

Abstract
The endocytic pathway plays an instrumental role in recycling internalized molecules back to the plasma membrane or in directing them to lysosomes for degradation. We recently reported a new role of endosomes–the delivery of components from extracellular vesicles (EVs) to the nucleoplasm of recipient cells. Using indirect immunofluorescence, FRET, immunoisolation techniques, and RNAi, we report here a tripartite protein complex (referred to as the VOR complex) that is essential for the nuclear transfer of EV-derived components by orchestrating the specific localization of late endosomes into nucleoplasmic reticulum. We found that the VOR complex contains the endoplasmic reticulum–localized vesicle-associated membrane protein (VAMP)-associated protein A (VAP-A), the cytoplasmic oxysterol-binding protein–related protein 3 (ORP3), and late endosome–associated small GTPase Rab7. The silencing of VAP-A or ORP3 abrogated the association of Rab7-positive late endosomes with nuclear envelope invaginations and, hence, the transport of endocytosed EV-derived components to the nucleoplasm of recipient cells. We conclude that the VOR complex can be targeted to inhibit EV-mediated intercellular communication, which can have therapeutic potential for managing cancer in which the release of EVs is dysregulated.
Keywords: nuclear envelope, endosome, nuclear transport, cancer biology, exosome (vesicle), extracellular vesicles, nucleus, late endosome, ORP3, oxysterol-binding related protein, VAP-A, VOR complex, endocytosis, vesicle-associated membrane protein;
Introduction
Extracellular vesicles (EVs),3 such as exosomes and microvesicles, play diverse roles as shuttles between cells in numerous physiological processes, notably cellular differentiation, inflammation, and immunity among others (1). Their functional impact on target cells is related to the delivered components, i.e. proteins, lipids, and nucleic acids, which reflect the status and source of donor cells (2). The heterogeneity of EVs in terms of surface antigens also suggests different cargoes and hence distinct biological roles associated with their action (3). Molecular mechanisms regulating EV biogenesis, their release, and subsequent uptake by target cells have emerged during the last 2 decades (4). How their cargo molecules are selectively delivered to the intracellular sites of action, including the intranuclear compartment (5–8), is still obscure (9). This issue is particularly important given that the biogenesis and functionality of EVs are dysregulated under pathological conditions (10). EVs have received great attention as noninvasive biomarkers in biological fluids for diagnostic use, given that their amount is significantly increased in various cancerous tissues (11). Furthermore, EVs might be of therapeutic relevance as they could act as nanosized drug delivery vehicles in oncology and possibly in regenerative medicine (12). Altogether, interference with the release of EVs, their uptake by recipient cells, and identification of new intracellular routes and/or mechanisms, including the molecular players that regulate the delivery of the endocytosed EV-derived components in host cells, are potential new targets for innovative cancer treatments (13, 14).
Recently, we described a novel subnuclear compartment that is created by the entry of small GTPase Rab7-containing late endosomes in the nucleoplasmic reticulum (15). The latter is shaped by superficial and deep nuclear envelope invaginations (NEI) penetrating into the nucleoplasm. Although the function of nucleoplasmic reticulum remains somehow enigmatic, various membrane-bound organelles and cytoskeletal components were identified therein (16, 17). Likewise, adeno-associated virus particles were also observed in nuclear invaginations following infection (18). Given that late endosomes in NEI had often an elongated appearance and looked as a sword in its scabbard, we proposed to name this dual-structure “spathasome” from the Greek/Latin words “spathi/spatha” for sword (15). This structure appears to act as an intermediate compartment for the delivery of the content of endocytosed EVs (e.g. CD9–CD133 protein complexes) to the nucleoplasm of their host cell (15). The NEI-associated late endosomes and nuclear localization of EV-derived proteins were observed in cancer cells and mesenchymal stromal cells in cultures and in breast cancer patient biopsies (15). Although the nuclear transfer of components derived from EVs represented solely a minute fraction of total endocytosed EV proteins, they nonetheless modified the gene expression profile (15). Little is known about the biogenesis and dynamics of NEI-associated late endosomes. Being composed of two distinct organelles (i.e. nucleus and late endosome), they can be regulated by several mechanisms involving intrinsic and extrinsic factors. For instance, factors influencing the formation and/or stabilization of NEI can control their organization (16, 19, 20). In transformed cells that contain a hyper-developed nucleoplasmic reticulum (16), nuclear delivery of EV components via NEI-associated late endosomes may contribute to cancer progression. Extracellular factors and regulators of endocytosis may also modulate their dynamics. The amount of EVs added to the recipient cells has been shown to influence their frequency, and hence, the nuclear delivery of EV components (15).
The membrane contact sites of organelles with the endoplasmic reticulum (ER) are recognized in various physiological processes (e.g. lipid transport and membrane traffic) and are conserved in all eukaryotic cells (21, 22). The ER network makes close contact with mitochondria, Golgi apparatus, endosomes, and plasma membrane. Tethering complexes that maintain close proximity between the apposing ER and organelle-specific membranes stabilize contact sites. The ER contacts could regulate the maturation of endosomes and their fission and transport along microtubules (23–26). One major player in these contact zones is the integral ER protein VAP-A (vesicle-associated membrane protein (VAMP)-associated protein-A) (27). VAP-A interacts in trans with the peripheral late endosomal multidomain oxysterol-binding protein (OSBP)–related protein 1L (ORP1L) via the FFAT (diphenylalanine in an acidic tract) domain, whereas the N-terminal ankyrin repeat region of ORP1L binds to small GTPase Rab7 (28, 29). The contact site of the ER-late endosome is regulated by cholesterol level in endosomes, which is sensed by ORP1L or other members of the highly conserved OSBP family (30, 31). Sterol binding leads to conformational changes of ORP1L, which modulate their interaction with VAP-A (28). Two other late endosomal proteins, STARD3 (MLN64) and STARD3 N-terminal like, which contain the FFAT motif, are also able to bind VAP-A and sense sterol at ER–endosome contact sites (32). In addition, complexes of VAP-A and OSBP or OSBP-related protein 3 (ORP3) are implicated in the tethers of ER with Golgi apparatus and plasma membrane, respectively (33, 34). The VAP-A homolog, VAP-B, is involved in the close physical association of ER with mitochondria (35).
Given the membrane continuity between ER and outer nuclear membrane (ONM), we sought whether VAP proteins are implicated in tethering late endosomes within NEI. Here, we report that VAP-A, ORP3, and Rab7 form a tripartite complex essential for the presence of late endosomes in the nucleoplasmic reticulum and the nuclear transfer of EV-derived components. The discovery of the VOR complex (an acronym of VAP-A, ORP3, and Rab7) may lead to therapeutic applications by interfering with the mechanism of intercellular communication.
Results
VAP-A is present in type II NEI–containing late endosomes
We recently described in various cell types the presence of Rab7-positive (hereafter Rab7+) late endosomes in NEI (15). Given that the ONM is a continuation of the ER membrane, we investigated whether the ER-associated protein VAP-A, a protein involved in the tethering of ER to late endosomes distributed in the cytoplasm, was present in NEI. We chose to use human FEMX-I melanoma cells as a primary model since we discovered NEI-associated Rab7+ late endosomes from that (15). VAP-A was visualized by expressing a fusion of this protein with green fluorescence protein (VAP-A–GFP), and NEI were observed with an antibody to the inner nuclear membrane (INM) protein SUN domain-containing protein 2 (SUN2). We found VAP-A–GFP in SUN2+ NEI (Fig. 1A), but not all of them, were positive (see below). The presence of late endosomes in VAP-A+ NEI was then investigated. Rab7 as a fusion protein with the red fluorescence protein (Rab7–RFP) was used to highlight late endosomes in VAP-A–GFP-transfected FEMX-I cells. As displayed in Fig. 1B, Rab7–RFP+ late endosomes were found in VAP-A–GFP+ NEI. Their dynamic entry therein was also observed live by time-lapse video microscopy (Fig. 1C). Similar data were obtained by applying anti-Rab7 and VAP-A antibodies instead of fluorescent fusion proteins, and the expression level of VAP-A–GFP was comparable with the endogenous protein (see below).
Figure 1.
