 20, 23, 75), fragment-based inhibitor design analogous to serine protease fluorogenic SAS libraries (37, 39, 40), and in vivo imaging of cell type–specific hydrolase activity (24, 33).
Experimental procedures
Synthesis of fluorogenic esterase substrates
The synthesis and characterization of compounds 1C, 2C/4C, and 3C/5C (31); 1O and 4O (41); and 3O, 6C, 8C, 9C, and 10O (49) has been described previously. Compounds 1S, 2O, 2S, 3S, 4S, 5O, 5S, 6O, 6S, 7C, 7O, 7S, 8O, 8S, 9O, 9S, 10C, 11C, 11O, 11S, 12O, and 12S were each produced in a single step from a common fluorescein di(chloromethyl) ether precursor using a procedure described previously (49). Carboxylic acids employed for the synthesis of 5O, 6S, 7C, 8O, 8S, 10C, 11C, 11O, 11S, and 12O were obtained from Sigma-Aldrich. Carboxylic acids employed for the synthesis of 1S, 2O, 2S, and 4S were obtained from Alfa Aesar. Carboxylic acids employed for the synthesis of 3S, 5S, 7S, and 9S were obtained from Enamine. Carboxylic acids employed for the synthesis of 6O, 7O, and 9O were obtained from Matrix Scientific. The carboxylic acid employed for the synthesis of 12S was obtained from Chem-Bridge. Compounds 7C, 7O, 7S, 8O, 8S, 9O, 9S, 12O, and 12S are mixtures of stereoisomers that presumably resemble the isomeric mixture of the corresponding carboxylic acid starting material in distribution. All other chemicals were purchased from Sigma-Aldrich and used without further purification. All reactions were monitored using Macherey–Nagel analytical thin-layer chromatography plates (POLYGRAM® SIL G/UV254, polyester back). NMR spectra were obtained using a Bruker Biospin Avance III HD 400 operating at 400.19 MHz for 1H and 100.64 MHz for 13C. High-resolution MS was performed with electrospray ionization (ESI) by the Mass Spectrometry Facility at the Department of Chemistry, Indiana University using an Agilent 1200 HPLC-6130 MSD mass spectrometer.
The following procedure is representative for the synthesis of all compounds. Detailed characterization of all compounds is provided in the supporting information.
Synthesis of fluorescein bis((2-methylsulfanyl)-acetyloxymethyl ether) (1S)
Fluorescein bis(chloromethyl ether) (30.0 mg, 69.9 μmol, 1.0 eq), (2-methylthio)acetic acid (29.7 mg, 279.6 μmol, 4.0 eq), and Cs2CO3 (91.1 mg, 279.6 μmol, 4.0 eq) were dissolved in dry CH3CN (1 ml). Molecular sieves (100 mg) were added, and the reaction was covered in foil and allowed to stir for 24 h at ambient temperature. The reaction mixture was adsorbed onto celite and purified via column chromatography.
Fluorescein bis((2-methylsulfanyl)acetyloxymethyl ether) (1S)
Data for 1S: (42%, white solid). 1H NMR (CDCl3, 250 MHz): δ = 8.05 (d, J = 6.7 Hz, 1H), 7.74–7.62 (m, 2H), 7.17 (d, J = 7.6 Hz, 1H), 6.99 (s, 2H), 6.75 (s, 4H), 5.84 (s, 4H), 3.25 (s, 4H), 2.20 ppm (s, 6H). 13C NMR (CDCl3, 250 MHz): δ = 169.4, 168.8, 158.4, 152.9, 152.3, 134.9, 130.5, 129.5, 126.3, 125.3, 123.9, 113.5, 112.4, 103.6, 85.2, 82.9, 35.9,16.8 ppm. High-resolution MS (ESI): calcd for C30H23O9S2: 591.0784; found: 591.0740.
