

Abstract
The relaxin family peptides have been shown to exert several beneficial effects on the heart, including anti-apoptosis, anti-fibrosis, and anti-hypertrophy activity. Understanding their regulation might provide new opportunities for therapeutic interventions, but the molecular mechanism(s) coordinating relaxin expression in the heart remain largely obscured. Previous work demonstrated a role for the orphan nuclear receptor Nur77 in regulating cardiomyocyte apoptosis. We therefore investigated Nur77 in the hopes of identifying novel relaxin regulators. Quantitative real-time PCR (qRT-PCR) and enzyme-linked immunosorbent assay (ELISA) data indicated that ectopic expression of orphan nuclear receptor Nur77 markedly increased the expression of latexin-3 (RLN3), but not relaxin-1 (RLN1), in neonatal rat ventricular cardiomyocytes (NRVMs). Furthermore, we found that the β-adrenergic agonist isoproterenol (ISO) markedly stimulated RLN3 expression, and this stimulation was significantly attenuated in Nur77 knockdown cardiomyocytes and Nur77 knockout hearts. We showed that Nur77 significantly increased RLN3 promoter activity via specific binding to the RLN3 promoter, as demonstrated by electrophoretic mobility shift assay (EMSA) and chromatin immunoprecipitation (ChIP) assays. Furthermore, we found that Nur77 overexpression potently inhibited ISO-induced cardiomyocyte apoptosis, whereas this protective effect was significantly attenuated in RLN3 knockdown cardiomyocytes, suggesting that Nur77-induced RLN3 expression is an important mediator for the suppression of cardiomyocyte apoptosis. These findings show that Nur77 regulates RLN3 expression, therefore suppressing apoptosis in the heart, and suggest that activation of Nur77 may represent a useful therapeutic strategy for inhibition of cardiac fibrosis and heart failure.
Keywords: transcription factor; nuclear receptor; cardiomyocyte; apoptosis; relaxin; isoprenoid; Nur77, relaxin-3; beta-adrenergic receptor; transcriptional regulator; promoter
Introduction
The relaxin peptide family in humans consists of seven members, relaxin-1 (H1 relaxin), relaxin-2 (H2 relaxin), relaxin-3 (H3 relaxin), and insulin-like peptides (1). Like insulin, relaxin consists of two peptide chains, A and B, covalently linked by disulfide bonds. Through binding to the relaxin family peptide receptors (RXFPs)3, the relaxin peptide has been shown to initiate a wide range of biological effects in various systems, including regulation of cell survival, proliferation, vessel relaxation, inflammation, and fibrosis (1–4). Humans (and higher primates) have three relaxin genes, designated H1, H2, and H3 relaxin, whereas rodents have two genes, relaxin (equivalent to H2 relaxin) and relaxin-3 (equivalent to H3 relaxin) (5). H2 relaxin is the major source of circulating relaxin. Four relaxin family peptide receptors (RXFP1–4) have been identified (6, 7). H2 relaxin is a ligand for receptors RXFP1 and RXFP2, whereas H3 relaxin is the ligand for RXFP3 but also cross-reacts with RXFP1 and RXFP4 (6). Despite the physiological significance of relaxin in the reproductive and central nervous systems, the cardiovascular effects of relaxin have recently received significant attention based on its vasodilatory, anti-apoptotic, anti-inflammatory, anti-fibrotic, and proangiogenic effects (3, 4, 8). It is becoming increasingly recognized that relaxin has important therapeutic applications in cardiac protection, fibrosis, and wound healing, as suggested in recent clinical trials (1, 4, 9). For instance, relaxin-2 has been shown to potently attenuate cardiac fibrosis, as evidenced by the cardiac fibrotic phenotype in relaxin-2–deficient mice and inhibition of established cardiac fibrosis following relaxin treatment (3, 10). In the animal models of ischemia and reperfusion, relaxin-2 has been reported to reduce myocardial injury and preserve ventricular function (11, 12). Of the three known relaxin genes, relaxin-2 is the only relaxin known to circulate in the blood. Indeed, the circulating and/or cardiac levels of relaxin-2 have been reported to be increased in patients with congestive heart failure, suggesting the pathological significance of relaxin peptide in the development and progression of heart disease (13–15).
The cardiac function of relaxin-3 is poorly understood relative to that known for relaxin-2, although it has been shown to be necessary for cardiomyocyte proliferation and heart regeneration in zebrafish (16). Relaxin-3 was initially discovered to be predominantly expressed in the brain and play a critical role in regulating arousal, feeding, learning, and central responses to physiological stressors (17–22). Recently, relaxin-3 has been shown to elicit potent anti-apoptotic, anti-fibrotic, and anti-hypertrophic effects in the heart (23–26). Importantly, through binding to RXFP1, relaxin-3 demonstrated a synergistic effect with relaxin-2 to inhibit cardiac fibrosis (23), further suggesting that relaxin-3 is an important peptide hormone implicated in the regulation of cardiac function.
Although the pathophysiological importance of relaxin-3 in the cardiovascular system is increasingly recognized, the molecular mechanism(s) underlying the regulation of relaxin-3 expression remains poorly understood. Recently, in an effort to identify the target genes of orphan nuclear receptor Nur77 by microarray analyses, we found that relaxin-3 is one of the genes most up-regulated by Nur77 in neonatal rat cardiomyocytes. Furthermore, we demonstrate that Nur77-mediated up-regulation of relaxin-3 expression represents an essential mechanism for the inhibition of isoproterenol-induced cardiomyocyte apoptosis.
Results
Up-regulation of relaxin-3 expression by Nur77 in cardiomyocytes
Accumulating evidence suggests that Nur77 plays essential roles in regulating cardiomyocyte hypertrophy and apoptosis (27–29). To further investigate the molecular mechanisms involved, we performed gene microarray to identify the molecular targets of Nur77 in cardiomyocytes. Our study identified relaxin-3 (RLN3) as one of the most up-regulated genes in NRVMs overexpressing Nur77 (Table 1). Although relaxin peptides have been shown to inhibit fibrosis and apoptosis in cardiomyocytes (15, 30), the expression of relaxin and its receptors in cardiomyocytes was less explored. To this end, we performed RT-PCR to examine the expression of relaxin/relaxin receptors in NRVMs. As shown in Fig. 1A, we found that NRVMs abundantly express relaxin-1 (RLN1), relaxin-3 (RLN3), and RXFP1–3, indicating that the relaxin peptides may exert their biological effects through an autocrine mechanism in cardiomyocytes. The expression of RLN3 is comparable with that of RLN1 in cardiomyocytes as determined by qRT-PCR (Fig. 1B). To further verify the microarray data, we measured the RLN3 expression in NRVMs transduced with adenovirus bearing either Nur77 (Ad-Nur77) or LacZ (Ad-LacZ). As shown in Fig. 1 (C and D), adenovirus-mediated overexpression of Nur77 robustly increased the expression of RLN3 in cardiomyocytes, as determined by both qRT-PCR and ELISA. Interestingly, the expression of RLN1 was barely affected, suggesting the specificity of Nur77 in the regulation of RLN3 in NRVMs. Moreover, NOR-1, another NR4A receptor expressed in the heart (31), also significantly increased RLN3 expression, albeit to a lesser extent, as compared with Nur77, in cardiomyocytes (Fig. 1E). Together, our results identified Nur77 as a potent stimulator of RLN3 expression in cardiomyocytes.
Table 1.
Genes up-regulated by Nur77 in cardiomyocytes
| Genes | Log2 (ratio) (Ad-Nur77/Ad-LacZ) | Putative function |
|---|---|---|
| Relaxin-3 (Rln3) | 6.22 | Anti-apoptosis and anti-fibrosis |
| Agouti-related protein (Agrp) | 6.10 | Food intake and energy metabolism |
| Hydroxysteroid 11-β-dehydrogenase 2 (Hsd11b2) | 5.20 | Glucorticoid metabolism |
| Transmembrane protease, serine 11d | 5.07 | Host defense |
| Family with sequence similarity 105, member A | 4.09 | Fat metabolism |
| Interleukin 27 receptor, α | 3.99 | Immune regulation |
| Creatine kinase, mitochondrial 2, sarcomeric | 3.70 | Mitochondrial function |
| Thrombomodulin | 3.35 | Anti-inflammation and anti-thrombosis |
| Coiled-coil domain–containing 3 | 3.30 | Fat metabolism |
| Integrin, β6 | 2.23 | Cell adhesion |
Figure 1.