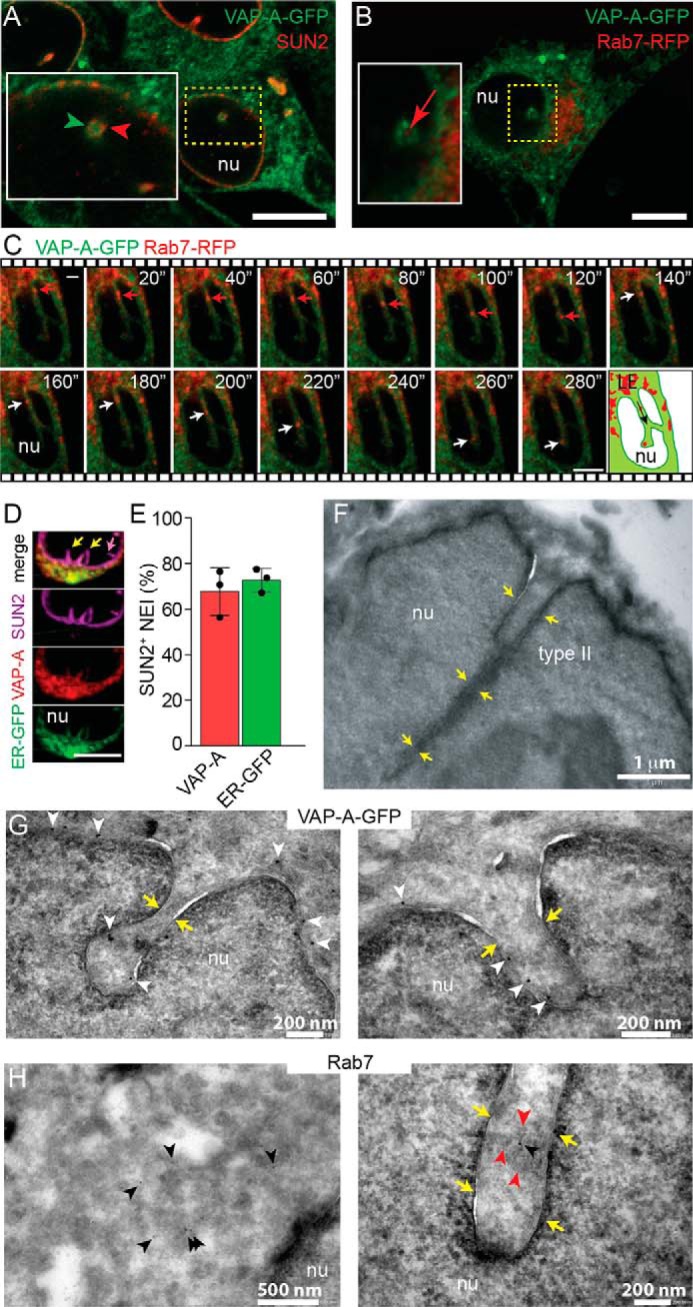
VAP-A is associated with type II NEI containing late endosomes. A and B, VAP-A–GFP-transfected FEMX-I cells were immunolabeled for SUN2 (A) or infected with Rab7–RFP baculovirus (B) and analyzed by CLSM. Area of a cross-section of NEI is magnified. A single x-y optical section is presented. C, FEMX-I cells expressing VAP-A–GFP and Rab7–RFP were analyzed by time-lapse video microscopy. Elapsed time is indicated in the top right corner. Arrowheads show the localization of VAP-A–GFP in SUN2-immunolabeled NEI (A), and arrows indicate Rab7–RFP+ late endosomes in NEI (B and C). Cartoon illustrates the direction (arrow) of Rab7+ late endosomes (LE) in NEI. D, cells expressing ER–GFP marker were double-immunolabeled for VAP-A and SUN2. A three-dimensional (3D) reconstruction of two adjacent sections (0.4-μm each) is presented. Only part of the nucleus is shown. Note the absence (purple arrow) or presence (yellow arrows) of VAP-A and ER–GFP in SUN2+ type I and II NEI, respectively. Additional information is provided in Fig. S1. E, percentage of SUN2+ NEI (type II) containing VAP-A or ER–GFP is presented. The means ± S.D. are shown (n = 3). The average of each experiment, where more than 50 cells were evaluated, is indicated. F, EM of FEMX-I cells shows that type II NEI are not only superficial indentations of the nuclear envelope but also appeared as deep recesses (yellow arrows). G and H, immunogold labeling on ultrathin cryosections reveals the presence of VAP-A–GFP in deep NEI of FEMX-I cells expressing the fusion protein (G, white arrowheads), whereas Rab7 proteins (H, black arrowheads) are found in the cytoplasm (left panel) and NEI (right panel) of FEMX-I cells. The presence of membrane-bound organelles in NEI is indicated with red arrowheads. nu, nucleoplasm. Scale bars, 5 μm (A–D) or as indicated (F–H).
Two types of NEI were reported (16). The first one contains solely the INM that is penetrating into the nucleoplasm, and the second involves both INM and ONM (Fig. S1A). Consequently, only type II NEI can host cytoplasmic organelles, notably late endosomes. To confirm the presence of VAP-A in type II NEI and to determine the proportion of type I versus type II NEI, we expressed a chimeric ER protein (i.e. GFP containing the ER signal sequence of calreticulin with a KDEL retention signal sequence) in FEMX-I cells and immunolabeled them for VAP-A and SUN2. Both types of NEI were detected (Fig. 1D and Fig. S1B and Video S1). Nonetheless, more than 70% of SUN2+ NEI were positive for chimeric ER protein and/or VAP-A, suggesting that the majority of NEI is of type II (Fig. 1E, data not shown). Originating from the nuclear surface, type II NEI can reach deep regions of the nucleoplasm, as observed by confocal laser-scanning microscopy (CLSM) or EM (Fig. 1, C and F). VAP-A–GFP proteins were also detected in deep NEI by immunogold EM (Fig. 1G), whereas Rab7+ late endosomes were observed in both NEI and cytoplasm (Fig. 1H). Of note, the entry of late endosomes in VAP-A+ NEI seems to be selective. Early endosomes or Golgi apparatus as highlighted by the expression of Rab5a and Golgi-resident enzyme N-acetylgalactosaminyltransferase 2 (GALNT2) fused in-frame to RFP, respectively, were excluded from there (Fig. S2, A and B). Altogether, these data indicate that VAP-A is found in NEI that contain late endosomes.
Entry of late endosomes in NEI requires VAP-A
To assess the requirement of VAP-A for the presence of late endosomes in NEI, its expression was silenced in melanoma cells using small hairpin (sh) RNAs (for technical details see under “Experimental procedures”). Scrambled shRNA was used as control. The overall expression of VAP-A was reduced by 65% as evaluated by immunoblotting (Fig. 2, A and B) and >95% at the single cell level by immunochemistry (Fig. S3A). No morphological alterations in VAP-A–deficient cells were observed by scanning EM (Fig. S3B). However, silencing VAP-A expression resulted in the absence of late endosomes in NEI (Fig. 2, C and D, and Videos S2 and S3), without affecting their distribution throughout the cytoplasm (see below).
Figure 2.

VAP-A is required for the entry of Rab7+ late endosomes in NEI. A–H, untransfected FEMX-I cells (control) or stably transfected with plasmids carrying scrambled shRNA, shVAP-A, or shVAP-B were analyzed by immunoblotting for VAP-A, VAP-B, and β-actin (A and E). Molecular mass markers (kDa) are indicated. Representative blots are shown. The relative VAP-A or VAP-B expression was quantified by comparison with control samples (B and F, red dotted line). The samples were normalized to β-actin. The means ± S.D. is shown with the individual value of each experience (n = 3). Alternatively, scrambled shRNA or shVAP-A/B transfected cells were infected with Rab7–RFP baculovirus and analyzed by CLSM after double-immunolabeling for VAP-A (C and D) or VAP-B (G and H) and SUN2. 3D reconstruction of 2–3 adjacent sections (0.4 μm each) is presented. Arrows indicate the presence of a given marker in NEI. Percentage of cells harboring either SUN2+ NEI-associated Rab7+ late endosomes, SUN2+ NEI only, or without NEI upon silencing VAP-A (D, see also Fig. S3) or VAP-B (H) were quantified. The means ± S.D. (D and H) are shown (n = 3). More than 50 cells were evaluated per experiment. p values are indicated. n.s., not significant. Images depicted in C are displayed in Videos S2 and S3. I, scrambled shRNA or shVAP-A–transfected FEMX-I cells were infected with ER–GFP and Rab7–RFP baculoviruses and analyzed by time-lapse video microscopy. Single x-y optical section (0.4 um) is presented. Elapsed time is indicated in the top right corner. Red and white arrows show the localization of Rab7–RFP in two distinct ER–GFP+ NEI (top, transverse section of NEI), and arrowheads indicate NEI without Rab7–RFP (bottom, sagittal section of three NEI). nu, nucleoplasm. Scale bars, 5 μm.
VAP-A has a homolog, VAP-B. Although the VAP-A silencing was not compensated by an up-regulation of VAP-B (Fig. 2, A and B), and Rab7+ late endosomes were still absent in VAP-B+ NEI of shVAP-A cells (Fig. S3C), we evaluated whether VAP-B substitutes VAP-A function by silencing its expression with shRNA. The overall VAP-B expression was reduced by 65% as observed by immunoblotting (Fig. 2, E and F), and by up to 95% at the single-cell level as detected by CLSM (Fig. 2G, data not shown). Silencing VAP-B did not affect VAP-A expression (Fig. 2, E and F) and had no effect on the entry of late endosomes in NEI (Fig. 2, G and H), indicating that VAP-A, but not VAP-B, is required for the presence of NEI-associated late endosomes.
Given the absence of late endosomes in NEI of cells specifically deficient in VAP-A, we evaluated whether their entry therein was also impaired. Scrambled shRNA and shVAP-A FEMX-I cells expressing the ER–GFP marker and Rab7–RFP were analyzed by time-lapse video microscopy. Although the movement of Rab7–RFP+ late endosomes in ER–GFP+ NEI was observed in control cells (Fig. 2I, top panels), none of them were observed going there upon VAP-A silencing (Fig. 2I, bottom panels). It is noteworthy that the traffic of late endosomes within the cytoplasm also seemed perturbed (Fig. 2I, data not shown). Altogether, these data suggest that the entry and retention of late endosomes in NEI requires VAP-A. Besides the data acquired with FEMX-I cells, a similar set of observations was made with another cell line, i.e. HeLa cells (Fig. S4), indicating that the current phenomenon is not restricted to melanoma cells.
ORP3 is required for the presence of late endosomes in NEI
To find out how VAP-A regulates the presence of late endosomes in NEI, we investigated whether known interacting partners of VAP-A (i.e. OSBP, ORP1L, STARD3, and ORP3) are expressed in FEMX-I cells and if they localize in NEI. Immunoblots of FEMX-I cell lysates revealed that they are all expressed, and the VAP-A silencing did not influence their expression level (Figs. S2C and S5, A and B). Indirect immunofluorescence analysis demonstrated that OSBP, ORP1L, or STARD3 was not associated with NEI (Fig. S2, C and D), whereas ORP3, a protein implicated in the tether of ER and plasma membrane, was (Fig. 3A). Moreover, ORP3 co-localized with VAP-A (Fig. S5C), and silencing VAP-A reduced the number of SUN2+ NEI containing ORP3, suggesting that VAP-A expression is necessary for the presence of ORP3 in NEI (Fig. 3, A and B).
Figure 3.

ORP3 is essential for the entry of Rab7+ late endosomes in NEI and co-localized with VAP-A and Rab7. A and B, FEMX-I cells stably transfected with plasmids carrying scrambled shRNA or shVAP-A were immunolabeled for ORP3 and SUN2 prior to CLSM. Single x-y optical section is presented. The amount of SUN2+ NEI containing ORP3 (A, square, see insets at right) was quantified (B). C and D, scrambled shRNA- or shORP3-transfected cells were infected with Rab7–RFP baculovirus and analyzed by CLSM after double-immunolabeling for ORP3 and SUN2 (C). Percentage of cells harboring either SUN2+ NEI-associated Rab7+ late endosomes, SUN2+ NEI only, or without NEI upon silencing ORP3 were quantified (D). The means ± S.D. are shown (n = 3). More than 30 cells were evaluated per experiment, and their average is presented (B). p values are indicated. E and F, FEMX-I cells expressing VAP-A–GFP were double-immunolabeled for ORP3 and Rab7 prior to CLSM. Rab7 antibody was directly conjugated to fluorophore Alexa Fluor 647, and an anti-mouse secondary antibody coupled to TRITC was used to detect ORP3 (E). The inset shows the merged panel with a 4.0-μm-long arrow indicating the area subjected to line scan analysis with RFI of each channel (F). Thick line indicates the position of late endosomes in NEI where three fluorescent signals overlaid. In each panel, arrows show the localization of a given protein in NEI. Images depicted in C (top panels) are displayed in Video S4. nu, nucleoplasm. Scale bars, 5 μm.
