

Abstract
Endoplasmic reticulum (ER) degradation-enhancing α-mannosidase–like 1 protein (EDEM1) is a protein quality control factor that was initially proposed to recognize N-linked glycans on misfolded proteins through its mannosidase-like domain (MLD). However, recent studies have demonstrated that EDEM1 binds to some misfolded proteins in a glycan-independent manner, suggesting a more complex binding landscape for EDEM1. In this study, we have identified a thiol-dependent substrate interaction between EDEM1 and the α1-antitrypsin ER-associated protein degradation (ERAD) clients Z and NHK, specifically through the single Cys residue on Z/NHK (Cys256), required for binding under stringent detergent conditions. In addition to the thiol-dependent interaction, the presence of weaker protein-protein interactions was confirmed, suggestive of bipartite client-binding properties. About four reactive thiols on EDEM1 were identified and were not directly responsible for the observed redox-sensitive binding by EDEM1. Moreover, a protein construct comprising the EDEM1 MLD had thiol-dependent binding properties along with its active glycan-trimming activities. Lastly, we identified an additional intrinsically disordered region (IDR) located at the C terminus of EDEM1 in addition to its previously identified N-terminal IDR. We also determined that both IDRs are required for binding to the ERAD component ERdj5 as an interaction with ERdj5 was not observed with the MLD alone. Together, our findings indicate that EDEM1 employs different binding modalities to interact with ERAD clients and ER quality control (ERQC) machinery partners and that some of these properties are shared with its homologues EDEM2 and EDEM3.
Keywords: endoplasmic reticulum (ER), endoplasmic reticulum-associated protein degradation (ERAD), protein folding, oxidation-reduction (redox), protein degradation, intrinsically disordered protein, α1 antitrypsin (A1AT), ER degradation-enhancing mannosidase-like 1 protein (EDEM1), ERdj5
Introduction
In the ER3 lumen, nascent polypeptides gain access to an array of molecular chaperones and foldases such as BiP, lectin molecular chaperones such as calnexin (CNX) and calreticulin (CRT), and oxidoreductases like PDI and ERp57, all of which comprise part of the ERQC machinery (1–4). The ERQC attempts to fold and rescue aberrantly folded proteins by protecting exposed hydrophobic regions (BiP), binding to N-linked glycans (CNX and CRT), and catalyzing the formation of the correct disulfide bonds (PDI, ERp57, etc.).
Terminally misfolded proteins are extracted from futile folding cycles, prevented from re-engaging the ERQC machinery, and ultimately targeted for degradation via the ER-associated degradation (ERAD) pathway (5, 6). Numerous ERAD components have previously been characterized that recognize exposed hydrophobic regions, mispaired disulfide bonds, or specific N-linked glycan structures. For instance, mannosidases contribute to the extensive glycan trimming that occurs on ERAD-bound clients, and oxidoreductases are proposed to facilitate retrotranslocation by unfolding and reducing ERAD clients to make them translocation-competent (7–9).
The ER degradation-enhancing α-mannosidase–like 1 protein (EDEM1) has been proposed to select and sequester terminally misfolded proteins away from productive folding cycles and target them for degradation. Evidence for these roles include preferential interaction with misfolded ERAD clients, direct interaction with ERAD machinery components such as Sel1L and ERdj5, and acceleration of the degradation of misfolded clients when overexpressed (10–14).
Although EDEM1 and its role in ERAD have been extensively studied and characterized since its discovery in 2001 (11, 13), many of the protein's properties remain enigmatic. For instance, early EDEM1 studies suggested that EDEM1 lacked mannosidase activity, although it shares a mannosidase-like domain similar to that of the active ER mannosidase Man1b1 (ER ManI) (11). However, several groups have demonstrated contradictory cellular findings showing that EDEM1 has weak mannosidase activity (15–17). Additionally, attempts to ascertain on which glycan branch EDEM1 acts have been conducted by determining the glycan profile of cells in which EDEM1 was overexpressed or knocked out, but in vitro analyses using recombinant EDEM1 or its MLD are lacking, likely due to difficulty in expressing recombinant EDEM1 and its MLD (17, 18).
EDEM1 has been implicated to play a role in ERAD and function in the ER lumen; however, how the protein is retained in the ER lumen remains undetermined. EDEM1 exhibits dual topology as a result of inefficient cleavage of its signal peptide, which yields a membrane-bound protein that is able to act on membrane-associated ERAD clients (19). However, EDEM1 also exists as a soluble luminal protein that remains in the ER but lacks a known ER retention/retrieval sequence. EDEM1 interacts with numerous ER-localized binding partners and is proposed to exist in a multiprotein complex, which may mediate its ER retention, although this has yet to be shown.
The client- or machinery-binding properties of EDEM1 appear multifaceted and specific to each binding partner. For instance, Lederkremer and co-workers (20) showed that N-terminal truncation of EDEM1 possessing a portion of the MLD is sufficient for binding to H2a. Petrescu and co-workers (21) identified an N-terminal IDR that is required for interaction with an unnatural soluble form of tyrosinase, further supporting the nonessential role of the MLD in the interaction between EDEM1 and H2a or soluble tyrosinase. Likewise, the role of glycans in EDEM1 client binding is perplexing and appears substrate-specific as in some cases (NHK, H2a, and SHH) interactions with EDEM1 were independent of substrate glycosylation; however, in other cases glycans appeared required (BACE457) (10, 12, 20, 22). Interactions between EDEM1 and ER machinery-binding partners have been reported and characterized, most notably that the EDEM1 MLD was involved in the interaction with Sel1L as mutating the putative catalytic triad or using mannosidase inhibitors abolished this interaction (10, 23).
Combined, these results illustrate a much more complex binding landscape of EDEM1 that involves distinct protein-dependent modes of interactions. In this study, we have identified bimodal interactions between EDEM1 and the α1-antitrypsin (A1AT) disease-associated and ERAD variants Z and NHK. The interactions between EDEM1 and Z/NHK involve covalent and weaker protein-protein interactions that were identified under different detergent conditions. We also provide evidence that the MLD of EDEM1 possesses glycosidase activity. Altogether, these findings broaden the scope of the EDEM substrate-binding properties and provide further evidence supporting cellular mannosidase activity of the EDEM1 MLD.
Results
The binding of EDEM1 to misfolded A1AT is bipartite and involves oxidation-dependent and weak protein-protein interactions
EDEM1 preferentially interacts with misfolded A1AT variants NHK and Z in a glycan-independent manner (10). Given the oxidative nature of the ER lumen, we determined whether this preferential interaction is oxidation-dependent. A pulse-chase experiment was performed to compare the protein interactions in the absence or presence of the reducing agent dithiothreitol (DTT) (Fig. 1A). EDEM1-FLAG was coexpressed in human embryonic kidney (HEK) 293T cells with wildtype (WT) A1AT, Z, or NHK. The proteins were radiolabeled for 30 min followed by a 15-min chase to allow EDEM1 to reach its mature folded state. A 30-min chase with or without 5 mm DTT was performed (+/− lanes) following the initial 15-min chase (Fig. 1A). Cells were lysed with 0.5% Triton X-100 (TX) in MNT buffer (0.5% Triton X-100, 20 mm MES, 100 mm NaCl, 20 mm Tris-HCl (pH 7.5)) or with 2% CHAPS (CH) in HBS (50 mm HEPES, 200 mm NaCl (pH 7.5)). Cell lysates were divided into equal fractions and subjected to anti-A1AT (Fig. 1A, lanes 1–12) and anti-FLAG (Fig. 1A, lanes 13–24) immunoprecipitations. Immunoprecipitations obtained from “TX” lysates were washed under stringent buffer conditions containing 0.1% SDS, whereas those obtained from “CH” lysates were washed under milder conditions using 0.5% CHAPS.