Purification of ybfF from V. cholera
WT ybfF from V. cholerae was purified in a manner similar to that described previously (41). The bacterial plasmid (pET-22b-Vc2097, which encodes a C-terminal His tag on ybfF) was transformed into Escherichia coli BL21 (DE3) RIPL cells (Agilent). A saturated overnight culture of E. coli BL21 (DE3) RIPL (pET-22b-Vc2097) in LB medium containing ampicillin (200 μg/ml) and chloramphenicol (30 μg/ml) was used to inoculate LB medium (1.0 liter) containing ampicillin (200 μg/ml) and chloramphenicol (30 μg/ml), and the bacterial culture was grown with constant shaking (225 rpm) at 37 °C. When the A600 reached 1.0–1.2, the temperature of the culture was decreased to 18 °C, and isopropyl β-d-thiogalactopyranoside was added to a final concentration of 1.0 mm. Protein induction proceeded for 16–20 h at 18 °C. Bacterial cultures were collected by centrifugation at 6,000 × g for 10 min at 4 °C. The bacterial cell pellet was resuspended in PBS (40 ml) and stored at −20 °C. To disrupt the bacterial cell wall, lysozyme (250 mg) and BugBuster detergent solution (4.0 ml of 10×; EMD Millipore) were added to the thawed cell pellet, and cell lysis proceeded with vigorous rotation on an orbital shaker for 2 h at 4 °C. To remove insoluble cell material, lysed cells were centrifuged at 16,000 × g for 10 min at 4 °C. Nickel-nitrilotriacetic acid–agarose (1.0 ml; Gold Biotechnology) was added to the soluble fraction and allowed to incubate at 4 °C for 15–30 min. The resin was washed six times with PBS containing increasing concentrations of ice-cold imidazole (three times with 40 ml of PBS + 10 mm imidazole, two times with 40 ml of PBS + 25 mm imidazole, and once with 40 ml of PBS + 50 mm imidazole) and recollected by centrifugation at 1,000 × g for 1 min at 4 °C. ybfF was eluted in PBS containing 250 mm imidazole (1.0 ml) and dialyzed against PBS overnight at 4 °C with constant stirring (10,000 molecular weight cut-off; Thermo Fisher Scientific). The purity of ybfF was confirmed by SDS-PAGE on a 4–20% gradient gel, and the purity was shown to be >95%. The concentration of ybfF was determined by measuring the absorbance at 280 nm and by calculating the extinction coefficient (ϵ280 = 23,950 m−1 s−1 with all free cysteines) using ExPASY ProtParam (41).
The Y16H variant of ybfF was produced by QuikChange II site-directed mutagenesis of pET-22b-Vc2097 template DNA using a derivation of the manufacturer's suggested procedure (Agilent) with the mutagenesis primer (5′-GGACATGTCACCTGTCGCGCATAGCCAACGGCGTCACG-3′, with the mutagenic codon underlined) and its reverse complement. Briefly, mutagenic PCR products were subjected to digestion with DpnI restriction endonuclease for 1 h at 37 °C to degrade WT template plasmid DNA. Mutated pET-22b-Vc2097 plasmid DNA was replicated by transformation into E. coli DH5α cells, followed by plasmid DNA isolation/purification from saturated overnight cultures using a commercial kit (IBI Scientific). The proper Y116H mutation in the ybfF DNA sequence was confirmed by DNA sequencing (Genewiz) using T7 sequencing primers. Plasmids coding for ybfF variants were transformed into E. coli BL21 (DE3) RIPL cells, and variants of ybfF were overexpressed and purified using the same procedure as for WT ybfF. For ybfF variants with tyrosine substitutions, the extinction coefficients were adjusted to correct for the loss of the phenol chromophore (ϵ280 = 22,460 m−1 s−1) (41).
Purification of Rv0045c from M. tuberculosis
Rv0045c protein was overexpressed in E. coli as an N-terminal His6 tag fusion using a bacterial expression plasmid (pET28a-Rv0045c) and purified identically to ybfF with the following changes (42, 55, 76). Bacterial plasmid (pET28-Rv0045c) was transformed into E. coli BL21 (DE3) RIPL cells (Agilent). A saturated overnight culture of E. coli BL21 (DE3) RIPL (pET28-Rv0045c) in LB medium containing kanamycin (40 μg/ml) and chloramphenicol (30 μg/ml) was used to inoculate LB medium (1.0 liter) containing kanamycin (40 μg/ml) and chloramphenicol (30 μg/ml), and the bacterial culture was grown with constant shaking (225 rpm) at 37 °C. The concentration of Rv0045c was determined by measuring the absorbance at 280 nm and converted to molarity units with an extinction coefficient of ϵ280 = 35,980 m−1 cm−1 calculated from the theoretical amino acid sequence using the ProtParam online proteomics tool on the ExPASy website (http://web.expasy.org/protparam) (42). For the H187Y variant Rv0045c, the extinction coefficient was adjusted to correct for the gain of a phenol chromophore (Rv0045c + Tyr ϵ280 = 37,470 m−1 cm−1) (42).