The latter observation prompted us to test whether ORP3 is also required for the localization and/or retention of late endosomes in NEI. Again, its expression was silenced using shRNA. Under these conditions, the expression level of VAP-A was not significantly changed as observed by immunoblotting (Fig. S6, A and B, p = 0.1717 (scrambled shRNA control) or 0.1243 (untransfected control)), and its localization in NEI was maintained as detected by CLSM (data not shown). Upon Rab7–RFP expression, Rab7 fluorescence was found associated with NEI (ORP3+/SUN2+) in scrambled shRNA cells, but not in NEI (ORP3−/SUN2+) of shORP3 cells (Fig. 3C, top and bottom panels, respectively, and Video S4), indicating that ORP3 is required for the presence of late endosomes in NEI (Fig. 3D).
The dual co-localizations of VAP-A/Rab7, VAP-A/ORP3, and ORP3/Rab7 in NEI indirectly suggest an interaction among all of them. This issue was substantiated by double-immunolabeling for ORP3 and Rab7 of FEMX-I cells expressing VAP-A–GFP. A triple co-localization of all proteins was observed in NEI (Fig. 3E). Line scan analysis with relative fluorescence intensities (RFI) of the green, red, and magenta channels along NEI revealed that the position of the Rab7+ late endosomes coincides with VAP-A and ORP3 (Fig. 3F, thick line). Although VAP-A and ORP3 signals followed a similar pattern along NEI, Rab7 immunoreactivity was nonetheless more restricted, suggesting that a potential interaction between VAP-A and ORP3 might occur independently of the presence of Rab7. Finding an equal incidence of ORP3 in Rab7+ and Rab7− NEI substantiated this issue (Fig. S6C).
Interaction of VAP-A, ORP3, and Rab7
To demonstrate an interaction between VAP-A and ORP3, a co-immunoisolation was performed using paramagnetic-bead technology. FEMX-I cells expressing VAP-A–GFP or parental cells were solubilized in a buffer containing 0.5% Triton X-100, and after centrifugation the resulting lysates were subjected to immunoisolation. VAP-A–GFP was immunoisolated from transfected cells by an anti-GFP antibody directly coupled to magnetic beads whereas ORP3 by an anti-ORP3 antibody followed by protein G–conjugated magnetic beads from both cell lines. Upon magnetic separation, an aliquot of the inputs and entire bound fractions were probed by immunoblotting. Interestingly, both VAP-A (VAP-A–GFP) and ORP3 were co-immunoisolated in both methods (Fig. 4A). Additionally, Rab7 was co-immunoisolated with them, suggesting the formation of a tripartite complex (Fig. 4A). In contrast, VAP-B failed to be co-immunoisolated from both cell lines. The ratio VAP-A(VAP-GFP)/ORP3 immunoreactivity was 0.651 ± 0.446, and only 0.002 ± 0.001 for VAP-B/ORP3 (n = 6). When a similar experiment was performed with VAP-B as bait, solely a small amount of ORP3 was co-immunoisolated, and no Rab7 (Fig. 4B). The latter data were observed either with VAP-A–GFP-transfected FEMX-I cells or untransfected cells (Fig. 4B, left and right panels, respectively). Of note, we did not detect the VAP-A/B heterodimer (data not shown).
Figure 4.

Tripartite complex of VAP-A, ORP3, and Rab7 in NEI. A and B, detergent lysates prepared from transfected FEMX-I cells expressing VAP-A–GFP (VAP-A–GFP+ FEMX-I) or untransfected cells (FEMX-I) were subjected to immunoisolation (IS) with either anti-GFP antibody-coupled magnetic beads or anti-ORP3 (A) or anti-VAP-B (B) antibody followed by protein G-coupled magnetic beads as indicated above each lane. An aliquot of the input (1:50) and the entire bound fractions were probed for ORP3, VAP-A/B, and Rab7 by immunoblotting. Molecular mass markers (kDa) are indicated. The antibody used is indicated in the top left corner of the blot. Arrows indicate the protein of interest, and the arrowhead indicates VAP-A–GFP protein. Representative blots are shown (n = 3–6). Note that the VAP-A–GFP expression level is similar to endogenous protein in VAP-A–GFP+ FEMX-I cells, and almost no VAP-B is co-immunoisolated with ORP3 in contrast to VAP-A.
Fluorescence resonance energy transfer reveals the proximity of VAP-A, ORP3, and Rab7 in NEI
Given that VAP-A, ORP3, and Rab7 are found not only in NEI but are also widely distributed in the cytoplasm, we used fluorescence resonance energy transfer (FRET), specifically the acceptor photobleaching method, to detect their molecular proximity within a given subcellular compartment. The principle of this technique is that energy transfer is eliminated (or reduced) when the acceptor is bleached, thus yielding an increase in donor fluorescence, which is a measure of FRET efficiency. To detect FRET, we monitored the donor and acceptor fluorescence intensities before, during, and after the acceptor photobleaching procedure (36). We used either RFP-fusion protein or a primary antibody followed by tetramethylrhodamine (TRITC)-conjugated secondary antibody as acceptor, whereas GFP-fusion protein or primary antibody/FITC-conjugated secondary antibody was used as donor. First, we examined the fluorescence intensity profiles of the FRET bleaching in NEI as a region of interest (ROI). Given the co-localization of VAP-A, ORP3, and Rab7 therein, we used the following three FRET pairs (acceptor/donor), i.e. ORP3–TRITC/VAP-A–GFP, Rab7–RFP/ORP3-FITC, and Rab7–RFP/VAP-A–GFP. Interestingly, increases in donor fluorescence intensity were detected with all three FRET pairs after acceptor bleaching, suggesting a physical proximity between ORP3, VAP-A, and Rab7 in NEI (Fig. 5, A, C, and D and Fig. S7A). The FRET pair Rab7–RFP/VAP-B-FITC did not reveal any increase in donor fluorescence upon acceptor photobleaching (Fig. 5, B and D), although VAP-B, just like VAP-A, is located in NEI. Second, we performed similar experiments on cytoplasmic areas or portions of nuclear membrane devoid of NEI as ROIs. As a positive control, the Rab7/VAP-A pair showed an increase in donor fluorescence (Fig. 5F and Fig. S7B, data not shown) in agreement with an earlier study that demonstrated the interaction of cytoplasmically-distributed late endosomes with ER (28). It is interesting to note that the FRET efficiencies of Rab7/VAP-A pair were lower in the cytoplasm by comparison with NEI. In contrast, neither Rab7/ORP3 nor ORP3/VAP-A pairs revealed a fluorescence transfer (Fig. 5, E and F, and Fig. S7, B and C). Overall, these data suggest that VAP-A, ORP3, and Rab7 form a tripartite complex (VOR complex) in NEI, which has a specific role in maintaining the late endosomes therein.
Figure 5.

FRET analysis of the interaction between ORP3, VAP-A/B, and Rab7 in NEI. A–F, potential interactions between ORP3, VAP proteins, and Rab7 were evaluated by FRET using the acceptor photobleaching method. When appropriate, FEMX-I cells expressing VAP-A–GFP and/or Rab7–RFP were immunolabeled for ORP3 or VAP-B followed by secondary antibody coupled to appropriate fluorophore (TRITC or FITC). Intensity profiles and FRET efficiencies of each pair in NEI (A–D) or cytoplasm (E and F) are presented. The means ± S.D. are shown (n = 3). At least five cells were evaluated per experiment, and all of them are plotted. Acceptor photobleaching with a 561-nm laser line starts at time 15 s. FRET efficiencies were measured in a given ROI in bleached and unbleached areas. Corresponding micrographs with ROI are shown in Fig. S7.
Microtubules are essential for the presence of late endosomes in NEI
Besides the implication of the VOR complex in the entry and/or maintenance of Rab7+ late endosomes in NEI, we found tubulin therein (Fig. 6A). This stimulated us to determine whether microtubules are essential for the ingress of Rab7+ late endosomes in NEI. Melanoma cells were treated with nocodazole (1 μm) for 45 min prior to double-immunolabeling for tubulin or VAP-A with Rab7. Although the presence of VAP-A in NEI was not perturbed after microtubule depolymerization, less than 5% of cells presented Rab7 immunoreactivity in VAP-A+ NEI versus ≈40% in untreated (control) and DMSO-treated cells, suggesting that the movement and/or retention of late endosomes within NEI requires an intact microtubule network (Fig. 6, B and C).
Figure 6.

Microtubules are required for the entry and/or maintenance of late endosomes in NEI. A, FEMX-I cells expressing Rab7–RFP were double-immunolabeled for α-tubulin and SUN2 prior to CLSM. B, FEMX-I cells were treated without (Control) or with nocodazole (Noc., 1 μm) for 45 min prior to double-immunolabeling for α-tubulin or VAP-A (top and bottom panels, respectively) and Rab7. DMSO-treated cells were used as solvent control. A single x-y optical section is presented. Area of NEI is magnified. Arrows indicate the presence of a given protein in NEI. C, number of cells containing VAP-A+ NEI-associated Rab7+ late endosomes under specific conditions was quantified. The means ± S.D. are shown (n = 3). More than 30 cells were evaluated per experiment, and their average is indicated. p value is indicated. D, drawing showing molecular interactions involved in the association of Rab7+ late endosomes (LE) with NEI. Player(s) involved in the interaction of late endosomes with microtubules remains to be identified (?). PH, pleckstrin homology domain of ORP3; INM, ONM, inner and outer nuclear membrane, respectively; nu, nucleoplasm. Scale bars, 5 μm.