Figure 1.

EDEM1 binding to ERAD clients A1AT Z and NHK is bipartite and involves Cys256 on Z/NHK. A, EDEM1-FLAG was coexpressed with A1AT WT, NHK, or Z in HEK293T cells. Cells were radiolabeled with [35S]Cys/Met for 30 min and chased for 15 min. DTT (5 mm) was added to the cells for 30 min where indicated (+DTT). Cells were lysed in MNT buffer containing TX or CH, and EDEM1 and A1AT were isolated using anti-FLAG (α-FLAG) and anti-A1AT (α-A1AT) antisera. The proteins were resolved by 9% reducing SDS-PAGE. B, cartoon representation of A1AT constructs depicting signal peptide (green), glycosylation sites (black), and Cys256 (yellow). Arrows denote point mutation E366K on Z and frameshift mutation at 342 on NHK. C, EDEM1-FLAG was coexpressed in HEK293T cells with A1AT WT, Z, Z C256S, NHK, or NHK C256S. The proteins were radiolabeled with [35S]Cys/Met for 30 min and chased for 1 h. Cells were lysed in 2% CHAPS or MNT buffer containing 0.1% SDS. Half of the cell lysate was subjected to anti-A1AT immunoprecipitation, and the other half was subjected to anti-FLAG immunoprecipitation and washed in 0.5% CHAPS or wash buffer containing 0.1% SDS, respectively. Proteins were resolved by 9% reducing SDS-PAGE. Gels are representative of three independent experiments.
The DTT treatment did not affect the maturation or secretion of WT A1AT as the protein appears to have reached its mature state as indicated by the presence of complex sugars acquired in the Golgi as demonstrated by the higher molecular weight species (Fig. 1A, lanes 1–4). Additionally, the DTT treatment did not appear to affect the inherent binding properties of EDEM1 as it did not bind to WT A1AT regardless of the of the addition of DTT using either TX/SDS or CHAPS washes (Fig. 1A, lanes 1–4 and 13–16).
Under mild detergent conditions (CHAPS), the interaction between EDEM1 and NHK (Fig. 1A, lanes 5, 6, 17, and 18) or Z (Fig. 1A, lanes 9, 10, 21, and 22) was maintained regardless of whether DTT was added. However, under stringent detergent conditions (TX/SDS), the interaction between EDEM1 and NHK (Fig. 1A, lanes 7, 8, 19, and 20) or Z (Fig. 1A, lanes 11, 12, 23, and 24) was disrupted upon addition of DTT, indicating the presence of an oxidation requirement as well as the involvement of weaker protein-protein interactions.
WT A1AT and the misfolded variants NHK and Z possess a single cysteine (Cys) residue at position 256 and three N-linked glycosylation sites (Fig. 1B). We have previously determined that the interaction between EDEM1 and NHK is glycan-independent under stringent detergent conditions (10). It was next determined whether the interaction between A1AT and EDEM1 involved A1AT Cys256.
C256S mutants of Z and NHK were generated and tested for their binding to EDEM1 (Fig. 1C). EDEM1-FLAG was coexpressed in HEK293T cells with WT A1AT, Z, Z C256S, NHK, or NHK C256S. The proteins were radiolabeled for 30 min followed by 1-h chase. Cells were lysed in MNT buffer and washed in buffer containing 0.1% SDS. EDEM1 no longer interacted with Z or NHK when the Cys was mutated to a Ser (Fig. 1C, lanes 21–24), suggesting that the previously observed covalent-like interaction between the misfolded A1AT ERAD variants and EDEM1 involved the single Cys256 from A1AT.
EDEM1-FLAG possesses multiple reactive maleimide-modifiable Cys
To determine whether the interaction between EDEM1 and NHK or Z involves reactive thiols or unpaired Cys on EDEM1, we conducted an in-cell maleimide modification assay using methoxy polyethylene glycol maleimide (PEG-maleimide) (Fig. 2A). HEK293T cells were transfected with EDEM1-FLAG, and a 1-h DTT pretreatment was added to designated dishes. Cells were lysed in sample buffer containing 5 mm PEG-maleimide (+PEG-maleimide) or 20 mm NEM (−PEG-maleimide) to modify reactive thiols.
Figure 2.

EDEM1 possesses ∼4 reactive thiols. A, EDEM1-FLAG was expressed in HEK293T cells. Cells were pretreated with DTT (5 mm) where indicated (+/−). Cells were lysed in sample buffer containing 5 mm PEG-maleimide (+PEG-maleimide) or 20 mm NEM (−PEG-maleimide). Proteins were resolved by 8.5% SDS-PAGE and immunoblotted (IB) with FLAG antibody. B, structural model (Phyre2.0) of the MLD (cyan) showing the three catalytic triad (red) and Cys residues (yellow). The cartoon representation of EDEM1 depicts signal peptide (green), MLD (cyan), putative catalytic residues (red), Cys residues (yellow), and predicted disulfides (*). C, FLAG-tagged EDEM1 (WT), C95S/C302S, C555S/C629S, C95S/C302S/C629S, and C95S/C302S/C555S/C629S were expressed in HEK293T cells and treated as in A. D, FLAG-tagged EDEM1 (WT), C95S/C302S/C629S, and C95S/C302S/C555S/C629S were coexpressed with A1AT WT, Z, and NHK in cells. Cells were radiolabeled with [35S]Cys/Met for 30 min and chased for 1 h. Cells were lysed in MNT buffer containing 0.1% SDS. Half of the cell lysate was subjected to anti-A1AT, and half was subjected to anti-FLAG immunoprecipitation. Proteins were resolved by 8.5% reducing SDS-PAGE. The asterisk denotes background bands. Gels are representative of three independent experiments.
Human EDEM1 possesses eight Cys residues that are well conserved across metazoan species (Fig. S1A). All eight Cys were modified upon treatment with PEG-maleimide following DTT pretreatment (Fig. 2A, compare mobility shift between lanes 3 and 4). However, under oxidizing conditions (−DTT pretreatment), approximately half of the Cys were accessible to maleimide modification (Fig. 2A, compare lane 2 with lanes 1, 3, and 4) as the modified protein migrates slower than the unmodified but faster than the fully modified sample, indicating that a subset of the Cys on EDEM1 was accessible and reactive at steady state.
The oxidized and maleimide-modified sample (Fig. 2A, lane 2) was compared with a series of EDEM1 constructs on which a single Cys-to-Ser mutation was generated to obtain EDEM1 comprising seven, six, five, and four total Cys residues (Fig. S1B). The number of accessible and reactive Cys on EDEM1 under oxidizing conditions can be estimated as 4 as the protein migrates at approximately the same level as EDEM1 with four Cys on SDS-PAGE (Fig. S1B, compare lane 2 with lane 3). Furthermore, modeling of the conserved EDEM1 MLD predicts that four of the Cys are in sufficient proximity to form two disulfide bonds (Cys160–Cys529 and Cys410–Cys457) (Fig. 2B, left panel). The two remaining Cys in the MLD (Cys302 and Cys555) might act as free thiols. There are two additional Cys found outside the predicted mannosidase domain, one N-terminal and the other C-terminal, which could pair up with each other or with Cys302 or Cys555 or remain free thiols. This information was used to derive a predicted disulfide map of EDEM1 (Fig. 2B, right panel).