Kinetic measurements with fluorogenic SAR library
The enzymatic activity of ybfF, Rv0045c, and their variants was measured against the fluorogenic SAR library (Fig. 1) using a 96-well microplate assay (31, 35, 77). Fluorogenic substrates were prepared as stock solutions in DMSO (10 mm) and were diluted into PBS containing acetylated BSA (PBS-BSA; 0.1 mg/ml) to a starting concentration of 100 μm. Acetylated BSA was added to reduce nonspecific adsorption to the plasticware common in microplate analysis (29). Eight serial substrate dilutions in triplicate (1:1; 120 μl into a 240-μl total volume) were made from one master substrate dilution (10 mm) using PBS-BSA. Fluorogenic substrate dilutions (95 μl) were then transferred to a black 96-well microplate (Corning, Inc.). Enzyme-catalyzed hydrolysis was initiated by individual triplicate addition of esterase from one master enzyme dilution (5 μl of 300 μg/ml; [final] = 15 μg/ml; [ybfF] = 523 nm, and [Rv0045c] = 423 nm) to the diluted fluorogenic substrates in the black 96-well microplate (100-μl final volume), and the fluorescence change (λex = 485 nm, λem = 528 nm) was measured for 7.5 min at 25 °C, collecting data every 50 s, on a Biotek Synergy H1 multimode plate reader (Biotek Instruments, Winooski, VT). The fluorescence change was converted to molar concentrations using a fluorescein standard curve (300 to 2.3 nm), whose fluorescence was measured simultaneously. The initial rates of the reactions were measured in triplicate and plotted versus fluorogenic substrate concentration. The saturation enzyme kinetic traces were fitted to a standard Michaelis–Menten equation using GraphPad Prism version 5.0 (GraphPad Software, La Jolla, CA), and values for kcat, Km, and kcat/Km were calculated (Fig. S1). Background hydrolysis rates (kuncat) were determined by measuring the hydrolysis of each fluorogenic substrate (10 μm) for 6 h in PBS at 25 °C, taking a measurement every 30 min, and solving for the rate of hydrolysis by finding the slope of the linear regression. The limited background photobleaching of fluorescein during the extended time course of the uncatalyzed rate measurement was corrected based on photobleaching of the fluorescein standard curve collected on the same plate under the same conditions.
Author contributions
A. W., L. D. L., G. C. H., and R. J. J. conceptualization; A. W., A. K., A. R., E. M. L., C. K., G. C. H., and R. J. J. data curation; A. W., A. K., A. R., E. M. L., C. K., G. C. H., and R. J. J. formal analysis; A. W., A. K., A. R., E. M. L., C. K., G. C. H., and R. J. J. investigation; A. W., A. K., A. R., E. M. L., C. K., G. C. H., and R. J. J. methodology; A. W., A. K., E. M. L., L. D. L., G. C. H., and R. J. J. writing-original draft; L. D. L., G. C. H., and R. J. J. supervision; L. D. L. and R. J. J. funding acquisition; L. D. L. validation; L. D. L., G. C. H., and R. J. J. project administration; L. D. L., G. C. H., and R. J. J. writing-review and editing.
Supplementary Material
Acknowledgments
We thank Perry Rabin for assistance with protein analysis as well as Dominique Stephens, Lindsey Drake, Stephanie Mitchell, and Luke Gallion for assistance with fluorogenic substrate synthesis.
The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supporting Methods, Tables S1–S34, and Fig. S1.
- EC
- Enzyme Commission
- PDB
- Protein Data Bank
- SAR
- structure activity relationship
- SAS
- substrate activity screening
- TS
- transition state
- ESI
- electrospray ionization
- LB
- lysogeny broth.