Delivery of EV-derived components in the nucleoplasm requires VAP-A and ORP3
We proposed that NEI-associated late endosomes participate in the transfer of proteins derived from EVs to the nucleoplasm of recipient cells (15). To substantiate this idea, FEMX-I cells expressing VAP-A–GFP were incubated with EVs labeled with membrane dye 1,1-dioctadecyl-3,3,3,3-tetramethylindocarbocyanine perchlorate (DiI). The EVs were produced by parental (untransfected) FEMX-I cells and enriched from conditioned media by differential centrifugation before labeling (see Fig. 7H) (15). Upon endocytosis, not only the EV-associated membrane would be stained but also the endosomal membranes per se given the potential fusion of EVs with them. After 5 h of EV–cell incubation, time-lapse video microscopy imaging revealed distinct DiI-labeled membrane structures penetrating in NEI (Fig. 7A, colored arrows) and tethering to VAP-A–GFP+ ONM (Fig. 7A, arrowhead, and Video S5). These data are in line with those observed with Rab7–RFP+ late endosomes (Fig. 1C) and suggest that EV-derived components can reach NEI. Next, FEMX-I cells expressing ER–RFP were incubated with fluorescent EVs that contain the tetraspanin membrane protein CD9 fused to GFP (CD9–GFP). They were produced by engineered CD9–GFP+ FEMX-I cells (15). Upon time-lapse video recording, EV-derived materials were observed, in addition to cytoplasm, in NEI (Fig. 7B, arrow). Interestingly, furtive punctate CD9–GFP appeared also in the nucleoplasm of host cells (Fig. 7B, circles).
Figure 7.

VOR complex is required for nuclear transfer of EV-derived proteins and nucleic acids. A and B, FEMX-I cells expressing either VAP-A–GFP (A) or ER–RFP (B) were incubated for 5 h with DiI-labeled EVs or CD9–GFP EVs, respectively, prior to analysis by time-lapse video microscopy. Elapsed time is indicated in the top right corner. Arrows indicate the entry of late endosomes containing DiI-labeled membranes (A) or CD9–GFP (B) in NEI and the arrowhead their tether to VAP-A–GFP+ ONM (A). GFP+ signals appearing in the nucleoplasm were highlighted (B, circles). Inset in B shows enlargement of NEI (dashed line) containing discrete punctate CD9–GFP signals. Still images (A) are from Video S5. C–E, scrambled shRNA (control, Ctl) or shVAP-A/B–transfected FEMX-I (C and D) or HeLa (E) cells were incubated with fluorescent EVs derived from CD9–GFP-expressing FEMX-I cells and then double-immunolabeled for VAP-A or VAP-B and SUN2 prior to CLSM. The amount of EV-derived CD9–GFP in the nuclear compartment was quantified using Fiji software. Micrographs are presented in Fig. S8. Independent values for each cell from three independent experiments (C, #1–3) and their average from three independent experiments (D and E) are presented. F and G, nuclei of FEMX-I cells incubated with CD9–GFP+ EVs were isolated before SUN2 immunolabeling and CLSM. Composite, single x-y section and z-projections are shown (F). GFP+ signals in the nucleoplasm were highlighted (circles) and quantified (G). H, scheme of isolation of EVs from FEMX-I cells and subsequent nucleic acid staining with SYTO 64 dye. I and J, scrambled shRNA or shVAP-A–transfected FEMX-I cells were incubated with SYTO 64-labeled EVs and immunolabeled as above. Nuclear SYTO 64 signals in a given cell (I, experiment #1) and their average from three independent experiments (J) were quantified. In all cases, means ± S.D. are shown (n = 3). More than 50 (C and D) and 30 (E, I, and J) cells or 30 isolated nuclei (G) were evaluated per experiment. p values are indicated. n.s., not significant. Scale bars, 5 μm.
The requirement of the VOR complex for the transport and tethering of late endosomes to ONM in NEI implied that the knockdown of VAP-A and/or ORP3 would inhibit the nuclear transfer of the EV-derived materials (membranous and/or soluble content). To explore this issue, the expression of VAP-A, VAP-B, or ORP3 was silenced in FEMX-I cells using shRNA prior to their incubation with in vivo labeled (i.e. CD9–GFP) EVs. After 5 h EV–cell incubation, cells were fixed, double-immunolabeled for VAP-A (VAP-B or ORP3) and SUN2, and analyzed by CLSM. Serial optical sections through the cell of interest were taken, and CD9–GFP associated with its nucleoplasm was quantified using Fiji. About 23 ± 0.4 discrete punctate GFP signals were detected in the nuclear compartment (Fig. 7, C and D, and Fig. S8A). Silencing VAP-A, but not VAP-B, resulted in ≈90% decrease in nuclear localization of EV-derived components (Fig. 7, C and D, and Fig. S8A). Similar data were observed with HeLa cells (Fig. 7E). Under these conditions, the general endocytosis of CD9–GFP+ EVs was not significantly affected (Fig. S8B, n = 10, p = 0.1734). The punctate CD9–GFP signal was also detected upon isolation of nuclei from cells exposed to EVs, indicating CD9–GFP were incorporated into nucleoplasm, and not solely associated with the nuclear periphery (Fig. 7, F and G). Interestingly, silencing the expression of ORP3 had a similar effect, suggesting that both VAP-A and ORP3 are required for the nuclear localization of proteins derived from EVs (Fig. S6D).
To investigate whether EV-derived components other than proteins were transported to the nucleoplasm of recipient cells by a mechanism involving the VOR complex, FEMX-I cell-derived EVs were stained with the nucleic acid dye SYTO 64. The excess of dye was removed prior to their incubation (5 h) with recipient cells (Fig. 7H). In scrambled shRNA cells, SYTO 64+ EV-derived nucleic acid fluorescence was detected in the nucleoplasm in a proportion similar to CD9–GFP, whereas their amount therein decreased by 90% in cells lacking VAP-A (Fig. 7, I and J). Collectively, these data suggest the implication of the VOR complex in the shuttling of biological information between EVs and the nuclear compartment of recipient cells.
Discussion
Understanding how cells interact by exchanging their content and how intracellular processes mediate this transfer bring a new dimension to our knowledge of tissue and cell homeostasis and how it can be disrupted in disease. In this context, deciphering the molecular details of the tethers of membrane-bound organelles, which facilitate the exchange of metabolites and signaling molecules, is of utmost interest (37). Here, we aimed to identify proteins involved in the localization of late endosomes in NEI and to determine their role in relying messages carried by EVs to nuclei of recipient cells.
By indirect immunofluorescence, immunoisolation techniques, FRET, and RNAi experiments, we found that the ER-associated protein VAP-A, but not its homolog VAP-B, forms a tripartite complex with ORP3 and Rab7 that is essential for the localization of late endosomes in NEI. VAP-A was already described to be instrumental for ER-late endosome tethers within the cytoplasm coordinated by ORP1L (28). Similarly, VAP-A together with ORP3 created a contact zone between ER and the plasma membrane and regulated the activity of small GTPase R-Ras and its downstream pathway (34). Although the interaction of ORP3 with VAP-A can be mediated by its FFAT motifs as demonstrated previously (38), the Rab7-binding domain remains to be identified. In contrast to ORP1L, ORP3 does not contain an ankyrin repeat region, which mediates Rab7 interaction (39). Nonetheless, a close contact of ORP3 with Rab7 can ensue when its pleckstrin homology domain binds to phosphoinositides associated with late endosomes (Fig. 6D) (28). It remains to be determined whether the R-Ras–binding site in ORP3 can be involved in the Rab7 interaction (40). Likewise, it will be of interest to reveal whether other players (proteins and membrane lipids) are engaged with this complex and/or promote its formation (see below).
The lack of ORP1L and STARD3 in NEI was surprising because both proteins were reported to be associated with late endosomes (32). Indirectly, this raises the following questions. How are the Rab7+ late endosomes or their subpopulations transported in NEI? Does a similar interplay mechanism between Rab7/Rab-interacting lysosomal protein (RILP)/ORP1L/dynactin subunit p150Glued and microtubules occur (28, 41)? The presence of tubulin in NEI and the impact of nocodazole treatment suggest it. The next step will be to determine whether RILP and p150Glued protein, which binds to dynein heavy chain, are associated with NEI and participate in the selective microtubule-dependent movement of late endosomes therein. As observed previously, early endosomes were excluded from VAP-A+ NEI (15). Alternatively, the kinesin-1–binding ER protein Protrudin that binds to VAP-A and contacts late endosomes via Rab7, allowing their movement along microtubules toward the plasma membrane (25, 42), might be involved in the transport of late endosomes in NEI. The lack of late endosome translocation to NEI as observed in shVAP-A cells by time-lapse videos would support it. However, we did not detect Protrudin in FEMX-I melanoma cells.4 In any scenario, the deficiency of VAP-A cannot be rescued by VAP-B, which is in line with other phenomena such as the promotion of neurite extensions in neurons (21, 42).
In addition to protein candidates per se, their post-translation modifications, membrane lipids (e.g. cholesterol and phosphoinositides), notably those associated with late endosomes, and the compartmentalization of NEI where tubular structures can constrain both cytosolic and membrane players should be considered. For instance, the hyper-phosphorylation of ORP3 has been shown to regulate its interaction with VAP-A and modulate its binding to phosphoinositides at the inner leaflet of the plasma membrane (34). Our FRET analyses indicate that the close association between ORP3 and VAP-A+ ONM occurs specifically in NEI and seems to be independent of the presence of Rab7+ late endosomes. Such information implies that the sequential events would involve the interaction of cytoplasmic ORP3 with NEI-associated VAP-A, which in turn will modulate their interaction with Rab7+ late endosomes (Fig. 8A). Nonetheless, the reduction of NEI containing ORP3 in shVAP-A cells is intriguing. Because ORP3 is composed of multiple functional domains, its targeting can be modulated by several determinants, which are most likely regulated by various stimuli, notably its lipid ligand(s) (43). Thus, VAP-A may predispose the ORP3 targeting signal by regulating the status and dynamics of cellular lipids (23–26), hence impacting the functional VAP-A–ORP3 interplay. It remains to be characterized whether the NEI-associated Rab7+ late endosome subpopulation is derived from STARD3+ “early” or ORP1L+ “late” late endosomes (44). The fusion of endocytosed EVs with the endosomal membrane suggests, however, that they might represent more mature, cholesterol-poor, and acidic late endosomes. Obviously, the stepwise assembly/disassembly of all proteins and lipids involved in the segregation of Rab7+ late endosomes in NEI will require further investigation to which our findings of the VOR complex have brought a molecular basis (Fig. 8B, model I).
