

Abstract
JIP1 was first identified as scaffold protein for the MAP kinase JNK and is a cargo protein for the kinesin1 molecular motor. JIP1 plays significant and broad roles in neurons, mainly as a regulator of kinesin1-dependent transport, and is associated with human pathologies such as cancer and Alzheimer disease. JIP1 is specifically recruited by the kinesin-light chain 1 (KLC1) of kinesin1, but the details of this interaction are not yet fully elucidated. Here, using calorimetry, we extensively biochemically characterized the interaction between KLC1 and JIP1. Using various truncated fragments of the tetratricopeptide repeat (TPR) domain of KLC1, we narrowed down its JIP1-binding region and identified seven KLC1 residues critical for JIP1 binding. These isothermal titration calorimetry (ITC)-based binding data enabled us to footprint the JIP1-binding site on KLC1-TPR. This footprint was used to uncover the structural basis for the marginal inhibition of JIP1 binding by the autoinhibitory LFP-acidic motif of KLC1, as well as for the competition between JIP1 and another cargo protein of kinesin1, the W-acidic motif–containing alcadein-α. Also, we examined the role of each of these critical residues of KLC1 for JIP1 binding in light of the previously reported crystal structure of the KLC1-TPR:JIP1 complex. Finally, sequence search in eukaryotic genomes identified several proteins, among which is SH2D6, that exhibit a motif similar to the KLC1-binding motif of JIP1. Overall, our extensive biochemical characterization of the KLC:JIP1 interaction, as well as identification of potential KLC1-binding partners, improves the understanding of how this growing family of cargos is recruited to kinesin1 by KLC1.
Keywords: isothermal titration calorimetry (ITC), protein-protein interaction, kinesin, protein engineering, site-directed mutagenesis, alcadein, MST, SH2D6, TPR domain, Y-acidic motif, JNK-interacting protein 1, kinesin light chain, TorsinA, autoinhibitory LFP-acidic motif, motor protein
Introduction
JIP13 and its close homolog JIP2 (JNK-interacting proteins 1 and 2, also known as islet brain (IB) 1 and 2, respectively) were first identified as scaffold proteins for JNK and p38 mitogen-activated protein kinases (1). In addition, they are adaptor proteins connecting the kinesin1 microtubule-based motor to cargo receptors such as p190 rhoGEF, ApoER2 (apolipoprotein E receptor 2), and APP (β-amyloid precursor protein) (2–6). In addition to its role as a kinesin1 adaptor, JIP1 regulates kinesin1-dependent transport: (i) it plays important roles in kinesin1 activation because its recruitment contributes to relieve motor domain inhibition for microtubule binding and motility (7, 8), and (ii) it acts as a coordinator for anterograde and retrograde transport by modulating association of vesicles between kinesin1 and another microtubule-based motor, the dynein:dynactin complex (8). Finally, the JIP1 binding to kinesin1 can favor or prevent the binding of other protein cargos, such as JIP3/4 (JNK-interacting proteins 3 and 4, which are structurally unrelated to JIP1/2) (9) or alcadein-α (ALCα, also known as calsyntenin) (10, 11), impacting their transport. Thus, JIP1/2, which are mainly expressed in pancreas and brain, play significant and broad roles that can be associated with human pathologies such as obesity, diabetes, cancer, or Alzheimer disease (12).
Kinesin1 is able to transport various cargos, like vesicles, organelles, or macromolecular assemblies, along microtubules (13). It functions as a heterotetramer composed of a homodimer of kinesin heavy chains (KHC) bound to two kinesin light chains (KLC). KHC consists of three parts: an N-terminal globular motor domain (head) that contains the ATP and microtubule binding sites, a central elongated coiled-coil (stalk) responsible for dimerization, and a C-terminal unstructured region (tail) that regulates motor motility and recruits cargos. KLC is composed of three parts: an N-terminal heptad repeat region that binds to the KHC stalk, a tetratricopeptide repeat (TPR) domain involved in cargo recruitment, and a variable C-terminal region. Within the flexible linker between the heptad repeat region and the TPR domain of KLC, there is a highly conserved leucine-phenylalanine-proline motif flanked by acidic residues (LFP-acidic motif) that can act like an autoinhibitory motif by folding back on the TPR domain to prevent cargo binding (14). The TPR domain of KLC consists of six TPR motifs (TPR1–6) with a non-TPR region of 40 residues inserted between the TPR5 and TPR6 motifs. Each motif repeat, involving two antiparallel α helices (A and B), stacks together in a parallel array to form an extended molecule with an overall right-handed superhelical architecture. The TPR domain adopts a cradle shape with helices A of each repeat lining the concave face (or groove) and helices B lining the convex face. In vertebrates, four KLC isoforms (KLC1–4) are identified, and crystal structures of two of these isoforms were determined. Thus, crystal structures of KLC1 and KLC2 showed that the TPR domain adopts a classical TPR fold consisting of 12 helices with the partially flexible non-TPR region extruding from the convex side at the C-terminal part of the TPR domain (15–17). Interestingly, the groove of KLC1 and KLC2 is composed of numerous asparagine and basic residues that allow binding of short and acidic segment sequences like those of the LFP-acidic autoinhibitory motif or protein cargos, such as ALCα, TorsinA, or JIP1.
Several experiments were reported concerning the JIP1 recruitment by KLC1. On one side, co-immunoprecipitation assays revealed that the extreme C terminus of JIP1 (JIP1-Cter, -TCPTEDIYLECOOH sequence) is required to bind to the TPR domain of KLC1, and the tyrosine residue (Tyr709) at position −3 from the end is critical for this interaction (6). Also, the last glutamic acidic residue (Glu711) of JIP1 was shown to be critical for KLC1 binding in vitro (15). On the other side, in vitro binding assays reported that JIP1 does not interact with KLC2 and is thus a specific cargo for KLC1 with the Asn343, located in the TPR groove of KLC1 responsible for this specificity (15). Despite there being no experimental information available concerning the specificity of KLC3 and KLC4 for JIP1, they share with KLC1 an asparagine residue at this determinant position, indicating that KLC3 and KLC4 probably recruit JIP1. Further, directed yeast two-hybrid investigations reveal that multiple mutations into the TPR groove of KLC1, especially on asparagine residues, abolished JIP1 binding (9). Altogether, these data reveal that the extreme C terminus of JIP1 binds into the groove of the TPR domain, but the details of this interaction are not yet elucidated.
Interestingly, JIP1 competes with ALCα for kinesin1 transport, revealing that kinesin1 cannot recruit at the same time JIP1 and ALCα. ALCα, as well as the lysosome adaptor SKIP (SifA-kinesin–interacting protein) belong to a kinesin1 cargo family that is recruited by KLC1/2 through a bipartite tryptophan-based binding motif flanked by acidic residues (W-acidic motif) (10, 11, 18–20). Such a competition can be due to an overlap of JIP1- and ALCα-binding sites into the TPR domain of KLC. Interestingly, KLC-binding motifs of JIP1 and W-acidic cargos exhibit similarities. In both, an aromatic residue is required to interact with KLC (6, 10, 11), and on each side of it, acidic residues produce negative charged stretches in these motifs. However, isothermal titration calorimetry (ITC) experiments reveal that the determinant Asn343 of KLC1, which is critical for JIP1 interaction, is not involved in ALCα binding (15). Further, Dodding and co-workers (14) showed that the autoinhibitory LFP-acidic motif of KLC affected binding of SKIP in vitro, whereas it only marginally reduced that of JIP1. Altogether, these observations suggest that despite the mode of binding of JIP1 and W-acidic cargos with KLC sharing similarities, the details of their interactions are distinct.
To characterize the JIP1-binding site on the TPR domain of KLC1, we performed isothermal titration calorimetry (ITC) binding assays between various truncated fragments as well as mutants of the TPR domain of KLC1 and a peptide covering the last 10 residues of JIP1 (C10). Sequence search in eukaryotic genomes identified the mouse SH2D6 protein, which exhibits a consensus motif that is very similar to the KLC1-binding motif of JIP1; microscale thermophoresis (MST) binding assays were also conducted to characterize this potential interaction. By comparing with available crystal structures of KLC1-TPR complexes, our biochemical data allow us to discuss the structural basis of the marginal inhibition of the LFP-acidic motif for JIP1 binding to KLC-TPR as well as the competition between JIP1 and W-acidic motif cargo binding. During the revision of this manuscript, the crystal structure of KLC1-TPR bound to JIP1 was released. Thus, in light of this new 3D structure, we discussed the role of each of the critical residues of KLC1-TPR for JIP1 binding identified here.