Nur77 increases relaxin-3 expression in neonatal rat cardiomyocytes. A, expression of RLN1, RLN3, and RXFPs in NRVMs, as determined by RT-PCR; B, effects of orphan nuclear receptor Nur77 and NOR-1 on the expression of relaxin-3 in NRVMs. NRVMs were transduced with the indicated adenoviruses at an MOI of 50. 48 h after transduction, the expression of RLN3 was then determined by real-time PCR (n = 4); *, p < 0.05 compared with Ad-LacZ. B, amplicons of RLN1, RLN3, and Nur77 were detected by real-time PCR. C, Nur77 increases RLN3, but not RLN1, expression in a dose-dependent manner. NRVMs were transduced with the indicated MOIs of adenoviruses. 48 h after transduction, expression of RLN1 and RLN3 was determined by real-time PCR (n = 4); *, p < 0.05 compared with Ad-LacZ at MOI = 50. Expression of Nur77 and GAPDH was determined by Western blotting. D, increased levels of RLN3 in the supernatants of NRVMs transduced with Ad-Nur77. NRVMs were transduced with the indicated adenoviruses at MOI of 30. 48 h after transduction, the levels of RLN3 in the supernatants were determined by ELISA (n = 4). *, p < 0.05 compared with Ad-LacZ at MOI = 50. E, effects of Nur77, NOR1, and DN-Nur77 on the RLN3 expression in cardiomyocytes. NRVMs were transduced with the indicated adenoviruses at MOI = 30. 48 h after transduction, expression of RLN3 was determined by qRT-PCR (n = 4). *, p < 0.05 compared with Ad-LacZ. Error bars, S.D.
Nur77 up-regulates RLN3 expression in cardiomyocytes at transcriptional levels
As shown in Fig. 1B, the dominant negative Nur77 (DN-Nur77), which lacks the N-terminal activation function-1 (AF-1) domain of Nur77 (32), had no effect on RLN3 expression, indicating that The AF-1 domain is critically involved in Nur77-mediated up-regulated RLN3 expression in cardiomyocytes. In the nucleus, Nur77 has been shown to function as a transcription factor to regulate expression of target genes through binding to an NGFI-B response element (NBRE; AAAGGTCA) as monomers or to a Nur77 response element (TGATATTTX6AAATGCCA) as homodimers (32, 33). Indeed, when we searched the relaxin-3 promoter region for the existence of the consensus binding sites of Nur77, we found a conserved NBRE site located between bp −117 and −110 of the rat relaxin-3 promoter, which is highly conserved among humans, rats, and mice (Fig. 2A). We then examined whether or not this region is indeed responsible for Nur77-induced relaxin-3 transcription in cardiomyocytes by constructing luciferase reporter vectors bearing rat relaxin-3 promoter and its mutants (Fig. 2B). We found that Nur77 overexpression markedly increased the rat RLN3 promoter activity, as determined by promoter-driven luciferase assays in cardiomyocytes. Deletion or mutation of this NBRE completely abolished the Nur77-induced RLN3 promoter activity (Fig. 2C). To investigate whether Nur77 is recruited to the RLN3 promoter, we performed ChIP assays in NRVMs overexpressing Nur77. As shown in Fig. 2D, Nur77 was found to bind specifically to the RLN3 promoter region. These data demonstrate that Nur77 increases RLN3 expression through its specific binding to the NBRE of the RLN3 promoter.
Figure 2.

Nur77 increases relaxin-3 expression at transcriptional levels. A, localization of the NBRE in the promoter region of human, rat, and mouse RLN3; B, schematic representation of relaxin-3 promoter and its mutants. C, Nur77 increases RLN3 promoter activity in a dose-dependent manner. NRVMs cultured in 12-well plates were transiently transfected with 200 ng of RLN3 promoter luciferase reporter vector and 20 ng of pCMV-R.Luc, together with the indicated amount of Nur77 expression vector. 48 h after transfection, NRVMs were washed once with ice-cold PBS and then analyzed for luciferase reporter activities using the dual luciferase reporter assay system (Promega) (n = 4). *, p < 0.05 compared with Ad-LacZ group. D, Nur77 binds to the RLN3 promoter. NRVMs were transduced with the indicated adenoviruses at an MOI of 50. 48 h after transduction, the recruitment of Nur77 to the RLN3 promoter was determined by ChIP assays. Error bars, S.D.
ISO increases RLN3 expression in cardiomyocytes in a Nur77-dependent manner
ISO has previously shown to up-regulate RLN3 expression in the heart through a yet unknown mechanism (11). Our previous data demonstrated that ISO can potently up-regulate Nur77 expression in the heart (28), which prompted us to speculate that Nur77 may be responsible for the ISO-induced RLN3 expression in cardiomyocytes. Consistent with previous reports (28, 29), our results demonstrated that ISO markedly increases expression of both Nur77 and RLN3 in a time- and dose-dependent manner (Fig. 3, A and B). To further determine the molecular signaling pathways involved in ISO-induced RLN3 expression, NRVMs were pretreated with various kinase inhibitors 1 h before the ISO stimulation. As shown in Fig. 3C, ISO-induced RLN3 expression was completely inhibited by PKA inhibitor PKI, the calcium blocker, and the β-AR antagonist, suggesting that both PKA and intracellular calcium pathways are involved in the expression of RLN3 induced by β-AR stimulation in cardiomyocytes, which is consistent with the signaling pathways involved in ISO-induced Nur77 expression in cardiomyocytes (28). Similarly, PE, which has been shown to stimulate nuclear Nur77 expression in our previous study, also markedly increases RLN3 expression in cardiomyocytes, whereas ET-1, which was shown to increase Nur77 expression in the cytoplasm of cardiomyocytes in our recent study (28), had no significant effect on the RLN3 expression (Fig. 3D), further indicating the importance of the nuclear transcriptional activity of Nur77 in up-regulating RLN3 expression in cardiomyocytes.
Figure 3.

ISO up-regulates expression of Nur77 and RLN3 in cardiomyocytes. A, NRVMs cultured in 6-well plates were starved in serum-free medium for 48 h and then stimulated with ISO (10 μm) for the indicated times. The expression of Nur77 and RLN3 was determined by real-time PCR (n = 4). *, p < 0.05 compared with time at 0 h; #, p < 0.05 compared with time at 0 h. B, NRVMs cultured in 6-well plates were starved in serum-free medium for 48 h and then stimulated with indicated concentrations of ISO. The expression of Nur77 and RLN3 was determined at 1 and 6 h, respectively, after ISO stimulation by real-time PCR (n = 4). *, p < 0.05 compared with ISO at 0 μm; #, p < 0.05 compared with ISO at 0 μm. C, NRVMs cultured in 6-well plates were starved in serum-free medium for 48 h and then pretreated with the indicated inhibitors for 1 h and then stimulated with either 10 μm ISO or sterile 0.9% saline for 6 h. The expression of RLN3 was determined by real-time PCR; *, p < 0.05 compared with saline; #, p < 0.05 compared with ISO alone at 10 μm. D, NRVMs cultured in 6-well plates were starved in serum-free medium for 48 h and then stimulated with either 0.9% saline solution, 10 μm ISO, 10 μm PE, or 100 nm ET-1 for 6 h. The expression of RLN3 was then determined by real-time PCR (n = 4). *, p < 0.05 compared with saline. Error bars, S.D.
To further substantiate the significance of Nur77 in ISO-induced RLN3 expression in cardiomyocytes, we examined the effects of ISO stimulation on RLN3 promoter activity. As shown in Fig. 4A, ISO treatment dose-dependently increases the RLN3 promoter activity, whereas the deletion of the NBRE located between bp −117 and −110 of the rat RLN3 promoter significantly abolished the stimulatory effect of ISO on RLN3 promoter activity. Furthermore, ISO treatment significantly increased the binding of Nur77 to the RLN3 promoter, as determined by both EMSA and ChIP assays (Fig. 4, B and C). Together, these results suggest that Nur77 is a transcriptional activator that is indeed responsible for the ISO-induced RLN3 expression in cardiomyocytes.
Figure 4.