Figure 8.
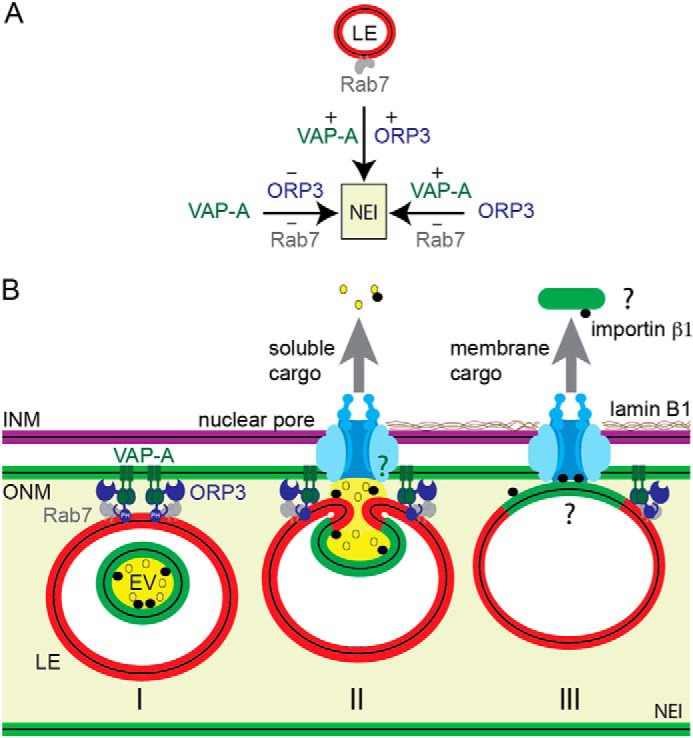
Model summarizing the VOR complex players and their interactions within NEI. A, presence of Rab7+ late endosomes (LE) in NEI requires VAP-A and ORP3, whereas ORP3 needs VAP-A, which is constitutively associated with ONM of type II NEI. B, VOR complex proteins (VAP-A, ORP3, Rab7) allow the tether of late endosomes to ONM in NEI (I). In this model, EVs are transported to late endosomes after their endocytosis. The lack of NEI-associated late endosomes and the inhibition of transfer of EV-derived components (soluble and membranous) into the nucleoplasm of host cells after importazole treatment (15) suggest that nuclear pores and importin β1 play a role in these processes (II). Their potential interaction (green question mark) with VOR complex and/or late endosomes should also be addressed. Other urgent questions remain open, notably the mechanism(s) allowing the extraction of EV-derived membrane proteins (green) from endosomal membrane (red) and their transfer into nucleoplasm through the nuclear pores, which are size-restricted (III, black question marks). EV, extracellular vesicle; LE, late endosome; INM, ONM, inner and outer nuclear membrane, respectively.
Based on current knowledge of nuclear transport, nuclear pores present in NEI seem to be required for the transfer of EV-derived components into the nucleoplasm. The inhibition of such nuclear cargo transfer and the lack of NEI-associated late endosomes after importazole treatment suggest it (15). Does a contact between NEI-associated late endosomes and nuclear pores occur? Theoretically, the function of nuclear pores and/or the associated proteins could be distinct within folds of the nuclear envelope (45). The high level of membrane curvature, particularly at the tip of NEI, could alter the molecular transport and other features of nuclear pores. In addition to nuclear import/export properties, they may contribute to the segregation and/or immobilization of late endosomes. After the fusion of EVs with the late endosomal membrane, the EV-associated importin β1 (15) may participate in these processes in addition to nuclear translocation of EV-derived components (soluble and membranous cargos) (see below, Fig. 8B, model II, question mark). These mechanisms can occur simultaneously. The intriguing interaction of another ER-located OSBP-related protein, i.e. ORP8, with nucleoporin Nup62 might be more than a coincidence in such context (46). ORP3 together with VAP-A might initiate the contacts of late endosomes to ONM and subsequently modulate their interaction with nuclear pore components. The latter issue will deserve further investigation.
Although the nuclear transfer of soluble, cytoplasmic cargo carried by EVs can be achieved through nuclear pores after EV–late endosome fusion (Fig. 8B, model II), it remains to be defined how EV-derived membrane proteins (or complex of them (15)) can be extracted from the endosomal membrane and translocated to nucleoplasm through the nuclear pore complexes, which are size-restricted (Fig. 8B, model III, question mark). The role of membrane microdomains (or lipid rafts) might be envisioned in this context, and the distinct punctate CD9–GFP signals suggest it (Fig. 7B). Tetraspanin CD9 is known to be associated with such microdomains (15). In addition to proteins, we report here that NEI-associated late endosomes shuttle nucleic acids to the nucleoplasm. EVs are rich sources of various types of nucleic acids (47), and their intercellular transfer can (dys)regulate the cellular microenvironment of producing cells (48). Such phenomenon can occur during development and normal physiological conditions, but also in diseases, leading to their progression. It will be of interest to decipher the targets of EV-derived nucleic acids in the nucleoplasm of host cells. The deep penetration of NEI in the nucleus (see also Fig. 1F) (15, 16), which often resulted in their close proximity to nucleoli, indirectly suggests that EV-derived components could play a role in biological events linked to it such as ribosomal biogenesis and/or protein sequestration (49). Long noncoding RNAs carried by EVs might be relevant with the latter post-translational regulation and will deserve particular attention.
Altogether, we have identified the key proteins that regulate the association of late endosomes with the NEI, allowing the nuclear transfer of components derived from endocytosed EVs. Knowing that mechanisms of intercellular communication are often hijacked in cancer to promote invasion and metastasis, the proteins identified here might represent new targets for therapeutic intervention. Finally, our findings warrant a re-evaluation of the intracellular trafficking of certain enveloped viruses given that they can use a similar strategy to deliver their contents into the host nuclei.
Experimental procedures
Chemicals
Nocodazole was purchased from Sigma. Stock solution was prepared at 5 mg/ml in DMSO and was used at 1 μm as working concentration.
Antibodies
The primary antibodies (Abs) used for immunodetection are listed in Table S1. The secondary Abs utilized were TRITC-conjugated anti-mouse IgG (715-025-150, Jackson ImmunoResearch, West Grove, PA) or anti-rabbit IgG (711-025-152), Cy5-conjugated anti-rabbit IgG (711-175-152) or anti-mouse IgG (715-025-150), and FITC-conjugated anti-mouse IgG (GTX26669, GeneTex, Irvine, CA) or anti-rabbit IgG (GTX27050). For immunoblotting, IRDye 680RD anti-mouse IgG (926-68070) and anti-rabbit IgG (926-68071) were purchased from LI-COR Biosciences (Lincoln, NE).
Cell culture
The FEMX-I cell line was originally derived from a lymph node metastasis of a patient with malignant melanoma (50). FEMX-I cells were highly metastatic in immunodeficient mice (50, 51) and were found to be WT for BRAF, PTEN, and NRAS (15). The HeLa cells (ATCC®CCL-2TM) were obtained from the American Type Culture Collection. FEMX-I and HeLa cells were cultured in RPMI 1640 medium (Corning Inc.) or Dulbecco's modified Eagle's medium (ThermoFisher Scientific, Waltham, MA), respectively, containing 10% fetal bovine serum (Atlanta Biologicals, Inc., Flowery Branch, GA), 2 mm l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin (Corning).
DNA plasmids and shRNAs
Vectors pCMV6-AC-GFP and pCMV3-C-GFPSpark were employed to generate C-terminal GFP-tagged fusion protein of CD9 (RG202000; OriGene, Rockville, MD) and VAP-A (HG11412-ACG; Sino Biological, Beijing, China), respectively. For knockdown expression, shRNA clones targeting VAP-A (HSH022333-nH1; accession no. NM_003574.5), VAP-B (HSH022331-nH1; accession no. NM_004738.3), and ORP3 (HSH006982-nH1; accession no. NM_015550.2) were purchased from GeneCopoeia (Rockville, MD). A scrambled shRNA (CSHCTR001-nH1; GeneCopoeia) was used as control. Four shRNA plasmids each with different target sequences were evaluated individually by both immunoblotting and CLSM, and those with the highest knockdown percentage were pooled together. The use of individual ones gave the same outcomes ruling out the possibility of off-target effects (data not shown). However, the number of cells showing >90% reduction of antigen immunoreactivity, as observed by CLSM, was more restricted limiting the number of cells of interest. Thus, two shRNAs with the VAP-A target sequences 5′-CCACACAGTGTTTCACTTAAT-3′ and 5′-GCACATTGAGTCCTTTATGAA-3′ were utilized. Likewise for VAP-B, 5′-GGATGACACCGAAGTTAAGAA-3′ and 5′-GGTAAATTGGATTGGTGGATC-3′ and four shRNAs with the ORP3 target sequences 5′-CCATGTTTCCACATGAAGTTA-3′, 5′-CCTCCAATCCTAATTTGTCAA-3′, 5′-GCCCATAAAGTTTACTTCACT-3′, and 5′-GGAGAAACATATGAATGTATT-3′ were selected for the entire study.
Transfection
FEMX-I cells were transfected with 10 μg of CD9–GFP plasmid using FuGENE® HD transfection reagents (Promega, Madison, WI) or VAP-A–GFP plasmid with Lipofectamine 3000 compounds (ThermoFisher Scientific). Cells were selected by introducing G418 (400 μg/ml, ThermoFisher Scientific) or hygromycin (200 μg/ml, Sigma), respectively, in the culture medium for 7 days resulting in >99% GFP-positive cells. To inhibit VAP-A, VAP-B, or ORP3 expression, FEMX-I cells were transfected with 500 ng of pooled shRNA plasmids containing the puromycin resistance gene using Lipofectamine 3000 reagents. After transfection, stable cell lines were selected by introducing 1 μg/ml puromycin (Sigma) in the culture medium. The same procedure was used on HeLa cells to inhibit VAP-A expression. All antibiotics were removed from the medium at least 1 week before experiments.