References
- 1. Sousa F. Sr., Ramos M. J., Lim C., and Fernandes P. A. (2015) Relationship between enzyme/substrate properties and enzyme efficiency in hydrolases. ACS Catal. 5, 5877–5887 10.1021/acscatal.5b00923 [DOI] [Google Scholar]
- 2. Long J. Z., and Cravatt B. F. (2011) The metabolic serine hydrolases and their functions in mammalian physiology and disease. Chem. Rev. 111, 6022–6063 10.1021/cr200075y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bachovchin D. A., and Cravatt B. F. (2012) The pharmacological landscape and therapeutic potential of serine hydrolases. Nat. Rev. Drug Discov. 11, 52–68 10.1038/nrd3620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lenfant N., Hotelier T., Bourne Y., Marchot P., and Chatonnet A. (2013) Proteins with an α/β hydrolase fold: relationships between subfamilies in an ever-growing superfamily. Chem. Biol. Interact. 203, 266–268 10.1016/j.cbi.2012.09.003 [DOI] [PubMed] [Google Scholar]
- 5. Lenfant N., Hotelier T., Velluet E., Bourne Y., Marchot P., and Chatonnet A. (2013) ESTHER, the database of the α/β-hydrolase fold superfamily of proteins: tools to explore diversity of functions. Nucleic Acids Res. 41, D423–D429 10.1093/nar/gks1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Holmquist M. (2000) α/β-Hydrolase fold enzymes: structures, functions and mechanisms. Curr. Protein Pept. Sci. 1, 209–235 10.2174/1389203003381405 [DOI] [PubMed] [Google Scholar]
- 7. Kourist R., Jochens H., Bartsch S., Kuipers R., Padhi S. K., Gall M., Böttcher D., Joosten H. J., and Bornscheuer U. T. (2010) The α/β-hydrolase fold 3DM database (ABHDB) as a tool for protein engineering. Chembiochem 11, 1635–1643 10.1002/cbic.201000213 [DOI] [PubMed] [Google Scholar]
- 8. Simon G. M., and Cravatt B. F. (2010) Activity-based proteomics of enzyme superfamilies: serine hydrolases as a case study. J. Biol. Chem. 285, 11051–11055 10.1074/jbc.R109.097600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rauwerdink A., and Kazlauskas R. J. (2015) How the same core catalytic machinery catalyzes 17 different reactions: the serine-histidine-aspartate catalytic triad of α/β-hydrolase fold enzymes. ACS Catal. 5, 6153–6176 10.1021/acscatal.5b01539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Busto E., Gotor-Fernández V., and Gotor V. (2010) Hydrolases: catalytically promiscuous enzymes for non-conventional reactions in organic synthesis. Chem. Soc. Rev. 39, 4504–4523 10.1039/c003811c [DOI] [PubMed] [Google Scholar]
- 11. Devamani T., Rauwerdink A. M., Lunzer M., Jones B. J., Mooney J. L., Tan M. A. O., Zhang Z.-J., Xu J.-H., Dean A. M., and Kazlauskas R. J. (2016) Catalytic promiscuity of ancestral esterases and hydroxynitrile lyases. J. Am. Chem. Soc. 138, 1046–1056 10.1021/jacs.5b12209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bornscheuer U. T. (2002) Microbial carboxyl esterases: classification, properties and application in biocatalysis. FEMS Microbiol. Rev. 26, 73–81 10.1111/j.1574-6976.2002.tb00599.x [DOI] [PubMed] [Google Scholar]
- 13. Carr P. D., and Ollis D. L. (2009) α/β hydrolase fold: an update. Protein Pept. Lett. 16, 1137–1148 10.2174/092986609789071298 [DOI] [PubMed] [Google Scholar]
- 14. Martínez-Martínez M., Coscolín C., Santiago G., Chow J., Stogios P. J., Bargiela R., Gertler C., Navarro-Fernández J., Bollinger A., Thies S., Méndez-García C., Popovic A., Brown G., Chernikova T. N., García-Moyáno A., et al. (2018) Determinants and prediction of esterase substrate promiscuity patterns. ACS Chem. Biol. 13, 225–234 10.1021/acschembio.7b00996 [DOI] [PubMed] [Google Scholar]
- 15. Lavis L. D., Chao T. Y., and Raines R. T. (2006) Fluorogenic label for biomolecular imaging. ACS Chem. Biol. 1, 252–260 10.1021/cb600132m [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lavis L. D., Chao T.-Y., and Raines R. T. (2011) Synthesis and utility of fluorogenic acetoxymethyl ethers. Chem. Sci. 2, 521–530 10.1039/C0SC00466A [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jochens H., Hesseler M., Stiba K., Padhi S. K., Kazlauskas R. J., and Bornscheuer U. T. (2011) Protein engineering of α/β-hydrolase fold enzymes. Chembiochem 12, 1508–1517 10.1002/cbic.201000771 [DOI] [PubMed] [Google Scholar]