Results and discussion
Sequence analysis of the KLC1-binding motif of JIP1/2
The multiple-sequence alignment of full-length homologs of JIP1 and JIP2 shows that JIP1/2-like homologs can be found in several clades of metazoans, including insects and nematodes. In invertebrates, a single homolog of JIP1/2 was found, hereafter referred to as JIP1-like. The first duplication giving rise to the two JIP1/2 subfamilies can be positioned at the origin of the vertebrate clade because a single version of the protein is found in species closely related to vertebrates such as lancelets (Branchiostoma floridae) and tunicates (Ciona intestinalis). The extreme C terminus covering the KLC1-binding motif is very well conserved in evolution in all species. Fig. S1 shows a phylogenetic tree for JIP1 and JIP2 homologs as well as a multiple-sequence alignment of the extreme C terminus containing the KLC1-binding motif of JIP1/2 homologs. In vertebrates, the sequence of the extreme 10 last residues of JIP1/2 is highly conserved, and the consensus sequence is (T/A)CPTED(I/M)YLECOOH. Thus, the critical tyrosine is flanked by isoleucine (or methionine) and leucine residues, forming a hydrophobic triplet. The hydrophobic triplet is surrounded by three acidic residues, a glutamate and an aspartate residue before and a glutamate after. Also, because the JIP1-Cter motif is found at the extreme C terminus of JIP1, the C-terminal carboxylate represents a fourth negative charge. Finally, before the hydrophobic acidic sequence, three other conserved residues are found, cysteine, proline, and threonine.
To identify proteins exhibiting a similar motif as the KLC1-binding motif of JIP1/2, we searched the consensus motif (E/D)(E/D)(I/M/L/V)Y(I/M/L/V)(E/D) against the UniProtKB database using the ScanProsite (21) tool. We first filtered the 599 identified motifs (in 598 sequences) by eliminating noneukaryotic sequences and keeping a single isoform per protein, which left 193 motifs (in 192 sequences) for further analysis. Interestingly, none of the proteins identified exhibit the potential KLC1-binding motif at the extreme C terminus, except JIP1/2. We then filtered this list by removing all groups of proteins for which the potential KLC1-binding motif is found mainly in a region with predicted secondary structure. Indeed, the KLC1-binding motif in JIP1 is found in an unstructured region that is certainly a prerequisite to bind into the TPR domain groove. Finally, nonmammalian sequences were filtered out for final analysis. Thus, only 16 groups (among which were six groups with at least two proteins and 10 singletons) of proteins were identified in addition to the JIP1 and JIP2 homologs (Table S1), among which the group with the highest number of proteins (22 proteins) consists of the α-subunit of the sodium/potassium voltage-gated channel proteins, whereas the group that shares the highest conservation with the KLC1-binding motif of JIP1/2 (strictly identical on the positions that define the search consensus motif) consists of a single protein: the mouse SH2 domain-containing protein 6 (SH2D6). Also, referred to as SLNK, SH2D6 is an SH2 domain–containing adaptor molecule identified as an ortholog of SLP76 (also known as LCP2; see the hOPMAP web server (22)), which is essential for signaling downstream of integrin and receptors containing immunoreceptor tyrosine-based activation motifs (23). The potential KLC1-binding motif of mouse SH2D6 (accession number Q9D413) is located in the unstructured N-terminal half part surrounded by proline residues, allowing its potential interaction into the TPR domain groove of KLC1. A multiple-sequence alignment of SH2D6 homologs for the region containing the potential KLC1-binding motif is shown in Fig. S2. In vertebrates, the consensus sequence of the motif in SH2D6 is (E/N)(D/N/G/S)(L/I/V/T)Y(L/V)(E/Q), which is well conserved, although less than in JIP1/2. Beyond the consensus motif, differences are, however, observed between JIP1 and SH2D6. In SH2D6, three acidic residues are found before the hydrophobic triplet in place of two in JIP1/2. In addition, a conserved two-basic residue (KK) patch is found at the N terminus of the consensus motif, whereas no basic residue is identified in JIP1/2 (Fig. S1). And finally, because the consensus motif in SH2D6 is located in the internal part of the protein, there is no C-terminal carboxylate at the end of the motif. Altogether, this search highlights that a consensus motif similar to the KLC1-binding motif of JIP1/2 can be found in the unstructured internal parts of other proteins. However, biochemical analysis is required to validate these potential candidates as KLC1-binding partners.
The KLC1-binding motif of JIP1 is highly specific
Using ITC, we characterized the interaction between the complete TPR domain of KLC1 (ranging from the first helix (A1) of the TPR1 motif to the last helix (B6) of the TPR6 motif; called hereafter fragment [A1-B6]; Table S2) and peptides covering the last 10 residues of human JIP1 (called hereafter C10; sequence Ac-TCPTEDIYLECOOH; Table S3). ITC data gave a dissociation constant (Kd) of 5.0 ± 0.5 μm for this reference measurement (Table 1 and Fig. 1A) that is in accordance with previously reported values (14, 15). The stoichiometry determined is N = 0.85 ± 0.01, close to 1, corresponding to one JIP1-C10 peptide bound to one KLC1-TPR molecule. This reference measurement was performed independently several times (n = 7), which shows that the dissociation constant ranges from 2.5 ± 0.4 to 8.3 ± 0.6 μm (mean ± S.D. of 5.8 ± 0.7 μm; Table S4), revealing an experimental 2-fold factor variation in Kd. Thermodynamic parameters exhibit negative enthalpy change and negative entropy contribution, supporting favorable hydrogen bonds and hydrophobic interactions. Further, measurements performed at variable ionic strengths showed the strong impact of salt concentration on the binding affinity (from 5 to 106 μm, respectively, for NaCl concentration ranging from 150 to 500 mm; Fig. 1B), revealing that electrostatics also play an important role in the interaction process. Altogether, these observations are in accordance with the presence of hydrophobic and acidic residues in the KLC1-binding motif of JIP1.
Table 1.
Thermodynamic parameters of the ITC experiments between various KLC1-TPR fragments and mutants and JIP1-C10 peptides
The factor fold is calculated considering KLC1-TPR-[A1-B6] fragment as the reference experiment (factor fold = 1) and represents the -fold decrease in Kd. The heat of dilution for injection was controlled with reference injections containing peptide alone before fitting. Each titration was performed in duplicates or triplicates, and the values reported here correspond to one titration. *, ITC measurement performed at 250 mm NaCl.

a Values were determined from fits of the ITC profile using the single-binding site model.
b Kd was determined from Ka derived from fits of the ITC profile using the single-binding site model. The indicated errors reflect the uncertainties calculated by Origin from the fit of the ITC profile.
Figure 1.

Mechanism of JIP1-Cter binding to KLC1-TPR domain. A, isothermal titration calorimetry measurements of the reference KLC1-TPR-[A1-B6] fragment with the JIP1-C10-wt peptide. This reference measurement was performed at 25 °C in a buffer containing 25 mm Hepes, pH 7.0, and 150 mm NaCl. B, superposition of ITC curves of the reference KLC1-TPR-[A1-B6] fragment with the JIP1-C10-wt peptide at 25 °C in a buffer containing variable ionic strength (150, 250, or 500 mm NaCl). C, superposition of ITC curves showing the interaction between the reference KLC1-TPR-[A1-B6] fragment and JIP1-C10-E706A/D707A double mutant (blue), JIP1-C10-I708A/L710A double mutant (green), and JIP1-C10-Y709A mutant (red). For comparison, the reference measurement between the KLC1-TPR-[A1-B6] fragment and the JIP1-C10-wt peptide is reported (black). Sequence alignment of the different JIP1-C10 peptides used for this experiment is shown below. The solid lines drawn through the data points match the best fit to the data.
To delineate the minimal region of JIP1 required to interact with KLC1-TPR, we conceived various mutations on the JIP1-C10 peptide (Table S3) and performed ITC binding experiments with the reference KLC1-TPR-[A1-B6] fragment (Table 1 and Fig. 1C). First, we examined a JIP1-C10 peptide carrying the Y709A mutation and confirmed the critical role of this residue for the interaction with KLC1-TPR because no binding is detected between JIP1-C10-Y709A and the KLC1-TPR-[A1-B6] fragment. Then we examined two JIP1-C10 peptides with a double mutation removing either the two hydrophobic residues flanking the critical tyrosine residue (JIP1-C10-I708A/L710A) or the two acidic residues before it (JIP1-C10-E706A/D707A). Both JIP1-C10-E706A/D707A and JIP1-C10-I708A/L710A peptides exhibit no binding for KLC1-TPR-[A1-B6] fragment (Table 1 and Fig. 1C). These data highlight that although residues Tyr709 and Glu711 of JIP1 are critical for KLC1-TPR binding (6, 15), their presence is not sufficient, and thus other residues among the EDIYLE sequence are also required. Altogether, these observations reveal that the KLC1-binding motif of JIP1 exploits several unique sequence-specific features that render its recognition by KLC1 highly specific.
A potential KLC1-binding motif in the internal unstructured part of SH2D6
Above, we identified a potential KLC1-binding motif in SH2D6 protein (Table S1), which we now examined for its ability to interact with KLC1. Two peptides covering the potential KLC1-binding motif of SH2D6 (sequences Ac-PDEDIYLECE-NH2 and Ac-KKPDEDIYLECE-NH2; Fig. 2A and Table S3) were conceived, and ITC-binding experiments were performed with the reference KLC1-TPR-[A1-B6] fragment. Unexpectedly, no binding was detected between these two SH2D6 peptides and KLC1-TPR-[A1-B6] (Fig. 2B and Table S5). Of note, interaction was also tested with an equivalent fragment of the KLC2 isoform, and still no binding was detected (Fig. 2B).