Baculoviral infection
To induce the expression of specific reporter genes that highlight organelles of interest, the baculovirus-based CellLight BacMam 2.0 Golgi–RFP, early endosomes–RFP, late endosomes–RFP, and ER–GFP or -RFP (ThermoFisher Scientific) encoding for the Golgi-resident enzyme N-acetylgalactosaminyltransferase 2 (GALNT2), Rab5a, Rab7, and ER signal sequence of calreticulin with KDEL sequence (ER retention signal)–GFP/RFP, respectively, were used. Viral particles were added at a concentration of 30 per cell for 24–48 h before immunocytochemistry.
Labeling of EVs and incubation with cells
EVs were prepared from parental or CD9–GFP-transfected FEMX-I cells cultured in serum-free medium supplemented with 2% B-27 supplement (ThermoFisher Scientific) on 6-well plates pre-coated with 20 μg/ml poly(2-hydroxyethyl methacrylate) (Sigma) to prevent their attachment as described (15). After 72 h of incubation, EVs were enriched by differential centrifugation as described (52). Briefly, after low-speed centrifugations (300 and 1200 × g), conditioned medium was centrifuged at 10,000 × g for 30 min at 4 °C, and the resulting supernatant was centrifuged at 200,000 × g for 60 min at 4 °C. The pellet was resuspended in 200 μl of PBS. Concentration of particles was determined by nanoparticle tracking analysis using the NanoSight LM10 unit (Malvern Instruments Ltd., Malvern, UK). Enriched EVs from parental cells were stained with 10 μm SYTO 64 red fluorescent nucleic acid stain (catalog no. S11346, ThermoFisher Scientific) or 5 μm of the membrane dye DiI (ThermoFisher Scientific) for 20 min at 37 °C. To remove excess dye, the EVs were transferred into a spin column (Exosome Spin Columns, Mr 3000; catalog no. 4484449, ThermoFisher Scientific) and centrifuged at 750 × g for 2 min (see Fig. 7H). The process was repeated twice. Stock solution was prepared at 2 × 1010 particles/ml. SYTO 64/DiI-labeled EVs or CD9–GFP+ EVs were then added to recipient cells (1 × 105) in a volume of 1 ml of culture medium at a final concentration of 109 particles/ml (1.5 μg of protein/ml) and incubated for 5 h at 37 °C as described previously (15).
Isolation of nuclei
Nuclei from cells incubated with CD9–GFP+ EVs were isolated using NE-PER nuclear extraction reagents in the presence of 1× Halt protease inhibitor mixture (both from ThermoFisher Scientific) to prevent protein degradation. Briefly, cells were trypsinized, washed with PBS, resuspended in ice-cold CER I solution, and incubated for 10 min on ice. CER II was added for 1 min on ice, and the samples were then centrifuged at 16,000 × g for 5 min. The supernatant was discarded, and the resulting pellet containing nuclei was resuspended in PBS and processed for SUN2 immunocytochemistry as described below. After immunolabeling, nuclei were allowed to settle on glass-bottom dishes and imaged by CLSM.
Immunocytochemistry and CLSM
Cells grown on 35-mm glass-bottom dishes coated with poly-d-lysine (MatTek Corp., Ashland, MA) were processed for immunocytochemistry after baculovirus infection, nocodazole treatment, and/or incubation with EVs. Cells (or isolated nuclei, see above) were washed with PBS, fixed in 4% paraformaldehyde (PFA) for 15 min at room temperature, or pre-chilled 100% methanol for 10 min at −20 °C for the nocodazole experiments, washed twice with PBS, permeabilized with 0.2% Tween 20 in PBS for 15 min at room temperature, and blocked with 1% bovine serum albumin (BSA) for 1 h at room temperature. They were then incubated with primary antibodies listed in Table S1 for 60 min at room temperature, washed twice with PBS, incubated with appropriate fluorescent secondary antibodies for 30 min, and washed twice prior to observation. All antibodies were diluted in permeabilization buffer containing 1% BSA prior to use. As a negative control, only secondary Ab was employed (data not shown). Cells or isolated nuclei were imaged in PBS by CLSM using a Nikon A1R+ inverted confocal microscope with a ×60 Apo-TIRF oil-immersion objective and a numerical aperture of 1.49 at either 512 × 512 or 1024 × 1024 pixel resolution. 405, 488, 561, and 638 nm solid-state lasers were used to excite 4′,6-diamidino-2-phenylindole (DAPI), GFP/FITC, TRITC/RFP/SYTO 64, and Cy5/Alexa Fluor 647, respectively, and corresponding fluorescence emissions were collected using 425–475-, 500–550-, 570–620-, and 662–737-nm long pass filters.
All images were acquired under the same microscope settings for subsequent calculations of mean fluorescence and recorded using NIS Elements software (Nikon). Raw images were processed using Fiji (53). Three-dimensional reconstruction was performed using 1–3 x-y optical sections (0.4 μm each) using the NIS Elements program.
Fluorescence quantification
Fluorescence signal was quantified using Fiji software (53). Data were processed and plotted with Excel (Microsoft). All bar graphs represent mean values of three independent experiments ± S.D. To count nuclear EV-derived fluorescent materials, ROIs were drawn along the nucleus, and an auto threshold generated by Fiji was applied. Positive signals were counted using the “analyze particle” function and presented as dot plots, generated by GraphPad Prism 5. In Fig. 7, C and I, each dot represents the value from a single cell with the black bars as the means ± S.D. To measure the mean fluorescence intensity, each cell was outlined by drawing an ROI around it, and the “measure” function on Fiji was used to calculate the fluorescence value, area, and integrated density. A background measurement of an area without cells was also taken. For each cell, the total cell fluorescence (TCF) was then calculated as: TCF = ID − (A × F), where ID and A are the integrated density and area of a given cell, respectively, and F is the fluorescence value of the background.
Time-lapse video microscopy
FEMX-I cells expressing VAP-A–GFP or ER–GFP and Rab7–RFP were imaged live under 37 °C and 5% CO2 condition. Time-lapse video was acquired every 20 s for 5 min using the Nikon A1R+ confocal microscope. Alternatively, DiI-labeled EVs or CD9 (5 × 108 particles/ml) were incubated with FEMX-I cells expressing either VAP-A–GFP or ER–RFP for 5 h prior to imaging live. Images were acquired every 10 or 20 s for 10 min, and videos were made at a playing speed of three frames/s. A single x-y optical section (0.2 μm) was acquired at a given time.
Scanning electron microscopy
FEMX-I cell lines were grown on coverslips coated with 0.01% poly-l-lysine (Sigma, Darmstadt, Germany). After 2 days, they were fixed in 2% glutaraldehyde for 1 h at room temperature and then overnight at 4 °C. Following a 2-h post-fixation in 1% osmium tetroxide at 4 °C, they were subjected to dehydration in an acetone gradient (25–100%) and critical point-drying in a CO2 system (Critical Point Dryer, Leica Microsystems, EM CPD 300, Wetzlar, Germany). Samples were then sputter-coated with gold (sputter coating device SCD 050; BAL-TEC GmbH, Witten, Germany) and examined at a 5-kV accelerating voltage in field emission-scanning electron microscope (Jeol JSM 7500F, Japan).
Immunoelectron microscopy
FEMX-I cell lines grown in Petri dishes were fixed for 1 h in PFA (4%) in 0.1 m phosphate buffer (PB), scraped, and processed for Tokuyashu cryosectioning and immunolabeling as described (54). Cell suspension was washed with PB, infiltrated stepwise with 10% gelatin at 37 °C, centrifuged, and then cooled on ice. Gelatin block was cut to cubes (<1 mm3), immersed in 2.3 m sucrose, and incubated for 24 h at 4 °C. Samples were mounted on specimen holders (catalog no. 16701950, Leica Microsystems), frozen in liquid nitrogen, and then cut in 70-nm ultrathin sections using an ultracryomicrotome (Leica Microsystems UC6+FC6, Wetzlar, Germany). Sections were picked-up with a mixture of 2% methylcellulose, 2.3 m sucrose (1:1) and placed on the Formvar-coated copper grids. For immunogold labeling, grids were washed in PBS at 37 °C and then further washed in PBS containing 0.1% glycine, blocked in PBS containing 1% BSA, and incubated for 1 h with anti-GFP or Rab-7 antibody (Table S1) diluted in PBS/BSA. After washing in PBS, sections were incubated for 30 min with protein A-conjugated 10-nm gold particles (Cell Microscopy Core, Utrecht, Netherlands). After washing in PBS, sections were post-fixed in 1% glutaraldehyde for 5 min and washed in PBS. Sections were contrasted with neutral uranyl oxalate solution (2% uranyl acetate in 0.15 m oxalic acid, pH 7.0) for 5 min, washed in water, and incubated for 5 min in methylcellulose containing 0.4% uranyl acetate and air-dried. Samples were examined by FEI Morgagni 268 transmission electron microscope (FEI, Eindhoven, The Netherlands) equipped with MegaView III digital camera (Olympus, Soft Imaging Systems, Münster, Germany) or with a JEM 1400Plus equipped with a Ruby digital camera (Jeol, Garching, Germany), both instruments running at 80 kV.
Immunoisolation of VAP and ORP3 proteins
Parental or FEMX-I cells expressing VAP-A–GFP were lysed in pre-chilled lysis buffer (0.5% Triton X-100, 150 mm NaCl, 50 mm Tris-HCl, pH 8.0, supplemented with Set III protease inhibitor mixture (Calbiochem)) on ice for 30 min, followed by centrifugation for 10 min at 4 °C at 12,000 × g. Immunoisolations were performed on cell lysates using anti-GFP antibody coupled to magnetic beads from μMACS epitope tag protein isolation kit (Miltenyi Biotec, Bergisch Gladbach, Germany) or antibodies directed against ORP3 or VAP-B followed by protein G-conjugated magnetic beads according to the manufacturer's protocols. Labeled samples were applied to μColumn for magnetic separation (Miltenyi Biotec). Materials retained in columns were washed (four times) with 1 ml of ice-cold lysis buffer and rinsed once with 20 mm Tris-HCl, pH 7.5. Pre-heated (95 °C) SDS buffer (1% SDS, 50 mm DTT, 1 mm EDTA, 10% glycerol, 0.005% bromphenol blue, 50 mm Tris-HCl, pH 6.8) was applied onto the column to elute the bound fractions. The protein samples were then separated onto a 4–12% BisTris precast gel (ThermoFisher Scientific) and analyzed by immunoblotting for various antigens.