- 18. Leroy E., Bensel N., and Reymond J. L. (2003) A low background high-throughput screening (HTS) fluorescence assay for lipases and esterases using acyloxymethylethers of umbelliferone. Bioorg. Med. Chem. Lett. 13, 2105–2108 10.1016/S0960-894X(03)00377-9 [DOI] [PubMed] [Google Scholar]
- 19. Grognux J., and Reymond J. L. (2004) Classifying enzymes from selectivity fingerprints. Chembiochem 5, 826–831 10.1002/cbic.200300779 [DOI] [PubMed] [Google Scholar]
- 20. Maillard N., Babiak P., Syed S., Biswas R., Mandrich L., Manco G., and Reymond J.-L. (2011) Five-substrate cocktail as a sensor array for measuring enzyme activity fingerprints of lipases and esterases. Anal. Chem. 83, 1437–1442 10.1021/ac102994n [DOI] [PubMed] [Google Scholar]
- 21. Romano D., Bonomi F., de Mattos M. C., de Sousa Fonseca T., de Oliveira Mda C. F., and Molinari F. (2015) Esterases as stereoselective biocatalysts. Biotechnol. Adv. 33, 547–565 10.1016/j.biotechadv.2015.01.006 [DOI] [PubMed] [Google Scholar]
- 22. Kim S., Kim H., Choi Y., and Kim Y. (2015) A new strategy for fluorogenic esterase probes displaying low levels of non-specific hydrolysis. Chem. Eur. J. 21, 9645–9649 10.1002/chem.201501127 [DOI] [PubMed] [Google Scholar]
- 23. Cai R., Yuan Y., Wang Z., Wang J., and Yue T. (2016) Discrimination of alicyclobacillus strains by lipase and esterase fingerprints. Food Anal. Methods 9, 1128–1133 10.1007/s12161-015-0290-8 [DOI] [Google Scholar]
- 24. Tian L., Yang Y., Wysocki L. M., Arnold A. C., Hu A., Ravichandran B., Sternson S. M., Looger L. L., and Lavis L. D. (2012) Selective esterase-ester pair for targeting small molecules with cellular specificity. Proc. Natl. Acad. Sci. U.S.A. 109, 4756–4761 10.1073/pnas.1111943109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Qian L., Liu J.-Y., Liu J.-Y., Yu H.-L., Li C.-X., and Xu J.-H. (2011) Fingerprint lipolytic enzymes with chromogenic p-nitrophenyl esters of structurally diverse carboxylic acids. J. Mol. Catal. B Enzym. 73, 22–26 [Google Scholar]
- 26. Komatsu T., Hanaoka K., Adibekian A., Yoshioka K., Terai T., Ueno T., Kawaguchi M., Cravatt B. F., and Nagano T. (2013) Diced electrophoresis gel assay for screening enzymes with specified activities. J. Am. Chem. Soc. 135, 6002–6005 10.1021/ja401792d [DOI] [PubMed] [Google Scholar]
- 27. Komatsu T., and Urano Y. (2015) Evaluation of enzymatic activities in living systems with small-molecular fluorescent substrate probes. Anal. Sci. 31, 257–265 10.2116/analsci.31.257 [DOI] [PubMed] [Google Scholar]
- 28. Malin-Berdel J., and Valet G. (1980) Flow cytometric determination of esterase and phosphatase activities and kinetics in hematopoietic cells with fluorogenic substrates. Cytometry 1, 222–228 10.1002/cyto.990010308 [DOI] [PubMed] [Google Scholar]
- 29. Macarron R., and Hertzberg R. P. (2011) Design and implementation of high throughput screening assays. Mol. Biotechnol. 47, 270–285 10.1007/s12033-010-9335-9 [DOI] [PubMed] [Google Scholar]
- 30. Yang Y. Z., Babiak P., and Reymond J. L. (2006) New monofunctionalized fluorescein derivatives for the efficient high-throughput screening of lipases and esterases in aqueous media. Helv. Chim. Acta 89, 404–415 10.1002/hlca.200690041 [DOI] [Google Scholar]
- 31. Hedge M. K., Gehring A. M., Adkins C. T., Weston L. A., Lavis L. D., and Johnson R. J. (2012) The structural basis for the narrow substrate specificity of an acetyl esterase from Thermotoga maritima. Biochim. Biophys. Acta 1824, 1024–1030 10.1016/j.bbapap.2012.05.009 [DOI] [PubMed] [Google Scholar]
- 32. Żądło-Dobrowolska A., Szczygieł M., Koszelewski D., Paprocki D., and Ostaszewski R. (2016) Self-immolative versatile fluorogenic probes for screening of hydrolytic enzyme activity. Org. Biomol. Chem. 14, 9146–9150 10.1039/C6OB01488G [DOI] [PubMed] [Google Scholar]