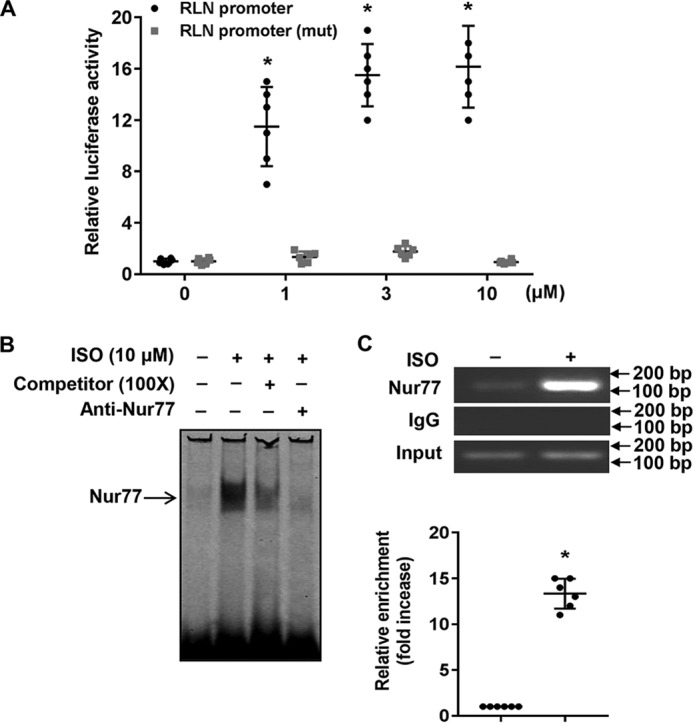
Nur77 is involved in the ISO-induced RLN3 expression in cardiomyocytes. A, NRVMs cultured in 12-well plates were transiently transfected with either 200 ng of RLN3 promoter or 400 ng of mutant luciferase reporter vector, 20 ng of pCMV-R.Luc. 48 h after transfection, cells were stimulated with the indicated concentration of ISO for 6 h. Cells were washed once with ice-cold PBS and then analyzed for luciferase activities using the Dual-Luciferase reporter assay system (Promega) (n = 6). *, p < 0.05 compared with ISO at 0 μm. B, NRVMs were stimulated with 10 μm ISO for 3 h, and the nuclear extracts (NE) were then isolated and used for EMSA to detect the interaction of Nur77 with the NBRE in the RLN3 promoter. A 100-fold excess of unlabeled competitor and anti-Nur77 antibody was used to demonstrate the specificity of the shifted complex. C, myocytes were stimulated with 10 μm ISO for 3 h, and the recruitment of Nur77 to the RLN3 promoter was determined by ChIP assays using anti-Nur77 antibody. *, p < 0.05 compared with ISO at 0 μm. Error bars, S.D.
Knockdown of Nur77 attenuates ISO-induced RLN3 expression both in vitro and in vivo
To further evaluate the role of the endogenous Nur77 in regulating RLN3 expression in cardiomyocytes, we performed a loss-of-function study by knockdown Nur77 expression. Indeed, transfection of cardiomyocytes with Nur77-specific siRNA (siNur77) markedly inhibited Nur77 expression, as determined by Western blotting (Fig. 5A). Accordingly, the ISO-induced RLN3 expression, as determined by qRT-PCR (Fig. 5B) and ELISA (Fig. 5C), was substantially attenuated in Nur77 knockdown cells. To examine whether Nur77 is involved in ISO-induced RLN3 expression in vivo, we collected the hearts from WT and Nur77 knockout mice at 12 h after a single intraperitoneal injection of ISO (1 mg/kg) to determine the expression of RLN3 by qRT-PCR. The mRNA expression of RLN3 in the hearts of WT mice was found to be substantially induced by ISO stimulation, with a maximal induction of ∼6-fold, whereas the ISO-induced RLN3 expression was markedly reduced in the hearts of Nur77-deficient mice (Fig. 5D). Taken together, these studies further highlight the importance of Nur77 in the ISO-induced RLN3 expression in the heart.
Figure 5.

Knockdown of Nur77 attenuates ISO-induced RLN3 expression in cardiomyocytes. A, NRVMs were transfected with 30 nm either Nur77 siRNA (siNur77) or control siRNA (siCTL). 72 h after transfection, the expression of Nur77 was determined by Western blotting (n = 4). *, p < 0.05 compared with siCTL. B, NRVMs were transfected with 30 nm either siNur77 or siCTL. 72 h after transfection, cells were stimulated with the indicated concentrations of ISO for 6 h. The expression of RLN3 was then determined by qRT-PCR (n = 4). *, p < 0.05 compared with ISO at 0 μm. C, Nur77 knockdown attenuated ISO-induced RLN3 levels in the supernatants of cardiomyocytes. NRVMs were transfected with 30 nm either siNur77 or siCTL. 48 h after transfection, cardiac cells were stimulated with 10 μm ISO for 48 h. The levels of RLN3 in the supernatants were determined by ELISA (n = 4). *, p < 0.05 compared with cardiomyocytes transfected with siCTL. D, adult WT and Nur77 knockout mice (Nur77 KO) were treated with a single intraperitoneal injection of ISO (1 mg/kg) or 0.9% saline (Veh), and 12 h after injection of ISO, the hearts were harvested for extraction of total RNAs. The expression of RLN3 was then determined by qRT-PCR. *, p < 0.05 versus saline-treated WT mice (Veh); #, p < 0.05 versus ISO-treated WT mice. The data represent three independent experiments with five mice in each group. Error bars, S.D.
RLN3 is essential for Nur77-mediated suppression of ISO-induced cardiomyocyte apoptosis
To substantiate the functional significance of the Nur77/RLN3 axis in cardiomyocyte biology, we investigated the effects of Nur77 and RLN3 on ISO-induced cardiomyocyte apoptosis by performing both gain- and loss-of-function studies. As shown in Fig. 6 (A and B), treatment of cardiomyocytes with ISO significantly induced cardiomyocyte apoptosis, as determined by both terminal deoxynucleotidyltransferase-mediated dUTP nick-end labeling (TUNEL) staining and cell death ELISA. Furthermore, knockdown of Nur77 markedly promoted ISO-induced cardiomyocyte apoptosis, suggesting the critical roles of endogenous Nur77 in suppressing cardiomyocyte apoptosis. Moreover, treatment of cardiomyocytes with recombinant RLN3 (100 ng/ml) substantially prevented ISO-induced apoptosis in both control siRNA– and Nur77 siRNA–transfected cardiomyocytes. To further investigate the role of RLN3 in suppressing ISO-induced cardiomyocyte apoptosis by Nur77, we performed a loss-of-function study by knockdown RLN3 in cardiomyocytes. As shown in Fig. 7A, transfection of cardiomyocytes with RLN3-specific siRNA inhibited RLN3 expression by ∼90%, without an effect on RLN1 expression (data not shown), as determined by qRT-PCR. As shown in Fig. 7 (B–D), adenovirus-mediated overexpression of Nur77 markedly attenuated ISO-induced cardiomyocyte apoptosis in control siRNA (siCTL)-transfected cells, as determined by TUNEL staining, cell death ELISA, and cleavage of caspase-3. The protective effect of Nur77 overexpression on ISO-induced cell apoptosis, however, was substantially weakened in RLN3 siRNA–transfected cardiomyocytes. Taken together, these findings further suggest that RLN3 is an essential mediator involved in Nur77-induced protection against ISO-induced cardiac injury.
Figure 6.