Immunoblotting
Cells were trypsinized and centrifuged at 200 × g for 5 min. The resulting pellet was resuspended in ice-cold lysis buffer (1% Triton X-100, 100 mm NaCl, 50 mm Tris-HCl, pH 7.5, supplemented with the Set III protease inhibitor mixture) and incubated on ice for 30 min. Detergent lysates were centrifuged at 12,000 × g for 20 min. Supernatant was collected, and Laemmli buffer was added. Protein samples were separated by a 4–12% BisTris precast gel along with the Trident pre-stained protein molecular weight ladder (GeneTex) and transferred to a nitrocellulose membrane (ThermoFisher Scientific) overnight at 4 °C. Membranes were incubated in a blocking buffer (PBS containing 1% BSA) for 60 min at room temperature, and then probed with a given primary Ab (Table S1) for 90 min at room temperature. After three washing steps for 10 min each with PBS containing 0.1% Tween 20 (washing buffer), membranes were incubated with an appropriate IRdye secondary Ab(LI-COR) for 30 min at room temperature. Membranes were rinsed three times with washing buffer and once with double-distilled H2O, and antigen–Ab complexes were visualized using an Odyssey CLx system (LI-COR).
Acceptor photobleaching FRET
FRET was performed using the acceptor photobleaching method (36, 55). FEMX-I cells expressing VAP-A–GFP and/or Rab7–RFP fusion proteins were fixed with 4% PFA for 15 min and permeabilized with 0.2% Tween 20 in PBS for 10 min. When appropriate, cells were immunolabeled for ORP3 followed by secondary antibody conjugated to either TRITC, when paired with VAP-A–GFP cells, or FITC, when paired with Rab7–RFP cells. Cells expressing Rab7–RFP were also immunolabeled for VAP-B followed by FITC-conjugated secondary antibody. The acceptor (red channel) was bleached for 1 s within a specific ROI using the 561-nm laser at 100% power. The donor and acceptor channels were collected using 500–550- and 570–620-nm long-pass filters, respectively. FRET efficiency was calculated using the Nikon NIS Elements analysis software as FRETeff = (Ipost − Ipre)/Ipost × 100, where Ipre and Ipost are the total fluorescence of the ROI before and after bleaching. Areas outside the labeling were chosen to subtract the background noise (data not shown).
Statistical analysis
All statistics were performed on at least three independent experiments utilizing the GraphPad Prism 5 software. A minimum of 30 cells was analyzed in each experiment. Unless indicated, statistical analysis was determined using a two-tailed Student's t test. p values inferior to 0.05 were considered statistically significant.
Author contributions
M. F. S., G. R., D. C., and A. L. conceptualization; M. F. S., D. C., and A. L. data curation; M. S., G. R., J. K., T. K., D. C., and A. L. investigation; M. F. S., G. R., J. K., D. C., and A. L. writing-original draft; M. F. S., G. R., J. K., T. K., D. C. and A. L. methodology; D. C. and A. L. supervision; D. C. and A. L. project administration; D. C. and A. L. writing-review and editing.
Supplementary Material
Acknowledgments
We thank Fabio Anzanello for skilled technical assistance; Kyle Choi and Matthew Stein for assistance with CLSM; Susanne Kretschmar from the Electron Microscopy/Histology (EMH) Facility at the BIOTEC (Dresden, Germany) for support with immunogold EM; Oystein Fodstad for FEMX-I cells; Eivind Hovig for sharing unpublished data; and Yuri Lazebnik for critical review of the manuscript. The European Regional Development Fund supports the EMH Facility.
United Kingdom patent application GB1810556.9 and United States provisional patent number 62/690,616 are pending.
This article contains Figs. S1–S8, Table S1, and Videos S1–S5.
M. F. Santos and A. Lorico, unpublished data.
- EV
- extracellular vesicle
- NEI
- nuclear envelope invagination
- PFA
- paraformaldehyde
- OSBP
- oxysterol-binding protein
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- CLSM
- confocal laser-scanning microscopy
- RFP
- red fluorescent protein
- RFI
- relative fluorescence intensity
- ER
- endoplasmic reticulum
- FFAT
- diphenylalanine in an acidic tract
- ONM
- outer nuclear membrane
- INM
- inner nuclear membrane
- shRNA
- small hairpin RNA
- TRITC
- tetramethylrhodamine
- ROI
- region of interest
- DiI
- 1,1-dioctadecyl-3,3,3,3-tetramethylindocarbocyanine perchlorate
- Ab
- antibody.
References
- 1. Tkach M., and Théry C. (2016) Communication by extracellular vesicles: where we are and where we need to go. Cell 164, 1226–1232 10.1016/j.cell.2016.01.043 [DOI] [PubMed] [Google Scholar]
- 2. Raposo G., and Stoorvogel W. (2013) Extracellular vesicles: exosomes, microvesicles, and friends. J. Cell Biol. 200, 373–383 10.1083/jcb.201211138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kowal J., Arras G., Colombo M., Jouve M., Morath J. P., Primdal-Bengtson B., Dingli F., Loew D., Tkach M., and Théry C. (2016) Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. U.S.A. 113, E968–E977 10.1073/pnas.1521230113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kowal J., Tkach M., and Théry C. (2014) Biogenesis and secretion of exosomes. Curr. Opin. Cell Biol. 29, 116–125 10.1016/j.ceb.2014.05.004 [DOI] [PubMed] [Google Scholar]
- 5. Nunukova A., Neradil J., Skoda J., Jaros J., Hampl A., Sterba J., and Veselska R. (2015) Atypical nuclear localization of CD133 plasma membrane glycoprotein in rhabdomyosarcoma cell lines. Int. J. Mol. Med. 36, 65–72 10.3892/ijmm.2015.2210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Waldenström A., Gennebäck N., Hellman U., and Ronquist G. (2012) Cardiomyocyte microvesicles contain DNA/RNA and convey biological messages to target cells. PLoS ONE 7, e34653 10.1371/journal.pone.0034653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dovrat S., Caspi M., Zilberberg A., Lahav L., Firsow A., Gur H., and Rosin-Arbesfeld R. (2014) 14-3-3 and β-catenin are secreted on extracellular vesicles to activate the oncogenic Wnt pathway. Mol. Oncol. 8, 894–911 10.1016/j.molonc.2014.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cai J., Han Y., Ren H., Chen C., He D., Zhou L., Eisner G. M., Asico L. D., Jose P. A., and Zeng C. (2013) Extracellular vesicle-mediated transfer of donor genomic DNA to recipient cells is a novel mechanism for genetic influence between cells. J. Mol. Cell. Biol. 5, 227–238 10.1093/jmcb/mjt011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kanada M., Bachmann M. H., Hardy J. W., Frimannson D. O., Bronsart L., Wang A., Sylvester M. D., Schmidt T. L., Kaspar R. L., Butte M. J., Matin A. C., and Contag C. H. (2015) Differential fates of biomolecules delivered to target cells via extracellular vesicles. Proc. Natl. Acad. Sci. U.S.A. 112, E1433–E1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Desrochers L. M., Antonyak M. A., and Cerione R. A. (2016) Extracellular vesicles: satellites of information transfer in cancer and stem cell biology. Dev. Cell 37, 301–309 10.1016/j.devcel.2016.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Soung Y. H., Ford S., Zhang V., and Chung J. (2017) Exosomes in cancer diagnostics. Cancers 9, 8 10.3390/cancers9010008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fais S., O'Driscoll L., Borras F. E., Buzas E., Camussi G., Cappello F., Carvalho J., Cordeiro da Silva A., Del Portillo H., El Andaloussi S., Ficko Trček T., Furlan R., Hendrix A., Gursel I., Kralj-Iglic V., et al. (2016) Evidence-based clinical use of nanoscale extracellular vesicles in nanomedicine. ACS Nano 10, 3886–3899 10.1021/acsnano.5b08015 [DOI] [PubMed] [Google Scholar]
- 13. Sarko D. K., and McKinney C. E. (2017) Exosomes: origins and therapeutic potential for neurodegenerative disease. Front. Neurosci. 11, 82 10.3389/fnins.2017.00082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ying W., Riopel M., Bandyopadhyay G., Dong Y., Birmingham A., Seo J. B., Ofrecio J. M., Wollam J., Hernandez-Carretero A., Fu W., Li P., and Olefsky J. M. (2017) Adipose tissue macrophage-derived exosomalmiRNAs can modulate in vivo and in vitro insulin sensitivity. Cell 171, 372–384.e12 10.1016/j.cell.2017.08.035 [DOI] [PubMed] [Google Scholar]
- 15. Rappa G., Santos M. F., Green T. M., Karbanová J., Hassler J., Bai Y., Barsky S. H., Corbeil D., and Lorico A. (2017) Nuclear transport of cancer extracellular vesicle-derived biomaterials through nuclear envelope invagination-associated late endosomes. Oncotarget 8, 14443–14461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Malhas A., Goulbourne C., and Vaux D. J. (2011) The nucleoplasmic reticulum: form and function. Trends Cell Biol. 21, 362–373 10.1016/j.tcb.2011.03.008 [DOI] [PubMed] [Google Scholar]