We next sought to identify the reactive thiols at steady state using the maleimide modification assay and monitoring the mobility shifts of each sample under oxidizing conditions compared with that of WT EDEM1 (Fig. 2C). Combinational Cys-to-Ser mutants of the predicted reactive Cys (C95S/C302S, C555S/C629S, C95S/C302S/C629S, and C95S/C302S/C555S/C629S) were generated and transfected in HEK293T cells. Proteins were subjected to PEG-maleimide modification under reducing (+DTT pretreatment) and nonreducing (−DTT pretreatment) conditions. Proteins were resolved by SDS-PAGE and subjected to immunoblotting.
The resulting blots were positioned such that the reduced protein samples in each condition were aligned (Fig. 2C, lanes 1, 2, 5, 7, 9, 10, 13, 15, 17, and 19). The mobility shifts of the oxidized and maleimide-labeled combinational mutants C95S/C302S and C95S/C302S/C629S were compared with WT EDEM1 (Fig. 2C, compare lanes 6 and 14 with lane 3), and in both instances the proteins migrated faster through the gel with the triple mutant migrating the fastest, indicating that these constructs possess fewer accessible thiols than the WT protein, likely two and one, respectively. The same analysis was performed with constructs in which Cys555 was mutated to Ser (C555S/C629S and C95S/C302S/C555S/C629S); however, both mutations resulted in a smear when labeled with PEG-maleimide under oxidizing conditions (Fig. 2C, lanes 11 and 18). The same result was obtained with the single C555S point mutant (data not shown), which may implicate Cys555 in forming transient intermediate disulfides, perhaps as the resolving Cys residue, which when mutated to a Ser results in mixed disulfide species.
The interaction between EDEM1 comprising single Cys-to-Ser mutations and NHK was maintained under stringent detergent conditions (Fig. S2), indicating that a single EDEM1 Cys residue was not solely responsible for maintaining the interaction. We then determined whether the reactive thiols contributed to the thiol-dependent interaction that was observed under stringent detergent conditions. To this end, WT EDEM1-FLAG, C95S/C302S/C629S, and C95S/C302S/C555S/C629S were coexpressed with A1AT WT, Z, and NHK (Fig. 2D).
The EDEM1-FLAG C95S/C302S/C629S and C95S/C302S/C555S/C629S constructs both coimmunoprecipitated with Z and NHK as observed by the presence of EDEM1-FLAG protein (Fig. 2D, lanes 5, 6, 8, and 9 compared with WT in lanes 1, 4, and 7) with the anti-A1AT immunoprecipitation. These interactions were verified using anti-FLAG immunoprecipitations where the presence of Z and NHK was observed with the EDEM1 pulldowns (Fig. 2D, lanes 11, 12, 14, 15, 17, and 18 compared with WT in lanes 10, 13, and 16). Similar to the WT EDEM1, EDEM1-FLAG C95S/C302S/C629S and C95S/C302S/C555S/C629S exhibit preferential interaction with ERAD clients as neither protein interacted with ATAT WT (Fig. 2D, lanes 10, 13, and 16). Taken together, EDEM1-FLAG possesses multiple reactive thiols as Cys at positions 95, 302, 555, and 629 appear unpaired and accessible to maleimide modification at steady state; however, these residues do not appear to mediate the thiol-dependent interaction that was observed between EDEM1 and NHK/Z.
Identification of EDEM1 IDRs
Previous studies by Petrescu and co-workers (21) have identified an IDR at the N terminus of EDEM1 that was required for soluble tyrosinase binding. To address whether other IDRs are present on EDEM1, the full-length protein sequence was queried using PONDR-FIT, DISOPRED, DisProt, and FoldIndex (24–27). Each algorithm, with the exception of one, identified two regions that are predicted as IDRs, located at the N and C termini of the protein (Fig. S3A, left panel). These IDRs constitute the bulk of the sequence flanking the MLD, making them potential modes of interaction between EDEM1 and ER quality control machinery or other possible ERAD clients.
ER localization of the EDEM1 constructs
To investigate the role of the EDEM1 IDRs in localization and to determine whether a construct comprised solely of the MLD lacking both IDRs remains ER-localized, we generated constructs lacking either IDRs (ΔIDR(N) and ΔIDR(C)) or both (MLD), which comprises the mannosidase-like domain alone (Fig. S3A, right panel). Each construct contains the putative WT EDEM1 signal sequence as well as the same FLAG epitope at the C terminus. To address whether the truncated constructs are targeted to the ER, their subcellular localization was monitored by confocal immunofluorescence microscopy (Fig. S3B) and by endoglycosidase sensitivity (Fig. 3A).
Figure 3.
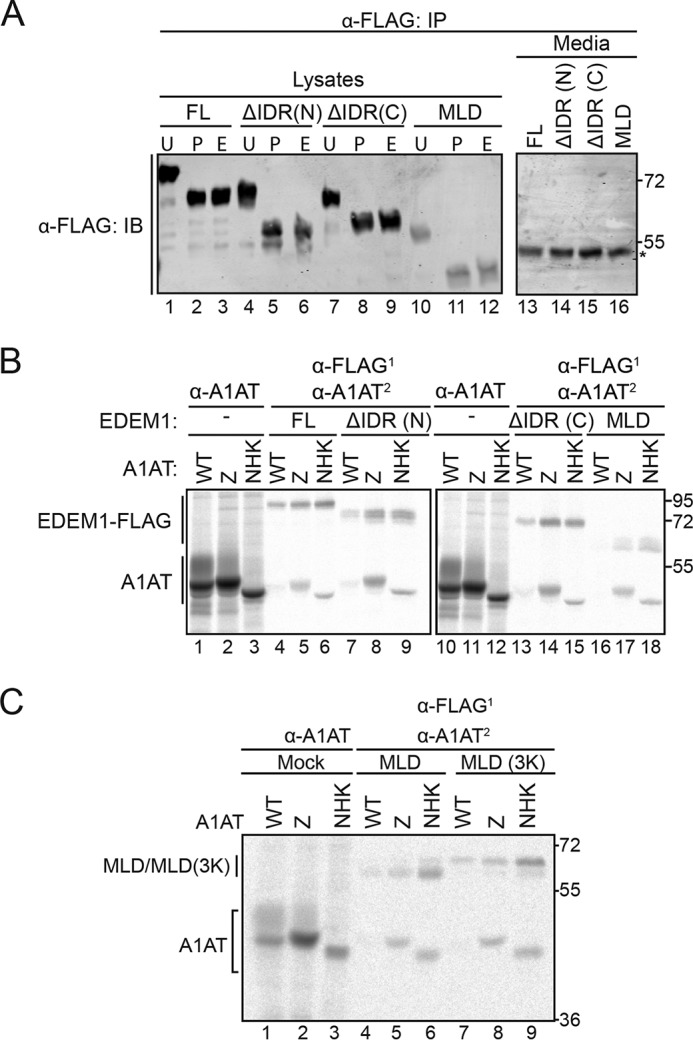
EDEM1 lacking IDRs is targeted to the ER and preferentially interacts with ERAD substrates. A, HEK293T cells were transfected with FLAG-tagged EDEM1(FL), ΔIDR(N), ΔIDR(C), and MLD. Cells were lysed in MNT buffer. Lysates and media were subjected to anti-FLAG immunoprecipitation (IP), and proteins samples were treated with PNGase F (P), Endo H (E), or left untreated (U). Proteins were resolved by 7.5% reducing SDS-PAGE and immunoblotted against FLAG. The asterisk denotes IgG heavy chain. B, empty plasmid (−), EDEM1(FL)-FLAG, ΔIDR(N)-FLAG, ΔIDR(C)-FLAG, and MLD-FLAG constructs were cotransfected with the A1AT WT, Z, and NHK in HEK293T cells. Proteins were radiolabeled for 30 min with [35S]Cys/Met and chased for 1 h. Lysates were subjected to sequential (first (1), anti-FLAG; second (2), anti-A1AT) or single anti-A1AT immunoprecipitation. Proteins were resolved by reducing 8% SDS-PAGE. C, MLD-FLAG, MLD(3K)-FLAG, and empty plasmid (Mock) were coexpressed with WT A1AT, Z, or NHK in HEK293T cells. Cells were radiolabeled with [35S]Cys/Met for 30 min and chased for 1 h. Cells were lysed in MNT buffer containing Triton X-100. 70% of the lysate was subjected to sequential immunoprecipitation (first, anti-FLAG; second, anti-A1AT), and 20% was subjected to anti-A1AT immunoprecipitation. The proteins were resolved by 8% reducing SDS-PAGE. Gels are representative of three independent experiments.