- 33. Dube S., Dube H., Green N. B., Larsen E. M., White A., Johnson R. J., and Kowalski J. R. (2017) In vivo delivery and activation of masked fluorogenic hydrolase substrates by endogenous hydrolases in C. elegans. Chembiochem 18, 1807–1813 10.1002/cbic.201700278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Filippova E. V., Weston L. A., Kuhn M. L., Geissler B., Gehring A. M., Armoush N., Adkins C. T., Minasov G., Dubrovska I., Shuvalova L., Winsor J. R., Lavis L. D., Satchell K. J., Becker D. P., Anderson W. F., and Johnson R. J. (2013) Large scale structural rearrangement of a serine hydrolase from Francisella tularensis facilitates catalysis. J. Biol. Chem. 288, 10522–10535 10.1074/jbc.M112.446625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Johnson R. J., Hoops G. C., Savas C. J., Kartje Z., and Lavis L. D. (2014) A sensitive and robust enzyme kinetic experiment using microplates and fluorogenic ester substrates. J. Chem. Educ. 92, 385–388 [Google Scholar]
- 36. Farberg A. M., Hart W. K., and Johnson R. J. (2016) The unusual substrate specificity of a virulence associated serine hydrolase from the highly toxic bacterium, Francisella tularensis. Biochem. Biophys. Rep. 7, 415–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wood W. J., Patterson A. W., Tsuruoka H., Jain R. K., and Ellman J. A. (2005) Substrate activity screening: a fragment-based method for the rapid identification of nonpeptidic protease inhibitors. J. Am. Chem. Soc. 127, 15521–15527 10.1021/ja0547230 [DOI] [PubMed] [Google Scholar]
- 38. Patterson A. W., Wood W. J., and Ellman J. A. (2007) Substrate activity screening (SAS): a general procedure for the preparation and screening of a fragment-based non-peptidic protease substrate library for inhibitor discovery. Nat. Protoc. 2, 424–433 10.1038/nprot.2007.28 [DOI] [PubMed] [Google Scholar]
- 39. Soellner M. B., Rawls K. A., Grundner C., Alber T., and Ellman J. A. (2007) Fragment-based substrate activity screening method for the identification of potent inhibitors of the Mycobacterium tuberculosis phosphatase PtpB. J. Am. Chem. Soc. 129, 9613–9615 10.1021/ja0727520 [DOI] [PubMed] [Google Scholar]
- 40. Breen M. E., Steffey M. E., Lachacz E. J., Kwarcinski F. E., Fox C. C., and Soellner M. B. (2014) Substrate activity screening with kinases: discovery of small-molecule substrate-competitive c-Src inhibitors. Angew. Chem. Int. Ed. Engl. 53, 7010–7013 10.1002/anie.201311096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ellis E. E., Adkins C. T., Galovska N. M., Lavis L. D., and Johnson R. J. (2013) Decoupled roles for the atypical, bifurcated binding pocket of the ybfF hydrolase. Chembiochem 14, 1134–1144 10.1002/cbic.201300085 [DOI] [PubMed] [Google Scholar]
- 42. Lukowski J. K., Savas C. P., Gehring A. M., McKary M. G., Adkins C. T., Lavis L. D., Hoops G. C., and Johnson R. J. (2014) Distinct substrate selectivity of a metabolic hydrolase from Mycobacterium tuberculosis. Biochemistry 53, 7386–7395 10.1021/bi501108u [DOI] [PubMed] [Google Scholar]
- 43. McKary M. G., Abendroth J., Edwards T. E., and Johnson R. J. (2016) Structural basis for the strict substrate selectivity of the mycobacterial hydrolase LipW. Biochemistry 55, 7099–7111 10.1021/acs.biochem.6b01057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lavis L. D., and Raines R. T. (2014) Bright building blocks for chemical biology. ACS Chem. Biol. 9, 855–866 10.1021/cb500078u [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tallman K. R., Levine S. R., and Beatty K. E. (2016) Small-molecule probes reveal esterases with persistent activity in dormant and reactivating Mycobacterium tuberculosis. ACS Infect. Dis. 2, 936–944 10.1021/acsinfecdis.6b00135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Deb C., Daniel J., Sirakova T. D., Abomoelak B., Dubey V. S., and Kolattukudy P. E. (2006) A novel lipase belonging to the hormone-sensitive lipase family induced under starvation to utilize stored triacylglycerol in Mycobacterium tuberculosis. J. Biol. Chem. 281, 3866–3875 10.1074/jbc.M505556200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rosenau F., Isenhardt S., Gdynia A., Tielker D., Schmidt E., Tielen P., Schobert M., Jahn D., Wilhelm S., and Jaeger K.-E. (2010) Lipase LipC affects motility, biofilm formation and rhamnolipid production in Pseudomonas aeruginosa. FEMS Microbiol. Lett. 309, 25–34 [DOI] [PubMed] [Google Scholar]