- 17. Jorgens D. M., Inman J. L., Wojcik M., Robertson C., Palsdottir H., Tsai W. T., Huang H., Bruni-Cardoso A., López C. S., Bissell M. J., Xu K., and Auer M. (2017) Deep nuclear invaginations are linked to cytoskeletal filaments- integrated bioimaging of epithelial cells in 3D culture. J. Cell Sci. 130, 177–189 10.1242/jcs.190967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lux K., Goerlitz N., Schlemminger S., Perabo L., Goldnau D., Endell J., Leike K., Kofler D. M., Finke S., Hallek M., and Büning H. (2005) Green fluorescent protein-tagged adeno-associated virus particles allow the study of cytosolic and nuclear trafficking. J. Virol. 79, 11776–11787 10.1128/JVI.79.18.11776-11787.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gehrig K., Cornell R. B., and Ridgway N. D. (2008) Expansion of the nucleoplasmic reticulum requires the coordinated activity of lamins and CTP:phosphocholine cytidylyltransferase α. Mol. Biol. Cell 19, 237–247 10.1091/mbc.e07-02-0179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Drozdz M. M., and Vaux D. J. (2017) Shared mechanisms in physiological and pathological nucleoplasmic reticulum formation. Nucleus 8, 34–45 10.1080/19491034.2016.1252893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Raiborg C., Wenzel E. M., and Stenmark H. (2015) ER-endosome contact sites: molecular compositions and functions. EMBO J. 34, 1848–1858 10.15252/embj.201591481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Phillips M. J., and Voeltz G. K. (2016) Structure and function of ER membrane contact sites with other organelles. Nat. Rev. Mol. Cell Biol. 17, 69–82 10.1038/nrm.2015.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Johansson M., Rocha N., Zwart W., Jordens I., Janssen L., Kuijl C., Olkkonen V. M., and Neefjes J. (2007) Activation of endosomal dynein motors by stepwise assembly of Rab7-RILP-p150Glued, ORP1L, and the receptor βlll spectrin. J. Cell Biol. 176, 459–471 10.1083/jcb.200606077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rowland A. A., Chitwood P. J., Phillips M. J., and Voeltz G. K. (2014) ER contact sites define the position and timing of endosome fission. Cell 159, 1027–1041 10.1016/j.cell.2014.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Raiborg C., Wenzel E. M., Pedersen N. M., Olsvik H., Schink K. O., Schultz S. W., Vietri M., Nisi V., Bucci C., Brech A., Johansen T., and Stenmark H. (2015) Repeated ER-endosome contacts promote endosome translocation and neurite outgrowth. Nature 520, 234–238 10.1038/nature14359 [DOI] [PubMed] [Google Scholar]
- 26. Dong R., Saheki Y., Swarup S., Lucast L., Harper J. W., and De Camilli P. (2016) Endosome-ER contacts control actin nucleation and retromer function through VAP-dependent regulation of PI4P. Cell 166, 408–423 10.1016/j.cell.2016.06.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lev S., Ben Halevy D., Peretti D., and Dahan N. (2008) The VAP protein family: from cellular functions to motor neuron disease. Trends Cell Biol. 18, 282–290 10.1016/j.tcb.2008.03.006 [DOI] [PubMed] [Google Scholar]
- 28. Rocha N., Kuijl C., van der Kant R., Janssen L., Houben D., Janssen H., Zwart W., and Neefjes J. (2009) Cholesterol sensor ORP1L contacts the ER protein VAP to control Rab7-RILP-p150 glued and late endosome positioning. J. Cell Biol. 185, 1209–1225 10.1083/jcb.200811005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zerial M., and McBride H. (2001) Rab proteins as membrane organizers. Nat. Rev. Mol. Cell Biol. 2, 107–117 10.1038/35052055 [DOI] [PubMed] [Google Scholar]
- 30. Suchanek M., Hynynen R., Wohlfahrt G., Lehto M., Johansson M., Saarinen H., Radzikowska A., Thiele C., and Olkkonen V. M. (2007) The mammalian oxysterol-binding protein-related proteins (ORPs) bind 25-hydroxycholesterol in an evolutionarily conserved pocket. Biochem. J. 405, 473–480 10.1042/BJ20070176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wyles J. P., McMaster C. R., and Ridgway N. D. (2002) Vesicle-associated membrane protein-associated protein-A (VAP-A) interacts with the oxysterol-binding protein to modify export from the endoplasmic reticulum. J. Biol. Chem. 277, 29908–29918 10.1074/jbc.M201191200 [DOI] [PubMed] [Google Scholar]
- 32. Alpy F., Rousseau A., Schwab Y., Legueux F., Stoll I., Wendling C., Spiegelhalter C., Kessler P., Mathelin C., Rio M. C., Levine T. P., and Tomasetto C. (2013) STARD3 or STARD3NL and VAP form a novel molecular tether between late endosomes and the ER. J. Cell Sci. 126, 5500–5512 10.1242/jcs.139295 [DOI] [PubMed] [Google Scholar]
- 33. Mesmin B., Bigay J., Moser von Filseck J., Lacas-Gervais S., Drin G., and Antonny B. (2013) A four-step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER-Golgi tether OSBP. Cell 155, 830–843 10.1016/j.cell.2013.09.056 [DOI] [PubMed] [Google Scholar]
- 34. Weber-Boyvat M., Kentala H., Lilja J., Vihervaara T., Hanninen R., Zhou Y., Peränen J., Nyman T. A., Ivaska J., and Olkkonen V. M. (2015) OSBP-related protein 3 (ORP3) coupling with VAMP-associated protein A regulates R-Ras activity. Exp. Cell Res. 331, 278–291 10.1016/j.yexcr.2014.10.019 [DOI] [PubMed] [Google Scholar]
- 35. Stoica R., De Vos K. J., Paillusson S., Mueller S., Sancho R. M., Lau K. F., Vizcay-Barrena G., Lin W. L., Xu Y. F., Lewis J., Dickson D. W., Petrucelli L., Mitchell J. C., Shaw C. E., and Miller C. C. (2014) ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 5, 3996 10.1038/ncomms4996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ivanusic D., Eschricht M., and Denner J. (2014) Investigation of membrane protein–protein interactions using correlative FRET-PLA. Biotechniques 57, 188–191, 193–198 [DOI] [PubMed] [Google Scholar]
- 37. Eisenberg-Bord M., Shai N., Schuldiner M., and Bohnert M. (2016) A tether is a tether is a tether: tethering at membrane contact sites. Dev. Cell 39, 395–409 10.1016/j.devcel.2016.10.022 [DOI] [PubMed] [Google Scholar]
- 38. Murphy S. E., and Levine T. P. (2016) VAP, a versatile access point for the endoplasmic reticulum: review and analysis of FFAT-like motifs in the VAPome. Biochim. Biophys. Acta 1861, 952–961 10.1016/j.bbalip.2016.02.009 [DOI] [PubMed] [Google Scholar]
- 39. Johansson M., Lehto M., Tanhuanpää K., Cover T. L., and Olkkonen V. M. (2005) The oxysterol-binding protein homolog ORP1L interacts with Rab7 and alters functional properties of late endocytic compartments. Mol. Biol. Cell 16, 5480–5492 10.1091/mbc.e05-03-0189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lehto M., Mäyränpää M. I., Pellinen T., Ihalmo P., Lehtonen S., Kovanen P. T., Groop P. H., Ivaska J., and Olkkonen V. M. (2008) The R-Ras interaction partner ORP3 regulates cell adhesion. J. Cell Sci. 121, 695–705 10.1242/jcs.016964 [DOI] [PubMed] [Google Scholar]
- 41. Cantalupo G., Alifano P., Roberti V., Bruni C. B., and Bucci C. (2001) Rab-interacting lysosomal protein (RILP): the Rab7 effector required for transport to lysosomes. EMBO J. 20, 683–693 10.1093/emboj/20.4.683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Saita S., Shirane M., Natume T., Iemura S., and Nakayama K. I. (2009) Promotion of neurite extension by protrudin requires its interaction with vesicle-associated membrane protein-associated protein. J. Biol. Chem. 284, 13766–13777 10.1074/jbc.M807938200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lehto M., Hynynen R., Karjalainen K., Kuismanen E., Hyvärinen K., and Olkkonen V. M. (2005) Targeting of OSBP-related protein 3 (ORP3) to endoplasmic reticulum and plasma membrane is controlled by multiple determinants. Exp. Cell Res. 310, 445–462 10.1016/j.yexcr.2005.08.003 [DOI] [PubMed] [Google Scholar]
- 44. van der Kant R., Zondervan I., Janssen L., and Neefjes J. (2013) Cholesterol-binding molecules MLN64 and ORP1L mark distinct late endosomes with transporters ABCA3 and NPC1. J. Lipid Res. 54, 2153–2165 10.1194/jlr.M037325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fischer A. H., Taysavang P., and Jhiang S. M. (2003) Nuclear envelope irregularity is induced by RET/PTC during interphase. Am. J. Pathol. 163, 1091–1100 10.1016/S0002-9440(10)63468-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhou T., Li S., Zhong W., Vihervaara T., Béaslas O., Perttilä J., Luo W., Jiang Y., Lehto M., Olkkonen V. M., and Yan D. (2011) OSBP-related protein 8 (ORP8) regulates plasma and liver tissue lipid levels and interacts with the nucleoporin Nup62. PLoS ONE 6, e21078 10.1371/journal.pone.0021078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Valadi H., Ekström K., Bossios A., Sjöstrand M., Lee J. J., and Lötvall J. O. (2007) Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 9, 654–659 10.1038/ncb1596 [DOI] [PubMed] [Google Scholar]
- 48. Maas S. L. N., Breakefield X. O., and Weaver A. M. (2017) Extracellular vesicles: unique intercellular delivery vehicles. Trends Cell Biol. 27, 172–188 10.1016/j.tcb.2016.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Audas T. E., Jacob M. D., and Lee S. (2012) Immobilization of proteins in the nucleolus by ribosomal intergenic spacer noncoding RNA. Mol. Cell 45, 147–157 10.1016/j.molcel.2011.12.012 [DOI] [PubMed] [Google Scholar]
- 50. Fodstad O., Kjønniksen I., Aamdal S., Nesland J. M., Boyd M. R., and Pihl A. (1988) Extrapulmonary, tissue-specific metastasis formation in nude mice injected with FEMX-I human melanoma cells. Cancer Res. 48, 4382–4388 [PubMed] [Google Scholar]
- 51. Rappa G., Fodstad O., and Lorico A. (2008) The stem cell-associated antigen CD133 (Prominin-1) is a molecular therapeutic target for metastatic melanoma. Stem Cells 26, 3008–3017 10.1634/stemcells.2008-0601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rappa G., Mercapide J., Anzanello F., Pope R. M., and Lorico A. (2013) Biochemical and biological characterization of exosomes containing prominin-1/CD133. Mol. Cancer 12, 62 10.1186/1476-4598-12-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J. Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., and Cardona A. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Slot J. W., and Geuze H. J. (2007) Cryosectioning and immunolabeling. Nat. Protoc. 2, 2480–2491 10.1038/nprot.2007.365 [DOI] [PubMed] [Google Scholar]
- 55. Karpova T. S., Baumann C. T., He L., Wu X., Grammer A., Lipsky P., Hager G. L., and McNally J. G. (2003) Fluorescence resonance energy transfer from cyan to yellow fluorescent protein detected by acceptor photobleaching using confocal microscopy and a single laser. J. Microsc. 209, 56–70 10.1046/j.1365-2818.2003.01100.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.