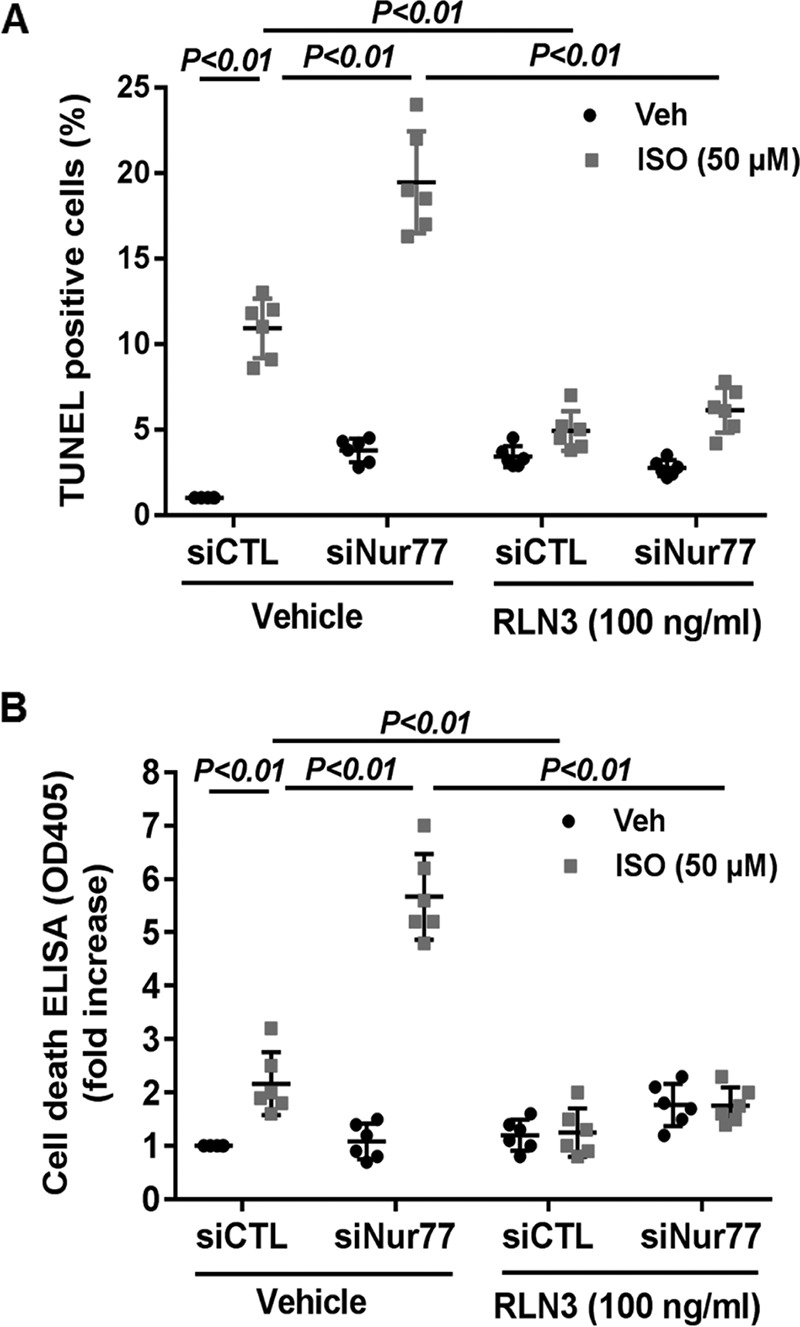
Nur77 knockdown augments ISO-induced cardiomyocyte apoptosis, which was prevented by RLN3. NRVMs were transfected with 30 nm either siNur77 or siCTL. 24 h after transfection, myocytes were cultured in the serum-free culture condition for 24 h and stimulated with 50 μm ISO in the presence and absence of 100 ng/ml recombinant RLN3 for 48 h. Cardiac myocyte apoptosis was then determined by TUNEL staining (A) and cell death ELISA kit (B). The data represent six independent experiments. Error bars, S.D.
Figure 7.

Nur77-mediated inhibition of ISO-induced cardiomyocyte apoptosis is attenuated in RLN3 knockdown cardiomyocytes. A, cardiomyocytes were transfected with 30 nm either RLN3 siRNA (siRLN) or siCTL. 72 h after transfection, expression of RLN3 was determined by qRT-PCR (n = 4). *, p < 0.05 compared with vehicle-treated siCTL-transfected cells; #, p < 0.05 compared with ISO-treated siCTL-transfected cells. B, cardiomyocytes were transfected with 30 nm either siRLN or siCTL. 24 h after transfection, myocytes were transduced with either Ad-LacZ or Ad-Nur77 at an MOI of 50 in the serum-free medium for 24 h and then stimulated with either 50 μm ISO or vehicle for 48 h. Myocyte apoptosis was then determined by TUNEL (B) and cell death detection ELISA (C). The data represent six independent experiments. D, cardiomyocytes were transfected with 30 nm either siRLN or siCTL. 24 h after transfection, myocytes were transduced with either Ad-LacZ or Ad-Nur77 at an MOI of 50 in the serum-free medium for 24 h and then stimulated with either 50 μm ISO or 0.9% saline for 48 h. The levels of cleaved caspase-3 (c-Casp 3) were determined by Western blotting and quantitated by densitometric analysis. The data represent three independent experiments. Error bars, S.D.
Discussion
NR4A receptors are immediate-early genes that are regulated by various physiological stimuli, including growth factors, hormones, and inflammatory signals in the cardiovascular system (34, 35). An increasing number of studies have demonstrated that NR4A receptors play important roles in the development of various cardiovascular diseases, including atherosclerosis, restenosis, angiogenesis, and heart failure (32, 34, 36). Previously, our studies identified Nur77 as a novel negative regulator for the β-AR–induced cardiac hypertrophy through inhibiting the NFATc3 and GATA4 transcriptional pathways (28). Here, we provide further evidence highlighting the critical importance of Nur77 in regulating cell survival and relaxin-3 expression in cardiomyocytes.
RLN3 was originally identified as an abundant neuropeptide in the brain, and it has been shown to possess a variety of biological functions, including regulation of arousal and behavioral activation, appetite regulation, stress responses, anxiety, memory, sleep, and circadian rhythm (18, 22, 37, 38). Recently, it has been increasingly recognized that RLN3 exerts cardiac protective effects via its anti-fibrotic, anti-hypertrophic, anti-inflammatory, and vasodilatory actions (23, 26). For instance, in zebrafish, relaxin-3 has been shown to be essentially involved in regulating cardiomyocyte proliferation and heart regeneration (16). Furthermore, pharmacological activation of the RLN3/RXFP3 pathway has been shown to exert potent anti-apoptotic, anti-fibrotic, and anti-hypertrophic effects in the heart (26). Indeed, relaxin peptides and their receptors have been shown to be expressed in the mouse and human heart. However, the molecular mechanism underlying the RLN3 expression in the heart remains largely unknown. Consistent with previous reports (2, 6), we found that RLN1, RLN3, and their receptors, such as RXFP1, -2, and -3, are highly expressed in neonatal rat cardiomyocytes, suggesting that the relaxin/relaxin receptor system may represent an important signaling pathway in the regulation of various biological effects, such as proliferation, hypertrophy, inflammation, and angiogenesis in the heart. Whether relaxin exerts biological function in the heart through autocrine and/or paracrine mechanisms, however, warrants further investigation.
The NR4A subfamily consists of three well-conserved members, Nur77 (NR4A1), Nurr1 (NR4A2), and NOR-1 (NR4A3), respectively (35). Like other nuclear receptors, NR4A receptors consist of an N-terminal transactivation domain, a central two-zinc-finger DNA-binding domain, and a C-terminal ligand-binding domain (39). So far, no ligands have been identified for these receptors, and therefore, they are classified as orphan receptors (35). Recently, NR4A receptors have been shown to play essential roles in the development of atherosclerosis, restenosis, fibrosis, and angiogenesis (34, 40–42). However, the functional role of NR4A receptors in cardiomyocyte biology remains largely unknown, although several studies have suggested that the NR4A receptors are highly expressed in the heart and that their expression is regulated by several external pathological stimuli, such as oxidative stress, pressure overload, and β-AR activation (27, 28, 32, 43). Previously, we have demonstrated that in response to ISO stimulation, Nur77 expression is substantially increased, and overexpression of Nur77 markedly attenuates β-AR–induced cardiac hypertrophy and heart failure through inhibiting the NFATc3 and GATA4 transcriptional pathways (28). In the present study, we add to our understanding of how Nur77 is able to circumvent cardiomyocyte apoptosis in response to the β-AR stimulation. Our results show that Nur77 functions as a potent inhibitor of cardiomyocyte apoptosis, at least in part, through increasing the expression of relaxin-3 in the heart. Depending on its intracellular localization, Nur77 can exert the biological effects through genomic and nongenomic effects. Previously, we have demonstrated that in the nucleus of cardiomyocytes, Nur77 specifically interacts with GATA4 and NFATc3, and this nongenomic effect is mainly responsible for the inhibition of both ISO- and PE-induced cardiac hypertrophy (28). In the present study, we found that Nur77 specifically binds to the NBRE located in the relaxin-3 promoter region, as demonstrated by both EMSA and ChIP assays, to increase RLN3 promoter activity. Furthermore, DN-Nur77, which is defective in its transactivation domain (32), failed to increase relaxin-3 expression, further suggesting that the transcriptional activity of Nur77 is mainly responsible for the stimulatory effects on relaxin-3 expression in cardiomyocytes. In this regard, our study for the first time identified Nur77 as an essential transcriptional factor implicated in the regulation of relaxin-3 in cardiac cells.