Single transfections of FLAG-tagged EDEM1(FL), ΔIDR(N), ΔIDR(C), and MLD were performed in HEK293A cells, and staining was compared against ER (CRT) or Golgi (Giantin) markers (Fig. S3B). Signals from each EDEM1 construct colocalized with CRT; however, no colocalization was observed with the Golgi marker, suggesting that all constructs were ER-resident proteins.
EDEM1 and the IDR constructs are predicted to possess multiple N-linked glycosylation consensus sites. EDEM1(FL) was previously shown to be N-glycosylated; thus, an N-glycosylation assay would reveal whether the EDEM1 constructs lacking the IDRs were targeted to and reside in the ER (19). The proteins were isolated by immunoprecipitation from cells expressing EDEM1(FL), ΔIDR(N), ΔIDR(C), and MLD; deglycosylated using PNGase F (P) or Endo H (E) or left untreated (U) (Fig. 3A); and visualized by immunoblotting. PNGase F removes mannose-rich N-glycans found in the ER as well as complex N-glycans that are acquired in the Golgi, whereas Endo H only trims high-mannose N-glycans encountered in the ER. Because the molecular mass of an N-linked glycan is ∼2.5 kDa, glycosylated proteins should migrate slower on an SDS-polyacrylamide gel compared with deglycosylated proteins. Mobility shifts upon PNGase F and Endo H treatment were observed for all EDEM1 IDR constructs (comparing “U” lanes with “P” and “E” lanes), indicating that ΔIDR(N), ΔIDR(C), and MLD were all targeted to the ER and glycosylated with high-mannose carbohydrates and did not receive complex N-glycans in the Golgi. Together, these observations are consistent with ΔIDR(N), ΔIDR(C), and MLD being targeted to and residing in the ER.
EDEM1 MLD preferentially interacts with ERAD substrates
To attempt to reduce EDEM1 down to a smaller functional unit and to determine whether the conserved N- and C-terminal MLD-flanking regions are required for ERAD client binding, the binding of the EDEM1 constructs to A1AT WT and its mutant variants was characterized using a radiolabel pulse-chase approach (Fig. 3B). The IDRs were not required for the interaction with Z and NHK as binding to Z and NHK, but not WT, occurred for both EDEM1 ΔIDR(N) and ΔIDR(C) (Fig. 3B, lanes 8, 9, 14, and 15 compared with lanes 7 and 13). The MLD construct, which lacks both the N- and C-terminal IDRs, also preferentially binds to misfolded variants Z and NHK over WT (Fig. 3B, lanes 16–18).
Moreover, the 3 acidic putative active-site residues were replaced with positively charged Lys residues (MLD(3K)) to abolish any potential carbohydrate binding (Fig. 3C, lanes 7–9). Interestingly, the interaction between MLD(3K) and both NHK and Z was maintained, whereas no interaction was observed with WT A1AT, which further validates the ERAD client-specific glycan-independent binding property of the MLD. The MLD also bound to a nonglycosylated NHK, indicating that binding was glycan-independent (Fig. S3C). Taken together, these results demonstrate that the EDEM1 MLD possesses the ability to preferentially interact with the nonnative ERAD clients NHK and Z in a glycan-independent manner.
MLD substrate binding is redox-sensitive
Because the EDEM1 MLD exhibits preferential interaction with ERAD clients, we next determined whether this interaction involves thiols by monitoring binding upon addition of DTT (Fig. 4A). A pulse-chase analysis was performed (as described in Fig. 1A) using a sequential immunoprecipitation of the protein with anti-FLAG followed by anti-A1AT (Fig. 4A, lanes 7–12). As expected, the MLD did not interact with WT A1AT (Fig. 4A, lanes 7 and 8). However, like full-length EDEM1, the interaction between Z and NHK was oxidation-dependent under stringent detergent conditions (Fig. 4A, compare lane 10 with lane 9 and lane 12 with lane 11).
Figure 4.

EDEM1 MLD interacts with Z/NHK through Cys256 and possesses reactive thiols. A, MLD-FLAG was coexpressed with A1AT WT, Z, or NHK in HEK293T cells. Cells were radiolabeled for 30 min and chased for 15 min. DTT (5 mm) was added to the cells for 30 min where indicated (+DTT). Cells were lysed in MNT buffer containing TX. A portion of the lysate (65%) was subjected to sequential immunoprecipitation (first (1), anti-FLAG; second (2), anti-A1AT), 20% was subjected to anti-A1AT, and 10% was subjected to anti-FLAG. The proteins were resolved by 8% reducing SDS-PAGE. B, MLD-FLAG was coexpressed with A1AT WT, Z, Z C256S, NHK, NHK C256S, NHK NOG, and NHK NOG C256S in HEK293T cells. Cells were radiolabeled for 30 min, chased for 1 h, and lysed in MNT buffer. Proteins were immunoprecipitated as in A and resolved by 8% reducing SDS-PAGE. The asterisk denotes a background band. C, MLD-FLAG was expressed in HEK293T cells. Cells were pretreated for 1 h with DTT (5 mm) where indicated (+/−). Cells were lysed in sample buffer containing 5 mm PEG-maleimide (+PEG-maleimide) or 20 mm NEM (−PEG-maleimide). Proteins were resolved by 8% SDS-PAGE and immunoblotted (IB) against FLAG antibody. The asterisk denotes a background band. D, MLD-FLAG, MLD C160S/C529S, MLD C410S/C457S, MLD C302S/C555S, and Cys-less MLD were coexpressed in HEK293T cells with WT A1AT, Z, or NHK. The proteins were radiolabeled for 30 min and chased for 1 h. Proteins were immunoprecipitated as in A and resolved by 8% reducing SDS-PAGE. Gels are representative of three independent experiments.