Figure 2.
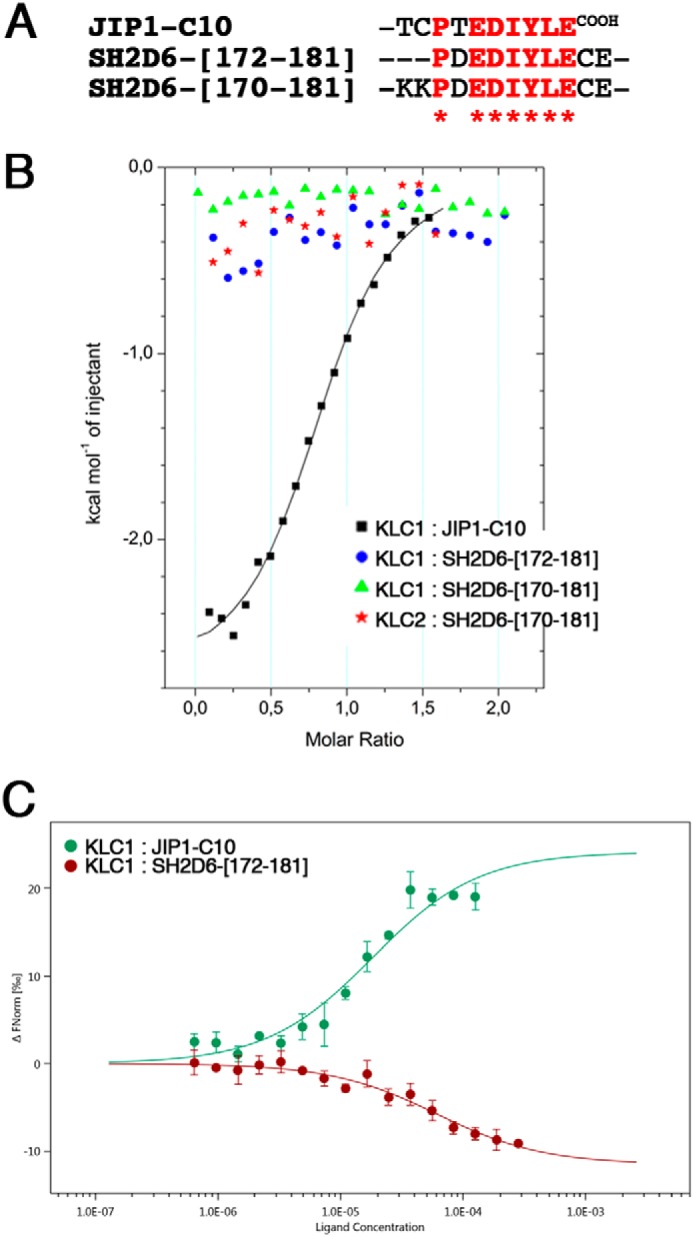
Binding experiments between SH2D6 peptides and the reference KLC1-TPR-[A1-B6] fragment. A, sequence alignment of the different SH2D6 and JIP1-C10 peptides used for these binding experiments. B, superposition of ITC curves showing the interaction between the reference KLC1-TPR-[A1-B6] fragment and JIP1-C10 WT (reference measurement; black), SH2D6-[172–181] peptide (blue) and SH2D6-[170–181] peptide (green), as well as the interaction between an equivalent KLC2-TPR-[A1-B6]-wt fragment and SH2D6-[170–181] peptide (red). The solid lines drawn through the data points match the best fit to the data. C, superposition of MST curves showing the interaction between the labeled KLC1-TPR-[A1-B6] fragment and JIP1-C10 WT (reference measurement; green) and the SH2D6-[172–181] (red) peptides. The curves represent an average of three independent measurements with the S.D. shown by error bars.
Due to this unexpected result, and because we cannot exclude the possibility that interaction between KLC1 and SH2D6 peptides might occur in the absence of observed enthalpy release, we used MST to reassess this interaction. In contrast to ITC, an interaction between the reference KLC1-TPR-[A1-B6] fragment and the SH2D6-[172–181] peptide (Fig. 2C) is detected by MST. Whereas the labeled KLC1-TPR-[A1-B6] fragment binds to JIP1-C10-wt peptide with an affinity of 18.2 ± 5.8 μm, it binds to the SH2D6-[102–112] peptide with an affinity of 60.9 ± 15.7 μm. Despite the fact that the affinity is lower by a 3-fold factor than for JIP1, it is however significant. The difference of binding affinity between KLC1-TPR-[A1-B6] fragment and JIP1-C10-wt peptide observed in ITC and MST is probably due to the use of lysine-labeled KLC1-TPR in MST experiments. Thus, these observations reveal that despite sequence differences at the N-terminal and C-terminal parts of the potential KLC1-binding motif in SH2D6, the central strictly conserved EDIYLE sequence, which contains critical residues for JIP1 interaction, is sufficient for KLC1-TPR binding.
Mapping of the minimal region of KLC1-TPR for JIP1-Cter binding
To identify precisely regions of the KLC1-TPR domain that are required for JIP1 binding, we designed several truncated fragments of the TPR domain of KLC1 (Fig. 3 and Table S2) based on available 3D structures (15, 17). They were conceived such that KLC1-TPR is shortened by one helix either at the N terminus or at the C terminus of the domain. To conserve a functional TPR domain groove, no fragments smaller than three consecutive TPR motifs were conceived (24). Thus, the first fragment truncated at the N terminus is deleted from the first helix (fragment [B1-B6]), the second one consists of the additional deletion of the second helix (fragment [A2-B6]), and so on. N-terminal truncations were done up to a minimal fragment consisting of the last three TPR motifs, including the non-TPR region ([A4-B6] fragment). Similarly, the first fragment truncated at the C terminus is deleted from the last helix (fragment [A1-A6]), the two next consist of the additional deletion of the next to last helix with or without the non-TPR region (fragments [A1-nonTPR] and [A1-B5], respectively), and so on. C-terminal truncations were done up to a fragment consisting of the first three TPR motifs ([A1-B3] fragment). Furthermore, a fragment consisting of the full TPR domain, but deleted from the non-TPR region, was conceived (fragment [A1-B6]-ΔnonTPR). Among these various KLC1-TPR fragments, some are not soluble or not stable during the purification process and precipitate, such as the [A1-A6], [A1-B4], and [A4-B6] fragments (Table S2). Thus, these fragments were not used for ITC binding experiments. To evaluate the structural integrity of soluble KLC1-TPR fragments, we performed MALS, CD, and DSC experiments (Table S2). SEC-MALS experiments revealed that all KLC1-TPR fragments studied here are monomeric in solution. CD experiments showed that all KLC1-TPR fragments are mainly α-helical with helix content ranging from 62.5 to 85%. DSC experiments gave melting temperatures (Tm) ranging from 47.2 to 61.6 °C. Altogether, these results demonstrate that these KLC1-TPR fragments are monomeric, well folded, and stable. However, because of significant differences in secondary structure helix content and Tm between these KLC1 fragments, we cannot exclude the possibility that truncations impact the overall dynamics of these TPR domain fragments.
Figure 3.

The truncated fragments of the TPR domain of KLC1. A, schema of full-length KLC1. B, 3D structure of the TPR domain of KLC1 (PDB code 3NF1 (15)). C, schematic representation of the different truncated fragments of the KLC1-TPR domain. The TPR domain of KLC1 is colored with a rainbow color code ranging from blue for the N terminus to red for the C terminus. Each TPR motif is indicated in a different color. The non-TPR region is shown as a gray loop.