Overall, the data reported herein provide evidence that the orphan nuclear receptor Nur77 is a novel inhibitor of cardiomyocyte apoptosis in response to β-AR stimulation, at least in part, through increasing RLN3 expression in the heart. Chronic stimulation of the β-AR has been shown to contribute significantly to the development of progressive cardiac dysfunction and cardiac remodeling in patients (44). Identification of novel regulators of β-AR signaling in cardiomyocytes will be of critical importance for a better treatment of patients with heart failure. In this regard, Nur77, which has been shown to inhibit both cardiomyocyte apoptosis and hypertrophy, may represent a novel potential therapeutic target for the treatment of ischemic heart disease and heart failure.
Experimental procedures
Primary culture of neonatal rat ventricular cardiomyocytes (NRVMs)
We obtained ventricles from 1-day-old Sprague–Dawley rats and isolated cardiac myocytes through digestion with trypsin-EDTA and type 2 collagenase as described previously (28). The protocol was approved by the institutional animal care and use committee of the Thomas Jefferson University. Briefly, the tissues were cut into small pieces and digested with 0.25% trypsin at 4 °C overnight. Collagenase (Worthington) (1 mg/ml in Hanks' balanced salt solution) was used to further digest tissues in a shaking bath at 37 °C for 20 min. The cell suspension was centrifuged at 1,000 rpm for 5 min and resuspended in Dulbecco's modified Eagle's medium with 1 g/liter glucose and 10% fetal bovine serum (Gibco). Cells were cultured for 2 h to allow fibroblast cells to attach to the flask. NRVMs were collected from the supernatants and cultured with Dulbecco's modified Eagle's medium containing 1 g/liter glucose plus 10% fetal bovine serum and 1% penicillin-streptomycin (Gibco).
Mice
Nur77 knockout mice were purchased from Jackson Laboratory (Bar Harbor, ME), and animals were maintained on a C57BL/6 and 129SvJ hybrid background. Nur77+/− mice were crossed to obtain the WT and knockouts. WT and Nur77 knockouts (10–12 weeks of age, 5–6 mice/group) were subjected to a single intraperitoneal injection of ISO (1 mg/kg) (Sigma). 12 h after injection, hearts were then collected, and total RNA was extracted by using TRIzol reagent (Gibco/BRL) according to the manufacturer's instruction. The expression of relaxin-3 was then quantitated by qRT-PCR.
Adenovirus construction
Adenoviruses harboring WT FLAG-tagged Nur77 (Ad-Nur77), dominant-negative FLAG-tagged Nur77 lacking the AF-1 domain (Ad-DN-Nur77), and NOR-1 (Ad-NOR1) were made using AdMax (Microbix) as described previously (32). Briefly, pBHGlox E1,3Cre, including the E1 adenoviral genome, was cotransfected with the pDC shuttle vector containing the gene of interest into Ad293 cells using FuGene 6 transfection reagent (Roche Applied Science). The viruses were propagated in Ad293 cells and purified using CsCl2 banding followed by dialysis against 20 mmol/liter TBS with 10% glycerol. Titering was performed on Ad293 cells using the Adeno-X Rapid Titer kit (Clontech) according to the instructions of the manufacturer.
Electrophoretic mobility shift assay (EMSA)
NRVMs were stimulated with 10 μm ISO for 3 h, and nuclear extracts were then prepared by differential centrifugation of cell homogenates as described previously (28). Briefly, cells were homogenized manually in hypotonic buffer (10 mmol/liter Tris-HCl (pH 7.5), 1 mmol/liter MgCl2, 1 mmol/liter EGTA, 1 mmol/liter EDTA, 1 mmol/liter phenylmethylsulfonyl fluoride, 5 μg/ml leupeptin, and 5 μg/ml aprotinin). The nuclear fraction was obtained from low-speed centrifugation (500 × g), followed by three washes with PBS. For EMSA, the nuclear fraction was dissolved in hypertonic buffer (20 mmol/liter HEPES (pH 7.9), 420 mmol/liter NaCl, 1.5 mmol/liter MgCl2, 0.2 mmol/liter EDTA, 25% glycerol, and the above-mentioned protease inhibitors). After high-speed centrifugation at 12,800 × g for 10 min, the supernatants (nuclear extracts) were isolated to detect the interaction of Nur77 with the NBRE in the rat RLN3 promoter by EMSA. The oligonucleotides corresponding to the binding sequence of Nur77 (NBRE; 5′-GGTAAAGGTCAGGTTGC-3′) in the rat RLN3 promoter were synthesized and labeled with IRDye 700 (IDT, Coralville, IA). EMSA was performed with Odyssey® IRDye® 700 IR dye–labeled double-stranded oligonucleotides coupled with the EMSA buffer kit (LI-COR Biosciences) according to the manufacturer's instructions. The specificity of the binding was examined using competition experiments, where a 100-fold excess of the unlabeled oligonucleotides was added to the reaction mixture before adding the IR dye–labeled oligonucleotide as we described previously (33).
siRNA experiments
Rat Nur77 siRNA was purchased from Santa Cruz Biotechnology, Inc. (catalog no. sc-108068), and rat RLN3 siRNAs and SilencerTM negative control siRNA were purchased from Thermo Fisher Scientific (catalog nos. AM16708, 1330001, and AM4611). Nur77 siRNA, RLN3 siRNA, and SilencerTM negative control siRNA were transfected into cardiomyocytes seeded in 6-well plates by using Lipofectamine RNAiMAX transfection reagent (Invitrogen) according to the recommendations of the manufacturer. In our studies, we routinely employed three siRNAs (or more) targeting different regions of genes to ensure the specificity of knockdown studies. The concentration of siRNAs used was 30 nm. 72 h after transfection of siRNAs, knockdown efficiency was verified by either Western blotting or qRT-PCR.
Immunoblotting
Cell lysates were made using radioimmune precipitation assay buffer (Thermo Scientific) containing 25 mm Tris-HCl, pH 7.6, 150 mm NaCl, 1% Nonidet P-40, 1% sodium deoxycholate, 0.1% SDS, and proteinase inhibitor mixture containing 2 mm phenylmethylsulfonyl fluoride, 20 μg/ml aprotinin, and 10 μg/ml leupeptin. Supernatants were resolved by SDS-PAGE and transferred to nitrocellulose (Bio-Rad). Blots were blocked with 5% nonfat milk in PBS with 0.1% Tween 20 (PBST) and then developed with diluted antibodies Nur77 (1:500 dilution; catalog no. sc-365113, Santa Cruz Biotechnology), cleaved caspase-3 (1:500 dilution; catalog no. 96645, Cell Signaling Technology), and GAPDH (1:2000 dilution; catalog no. sc-47724, Santa Cruz Biotechnology) at 4 °C overnight, followed by incubation with goat anti-rabbit IgG (H + L) (DyLight 680–conjugated, Thermo Scientific) or goat anti-mouse IgG (H + L) (DyLight 800–conjugated, Thermo Scientific) for 1 h. Blots were visualized on an Odyssey imaging system (LI-COR Biosciences). The intensity of the bands was quantified by using the Odyssey software.
qRT-PCR
Total RNA was extracted from cells and treated with DNase I using the RNeasy® Micro kit (Qiagen). mRNA levels of RLN3 and Nur77 were determined by qRT-PCR using cDNA obtained from the reverse transcription reactions as template, with the MyiQTM single-color real-time PCR detection system (Bio-Rad) and HotStart-IT SYBR Green one-step qRT-PCR master mix kit (Affymetrix). The primer sequences were as follows: rat RLN3 (NM_170667), 5′-GCGGTCGGGAGTTCATC-3′ (forward) and 5′-GGTGGTCTGTATTGGCTTCT-3′ (reverse); rat Nur77 (NM_024388), 5′-CTAACACTGCCAAGTTGGACTA-3′ (forward) and 5′-GAGCCAGAGAGCAAGTCATAAA-3′ (reverse); mouse RLN3 (NM_173184), 5′-CGCTGATGGAGAAGCCAATA-3′ (forward) and 5′-GCTCGACCACCGTAGAAAG-3′ (reverse); and 18S, 5′-TCAAGAACGAAAGTCGGAGG-3′ (forward) and 5′-GGACATCTAAGGGCATCAC-3′ (reverse). 18S mRNA served as a control for the amount of cDNA present in each sample. Data were analyzed using the comparative difference in cycle number (ΔCT) method according to the manufacturer's instructions. Protein kinase A inhibitor PKI (Santa Cruz Biotechnology), protein kinase C inhibitor GF109203X (Tocris), phospholipase C inhibitor U-73122 (Tocris), calcium channel blocker nifedipine (Tocris), and β-adrenergic receptor antagonist propranolol (Sigma) were used to define the molecular signaling pathways mediating ISO-induced RLN3 expression in cardiomyocytes.