We next determined whether the thiol-dependent interaction involves Cys256 for Z/NHK (Fig. 4B). The MLD was coexpressed in HEK293T cells with A1AT WT, Z, Z C256S, NHK, NHK C256S, NHK no glycans (NOG), and NHK NOG C256S. The proteins were radiolabeled for 30 min and chased for 1 h. The cells were lysed under stringent detergent conditions (TX/SDS), and protein complexes were isolated by single anti-A1AT and anti-FLAG immunoprecipitation as well as sequential immunoprecipitation. The Cys256 residue on NHK/Z also plays a role in the interaction with the MLD as the Cys-to-Ser mutations abolished the interaction (Fig. 4B, compare lane 6 with lane 5, lane 15 with lane 14, and lane 17 with lane 16).
The EDEM1 Cys at positions 160, 410, 457, and 529 are predicted to be involved in disulfides (Fig. 2B) and are all found in the MLD. Additionally, our PEG-maleimide analysis revealed that these Cys do not appear modified under oxidizing conditions, suggesting that they are either involved in disulfide interactions or not accessible to maleimide modification.
The modeled structure of the MLD predicts two potential disulfides and two unpaired Cys (Fig. 2B). Our PEG-maleimide analysis on full-length EDEM1 identified approximately four reactive thiols and approximately four thiols that are inaccessible to maleimide modification (Fig. 2, A and C). Based on these results, we expect that a subset (∼2) of the Cys on the MLD will be accessible to maleimide modification, whereas the remaining four would not be. Like the full-length EDEM1, a subset of the Cys residues on MLD is reactive at steady state (Fig. 4C, compare lane 3 with lanes 1, 2, and 4).
Next, we determined whether the Cys that are predicted to be paired and experimentally inaccessible to PEG-maleimide (Cys160, Cys529, Cys410, and Cys457) are involved in the thiol-dependent interaction between MLD and Z/NHK (Fig. 4D). Each of the Cys/Ser combinational mutants were coexpressed in HEK293T cells with WT A1AT, Z, and NHK. Radiolabeled proteins were isolated using single immunoprecipitation with anti-A1AT antibody (Fig. 4D, lanes 1–3, 10–12, 19–21, 28–30, and 37–39) and anti-FLAG (Fig. 4D, lanes 7–9, 16–18, 25–27, 34–36, and 43–45) and by sequential immunoprecipitation (Fig. 4D, lanes 4–6, 13–15, 22–24, 31–33, and 40–42). Binding to WT A1AT was not observed regardless of the mutation (Fig. 4D, compare lanes 13, 22, 31, and 40 with lane 4). Binding to Z and NHK persisted when either predicted disulfide pairs or free thiols were mutated (Fig. 4D, compare lanes 14, 15, 23, 24, 32, and 33 with lanes 5 and 6). However, the Cys-less construct did not exhibit any binding to Z or NHK (Fig. 4D, compare lanes 41 and 42 with lanes 5 and 6), which is likely due to the resulting compromised structural integrity of the Cys-less MLD.
Altogether, the MLD possesses the ability to bind NHK and Z in a thiol-dependent manner, involving Cys256 on Z/NHK. However, the MLD Cys that are predicted to be involved in disulfides, like the predicted unpaired Cys, are not directly involved in this interaction. Cys residues appear to function as structural components of the MLD, and at least one of these disulfides pairs is required for ERAD client binding.
EDEM1 MLD has glycosidase activity
To further characterize the MLD and elucidate whether it possesses mannose-trimming properties, the mobility of NHK on SDS-PAGE was monitored (Fig. 5). A quadruple knockout clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9 (CRISPR/Cas9) CHO cell line was used where four mannosidases were knocked out (Man1a1/a2/b1/c1). The only putative mannosidases that are present in these knockout cell lines are EDEM1, -2, and -3. Thus, overexpressing the MLD in these cell lines might enhance any trimming effect on NHK as the presence and effect of other functional mannosidases are minimized. NHK was coexpressed in the quadruple mannosidase knockout CHO cells with a mock empty plasmid (Fig. 5, lanes 1–6), the catalytically active Man1b1-FLAG (Fig. 5, lanes 7–12), EDEM1-FLAG (Fig. 5, lanes 13–18), EDEM1(3K)-FLAG (Fig. 5, lanes 19–24), MLD-FLAG (Fig. 5, lanes 25–30), MLD(3K)-FLAG (Fig. 5, lanes 31–36), and MLD(Cys-less)-FLAG (Fig. 5, lanes 37–42) in addition to NHK with the mannosidase inhibitor kifunensine (Fig. 5, lanes 43–45). Proteins were radiolabeled for 30 min and chased for the indicated times. Consistent with previous data, mannose trimming was abolished upon addition of kifunensine (Fig. 5, lanes 43–45) and mutation of the acidic putative active-site residues on EDEM1 (Fig. 5, compare lanes 22–24 with lanes 16–18). Importantly, the MLD also possesses glycan-trimming ability as observed by the increase in mobility of NHK upon coexpression with MLD (Fig. 5, lanes 28–30) compared with MLD(3K) (Fig. 5, lanes 34–36). These results indicate that the MLD is properly folded and possesses glycosidase activity.
Figure 5.

EDEM1 MLD promotes glycan trimming. Top and bottom left panels, empty plasmid (Mock), Man1b1-FLAG, EDEM1-FLAG, EDEM1(3K)-FLAG, MLD-FLAG, MLD(3K)-FLAG, and MLD(Cys-less)-FLAG were co-expressed with NHK in Man1a1/a2/b1/c1 KO CHO cells. 150 mm kifunensine (KIF) was added for 5 h prior to the pulse where indicated. Proteins were radiolabeled for 30 min with [35S]Cys/Met and chased for 0, 1, and 2 h. Cells were lysed in MNT buffer, and lysates were divided equally and subjected to anti-FLAG and anti-A1AT immunoprecipitation. Proteins were resolved by reducing 8% SDS-PAGE. Bottom right panels, the NHK bands in the designated lanes were magnified, and a red line was drawn through the center of each 0-h time point and through the 4-h time point to illustrate mobility shifts. Gels are representative of three independent experiments.
Role of IDRs in EDEM1 stability and function
Given that the MLD exhibited similar substrate-binding properties as that of the full-length EDEM1, we wanted to determine whether it possessed similar inherent properties of EDEM1, specifically its short half-life and the ability to associate with another known EDEM1 binding partner, ERdj5.
Half-lives
EDEM1 is reported to have a rapid turnover rate with a half-life of ∼2 h (28), especially compared with other ER-resident proteins such as BiP, a key member of the ER quality control machinery, which is in the range of 28–33 h (29). Furthermore, proteins that are intrinsically disordered or contain IDRs possess, on average, a shorter half-life than proteins lacking any IDRs (30). Specifically, proteins containing long stretches of disordered regions (>30 residues) appear to have the shortest half-lives. The EDEM1 N- and C-terminal IDR regions are predicted to be 71 and 69 amino acids in length, respectively (Fig. S3A, right panel).
We thus postulated that the MLD construct, which lacks both IDRs, would have the longest half-life. To this end, each construct bearing a C-terminal FLAG epitope was expressed in HEK293T cells and subjected to a pulse-chase experiment after radiolabeling. Following lysis, the proteins were isolated through immunoprecipitation and resolved by SDS-PAGE (Fig. 6A, left panel). The amount of protein remaining at the indicated time points was quantified and normalized to the starting amount (Fig. 6A, right panel).
Figure 6.