We performed ITC binding experiments between the WT JIP1-C10 peptide and various truncated fragments of the KLC1-TPR domain (Table 1 and Table S4) and compared binding affinities with the reference [A1-B6] fragment of KLC1-TPR. On the one side, JIP1-C10 peptide binds to the [B1-B6] fragment with a Kd of 6.0 ± 1.0 μm, whereas it binds to the [A2-B6] and [B2-B6] fragments with Kd of 12.5 ± 2.3 and 33.4 ± 7.5 μm, respectively (Table 1 and Fig. 4A). Thus, deletion of the first helix (A1) of the TPR domain does not affect the binding affinity for JIP1-C10, whereas the additional deletion of B1 and A2 helices affects JIP1-C10 binding by a factor of 2.5 and 6.7, respectively. Accordingly, a negative enthalpy change decrease is observed for these interactions compared with the reference KLC1-TPR fragment, indicating a loss of contacts (Table 1). KLC1-TPR fragments with additional helix truncations (B2, A3, and B3 helices) exhibit no binding to JIP1-C10 (Table 1 and Fig. 4A). Because the presence of the histidine tag at the N terminus of the KLC1-TPR can impact or even prevent JIP1-C10 interaction, we performed ITC binding experiment with both [B2-B6] and [A3-B6] fragments cleaved from their histidine tag. Both [B2-B6] and [A3-B6] fragments showed the same binding behavior in the absence or presence of the histidine tag (Tables S4 and S6, and Fig. S3), which indicates that the presence of the histidine tag does not perturb JIP1-C10 binding. On the other side, JIP1-C10 peptide binds to the [A1-nonTPR] fragment with a Kd of 4.2 ± 1.1 μm, whereas it binds to [A1-B5] and [A1-A5] fragments with a Kd of 14.0 ± 1.2 and 35.5 ± 3.0 μm, respectively (Table 1 and Fig. 4B). Thus, deletion of the two last helices (A6 and B6) of the TPR6 motif does not affect the binding affinity for JIP1-C10, whereas the additional deletion of the non-TPR region affects JIP1-C10 binding by a factor of 2.8. This observation suggests that the non-TPR region moderately impacts JIP1 binding. This is confirmed by the fact that the complete [A1-B6] fragment deleted from the non-TPR region ([A1-B6]-ΔnonTPR fragment) similarly affects JIP1-C10 binding by a factor of 2.7 (Table 1 and Fig. 4B). The residues from the non-TPR region do not participate in the groove surface; thus, we suspect its contribution for JIP1 binding to be related to the dynamics of the TPR domain. This is supported by a decrease in the negative entropy contribution observed when the non-TPR region is deleted (−2.6 kcal/mol−1) compared with the reference fragment (−4.5 kcal/mol−1) (Table 1). Because the non-TPR region is disordered, especially in KLC1 (presence of five glycine residues and absence of a cysteine residue that is involved in a disulfide bridge in KLC2) (15–17), its deletion should decrease the overall KLC1-TPR flexibility. This is also supported with the [A1-B5] fragment that exhibits an important entropy contribution decrease compared with the [A1-nonTPR] fragment, which differs only by the presence of the non-TPR region (Table 1). Then the additional deletion of B5 helix significantly affects JIP1-C10 binding by a factor of 7.1 (Table 1). As a reminder, the [A1-B4] fragment is not soluble, and thus ITC experiments cannot be performed with this fragment. Finally, the [A1-A4] fragment exhibits no binding to JIP1-C10 (Table 1 and Fig. 4B). Altogether, these experiments narrow down the minimal region of KLC1-TPR for JIP1-C10 binding from A2 to B5 helices (Fig. 4C). Thus, both TPR1 and TPR6 motifs are dispensable for JIP1 recruitment; this observation agrees with previous cellular experiments (18). Finally, because JIP1 binds into the TPR groove of KLC1 and the latter is defined by A helices, we can delineate the JIP1-binding site at the surface of A2/A3/A4/A5 helices of KLC1-TPR (Fig. 4C).
Figure 4.

Minimal region of KLC1-TPR for JIP1-C10 binding. Superposition of ITC curves shows the interaction between the JIP1-C10-wt peptide and N-terminal truncated fragments (A) and C-terminal truncated fragments (B) of KLC1-TPR domain. For comparison, the reference measurement between the JIP1-C10-wt peptide and the complete KLC1-TPR-[A1-B6] fragment is reported (black). C, color-based representation of the 3D template structure of the KLC1-TPR domain (PDB code 3NF1) according to the -fold decrease in binding affinity (Kd) measured in ITC. The solid lines drawn through the data points match the best fit to the data.
Identification of critical residues of KLC1-TPR for JIP1-C10 binding
To better detail the JIP1-binding site of KLC1, we identified five asparagine and four basic residues that are located in the groove of the minimal region of the TPR domain and protrude at the surface accessible to interact with residues of JIP1 (Fig. 3A and Table S2). Of note, asparagine residues were targeted because (i) multiple mutations on asparagine residues of KLC1-TPR abolished JIP1 binding in a yeast two-hybrid assay (9), and (ii) the groove of the TPR domain of KLC1 includes numerous asparagine residues. Basic residues were targeted to complement the negative charges found in the KLC1-binding motif of JIP1. The five asparagine residues identified consist of Asn302, which is located on the A3 helix; Asn344 and Asn351 on the A4 helix; and Asn379 and Asn386 on the A5 helix (Fig. 5A). The four basic residues are the Arg266 located on the A2 helix, Arg327 on the B3 helix, Lys340 on the A4 helix, and Lys382 on the A5 helix (Fig. 5A). Based on the reference KLC1-TPR-[A1-B6] fragment, we conceived 11 KLC1-TPR mutants, each carrying a single point mutation on the positions mentioned above (Table S2). Most of these positions were replaced by alanine, except position Asn302 (Asn287 in KLC2), which was replaced by a leucine to allow consistent comparison with KLC2-TPR:SKIP-WD binding experiments reported previously (16). The structural integrity of each KLC1-TPR mutant was evaluated using MALS, CD, and DSC (Table S2), demonstrating that they are monomeric, well folded, and stable.
Figure 5.

Identification of critical residues of KLC1-TPR for JIP1-C10 binding. A, KLC1-TPR residues examined by ITC for their binding to JIP1-C10-wt peptide are indicated by white spheres and labeled on the 3D template structure of the KLC1-TPR domain. The critical Asn343 position is indicated with a red star (15). KLC1-TPR is shown in a cartoon and colored according to the -fold decrease in binding affinity (Kd) measured in ITC, as shown in Fig. 4C. B and C, superposition of ITC curves showing the interaction of the JIP1-C10-wt peptide with the asparagine mutants (B) and the arginine/lysine mutants (C) of the reference KLC1-TPR-[A1-B6] fragment. For comparison, the reference measurement between the JIP1-C10-wt peptide and the complete KLC1-TPR-[A1-B6] fragment is reported (black). The solid lines drawn through the data points match the best fit to the data. D, surface representation of the 3D structure of KLC1-TPR template (PDB code 3NF1; same orientation as in A) colored in white with critical residues indicated in red and residues in close proximity indicated in pink.
We performed ITC binding experiments between the WT JIP1-C10 peptide and various mutants of KLC1-TPR (Table 1 and Table S4) and compared binding affinities with the WT KLC1-TPR. Binding experiments show that the KLC1-TPR-N379A mutant exhibits similar binding affinity (Kd = 6.3 ± 0.9 μm) for JIP1-C10 as the WT KLC1-TPR (less than 2-fold decrease in Kd), whereas other asparagine mutations (N302L, N344A, N351A, and N386A) abolish JIP1-C10 binding (Table 1 and Fig. 5B). Also, ITC experiments show that KLC1-TPR-K340A and -K382A mutants exhibit similar binding affinity (Kd of 8.3 ± 2.0 and 3.6 ± 0.5 μm, respectively) for JIP1-C10 as the WT KLC1-TPR, whereas the R266A and R327A mutations abolish JIP1-C10 binding (Table 1 and Fig. 4C). Of note, all of these positions are conserved between the different isoforms of KLC. Interestingly, whereas Lys340 and Lys382 mutations to alanine did not impact JIP1-C10 binding, their mutation to glutamate abolished JIP1-C10 binding (Table 1 and Fig. 5C). These findings suggest that even if Lys340 and Lys382 do not make direct contact with JIP1-C10, they are in close proximity. Thus, in addition to the previously identified Asn343 (located on the A4 helix; Fig. 5A) (15), this work identified six residues of KLC1 that are critical for JIP1 binding (Arg266, Asn302, Asn344, Asn351, Asn386, and Arg327), as well as two residues that have indirect impact (Lys340 and Lys382). These results provide a footprint of the JIP1-binding site into the TPR domain groove of KLC1 (Fig. 5D).
Structural basis for the marginal inhibition of JIP1 binding by the LFP-acidic motif of KLC1
The footprint of the JIP1-binding site into the TPR domain groove of KLC1 provided by our biochemical data allows us to rationalize the marginal inhibition of JIP1 binding by the LFP-acidic motif of KLC1 (14). The crystal structure of KLC2-TPR bound to its LFP-acidic motif (PDB code 5FJY (14)) shows that the autoinhibitory motif lies along A1/A2/A3 helices into the groove of the TPR domain (Fig. 6A). Of note, no TPR domain closure is induced by the binding of the LFP-acidic motif, with a distance between A2 and A6 helix axes of 31.0 Å. Two critical residues for JIP1 binding, Arg266 and Asn302 (equivalent to Arg251 and Asn287 in KLC2, respectively; Table 1 and Fig. 5 (B and C)) are in close proximity to the LFP-acidic motif (Fig. 6A), whereas other residues of KLC1-TPR that are also critical for JIP1 binding (Arg327, Lys340, Asn344, Asn343, Asn351, Lys382, and Asn386) are not in proximity to the LFP-acidic motif (Fig. 6A) and thus not involved in the LFP-acidic motif interaction. Thus, we anticipate that JIP1 can interact at the C-terminal part of its binding site (B3/A4/B4/A5 helices) without being impeded by the LFP-acidic motif. This is further supported by the fact that the A2 helix, although important, is not critical for JIP1 binding (Table 1 and Fig. 4C). Then this initial docking of JIP1 to the B3/A4/B4/A5 helices of KLC1 might be sufficient to trigger the release of the LFP-acidic motif, for instance by inducing conformational changes, like a TPR domain closure, which will weaken the LFP-acidic motif interaction. Finally, the release of the LFP-acidic motif will facilitate access to the Arg266 and Asn302 residues, allowing the completion of the JIP1 binding process.