Reverse transcription-PCR
Total RNA was isolated from cardiomyocytes, and cDNAs were synthesized using random hexamers and Moloney murine leukemia virus reverse transcriptase (Invitrogen) (33). The cDNAs obtained were then amplified by PCR using gene-specific primers for rat RLN1, RLN3, and RXFP1–3. The primer sequences were as follows: rat RLN1 (NM_013413), 5′-GAGCCTTTCGATATGACGTTGA-3′ (forward) and 5′-GGATCTTCTGGTACAACCGATG-3′ (reverse); rat RLN3 (NM_170667), 5′-GCTGATGGAGAAGCCAATACA-3′ (forward) and 5′-GATCCTAGCACAAGCTGCTAAT-3′ (reverse); rat RXFP1 (NM_201417), 5′-GACAACAATGGGTGGTCTCTAC-3′ (forward) and 5′-CCCTGGCTTTAGGAAGGTTATT-3′ (reverse); rat RXFP2 (NM_001012475), 5′-CGTCCAACCCTCTTCTGTATG-3′ (forward) and 5′-GGACATAGCGTGAGTCGTATTC-3′ (reverse); and rat RXFP3 (NM_001008310), 5′-GACCATCGTAGTCCTTTCCTTC-3′ (forward) and 5′-ACACCGGGTGGATAGTAGAT-3′ (reverse).
Measurement of rat RLN3 by ELISA
Neonatal rat cardiomyocytes were transduced with Ad-LacZ and Ad-Nur77 for 48 h as described above. Supernatants were then collected and concentrated, and the levels of rat RLN3 in the supernatants were quantitated by the rat relaxin 3 (RLN3) ELISA kit supplied by MyBioSource. The assay was performed essentially according to the protocol provided by the manufacturer.
Transient transfection and luciferase assay
The rat RLN3 promoter was amplified by PCR using rat genomic DNA (Clontech) with primers (forward, 5′-GAGACTCGAGACTCCTATAATCCCAGCAC-3′; reverse, 5′-GAGAAAGCTTCGAGGAGAGCCCAGGAA-3′) and then cloned into the luciferase reporter plasmid pGL3-Basic (Promega) XhoI and HindIII sites. The rat RLN3 promoter deletion mutant (−110 to +70) pGL3-Basic was constructed by using flowing primers: forward, 5′-GAGACTCGAGGGTTGCCAACTGCCTCTCCTC-3′; reverse, 5′-GAGAAAGCTTCCGAGGAGAGCCCAGGAA-3′. The rat RLN3 promoter mutant cloned into pGL3-Basic was constructed using QuikChange II site-directed mutagenesis kit (Agilent Technologies), using the following PCR primers: 5′-GGCAAGGAGATGGTAAAAATCAGGTTGCCAAC-3′ and 5′-GTTGGCAACCTGATTTTTACCATCTCCTTGCC-3′. NRVMs seeded in 12-well plates were transfected with 200 ng of pGL3-RLN3 promoter luciferase reporter and 20 ng of pCMV-R.Luc transfection control plasmid with increasing concentrations of Nur77 expression vector using LipofectamineTM LTX (Invitrogen) transfection reagent. 36 h after transfection, cells were then treated with or without 10 μm ISO for 24 h and then directly lysed in the lysis buffer (Promega). 20 μl of the cell lysates were assayed for luciferase activity with a luciferase reporter assay system (Promega) and determined with a Synergy 2 multimode microplate reader (Bio-Tek) according to the manufacturer's instructions.
ChIP assays
ChIP assays were performed using a ChIP assay kit (Upstate) according to the instructions of the manufacturer. Soluble chromatin was prepared from neonatal rat cardiomyocytes stimulated with ISO (10 μm) for 3 h or cells transduced with either Ad-LacZ or Ad-Nur77 (MOI = 100) for 48 h. DNA-bound Nur77 subunit was immunoprecipitated by incubating with antibody directed against Nur77 or normal IgG (5 μg; Cell Signaling) overnight at 4 °C with rotation. After reversal of cross-links and digestion of bound proteins, the recovered DNA was quantified by PCR amplified using primer pairs that cover NBRE in the rat RLN3 promoter as follows: sense, 5′-GGAAGATGACAGGGAGCTG-3′; antisense, 5′-CTGACCTGGGTTATCTTCTGAG-3′.
TUNEL staining and cell death ELISA
A TUNEL assay was performed using the In Situ Cell Death Detection kit (Roche Applied Science) as we described previously. Briefly, after drug treatment, cardiomyocytes were fixed with 4% paraformaldehyde solution for 30 min at room temperature. After washing with PBS, cells were incubated with 0.3% H2O2 for 30 min at room temperature to block endogenous peroxidase activity. After incubation in the TUNEL reaction mixture for 60 min, the cells were visualized by microscopy (Nikon, Tokyo, Japan). Cell death ELISA was performed by using Cell Death Detection ELISA kit (Roche Applied Science). Briefly, the cytoplasmic fractions were added to the 96-well ELISA plates precoated with the antihistone mAb and incubated for 90 min at room temperature. After washing, bound nucleosomes were detected by the addition of anti-DNA peroxidase mAb and reacted for 60 min at room temperature. After the addition of substrate, absorbance was read with an ELISA reader at 405 nm.
Statistical analyses
All experiments were performed at least three times. All statistical analysis was performed using the SPSS statistical analysis program (version 17.0), and graphs were generated using GraphPad Prism software (version 5.0.1). All data are presented as mean ± S.D. For relative data analysis, the mean value of the control group is defined as 1 or as 100%. Differences between two groups were compared with the unpaired Student's t test. Differences between multiple groups were compared with one-way analysis of variance. A p value < 0.05 was considered to be significant.
Author contributions
X. Y., Z.-F. G., F. C., B. Y., N. Z., X. Z., X.-L. M., and J. S. designed the experiments. X. Y., Z.-F. G., F. C., B. Y., F. Y., X. L., N. Z., and G. Y. executed the experiments and analyzed the data. X. Y., Z.-F. G., and J. S. conceived the study. X. Y., Z.-F. G., and J. S. wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dr. Ross Summer and Xiaobo Sun for comments on the manuscript.
This work was supported by National Institutes of Health Grants R01HL103869 and R01GM123047, American Heart Association Established Investigator Award 16EIA27710023, and Chinese Natural Science Foundation Grant 81370418 (to J. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- RXFP
- relaxin family peptide receptor
- RLN
- relaxin
- β-AR
- β-adrenergic receptor
- NRVMs
- neonatal rat ventricular cardiomyocytes
- ISO
- isoproterenol
- PE
- phenylephrine
- ET-1
- endothelin-1
- EMSA
- electrophoretic mobility shift assay
- TUNEL
- terminal deoxynucleotidyltransferase dUTP nick-end labeling
- NBRE
- NGFI-B response element
- qRT-PCR
- quantitative real-time polymerase chain reaction
- GAPDH
- glyceraldehyde 3-phosphate dehydrogenase
- DN
- dominant negative
- AF-1
- activation function-1
- MOI
- multiplicity of infection
- siCTL
- control siRNA
- siNur77
- Nur77-specific siRNA
- siRLN
- RLN3 siRNA.