- 48. Johnson T. L., Waack U., Smith S., Mobley H., and Sandkvist M. (2015) Acinetobacter baumannii is dependent on the type II secretion system and its substrate LipA for lipid utilization and in vivo fitness. J. Bacteriol. 198, 711–719 10.1128/JB.00622-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bassett B., Waibel B., White A., Hansen H., Stephens D., Koelper A., Larsen E. M., Kim C., Glanzer A., Lavis L. D., Hoops G. C., and Johnson R. J. (2018) Measuring the global substrate specificity of mycobacterial serine hydrolases using a library of fluorogenic ester substrates. ACS Infect. Dis. 4, 904–911 10.1021/acsinfecdis.7b00263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Oosterhoff D., Pinedo H. M., Witlox M. A., Carette J. E., Gerritsen W. R., and van Beusechem V. W. (2005) Gene-directed enzyme prodrug therapy with carboxylesterase enhances the anticancer efficacy of the conditionally replicating adenovirus AdDelta24. Gene Ther. 12, 1011–1018 10.1038/sj.gt.3302492 [DOI] [PubMed] [Google Scholar]
- 51. Ferreira de Freitas R., and Schapira M. (2017) A systematic analysis of atomic protein-ligand interactions in the PDB. Medchemcomm 8, 1970–1981 10.1039/C7MD00381A [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Valley C. C., Cembran A., Perlmutter J. D., Lewis A. K., Labello N. P., Gao J., and Sachs J. N. (2012) The methionine-aromatic motif plays a unique role in stabilizing protein structure. J. Biol. Chem. 287, 34979–34991 10.1074/jbc.M112.374504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bissantz C., Kuhn B., and Stahl M. (2010) A medicinal chemist's guide to molecular interactions. J. Med. Chem. 53, 5061–5084 10.1021/jm100112j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chapelat J., Berst F., Marzinzik A. L., Moebitz H., Drueckes P., Trappe J., Fabbro D., and Seebach D. (2012) The substrate-activity-screening methodology applied to receptor tyrosine kinases: a proof-of-concept study. Eur. J. Med. Chem. 57, 1–9 10.1016/j.ejmech.2012.08.038 [DOI] [PubMed] [Google Scholar]
- 55. Zheng X., Guo J., Xu L., Li H., Zhang D., Zhang K., Sun F., Wen T., Liu S., and Pang H. (2011) Crystal structure of a novel esterase Rv0045c from Mycobacterium tuberculosis. PLoS One 6, e20506 10.1371/journal.pone.0020506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Park S. Y., Lee S. H., Lee J., Nishi K., Kim Y. S., Jung C. H., and Kim J. S. (2008) High-resolution structure of ybfF from Escherichia coli K12: a unique substrate-binding crevice generated by domain arrangement. J. Mol. Biol. 376, 1426–1437 10.1016/j.jmb.2007.12.062 [DOI] [PubMed] [Google Scholar]
- 57. Li P. Y., Ji P., Li C. Y., Zhang Y., Wang G. L., Zhang X. Y., Xie B. B., Qin Q. L., Chen X. L., Zhou B. C., and Zhang Y. Z. (2014) Structural basis for dimerization and catalysis of a novel esterase from the GTSAG motif subfamily of the bacterial hormone-sensitive lipase family. J. Biol. Chem. 289, 19031–19041 10.1074/jbc.M114.574913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wolfenden R. (2011) Benchmark reaction rates, the stability of biological molecules in water, and the evolution of catalytic power in enzymes. Annu. Rev. Biochem. 80, 645–667 10.1146/annurev-biochem-060409-093051 [DOI] [PubMed] [Google Scholar]
- 59. Olguin L. F., Askew S. E., O'Donoghue A. C., and Hollfelder F. (2008) Efficient catalytic promiscuity in an enzyme superfamily: an arylsulfatase shows a rate acceleration of 1013 for phosphate monoester hydrolysis. J. Am. Chem. Soc. 130, 16547–16555 10.1021/ja8047943 [DOI] [PubMed] [Google Scholar]
- 60. Eisenthal R., Danson M. J., and Hough D. W. (2007) Catalytic efficiency and kcat/KM: a useful comparator? Trends Biotechnol. 25, 247–249 10.1016/j.tibtech.2007.03.010 [DOI] [PubMed] [Google Scholar]
- 61. Radzicka A., and Wolfenden R. (1995) A proficient enzyme. Science 267, 90–93 10.1126/science.7809611 [DOI] [PubMed] [Google Scholar]