Figure 6.

Comparison of the binding sites of the LFP-acidic motif and the SKIP-WD motif with the JIP1-binding site footprint. A, 3D structure of KLC2-TPR bound to its LFP-acidic motif (PDB code 5FJY (14)). B, 3D structure of KLC2-TPR bound to SKIP-WD motif (PDB code 3ZFW (16)). In the KLC2-TPR:SKIP-WD motif complex structure, the A1 helix is not present in the KLC2-TPR fragment crystallized, whereas the B1 helix is not modeled due to electron density absence; thus, the TPR1 motif is absent from the 3D structure. Both KLC2-TPR molecules are shown with a surface representation in gray. KLC1-TPR residues that are critical for JIP1 binding are highlighted in red (residues in proximity to JIP1 are indicated in pink) and labeled with KLC1 numbering. The LFP-acidic motif and the SKIP-WD motif are shown in sticks and colored in green and yellow, respectively. Orientation is conserved with that of Fig. 5D (superposition is done on the TPR2 motif), but because of differences in TPR domain closure, residue positions are slightly different with respect to each other.
Structural basis for the competition between JIP1 and W-acidic motif cargos for KLC binding
JIP1 and ALCα compete for their transport by kinesin1-KLC (11), suggesting that both cargos cannot be recruited at the same time by KLC. The structural basis of this competition is, however, not elucidated. The competition can be direct with an overlap of both cargo-binding sites that would induce steric hindrances between cargos, preventing their co-binding. But the competition can also be indirect, with the binding of one cargo inducing KLC1-TPR conformational changes that will prevent the other cargo from recognizing its binding site. Indeed, the crystal structure of KLC2-TPR bound to the WD motif of SKIP showed that upon SKIP-WD binding, the TPR domain undergoes a closure (distance between A2 and A6 helix axes is 26.1 Å) that engenders formation of one hydrophobic pocket, which is required for the critical tryptophan residue of SKIP-WD to bind (PDB code 3ZFW (16)).
To better understand the structural basis of the competition between JIP1 and the W-acidic motif cargo family, we compared information from the crystal structure of KLC2 bound to the SKIP-WD motif with that provided by the footprint of the JIP1-binding site. The crystal structure of KLC2-TPR bound to the SKIP-WD motif shows that the W-acidic motif lies on a binding site formed by A2/A3/A4 helices of KLC2-TPR, whereas the JIP1-binding site covers A2/A3/A4/A5 helices of KLC1-TPR (Fig. 6B). Thus, the JIP1- and W-acidic motif cargo–binding sites overlap on the A2/A3/A4 helices. On this shared binding site, Arg251, Asn287, Arg312, Asn329, and Asn336 in KLC2 (equivalent to Arg266, Asn302, Arg327, Asn344, and Asn351, respectively, in KLC1) make hydrogen bonds with side chains and main chains of SKIP-WD motif (PDB code 3ZFW (16)). These five residues, which are involved in W-acidic motif cargo binding, are also critical for JIP1 binding (Table 1 and Fig. 5 (B and C)). Thus, at least five residues of KLC1 are involved in both JIP1 and W-acidic motif cargo interaction, preventing their co-binding to KLC1-TPR. Altogether, these observations support a direct competition between JIP1 and W-acidic motif cargos. However, we cannot exclude the possibility that additional indirect impact can take place due to conformational change of the TPR domain of KLC upon cargo binding that would modify the binding surface recognition, like formation of hydrophobic pockets.
Interestingly, despite the JIP1- and W-acidic motif cargo-binding sites largely overlapping on the TPR domain of KLC, they are not strictly equivalent. Indeed, the JIP1-binding site involved the A5 helix, which is not part of the W-acidic motif cargo-binding site. The residue Asn386 of KLC1 (equivalent to KLC2-Asn371), which is located on the A5 helix, is critical for JIP1-C10 binding (Table 1 and Fig. 5B), but it makes no interaction with the SKIP-WD motif (PDB code 3ZFW (16)). Using ITC binding experiments, we confirmed that the Asn386 of KLC1-TPR has no impact on ALCα-WD1 motif binding (Tables S4 and S7 and Fig. S4). Of note, KLC1-Asn343, which is located on the A4 helix, is critical for JIP1 binding but not for ALCα binding (15). Thus, these differences highlight that despite the fact that the KLC1-binding motifs of JIP1 and W-acidic motif cargos share similarities (aromatic acid charge sequence) and their binding site largely overlap, they exhibit distinct modes of binding.
Relationship between biochemical and structural data for the KLC1-TPR:JIP1 interaction
During the revision of this manuscript, the 3D structure of KLC1-TPR bound to the last 11 residues of JIP1 was released (PDB code 6FUZ (25)). We compared these structural data with the biochemical data presented in this study. First, in the crystal structure, the TPR domain of KLC1 exhibits a close conformation (distance between A2 and A6 helix axes is 20.7 Å) that allows JIP1 to bind KLC1-TPR from the TPR2 to the TPR5 motifs (Fig. 7A), as confirmed by ITC binding experiments using the truncated fragments of KLC1-TPR (Fig. 4). Then most of the KLC1-TPR residues required for JIP1 binding (Fig. 5) are in direct interactions with JIP1 (Fig. 7, B and C). KLC1-Asn351 makes one hydrogen bond with the carboxylate group of Asp707 and contributes to the formation of the hydrophobic pocket located at the interface of A3-A4 helices in which JIP1-Tyr709 lies (Fig. 7B). KLC1-Asn344 and Asn386 each make a double hydrogen bond with the backbone groups of Leu710 and Ile708 of JIP1, respectively (Fig. 7B), whereas KLC1-Asn343 contributes to this network of interactions with (i) one hydrogen bond to the backbone carbonyl group of Ile708 and (ii) one hydrogen bond to the carboxamide group of KLC1-Asn386 (Fig. 7B). Both KLC1-Arg266 and KLC1-Asn302 interact with the Glu711 of JIP1; the first participates in a salt bridge, whereas the second contributes with one hydrogen bond (Fig. 7C). Finally, KLC1-Lys340 and -Lys382 residues, which are not critical for JIP1 binding (Table 1 and Fig. 5C), make no direct interaction with JIP1 (Fig. 7, B and C), but both are in the vicinity of JIP1, which correlates with the fact that their mutation to the reverse charge glutamate abolishes JIP1 binding (Table 1 and Fig. 5C). KLC1-Asn379 which is not critical for JIP1 binding (Table 1 and Fig. 5B) locates in the van der Waals sphere of JIP1-Leu710 (Fig. 7B). Interestingly, KLC1-Arg327, which was identified as a critical residue for JIP1 binding (Table 1 and Fig. 5 (B and C)), is not in direct contact with JIP1. However, it participates, together with Asn301 and Gln341, in a network of hydrogen bonds that directs Asn344 to JIP1. Thus, this reveals that residues Asn301 and Gln341, together with Arg327, Asn343, Asn343, and Asn386, are required for the formation of a network of interactions that is critical for JIP1 binding. Altogether, our binding ITC experiments strongly support the KLC1:JIP1 complex structure recently determined (PDB code 6FUZ (25)).
Figure 7.

Relationship between biochemical and structural data for the KLC1-TPR:JIP1 interaction. A, 3D structure of KLC1-TPR bound to JIP1-C10 (PDB code 6FUZ (25)). KLC1-TPR is shown with a white surface representation, and critical residues for JIP1 binding are highlighted in red (residues in proximity to JIP1 are indicated in pink). The JIP1-C10 motif is shown in sticks and colored in teal. Orientation is conserved with that of Figs. 5D and 6 (superposition is done on the TPR2 motif), but because of differences in TPR domain closure, the positions of residues involved in JIP1-binding are slightly different with respect to each other. B and C, detailed views of the interaction between JIP1 and the critical residues of KLC1-TPR. The color code is as in A.