IDRs contribute to EDEM1 half-life but not to the ERjd5 association. A, left panel, FLAG-tagged EDEM1 ΔIDR(N)/(C), and MLD were transfected into HEK293T. Proteins were radiolabeled for 30 min with [35S]Cys/Met, and cells were lysed in MNT buffer. Time points were collected at 0, 2, and 4 h after the pulse. Proteins were resolved by reducing 8% SDS-PAGE. Right panel, the amount of EDEM1 protein remaining at 2 and 4 h was quantified and normalized to the starting material (0 h) and averaged from three independent experiments. The starting material includes both bands of the observed doublet. Statistical significance between MLD, EDEM1 ΔIDR(N), or EDEM1 ΔIDR(C) and EDEM1(FL) at 4 h was determined by an unpaired t test; the measurement designated ** for MLD has a p value of 0.005, and that designated * for EDEM1 ΔIDR(C) is 0.012. Error bars represent mean ± S.E. B, FLAG-tagged EDEM1(FL), EDEM1 ΔIDR(N), EDEM1 ΔIDR(C), and MLD and empty plasmid (Mock) were coexpressed with ERdj5-MYC in HEK293T cells. Cells were lysed in HBS buffer containing CHAPS. 10% of the lysate was collected for whole-cell lysate (WCL), and 40% was collected for anti-FLAG and anti-MYC immunoprecipitation (IP). Proteins were resolved by 8% reducing SDS-PAGE and immunoblotted against MYC, FLAG, and glucosidase II (GlucII). Gels are representative of three independent experiments.
Approximately 50% of EDEM1(FL) remained at the 2-h time point, whereas only ∼20% remained at 4 h. These results are consistent with previous turnover rates of EDEM1 in cell analyses (28). However, the half-life of the MLD construct, which lacks both IDRs, was ∼4 h (∼70% remains at 2 h). Similarly, the constructs lacking each IDR individually (ΔN/ΔC) have longer half-lives than the full-length construct. These observations further validated the identity and presence of two IDRs on EDEM1 that contribute to the previously observed short half-life of EDEM1.
ERdj5 binding
Because the IDRs were not required for substrate interaction with A1AT misfolded variants (Figs. 3, B and C, and S3C), we next determined whether the IDRs of EDEM1 were required for binding to the ER oxidoreductase ERdj5.
To this end, FLAG-tagged full-length EDEM1, the EDEM1 IDR deletion constructs, and the MLD were coexpressed with MYC-tagged ERdj5 in HEK293T cells. The cells were lysed 24 h post-transfection, and proteins were isolated through coimmunoprecipitation. Binding interactions were monitored through immunoblotting.
An interaction with ERdj5 was recovered with EDEM1 lacking either one of its IDRs (Fig. 6B, compare lanes 5, 6, 8, and 9 with lanes 2 and 3). However, the interaction with ERjd5 was lost with EDEM1 lacking both IDRs as displayed by the MLD (Fig. 6B, compare lanes 11 and 12 with lanes 2 and 3). This indicated that the presence of either of the IDRs was sufficient to interact with ERdj5, but the interaction was abolished when both IDRs were removed with the EDEM1 MLD. Taken together, these studies with EDEM1 MLD established its ER localization, determined its substrate binding properties, identified components that support binding to ERdj5, and confirmed it possesses catalytic glycosidase activity in cells.
Conservation of EDEM1 properties in EDEM2 and EDEM3
To determine whether EDEM2 and EDEM3 contain similar properties to EDEM1, EDEM2 and EDEM3 were characterized for substrate-binding properties and free thiol content and analyzed for IDRs. Human EDEM2 and EDEM3 were coexpressed in HEK293T cells with WT A1AT, Z, Z C256S, NHK, NHK C256S, NHK NOG, and NHK NOG C256S. The proteins were radiolabeled for 30 min and chased for 1 h. Cells were lysed under stringent detergent conditions (TX/SDS), lysates were split into equal portions, and protein complexes were isolated by anti-FLAG or anti-A1AT immunoprecipitation. Neither EDEM2 nor EDEM3 associated with WT A1AT (Fig. 7A and B, lane 6).
Figure 7.

EDEM2 and EDEM3 exhibit thiol-dependent binding to Z/NHK and expose reactive thiols. A, EDEM2-FLAG was coexpressed with WT A1AT, Z, Z C256S, NHK, NHK C256S, NHK NOG, and NHK NOG C256S in HEK293T cells. Cells were radiolabeled with [35S]Cys/Met for 30 min and chased for 1 h. Cells were lysed in MNT buffer containing Triton X-100. EDEM2 and A1AT variants were isolated using anti-FLAG and anti-A1AT immunoprecipitations, respectively. Proteins were resolved by 9% reducing SDS-PAGE. Asterisks denote background bands. B, same as in A except using EDEM3-FLAG. C, right panel, EDEM2-FLAG was expressed in HEK293T cells. Cells were pretreated with DTT (5 mm) where indicated (+/−). Cells were lysed in sample buffer containing 5 mm PEG-maleimide (+PEG-maleimide) or 20 mm NEM (−PEG-maleimide). Proteins were resolved by 8.5% SDS-PAGE and immunoblotted (IB) with FLAG antibody. Left panel, cartoon representation of EDEM2 depicting signal peptide (green), MLD (cyan), putative catalytic residues (red), Cys residues (yellow), predicted disulfides (*), and predicted IDRs (red dashed box). D, left panel, same as in C except using EDEM3-FLAG. Right panel, cartoon representation of EDEM3 depicting signal peptide (green), MLD (cyan), protease-associated domain (purple), putative catalytic residues (red), Cys residues (yellow), predicted disulfides (*), and predicted IDRs (red dashed box). Gels are representative of three independent experiments.
Like EDEM1 and the MLD, EDEM2 and EDEM3 coimmunoprecipitated with the ERAD clients Z and NHK (Fig. 7, A and B, lanes 7 and 9) in a glycan-independent manner (Fig. 7, A and B, lane 13). However, upon mutating Cys256 to Ser, the interaction was lost (Fig. 7, A and B, compare lane 8 with lane 7, lane 10 with lane 9, and lane 14 with lane 13), suggesting that the thiol dependence that was observed with EDEM1 and EDEM1 MLD is a conserved trait among all human EDEM homologues.
Human EDEM2 and EDEM3 possess numerous Cys residues, nine and 11, respectively (Fig. 7, C and D). When subjected to PEG-maleimide modification, both constructs displayed reactive thiols at steady state (Fig. 7, C and D, compare lane 3 with lanes 1, 2, and 4). Additionally, the EDEM2 and EDEM3 protein sequences were queried for IDRs, and like EDEM1, both proteins possess IDRs in regions outside the MLD, specifically at the C terminus (Fig. 7, C and D).
Together, these results indicate that EDEM1, -2, and -3 possess a conserved redox sensitivity involved in binding to A1AT ERAD substrates. The proteins are predicted to contain IDRs; however, in the case of EDEM1, these regions do not contribute to Z/NHK binding as the catalytically active EDEM1 MLD alone is capable of selectively binding to ERAD clients.
Discussion
Protein-to-protein interactions have been observed between EDEM1 and multiple ERAD clients, including BACE457, cystic fibrosis transmembrane conductance regulator (CFTR), Rhodopsin, H2a, and A1AT (10–12, 14, 20, 31). However, the basis for these substrate-binding interactions is uncertain. Here, we studied the interaction between EDEM1 and the misfolded A1AT variants Z and NHK. An additional mode of interaction that is redox-sensitive was revealed and provided further evidence that the MLD is active and sufficient to selectively bind the A1AT ERAD clients Z and NHK.