References
- 1. Samuel C. S., Du X. J., Bathgate R. A., and Summers R. J. (2006) “Relaxin” the stiffened heart and arteries: the therapeutic potential for relaxin in the treatment of cardiovascular disease. Pharmacol. Ther. 112, 529–552 10.1016/j.pharmthera.2005.05.012 [DOI] [PubMed] [Google Scholar]
- 2. Nistri S., Bigazzi M., and Bani D. (2007) Relaxin as a cardiovascular hormone: physiology, pathophysiology and therapeutic promises. Cardiovasc. Hematol. Agents Med. Chem. 5, 101–108 10.2174/187152507780363179 [DOI] [PubMed] [Google Scholar]
- 3. Samuel C. S., Bodaragama H., Chew J. Y., Widdop R. E., Royce S. G., and Hewitson T. D. (2014) Serelaxin is a more efficacious antifibrotic than enalapril in an experimental model of heart disease. Hypertension 64, 315–322 10.1161/HYPERTENSIONAHA.114.03594 [DOI] [PubMed] [Google Scholar]
- 4. Teerlink J. R., Cotter G., Davison B. A., Felker G. M., Filippatos G., Greenberg B. H., Ponikowski P., Unemori E., Voors A. A., Adams K. F. Jr., Dorobantu M. I., Grinfeld L. R., Jondeau G., Marmor A., Masip J., et al. (2013) Serelaxin, recombinant human relaxin-2, for treatment of acute heart failure (RELAX-AHF): a randomised, placebo-controlled trial. Lancet 381, 29–39 10.1016/S0140-6736(12)61855-8 [DOI] [PubMed] [Google Scholar]
- 5. Bathgate R. A., Samuel C. S., Burazin T. C., Gundlach A. L., and Tregear G. W. (2003) Relaxin: new peptides, receptors and novel actions. Trends Endocrinol. Metab. 14, 207–213 10.1016/S1043-2760(03)00081-X [DOI] [PubMed] [Google Scholar]
- 6. van der Westhuizen E. T., Halls M. L., Samuel C. S., Bathgate R. A., Unemori E. N., Sutton S. W., and Summers R. J. (2008) Relaxin family peptide receptors: from orphans to therapeutic targets. Drug Discov. Today 13, 640–651 10.1016/j.drudis.2008.04.002 [DOI] [PubMed] [Google Scholar]
- 7. Kamat A. A., Feng S., Bogatcheva N. V., Truong A., Bishop C. E., and Agoulnik A. I. (2004) Genetic targeting of relaxin and insulin-like factor 3 receptors in mice. Endocrinology 145, 4712–4720 10.1210/en.2004-0515 [DOI] [PubMed] [Google Scholar]
- 8. Teichman S. L., Unemori E., Dschietzig T., Conrad K., Voors A. A., Teerlink J. R., Felker G. M., Metra M., and Cotter G. (2009) Relaxin, a pleiotropic vasodilator for the treatment of heart failure. Heart Fail. Rev. 14, 321–329 10.1007/s10741-008-9129-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Felker G. M., Teerlink J. R., Butler J., Hernandez A. F., Miller A. B., Cotter G., Davison B. A., Filippatos G., Greenberg B. H., Ponikowski P., Voors A. A., Hua T. A., Severin T. M., Unemori E., and Metra M. (2014) Effect of serelaxin on mode of death in acute heart failure: results from the RELAX-AHF study. J. Am. Coll. Cardiol. 64, 1591–1598 10.1016/j.jacc.2014.05.071 [DOI] [PubMed] [Google Scholar]
- 10. Samuel C. S., Zhao C., Bathgate R. A., DU X. J., Summers R. J., Amento E. P., Walker L. L., McBurnie M., Zhao L., and Tregear G. W. (2005) The relaxin gene-knockout mouse: a model of progressive fibrosis. Ann. N.Y. Acad. Sci. 1041, 173–181 10.1196/annals.1282.025 [DOI] [PubMed] [Google Scholar]
- 11. Zhang J., Qi Y. F., Geng B., Pan C. S., Zhao J., Chen L., Yang J., Chang J. K., and Tang C. S. (2005) Effect of relaxin on myocardial ischemia injury induced by isoproterenol. Peptides 26, 1632–1639 10.1016/j.peptides.2005.02.008 [DOI] [PubMed] [Google Scholar]
- 12. Nonhoff J., Ricke-Hoch M., Mueller M., Stapel B., Pfeffer T., Kasten M., Scherr M., von Kaisenberg C., Bauersachs J., Haghikia A., and Hilfiker-Kleiner D. (2017) Serelaxin treatment promotes adaptive hypertrophy but does not prevent heart failure in experimental peripartum cardiomyopathy. Cardiovasc. Res. 113, 598–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dschietzig T., Richter C., Bartsch C., Laule M., Armbruster F. P., Baumann G., and Stangl K. (2001) The pregnancy hormone relaxin is a player in human heart failure. FASEB J. 15, 2187–2195 10.1096/fj.01-0070com [DOI] [PubMed] [Google Scholar]
- 14. Xie J., Chen Y., Li L., and Zhang S. (2015) H2 relaxin expression and its effect on clinical outcomes in patients with chronic heart failure. Int. J. Clin. Exp. Med. 8, 4420–4424 [PMC free article] [PubMed] [Google Scholar]
- 15. Ghosh R. K., Banerjee K., Tummala R., Ball S., Ravakhah K., and Gupta A. (2017) Serelaxin in acute heart failure: most recent update on clinical and preclinical evidence. Cardiovasc. Ther. 35, 55–63 10.1111/1755-5922.12231 [DOI] [PubMed] [Google Scholar]
- 16. Fang Y., Gupta V., Karra R., Holdway J. E., Kikuchi K., and Poss K. D. (2013) Translational profiling of cardiomyocytes identifies an early Jak1/Stat3 injury response required for zebrafish heart regeneration. Proc. Natl. Acad. Sci. U.S.A. 110, 13416–13421 10.1073/pnas.1309810110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Soloff M. S., Gal S., Hoare S., Peters C. A., Hunzicker-Dunn M., Anderson G. D., and Wood T. G. (2003) Cloning, characterization, and expression of the rat relaxin gene. Gene 323, 149–155 10.1016/j.gene.2003.09.015 [DOI] [PubMed] [Google Scholar]
- 18. Bathgate R. A., Samuel C. S., Burazin T. C., Layfield S., Claasz A. A., Reytomas I. G., Dawson N. F., Zhao C., Bond C., Summers R. J., Parry L. J., Wade J. D., and Tregear G. W. (2002) Human relaxin gene 3 (H3) and the equivalent mouse relaxin (M3) gene: novel members of the relaxin peptide family. J. Biol. Chem. 277, 1148–1157 10.1074/jbc.M107882200 [DOI] [PubMed] [Google Scholar]
- 19. McGowan B. M., Stanley S. A., Smith K. L., White N. E., Connolly M. M., Thompson E. L., Gardiner J. V., Murphy K. G., Ghatei M. A., and Bloom S. R. (2005) Central relaxin-3 administration causes hyperphagia in male Wistar rats. Endocrinology 146, 3295–3300 10.1210/en.2004-1532 [DOI] [PubMed] [Google Scholar]
- 20. Sudo S., Kumagai J., Nishi S., Layfield S., Ferraro T., Bathgate R. A., and Hsueh A. J. (2003) H3 relaxin is a specific ligand for LGR7 and activates the receptor by interacting with both the ectodomain and the exoloop 2. J. Biol. Chem. 278, 7855–7862 10.1074/jbc.M212457200 [DOI] [PubMed] [Google Scholar]
- 21. McGowan B. M., Stanley S. A., Smith K. L., Minnion J. S., Donovan J., Thompson E. L., Patterson M., Connolly M. M., Abbott C. R., Small C. J., Gardiner J. V., Ghatei M. A., and Bloom S. R. (2006) Effects of acute and chronic relaxin-3 on food intake and energy expenditure in rats. Regul. Pept. 136, 72–77 10.1016/j.regpep.2006.04.009 [DOI] [PubMed] [Google Scholar]