Modeling of KLC1-TPR:SH2D6 peptide supports MST binding experiments
The modeling of the potential KLC1-binding motif of mouse SH2D6 (172PDEDIYLECE181 sequence) into the TPR domain of KLC1 (Fig. 8A) represents a valuable structural basis to verify whether SH2D6 can accommodate into the KLC1-TPR groove. Because it is strictly identical to JIP1, the central part of the SH2D6 peptides (the EDIYLE sequence) should be able to interact with KLC1-TPR in the same manner as JIP1. The N-terminal part of the SH2D6 peptide (-KKPD- and -PD- sequences) should not prevent interaction with KLC1-TPR. Indeed, the modeling shows that Thr705 of JIP1 can be replaced by the Asp173 of SH2D6 without inducing steric hindrance or repulsion with KLC1-TPR (Fig. 8B). Moreover, the Asp173 of SH2D6 is close to two basic residues of KLC1-TPR, Lys393 and Arg476, respectively (Fig. 8B). In SH2D6, the two conserved lysine residues before the Asp173 should not be in interaction with KLC1-TPR, because in the crystal structure, residues 701–703 (-YTC- sequence) of JIP1 are not modeled, suggesting that no electron density is observed for these residues, probably due to their flexibility. Thus, this suggests that the N-terminal part of the SH2D6 that differs in sequence with JIP1 should not impede the binding to KLC1-TPR (Fig. 2C). Furthermore, in addition to the crystal structure of KLC1-TPR:JIP1 complex (PDB code 6FUZ (25)), that of KLC1-TPR bound to the extreme C terminus of TorsinA (PDB code 6FV0 (25)), another KLC1-binding partner (26), has been released at the same time. Of note, both crystal structures reveal that JIP1 and TorsinA share the same mode of binding to KLC1-TPR. In TorsinA, the sequence of the N-terminal part of the KLC1-binding motif (Fig. 8A) is even more divergent from that of JIP1 than that of SH2D6. Both the N terminus of JIP1 and that of TorsinA accommodate differently into the TPR domain of KLC1. This observation further supports that the sequence differences in the N-terminal part of the potential KLC1-binding motif of SH2D6 should not prevent its binding. At the C-terminal part of the SH2D6 peptide, differences are also observed compared with JIP1 with (i) the presence of two additional residues (-CE- sequence) and (ii) the absence of the C-terminal carboxylate (Fig. 8A). One difference is also observed between JIP1 and TorsinA, with the latter exhibiting one additional residue (Asp712) at its extreme C terminus (Fig. 8A). Both the C terminus of JIP1 and that of TorsinA accommodate differently into the TPR domain of KLC1. Thus, even if the SH2D6-Cys180 does not make the same network of hydrogen bonds with KLC1-TPR as TorsinA-Asp712 (located at the same position; Fig. 8A), it should not prevent SH2D6 from interacting with KLC1-TPR (Fig. 8C). Furthermore, similarly to the Asp712 of TorsinA, backbone carbonyl and amide groups of Cys180 of SH2D6 will contribute by a double hydrogen bond with the Asn302 of KLC1-TPR. Finally, the following Glu181 in SH2D6 should be able to accommodate into the TPR domain groove of KLC1-TPR and should even be stabilized by direct interactions with His217 and Arg285 of KLC1-TPR (Fig. 8C). Thus, the modeling of the potential KLC1-binding motif of SH2D6 into the TPR domain of KLC1 suggests that SH2D6 can bind into the TPR domain of KLC1, as shown by MST binding experiments (Fig. 2C). Difference in affinity (3-fold) observed between JIP1 and SH2D6 for KLC1-TPR binding can be explained by the sequence differences at the N terminus and/or the C terminus of their KLC1-binding motif. However, further experiments are required to confirm that SH2D6 might be a KLC1-binding partner in a cellular context and whether it is transported by kinesin1.
Figure 8.

Modeling of SH2D6 binding to KLC1-TPR domain. A, model of KLC1-TPR bound to the potential KLC1-binding motif of SH2D6 (model built from PDB code 6FUZ (25)). The KLC1-TPR domain is shown with a white surface representation, and residues critical for JIP1 binding are highlighted in red (and pink for residues in proximity). A second orthogonal view is shown to allow the visualization of the extreme C terminus (last three residues) of the SH2D6 peptide. B and C, detailed views of the interaction between SH2D6 and the KLC1-TPR domain. The potential KLC1-binding motif of SH2D6 is shown in sticks and colored in purple. The color code is as in A.
Conclusion
Using a protein engineering approach, we conceived various fragments of KLC1, truncated helix by helix either at the N terminus or the C terminus of its TPR domain. All of these KLC1-TPR fragments were assessed for their ability to bind to JIP1 by calorimetry. In that way, the JIP1-binding site of KLC1-TPR was narrowed down to the region extending from the TPR2 to TPR5 motifs, revealing that TPR1 and TPR6 motifs are dispensable for JIP1 binding. Then we identified nine residues located in the groove of the TPR domain of KLC1 that were independently examined for their ability to bind to KLC1-TPR. This allowed us to identify seven critical residues for JIP1 binding, as well as two additional residues that despite not being critical for the interaction are located in the vicinity of the JIP1 binding site. Altogether, this biochemical characterization provides a detailed footprint of the JIP1-binding site in the TPR domain groove of KLC1. Finally, this JIP1-binding site footprint gives insights to better understand the structural basis for (i) the marginal inhibition of JIP1 binding by the LFP-acidic motif of KLC1 and (ii) the competition between JIP1 and W-acidic motif cargos for KLC1 binding. During the revision of this manuscript, the 3D structure of KLC1-TPR bound to JIP1 was released (PDB code 6FUZ) and supports the JIP1-binding footprint identified in this study. Altogether, these complementary biochemical and structural data contribute to a detailed understanding of how JIP1 is recognized and interacts with KLC1-TPR.
We identified new potential KLC1-binding partners that will exhibit a similar motif as the KLC1-binding motif of JIP1. All of them possess this motif into their internal part and not at their extreme C terminus. One of them, the SH2D6 protein, shares strongly similar sequence with JIP1 for this motif. Binding experiments and modeling suggest that SH2D6 might be a KLC1-binding partner. Altogether, JIP1, TorsinA, and SH2D6 accommodate into the TPR domain of KLC1 sharing the central consensus sequence [−][Φ]Y[Φ][−] (with [−] a negative residue and [Φ] a hydrophobic/aromatic residue). Pernigo et al. (25) rightly named this latter the “Y-acidic” motif. However, differences are observed among these three proteins. First, whereas the Y-acidic motif of SH2D6 is in an internal unstructured part of the molecule, those of JIP1 and TorsinA are located at their extreme C terminus. Also, sequence differences are observed before and after the Y-acidic motif that lead to structural differences in KLC1-TPR interactions. This reveals that KLC1 can accommodate sequence and structural variations at the N terminus and C terminus of the Y-acidic motif. Such differences outside the Y-acidic motif probably represent a way to modulate the binding affinity of the cargo itself, but also a way to modulate competition with co-binding partners, as the KLC1 autoinhibitory motif or W-acidic motif cargos. Altogether, these observations reveal that the KLC1-binding motif observed in JIP1 is found in several other proteins: TorsinA and SH2D6 as well as potential KLC1-binding partners identified here using sequence search (Table S1). Interestingly, these also suggest that the Y-acidic motif is not only found at the extreme C terminus, but also in the internal part of KLC1 cargos, as observed for the W-acidic motif. Overall, this highlights that JIP1 belongs to a growing kinesin1 cargo family whose members share the same mode of binding, allowing some variations.
Experimental procedures
Primary sequence analysis
For sequence analysis, human JIP1 and JIP2, with accession numbers NP_005447.1 and NP_036456.1, respectively, were used. The phylogenetic study was performed using full-length sequences of JIP1 and JIP2 homologs retrieved using two iterations of InterEvolAlign (27): one iteration against the OMA 2011 “Entire genomes” database (28) and the other one against the RefSeq database. All homologous sequences were updated to the latest RefSeq sequences (29). Those full-length sequences were realigned using the MAFFT E-INS-i algorithm (30). A phylogenetic tree was calculated using the PhyML program (version 3.1) (31) with standard parameters (LG substitution model with four substitution rate categories). The tree was visualized with Dendroscope (32) using a cladogram representation. The associated multiple sequence alignment was visualized with Jalview (33) after removing columns containing more than 50% gaps.
For the KLC1-binding motif search, we used the consensus motif (E/D)(E/D)(I/M/L/V)Y(I/M/L/V)(E/D) against the UniProtKB (SwissProt including splice sequences + TrEMBL) database using the ScanProsite (21) tool. Predicted secondary structure filtering was performed using PSIPRED (34) on a profile built by one iteration of HHblits (35) against the Uniprot20 2016_02 database; groups where all proteins with confident secondary structure predictions have at least one position in the motif predicted as helix or strand were eliminated.
The multiple sequence alignment for SH2D6 (shown in Fig. S2) was built starting from the mouse sequence (Uniprot identifier Q9D413, SH2D6_MOUSE). HHblits (35) was used to search for homologous sequences in the Uniprot database (version 2016_02). The alignment was filtered to keep only sequences that have more than 40% sequence identity and more than 60% coverage with the query sequence (to avoid sequences matching only the C-terminal SH2 domain). Redundancy was removed by filtering the alignment down to maximum 95% sequence identity. No human sequences were present in the alignment because the human isoforms described in the Uniprot database are missing the region containing the JIP1-Cter motif. Therefore, human sequences were added by querying the NCBI RefSeq database (29) (which contains 17 predicted isoforms of human SH2D6) and filtering to keep only isoforms with maximum 95% mutual sequence identity. All sequences were realigned globally using the MAFFT E-INS-i algorithm (30). The multiple-sequence alignment was visualized with Jalview (32).