EDEM1 preferentially interacts with Z and NHK but does not bind to the WT A1AT, suggesting it may recognize elements on these misfolded proteins that are not present on the properly folded substrate (10). In the presence of a reducing agent, the interactions between EDEM1 and Z/NHK were lost under stringent detergent conditions (Fig. 1A). However, under milder detergent condition, the interactions persisted (Fig. 1A), demonstrating the involvement of two types of interactions: a strong covalent-like, oxidation-dependent interaction as well as a weak, protein-protein interaction as demonstrated by the recovered interaction under milder detergent conditions. The latter protein-protein interactions are possibly mediated through hydrophobic interactions as such interactions have been demonstrated between EDEM1 and the ERAD client BACE457 (32). A model of the EDEM1 MLD structure shows extensive surface-exposed hydrophobic patches on EDEM1 (Fig. S4), all of which may mediate the observed weak protein-protein interactions. Identifying the precise hydrophobic patch(es) involved in this interaction or determining whether other ERAD clients or machinery interactions involve hydrophobic interactions with EDEM1 will require further investigation.
The lone cysteine residue located on Z and NHK (Cys256) contributed to the observed oxidation-dependent interaction with EDEM1 (Fig. 1, B and C) as the interaction was lost upon mutating the Cys to Ser under strong detergent conditions but persisted under mild detergent conditions. This supports the observed oxidation sensitivity, presumably through a disulfide bond (Fig. 1C). Interestingly, Cys256 is located in the WT structure in a position that is partially exposed or buried (Fig. S5). Mutations such as those observed with NHK and Z would be expected to perturb the structure sufficiently to support increased accessibility of Cys256 to quality control factors and ERAD machinery. Additionally, aberrant Cys256-mediated dimers are formed between Z monomers and NHK monomers, whereas WT A1AT remains monomeric (33). Mutating the Cys residue to Ser on Z or reducing disulfides using DTT increased the secretion of Z as well as increased the complex carbohydrates observed by gel shifts, implicating a role for the Cys in retention of Z in the ER (33).
The presence of aberrant Cys256-mediated dimers that are formed between Z monomers and NHK monomers, which do not occur with WT A1AT, and increased secretion of Z upon Cys256-to-Ser mutation implicate a role for the Cys in ER retention of aberrantly folded Z, and perhaps NHK, as well as further provide evidence supporting the increased accessibility of Cys256 on Z compared with WT A1AT.
This suggests that Cys256 is suitably positioned to act as a quality control signal when exposed in aberrant structures. Although a heterodimer between the two proteins (Z and EDEM1 or NHK and EDEM1) was not readily observable (data not shown), it is likely that, because EDEM1 is proposed to exist in the ER as a member of a multiprotein complex, the presence of monomeric or dimeric Z or NHK would not result in an observable mobility shift on nonreducing SDS-PAGE.
A putative model of the EDEM1 MLD showed a conserved barrel structure with the three putative acidic catalytic residues centered at the core, similar to that of Man1b1 (Fig. 2B) (34). The model also revealed two potential disulfides as indicated by their predicted proximities and two unpaired Cys. The predicted map and the presence of reactive thiols were further tested upon subjection of combinational Cys-to-Ser mutants of the predicted unpaired Cys residues to PEG-maleimide modification (Fig. 2C). Given the oxidative nature of the ER and the role of EDEM1 in ERAD, it is surprising that four of the eight Cys on EDEM1 appear to be accessible to maleimide modification at steady state and suggests that these Cys are likely involved in a functional aspect of EDEM1, either to form direct stable or transient bonds with ERAD clients, ERAD machinery, or other EDEM1 proteins. However, none of the individual Cys were required to interact with Z or NHK (Fig. S2). Given the Cys-rich content of EDEM1, it is possible that there is a redundancy in Cys function. The interaction was only abolished when all Cys were mutated to Ser (Cys-less) in which case the protein was likely either structurally compromised due to the absence of disulfide bonds, as one disulfide was sufficient to maintain an interaction with NHK/Z, or a disulfide is required to act as an electron acceptor for a free thiol attack by the ERAD substrate. These results further support the absence of direct involvement of the Cys of EDEM1 in the apparent redox-sensitive interaction with Z/NHK.
The redox sensitivity of EDEM1 substrate selection likely involves an oxidoreductase other than ERdj5 because the MLD exhibits redox-sensitive binding to Z/NHK (Fig. 4, A and B) and does not interact with ERdj5 (Fig. 6B). It is likely that the interaction between EDEM1 and ERdj5 does not require the MLD as EDEM1 appears to interact with ERdj5 through its C-terminal thioredoxin cluster, a region that is not glycosylated (9). Several groups have demonstrated an interaction between EDEM1 and multiple oxidoreductases, including P5, PDI, ERp57, and ERp72 (35, 36). Likewise, ERdj3 was recently implicated in the degradation of the Z variant (37). Additionally, the EDEM3 MLD was recently discovered to form a disulfide bond with the oxidoreductase ERp46, which promoted the catalytic activity of EDEM3 (38). Whether these oxidoreductases or others are involved in the redox-sensitive substrate-binding properties of EDEM1 remains to be determined.
The truncated MLD construct, in which both regions flanking the EDEM1 MLD were removed, was used to assess the requirement of paired Cys residues in the EDEM1-Z/NHK interaction. The fundamental properties of the MLD were characterized. Although the EDEM2 MLD was expressed and characterized both in cells and as a recombinant protein, the properties of the EDEM1 MLD alone are uncharacterized. The EDEM1 MLD construct is targeted to and retained in the ER in contrast to the EDEM2 MLD (Figs. 3A and S3B) (39). Importantly, the MLD alone retained the ability to preferentially interact with ERAD clients in a glycan-independent manner, indicating that the mannosidase-like domain is sufficient to interact and preferentially select A1AT ERAD substrates, further reinforcing the MLD requirement in binding to a subset of ERAD clients (Figs. 3C and S3[[[[[[]C).
The MLD also exhibited the same oxidation-dependent binding and sensitivity to Z/NHK Cys256 upon stringent detergent condition, indicating that the ability and selectivity for Z and NHK binding is inherent to the MLD and did not require the flanking IDRs (Fig. 4, A and B). However, the covalent interaction is likely mediated through a cofactor as neither of the predicted disulfides was involved. Although the eight Cys residues on EDEM1 are conserved across species, at least a subset of them appear to function as structural elements of the MLD as indicated by loss of ERAD client binding in the absence of all Cys residues (Fig. 4C).
In addition, the MLD expressed in a cell line deficient for four mannosidases, Man1a1/a2/b1/c1, exhibited mannose-trimming properties as observed by the mobility shift of NHK on SDS-PAGE (Fig. 5). The mobility shift was lost upon mutation of the putative catalytic residues (MLD(3K)), suggesting that the catalytically inactive MLD(3K) exhibited a dominant negative effect, one in which NHK binds the inactive MLD, thus preventing it from interacting with the endogenous mannosidases EDEM1/2/3 (Fig. 5) (18). Overexpressing MLD(3K) and EDEM1(3K) increases their local concentrations to a higher degree than that of the endogenous EDEM proteins; thus, the binding of NHK to the exogenous proteins appears to be persistent, possibly not allowing access to the endogenous EDEM proteins. EDEM1 does not appear to act solely as a mannosidase as selective interactions with ERAD clients survive prolonged binding required with the coimmunoprecipitation protocol, unlike Man1b1 for which an interaction is not recovered. It is possible that this prolonged interaction indicates a role for EDEM1 in delivering ERAD clients to the retrotranslocation machinery. Altogether, these novel findings may facilitate purification of recombinant EDEM1 and its MLD for in vitro biochemical characterization, which has previously been unsuccessful, and potentially help identify the specific glycan branch on which EDEM1 acts.