- 22. McGowan B. M., Stanley S. A., Donovan J., Thompson E. L., Patterson M., Semjonous N. M., Gardiner J. V., Murphy K. G., Ghatei M. A., and Bloom S. R. (2008) Relaxin-3 stimulates the hypothalamic-pituitary-gonadal axis. Am. J. Physiol. Endocrinol. Metab. 295, E278–E286 10.1152/ajpendo.00028.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hossain M. A., Man B. C., Zhao C., Xu Q., Du X. J., Wade J. D., and Samuel C. S. (2011) H3 relaxin demonstrates antifibrotic properties via the RXFP1 receptor. Biochemistry 50, 1368–1375 10.1021/bi1013968 [DOI] [PubMed] [Google Scholar]
- 24. Zhang X., Ma X., Zhao M., Zhang B., Chi J., Liu W., Chen W., Fu Y., Liu Y., and Yin X. (2015) H2 and H3 relaxin inhibit high glucose-induced apoptosis in neonatal rat ventricular myocytes. Biochimie 108, 59–67 10.1016/j.biochi.2014.11.004 [DOI] [PubMed] [Google Scholar]
- 25. Zhang X., Fu Y., Li H., Shen L., Chang Q., Pan L., Hong S., and Yin X. (2018) H3 relaxin inhibits the collagen synthesis via ROS- and P2X7R-mediated NLRP3 inflammasome activation in cardiac fibroblasts under high glucose. J. Cell. Mol. Med. 22, 1816–1825 10.1111/jcmm.13464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang X., Pan L., Yang K., Fu Y., Liu Y., Chi J., Zhang X., Hong S., Ma X., and Yin X. (2017) H3 relaxin protects against myocardial injury in experimental diabetic cardiomyopathy by inhibiting myocardial apoptosis, fibrosis and inflammation. Cell. Physiol. Biochem. 43, 1311–1324 10.1159/000481843 [DOI] [PubMed] [Google Scholar]
- 27. Cheng Z., Völkers M., Din S., Avitabile D., Khan M., Gude N., Mohsin S., Bo T., Truffa S., Alvarez R., Mason M., Fischer K. M., Konstandin M. H., Zhang X. K., Heller Brown J., and Sussman M. A. (2011) Mitochondrial translocation of Nur77 mediates cardiomyocyte apoptosis. Eur. Heart J. 32, 2179–2188 10.1093/eurheartj/ehq496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yan G., Zhu N., Huang S., Yi B., Shang X., Chen M., Wang N., Zhang G. X., Talarico J. A., Tilley D. G., Gao E., and Sun J. (2015) Orphan nuclear receptor Nur77 inhibits cardiac hypertrophic response to β-adrenergic stimulation. Mol. Cell. Biol. 35, 3312–3323 10.1128/MCB.00229-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Medzikovic L., Schumacher C. A., Verkerk A. O., van Deel E. D., Wolswinkel R., van der Made I., Bleeker N., Cakici D., van den Hoogenhof M. M., Meggouh F., Creemers E. E., Remme C. A., Baartscheer A., de Winter R. J., de Vries C. J., et al. (2015) Orphan nuclear receptor Nur77 affects cardiomyocyte calcium homeostasis and adverse cardiac remodelling. Sci. Rep. 5, 15404 10.1038/srep15404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Patil N. A., Rosengren K. J., Separovic F., Wade J. D., Bathgate R. A. D., and Hossain M. A. (2017) Relaxin family peptides: structure-activity relationship studies. Br. J. Pharmacol. 174, 950–961 10.1111/bph.13684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Feng X. J., Gao H., Gao S., Li Z., Li H., Lu J., Wang J. J., Huang X. Y., Liu M., Zou J., Ye J. T., and Liu P. Q. (2015) The orphan receptor NOR1 participates in isoprenaline-induced cardiac hypertrophy by regulating PARP-1. Br. J. Pharmacol. 172, 2852–2863 10.1111/bph.13091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. You B., Jiang Y. Y., Chen S., Yan G., and Sun J. (2009) The orphan nuclear receptor Nur77 suppresses endothelial cell activation through induction of IκBα expression. Circ. Res. 104, 742–749 10.1161/CIRCRESAHA.108.192286 [DOI] [PubMed] [Google Scholar]
- 33. Qin Q., Chen M., Yi B., You X., Yang P., and Sun J. (2014) Orphan nuclear receptor Nur77 is a novel negative regulator of endothelin-1 expression in vascular endothelial cells. J. Mol. Cell. Cardiol. 77, 20–28 10.1016/j.yjmcc.2014.09.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhao Y., and Bruemmer D. (2009) NR4A orphan nuclear receptors in cardiovascular biology. Drug Discov. Today Dis. Mech. 6, e43–e48 10.1016/j.ddmec.2009.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wenzl K., Troppan K., Neumeister P., and Deutsch A. J. (2015) The nuclear orphan receptor NR4A1 and NR4A3 as tumor suppressors in hematologic neoplasms. Curr. Drug Targets 16, 38–46 10.2174/1389450115666141120112818 [DOI] [PubMed] [Google Scholar]
- 36. Ranhotra H. S. (2015) The NR4A orphan nuclear receptors: mediators in metabolism and diseases. J. Receptor Signal Transduct. Res. 35, 184–188 10.3109/10799893.2014.948555 [DOI] [PubMed] [Google Scholar]
- 37. Bathgate R. A., Lin F., Hanson N. F., Otvos L. Jr., Guidolin A., Giannakis C., Bastiras S., Layfield S. L., Ferraro T., Ma S., Zhao C., Gundlach A. L., Samuel C. S., Tregear G. W., and Wade J. D. (2006) Relaxin-3: improved synthesis strategy and demonstration of its high-affinity interaction with the relaxin receptor LGR7 both in vitro and in vivo. Biochemistry 45, 1043–1053 10.1021/bi052233e [DOI] [PubMed] [Google Scholar]
- 38. McGowan B. M., Minnion J. S., Murphy K. G., Roy D., Stanley S. A., Dhillo W. S., Gardiner J. V., Ghatei M. A., and Bloom S. R. (2014) Relaxin-3 stimulates the neuro-endocrine stress axis via corticotrophin-releasing hormone. J. Endocrinol. 221, 337–346 10.1530/JOE-13-0603 [DOI] [PubMed] [Google Scholar]
- 39. Wansa K. D., Harris J. M., and Muscat G. E. (2002) The activation function-1 domain of Nur77/NR4A1 mediates trans-activation, cell specificity, and coactivator recruitment. J. Biol. Chem. 277, 33001–33011 10.1074/jbc.M203572200 [DOI] [PubMed] [Google Scholar]
- 40. Mullican S. E., Zhang S., Konopleva M., Ruvolo V., Andreeff M., Milbrandt J., and Conneely O. M. (2007) Abrogation of nuclear receptors Nr4a3 and Nr4a1 leads to development of acute myeloid leukemia. Nat. Med. 13, 730–735 10.1038/nm1579 [DOI] [PubMed] [Google Scholar]
- 41. Hanna R. N., Shaked I., Hubbeling H. G., Punt J. A., Wu R., Herrley E., Zaugg C., Pei H., Geissmann F., Ley K., and Hedrick C. C. (2012) NR4A1 (Nur77) deletion polarizes macrophages toward an inflammatory phenotype and increases atherosclerosis. Circ. Res. 110, 416–427 10.1161/CIRCRESAHA.111.253377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. De Paoli F., Eeckhoute J., Copin C., Vanhoutte J., Duhem C., Derudas B., Dubois-Chevalier J., Colin S., Zawadzki C., Jude B., Haulon S., Lefebvre P., Staels B., and Chinetti-Gbaguidi G. (2015) The neuron-derived orphan receptor 1 (NOR1) is induced upon human alternative macrophage polarization and stimulates the expression of markers of the M2 phenotype. Atherosclerosis 241, 18–26 10.1016/j.atherosclerosis.2015.04.798 [DOI] [PubMed] [Google Scholar]
- 43. Myers S. A., Eriksson N., Burow R., Wang S. C., and Muscat G. E. (2009) β-Adrenergic signaling regulates NR4A nuclear receptor and metabolic gene expression in multiple tissues. Mol. Cell. Endocrinol. 309, 101–108 10.1016/j.mce.2009.05.006 [DOI] [PubMed] [Google Scholar]
- 44. Bristow M. R. (2000) β-Adrenergic receptor blockade in chronic heart failure. Circulation 101, 558–569 10.1161/01.CIR.101.5.558 [DOI] [PubMed] [Google Scholar]