- 62. Miller B. G., Hassell A. M., Wolfenden R., Milburn M. V., and Short S. A. (2000) Anatomy of a proficient enzyme: the structure of orotidine 5′-monophosphate decarboxylase in the presence and absence of a potential transition state analog. Proc. Natl. Acad. Sci. 97, 2011–2016 10.1073/pnas.030409797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Fox R. J., and Clay M. D. (2009) Catalytic effectiveness, a measure of enzyme proficiency for industrial applications. Trends Biotechnol. 27, 137–140 10.1016/j.tibtech.2008.12.001 [DOI] [PubMed] [Google Scholar]
- 64. Ceccarelli E. A., Carrillo N., and Roveri O. A. (2008) Efficiency function for comparing catalytic competence. Trends Biotechnol. 26, 117–118 10.1016/j.tibtech.2007.11.008 [DOI] [PubMed] [Google Scholar]
- 65. Wolfenden R. (2006) Degrees of difficulty of water-consuming reactions in the absence of enzymes. Chem. Rev. 106, 3379–3396 10.1021/cr050311y [DOI] [PubMed] [Google Scholar]
- 66. Kirby A. J., and Hollfelder F. (2009) From Enzyme Models to Model Enzymes, pp. 29–41, Royal Society of Chemistry, Cambridge [Google Scholar]
- 67. Wolfenden R. (1972) Analog approaches to the structure of the transition state in enzyme reactions. Acc. Chem. Res. 5, 10–18 10.1021/ar50049a002 [DOI] [Google Scholar]
- 68. Bar-Even A., Noor E., Savir Y., Liebermeister W., Davidi D., Tawfik D. S., and Milo R. (2011) The moderately efficient enzyme: evolutionary and physicochemical trends shaping enzyme parameters. Biochemistry 50, 4402–4410 10.1021/bi2002289 [DOI] [PubMed] [Google Scholar]
- 69. Lewis A. K., Dunleavy K. M., Senkow T. L., Her C., Horn B. T., Jersett M. A., Mahling R., McCarthy M. R., Perell G. T., Valley C. C., Karim C. B., Gao J., Pomerantz W. C., Thomas D. D., Cembran A., et al. (2016) Oxidation increases the strength of the methionine-aromatic interaction. Nat. Chem. Biol. 12, 860–866 10.1038/nchembio.2159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kozakov D., Hall D. R., Jehle S., Luo L., Ochiana S. O., Jones E. V., Pollastri M., Allen K. N., Whitty A., and Vajda S. (2015) Ligand deconstruction: why some fragment binding positions are conserved and others are not. Proc. Natl. Acad. Sci. U.S.A. 112, E2585–E2594 10.1073/pnas.1501567112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Segal E., Prestwood T. R., van der Linden W. A., Carmi Y., Bhattacharya N., Withana N., Verdoes M., Habtezion A., Engleman E. G., and Bogyo M. (2015) Detection of intestinal cancer by local, topical application of a quenched fluorescence probe for cysteine cathepsins. Chem. Biol. 22, 148–158 10.1016/j.chembiol.2014.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Váradi L., Luo J. L., Hibbs D. E., Perry J. D., Anderson R. J., Orenga S., and Groundwater P. W. (2017) Methods for the detection and identification of pathogenic bacteria: past, present, and future. Chem. Soc. Rev. 46, 4818–4832 10.1039/C6CS00693K [DOI] [PubMed] [Google Scholar]
- 73. Kolbe K., Veleti S. K., Johnson E. E., Cho Y. W., Oh S., and Barry C. E. 3rd (2018) Role of chemical biology in tuberculosis drug discovery and diagnosis. ACS Infect. Dis. 4, 458–466 10.1021/acsinfecdis.7b00242 [DOI] [PubMed] [Google Scholar]
- 74. Reymond J. L., Fluxa V. S., and Maillard N. (2009) Enzyme assays. Chem. Commun., 34–46 10.1039/b813732c [DOI] [PubMed] [Google Scholar]
- 75. Larsen E. M., Stephens D. C., Clarke N. H., and Johnson R. J. (2017) Ester-prodrugs of ethambutol control its antibacterial activity and provide rapid screening for mycobacterial hydrolase activity. Bioorg. Med. Chem. Lett. 27, 4544–4547 10.1016/j.bmcl.2017.08.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Guo J., Zheng X., Xu L., Liu Z., Xu K., Li S., Wen T., Liu S., and Pang H. (2010) Characterization of a novel esterase Rv0045c from Mycobacterium tuberculosis. PLoS One 5, e13143 10.1371/journal.pone.0013143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kowalski J. R., Hoops G. C., and Johnson R. J. (2016) Implementation of a collaborative series of classroom-based undergraduate research experiences spanning chemical biology, biochemistry, and neurobiology. CBE Life Sci Educ 15, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.