Gene constructs, protein expression, and purification
cDNAs encoding the complete TPR domain fragment of human KLC1 (residues 206–502; accession number NP_005543) were cloned into the pET-HTb plasmid in NcoI/KpnI restriction sites. All mutants of KLC1 were generated from the complete TPR domain fragment into the pET-HTb plasmid by ProteoGenix. All truncated fragments of the KLC1-TPR domain were cloned into the pET28a plasmid in NdeI/BamHI or NdeI/EcoRI restriction sites. The KLC1-TPR-[A1-B6]-nonTPR fragment is a synthetic gene (Proteogenix) cloned in the pET-28a plasmid in NCoI/XhoI restriction sites. The non-TPR region (residues 419–458) was deleted, and a short flexible linker (G4S)1 was inserted between Ser418 and Asp459. cDNAs encoding the complete TPR domain fragment of mouse KLC2 (residues 190–484; accession number NP_032477) were cloned into the pET28a plasmid in NdeI/EcoRI restriction sites. Table S2 provides information on the sequence boundaries of each of these KLC-TPR fragments and mutants.
All KLC fragments were produced in Escherichia coli BL21-Gold (DE3) as N terminus His tag fusion proteins. Cells were collected after induction with 0.4 mm isopropyl 1-thio-β-d-galactopyranoside for 4 h at 25 °C. Frozen bacteria were suspended in 25 mm Hepes, pH 7.0, containing 500 mm NaCl, 5% glycerol, 1 mm DTT, 20 mm imidazole, pH 7.0, 0.05% Triton, protease inhibitor mixture (1 mini-tablet/liter, Biotool), and 0.7 mg/ml lysozyme. The lysate was incubated for 1 h at 4 °C, disrupted by sonication, and ultracentrifuged at 40,000 rpm for 40 min at 4 °C. The soluble lysate was loaded onto a His-Trap 5-ml column (GE Healthcare). The column was equilibrated with 25 mm Hepes, pH 7.0, 500 mm NaCl, 5% glycerol, 1 mm DTT, and 20 mm imidazole, pH 7.0. The column was washed with 5 column volumes of the same buffer and eluted by one step with 25 mm Hepes, pH 7.0, 500 mm NaCl, 5% glycerol, 1 mm DTT, and 500 mm imidazole, pH 7.0. Then it was loaded on a HiLoad 16/60 Superdex 75 column (GE Healthcare) using elution buffer containing 25 mm Hepes, pH 7.0, and 250 mm NaCl. All KLC fragments and mutants were stored at −80 °C. Table S2 provides information on the stability of each of these KLC-TPR fragments and mutants (results from MALS, CD, and DSC experiments are reported; see supporting Experimental procedures).
Isothermal titration calorimetry
Binding affinities of the JIP1-C10 peptides, ALCα-WD1 peptide, and SH2D6 peptides (all synthesized by Proteogenix; Table S3) to all KLC1-TPR fragments and mutants and KLC2-TPR-wt were measured by ITC (ITC200 microcalorimeter, MicroCal Inc., Malvern Panalytical). Measurements were performed at 25 °C in a buffer containing 25 mm Hepes, pH 7.0, and 150 mm NaCl. All KLC-TPR was set between 40 and 90 μm concentration in the cell, whereas JIP1-C10, ALCα-WD1, and SH2D6 peptides were set in the syringe at 0.5–1 mm concentration. The JIP1-C10 peptides were purified on a Superdex Peptide gel filtration column before ITC experiments to improve the quality of the titration measurements. Due to protein stability concerns, the histidine tag was kept for KLC1-TPR. A first injection of 0.4 μl, which was not taken into account for the fitting, was followed by 20 injections of 2 μl at intervals of 180 s. Data were analyzed using the MicroCal Origin software provided by the manufacturer.
Microscale thermophoresis
The reference KLC1-TPR-[A1-B6] fragment was labeled using the GREEN-NHS Labeling kit (NanoTemper Technologies). The labeling reaction was performed according to the manufacturer's instructions. The labeled KLC1-TPR-[A1-B6] fragment was stored in 50 mm Hepes, pH 7, and 200 mm NaCl. For experiments, the labeled sample was then adjusted to 40 nm with the ITC buffer containing 25 mm Hepes, pH 7.0, and 150 mm NaCl supplemented with 0.05% Tween 20 (NanoTemper Technologies). The JIP1-C10-wt and the SH2D6-[172–181] peptides were dissolved in the same buffer to a concentration of 564 μm. The peptides were diluted 16 times in a 2:1 serial dilution. For thermophoresis, each peptide dilution was mixed with one volume of labeled KLC1-TPR-[A1-B6]. Each dilution was filled into Monolith NT standard treated capillaries (NanoTemper Technologies GmbH). Thermophoresis was measured using a Monolith NT.115 instrument (NanoTemper Technologies GmbH) at an ambient temperature of 22 °C with 3-s/20-s/1-s laser off/on/off times, respectively. Instrument parameters were adjusted with 80% LED power and 40% MST power. Data of three independently pipetted measurements were analyzed (MO.Affinity Analysis software, NanoTemper Technologies) using the signal from thermophoresis.
Modeling and structural analysis
Helix axis distances were calculated using helix_angles.py (R. L. Campbell, Queen's University) using A2 and A6 helices, which take into account the absence of A1 and B1 helices from the KLC2:SKIP-WD complex structure (PDB code 3ZFW). The model of KLC-TPR:SH2D6 complex was performed using the KLC1-TPR:JIP1 complex structure (PDB code 6FUZ) as guide. First, the lama glama nanobody (chain F) was removed. Then the eight residues of JIP1 (-PTEDIYLECOOH sequence) were replaced by the 10 residues of SH2D6 sequence (-PDEDIYLECE- sequence) based on (i) sequence alignment of both proteins and (ii) the two additional C-terminal residues of SH2D6 (-CE- sequence) were modeled guided by the KLC1-TPR:TorsinA structure (PDB code 6FV0). These modifications were performed manually using COOT (36). Then a geometry minimization was done using Phenix software (37) to take into account the new sequence of the peptide bound to KLC1-TPR. Structure and protein–protein interaction analysis were performed using the PyMOL graphical program (38) and the interactive tool PDBePISA (39). Figures were computed using PyMOL (38).
Author contributions
T. Q. N., M. A.-N., J. A., C. V., P. L., and J. M. conceptualization; T. Q. N., M. A.-N., C. V., M. C., F. V., P. L., and J. M. data curation; T. Q. N., M. A.-N., J. A., C. V., F. V., C. G., and J. M. formal analysis; T. Q. N., M. A.-N., J. A., P. F. V., P. L., and J. M. validation; T. Q. N., M. A.-N., J. A., C. V., M. C., F. V., and C. G. methodology; M. A.-N., M. C., P. F. V., P. L., and J. M. supervision; P. L. and J. M. writing-review and editing; J. M. funding acquisition; J. M. writing-original draft.
Supplementary Material
Acknowledgments
This work has benefited from the expertise of the Macromolecular Interaction Measurements Platform of I2BC. We are grateful to Drs. Davy Martin and Human Rezaei (INRA, VIM-UR0892, Jouy-en-Josas) for access to their CD equipment. We thank Dr. Benoît Gigant for useful discussions, as well as for careful reading and comments on the manuscript.
This work was supported by French Infrastructure for Integrated Structural Biology (FRISBI) Grant ANR-10-INSB-05-01 and by ARC Grant SFI20121205592 (to J. M.) as well as Cancéropole Ile-de-France Grant 2013-2-AD-06-UP 6-1 and ARC Grant DOC20160603834 (to T. Q. N.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supporting Experimental procedures, Tables S1–S7, and Figs. S1–S4.
- JIP
- JNK-interacting protein
- ALCα
- alcadein-α
- DSC
- differential scanning calorimetry
- ITC
- isothermal titration calorimetry
- KHC
- kinesin heavy chain
- KLC
- kinesin light chain
- MST
- microscale thermophoresis
- PDB
- Protein Data Bank
- SEC
- size-exclusion chromatography
- MALS
- multiangle light scattering
- SH2
- Src homology 2
- SH2D6
- SH2 domain–containing 6 protein
- TPR
- tetratricopeptide repeat
- JNK
- c-Jun N-terminal kinase
- 3D
- three-dimensional.
References
- 1. Whitmarsh A. J. (2006) The JIP family of MAPK scaffold proteins. Biochem. Soc. Trans. 34, 828–832 10.1042/BST0340828 [DOI] [PubMed] [Google Scholar]
- 2. Meyer D., Liu A., and Margolis B. (1999) Interaction of c-Jun amino-terminal kinase interacting protein-1 with p190 rhoGEF and its localization in differentiated neurons. J. Biol. Chem. 274, 35113–35118 10.1074/jbc.274.49.35113 [DOI] [PubMed] [Google Scholar]
- 3. Stockinger W., Brandes C., Fasching D., Hermann M., Gotthardt M., Herz J., Schneider W. J., and Nimpf J. (2000) The reelin receptor apoER2 recruits JNK-interacting proteins-1 and -2. J. Biol. Chem. 275, 25625–25632 10.1074/jbc.M004119200 [DOI] [PubMed] [Google Scholar]
- 4. Matsuda S., Yasukawa T., Homma Y., Ito Y., Niikura T., Hiraki T., Hirai S., Ohno S., Kita Y., Kawasumi M., Kouyama K., Yamamoto T., Kyriakis J. M., and Nishimoto I. (2001) c-Jun N-terminal kinase (JNK)-interacting protein-1b/islet-brain-1 scaffolds Alzheimer's amyloid precursor protein with JNK. J. Neurosci. 21, 6597–6607 10.1523/JNEUROSCI.21-17-06597.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Matsuda S., Matsuda Y., and D'Adamio L. (2003) Amyloid protein precursor (AβPP), but not AβPP-like Protein 2, is bridged to the kinesin light chain by the scaffold protein JNK-interacting protein 1. J. Biol. Chem. 278, 38601–38606 10.1074/jbc.M304379200 [DOI] [PubMed] [Google Scholar]
- 6. Verhey K. J., Meyer D., Deehan R., Blenis J., Schnapp B. J., Rapoport T. A., and Margolis B. (2001) Cargo of kinesin identified as JIP scaffolding proteins and associated signaling molecules. J. Cell Biol. 152, 959–970 10.1083/jcb.152.5.959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blasius T. L., Cai D., Jih G. T., Toret C. P., and Verhey K. J. (2007) Two binding partners cooperate to activate the molecular motor kinesin-1. J. Cell Biol. 176, 11–17 10.1083/jcb.200605099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fu M. M., and Holzbaur E. L. F. (2013) JIP1 regulates the directionality of APP axonal transport by coordinating kinesin and dynein motors. J. Cell Biol. 202, 495–508 10.1083/jcb.201302078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hammond J. W., Griffin K., Jih G. T., Stuckey J., and Verhey K. J. (2008) Co-operative versus independent transport of different cargoes by kinesin-1. Traffic 9, 725–741 10.1111/j.1600-0854.2008.00722.x [DOI] [PubMed] [Google Scholar]
- 10. Konecna A., Frischknecht R., Kinter J., Ludwig A., Steuble M., Meskenaite V., Indermühle M., Engel M., Cen C., Mateos J.-M., Streit P., and Sonderegger P. (2006) Calsyntenin-1 docks vesicular cargo to kinesin-1. Mol. Biol. Cell 17, 3651–3663 10.1091/mbc.e06-02-0112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Araki Y., Kawano T., Taru H., Saito Y., Wada S., Miyamoto K., Kobayashi H., Ishikawa H. O., Ohsugi Y., Yamamoto T., Matsuno K., Kinjo M., and Suzuki T. (2007) The novel cargo alcadein induces vesicle association of kinesin-1 motor components and activates axonal transport. EMBO J. 26, 1475–1486 10.1038/sj.emboj.7601609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zeke A., Misheva M., Reményi A., and Bogoyevitch M. A. (2016) JNK signaling: regulation and functions based on complex protein-protein partnerships. Microbiol. Mol. Biol. Rev. 80, 793–835 10.1128/MMBR.00043-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Morfini G., Schmidt N., Weissmann C., Pigino G., and Kins S. (2016) Conventional kinesin: biochemical heterogeneity and functional implications in health and disease. Brain Res. Bull. 126, 347–353 10.1016/j.brainresbull.2016.06.009 [DOI] [PubMed] [Google Scholar]
- 14. Yip Y. Y., Pernigo S., Sanger A., Xu M., Parsons M., Steiner R. A., and Dodding M. P. (2016) The light chains of kinesin-1 are autoinhibited. Proc. Natl. Acad. Sci. 113, 2418–2423 10.1073/pnas.1520817113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhu H., Lee H. Y., Tong Y., Hong B.-S., Kim K.-P., Shen Y., Lim K. J., Mackenzie F., Tempel W., and Park H.-W. (2012) Crystal structures of the tetratricopeptide repeat domains of kinesin light chains: insight into cargo recognition mechanisms. PLoS One 7, e33943 10.1371/journal.pone.0033943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pernigo S., Lamprecht A., Steiner R. A., and Dodding M. P. (2013) Structural basis for kinesin-1: cargo recognition. Science 340, 356–359 10.1126/science.1234264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nguyen T. Q., Chenon M., Vilela F., Velours C., Aumont-Nicaise M., Andreani J., Varela P. F., Llinas P., and Ménétrey J. (2017) Structural plasticity of the N-terminal capping helix of the TPR domain of kinesin light chain. PLoS One 12, e0186354 10.1371/journal.pone.0186354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kawano T., Araseki M., Araki Y., Kinjo M., Yamamoto T., and Suzuki T. (2012) A small peptide sequence is sufficient for initiating kinesin-1 activation through part of TPR region of KLC1. Traffic 13, 834–848 10.1111/j.1600-0854.2012.01350.x [DOI] [PubMed] [Google Scholar]
- 19. Boucrot E., Henry T., Borg J. P., Gorvel J. P., and Méresse S. (2005) The intracellular fate of Salmonella depends on the recruitment of kinesin. Science 308, 1174–1178 10.1126/science.1110225 [DOI] [PubMed] [Google Scholar]
- 20. Dodding M. P., Mitter R., Humphries A. C., and Way M. (2011) A kinesin-1 binding motif in vaccinia virus that is widespread throughout the human genome. EMBO J. 30, 4523–4538 10.1038/emboj.2011.326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. de Castro E., Sigrist C. J. A., Gattiker A., Bulliard V., Langendijk-Genevaux P. S., Gasteiger E., Bairoch A., and Hulo N. (2006) ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 34, W362–W365 10.1093/nar/gkl124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dey G., Jaimovich A., Collins S. R., Seki A., and Meyer T. (2015) Systematic discovery of human gene function and principles of modular organization through phylogenetic profiling. Cell Rep. 10, 993–1006 10.1016/j.celrep.2015.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Koretzky G. A., Abtahian F., and Silverman M. A. (2006) SLP76 and SLP65: complex regulation of signalling in lymphocytes and beyond. Nat. Rev. Immunol. 6, 67–78 10.1038/nri1750 [DOI] [PubMed] [Google Scholar]
- 24. D'Andrea L. D., and Regan L. (2003) TPR proteins: the versatile helix. Trends Biochem. Sci. 28, 655–662 10.1016/j.tibs.2003.10.007 [DOI] [PubMed] [Google Scholar]
- 25. Pernigo S., Chegkazi M., Yip Y., Treacy C., Glorani G., Hansen K., Politis A., Dodding M. P., and Steiner R. A. (2018) Structural basis for isoform-specific kinesin-1 recognition of Y-acidic cargo adaptors. bioRxiv 10.1101/322057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kamm C., Boston H., Hewett J., Wilbur J., Corey D. P., Hanson P. I., Ramesh V., and Breakefield X. O. (2004) The early onset dystonia protein TorsinA interacts with kinesin light chain 1. J. Biol. Chem. 279, 19882–19892 10.1074/jbc.M401332200 [DOI] [PubMed] [Google Scholar]
- 27. Faure G., Andreani J., and Guerois R. (2012) InterEvol database: exploring the structure and evolution of protein complex interfaces. Nucleic Acids Res. 40, D847–D856 10.1093/nar/gkr845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Altenhoff A. M., Schneider A., Gonnet G. H., and Dessimoz C. (2011) OMA 2011: orthology inference among 1000 complete genomes. Nucleic Acids Res. 39, D289–D294 10.1093/nar/gkq1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. O'Leary N. A., Wright M. W., Brister J. R., Ciufo S., Haddad D., McVeigh R., Rajput B., Robbertse B., Smith-White B., Ako-Adjei D., Astashyn A., Badretdin A., Bao Y., Blinkova O., Brover V., et al. (2016) Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745 10.1093/nar/gkv1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Katoh K., and Standley D. M. (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Guindon S., Dufayard J. F., Lefort V., Anisimova M., Hordijk W., and Gascuel O. (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 10.1093/sysbio/syq010 [DOI] [PubMed] [Google Scholar]
- 32. Huson D. H., and Scornavacca C. (2012) Dendroscope 3: an interactive tool for rooted phylogenetic trees and networks. Syst. Biol. 61, 1061–1067 10.1093/sysbio/sys062 [DOI] [PubMed] [Google Scholar]
- 33. Waterhouse A. M., Procter J. B., Martin D. M. A., Clamp M., and Barton G. J. (2009) Jalview version 2: a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 10.1093/bioinformatics/btp033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jones D. T. (1999) Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292, 195–202 10.1006/jmbi.1999.3091 [DOI] [PubMed] [Google Scholar]
- 35. Remmert M., Biegert A., Hauser A., and Söding J. (2011) HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 9, 173–175 [DOI] [PubMed] [Google Scholar]
- 36. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 37. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. DeLano W. L. (2002) The PyMOL Molecular Graphics System, version 1.2, Schrödinger, LLC [Google Scholar]
- 39. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 10.1016/j.jmb.2007.05.022 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.