In addition to identifying novel redox-sensitive properties between EDEM1 or the MLD and two ERAD substrates, we have identified a second IDR located at the C terminus of EDEM1 (Fig. S3A). Removing either or both IDRs did not affect the ER targeting or localization of EDEM1 as all constructs received N-linked glycans and exhibited glycoforms that are typically found on ER-resident proteins (Fig. 3A). None of the constructs were recovered in the media fractions, confirming their cellular retention (Fig. 3A). The ER retention of the MLD alone appears to require neither a retention sequence like KDEL nor the N- or C-terminal regions flanking it. Instead, the ER retention of soluble EDEM1 is a property of the MLD unlike that of EDEM2, demonstrating a key difference between the proteins (Fig. 3A and S3B) (39). The construct lacking either of the IDRs also retained the glycan-independent preferential interaction with Z and NHK, which is expected as both constructs display intact MLDs (Fig. 3B and S3C).
The present study provides evidence that EDEM1 possesses two IDRs that contribute to its observed short half-life. Unlike other ER-resident proteins such as BiP, EDEM1 is rapidly turned over and exhibits a half-life of ∼2 h. The stability of EDEM1 upon removal of both IDRs (EDEM1 MLD) appears to double the protein half-life from 2 to 4 h, which further supports the regions flanking the EDEM1 MLD as intrinsically disordered (Fig. 6A). Although a 4-h half-life is still considered short when compared with other ER-resident proteins, the short half-life of EDEM1 is likely linked to its function in ERAD to bind misfolded clients as they accumulate in the ER and target them for degradation. The short half-life is also proposed as a function of ERAD tuning to return the ER to basal EDEM1 levels once the stress is alleviated (40). The conservation of the IDRs across EDEM1 in various species further supports their necessity and role in its function (data not shown).
Although neither the N- nor C-terminal IDRs were required for binding to Z or NHK, unlike H2a or soluble tyrosinase, either of the IDRs appears necessary for binding to ERdj5 (Fig. 6B) (20, 21). Like EDEM1, other proteins such as the E3 ligase San1 and the deubiquitinating enzyme Ubp10 possess IDRs at both their N and C termini (41, 42). In both cases, the conformational flexibility of the IDRs allows San1 and Ubp10 to bind multiple partners. EDEM1 may use an analogous mechanism to bind partners such as ERdj5.
EDEM2 and EDEM3 interact with and accelerate the degradation of Z and NHK (18, 43, 44). This interaction also appears to be sensitive to the presence of Cys256 on NHK and Z (Fig. 7, A and B). EDEM2 and EDEM3 also possess reactive thiols at steady state (Fig. 7, C and D) and interact with the oxidoreductase TXNDC11 (45). Like EDEM1, EDEM2 and EDEM3 are predicted to possess IDRs (Fig. 7, C and D). These findings further support conserved properties among the mammalian EDEM protein family. Together, these findings uncover new modes of interaction that EDEM1 possesses and broaden the binding landscape of EDEM1. Specifically, EDEM1 does not interact with its binding partners or ERAD clients in a “one size fits all” manner. Instead, EDEM1 binding is multifaceted as different components are utilized depending on the properties or requirement of the specific binding partner, increasing the versatility of its substrate selectivity.
Experimental procedures
Reagents and plasmids
Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), and penicillin and streptomycin (P/S) were purchased from Invitrogen. Endo H, PNGase F, and all cloning reagents were acquired from New England Biolabs. HEK293 cells were purchased from ATCC (lot number 6322631), and HEK293A cells were purchased from Invitrogen (lot number 1806222). Cells were authenticated by a universal Mycoplasma detection kit (catalog number 30-1012K, ATCC). [35S]Met/Cys was acquired from PerkinElmer Life Sciences. Protein A–Sepharose 4B was from GE Healthcare. The following antibodies were used: α-FLAG (catalog number F3165, Sigma), α-A1AT (catalog number A0012, Dako), α-MYC (catalog number 9B11, Cell Signaling Technology), and α-glucosidase II (catalog number PA5-21431, Thermo Fisher). The following C-terminally tagged EDEM1 truncations were generated by PCR amplification and subcloned into p3XFLAG-CMV-14 (Addgene): EDEM1 ΔIDR(N) (Δ48–119), EDEM1 ΔIDR(C) (Δ583–652), and EDEM1 MLD (Δ48–119 and Δ583–652). EDEM3-MycFLAG was purchased from Origene Technologies (catalog number RC222885). Transfections were carried out with polyethyleneimine (PEI MAX, catalog number 24765, Polysciences, Inc.).
Metabolic labeling, affinity purification, and SDS-PAGE
HEK293 or CHO (knockout (KO) man1a1/1a2/1b1/1c1) cells were grown in 35- or 60-mm dishes where indicated, and all transfections were carried out for 18 h in DMEM (or minimum Eagle's medium for CHO cells) supplemented with 10% FBS and 1% P/S and incubated at 37 °C in 5% CO2. Cells were lysed on ice with either CHAPS buffer (2% CHAPS in HBS) or MNT buffer. The postnuclear supernatant was isolated by centrifugation followed by preclearing with unbound protein A–Sepharose beads and subsequent incubation with the corresponding antibody and Sepharose beads overnight. Immunopellets were isolated and washed with 0.5% CHAPS in HBS or Connie's wash buffer (100 mm Tris-HCl, 300 mm NaCl, 0.1% SDS, 0.05% Triton X-100 (pH 8.6)) where indicated. Proteins were eluted from beads with reducing sample buffer followed by SDS-PAGE. Radiolabeled samples were visualized using phosphorimaging (FLA-500, Fujifilm) and quantified using ImageQuant TL 1D gel analysis software (GE Healthcare).
Sequential immunoprecipitation
Immunopellets were washed with 0.5% CHAPS in HBS or Connie's wash buffer and eluted from beads with reducing sample buffer. When followed by a second immunoprecipitation, samples were eluted with 1% SDS in 10 mm Tris (pH 7.5), 150 mm NaCl at 100 °C and quenched with excess 2% CHAPS in HBS followed by incubation with the corresponding antibody and Sepharose beads overnight and washing with 0.5% CHAPS in HBS or Connie's wash buffer where indicated.
Affinity purification, SDS-PAGE, and immunoblotting
HEK293T cells were grown in 35- or 60-mm dishes, and all transfections were carried out for 18 h in DMEM supplemented with 10% FBS and 1% P/S and incubated at 37 °C in 5% CO2. Cells were lysed on ice with either CHAPS buffer or MNT buffer. The postnuclear supernatant was isolated by centrifugation followed by preclearing with unbound protein A–Sepharose beads and subsequent incubation with the corresponding antibody and Sepharose beads overnight. A fraction of the postnuclear supernatant (10%) was treated with 10% TCA to collect the whole-cell lysate where indicated. Immunopellets were isolated and washed with 0.5% CHAPS in HBS or Connie's wash buffer where indicated. Proteins were either subjected to glycosidase digestion (following the New England Biolabs protocol) or eluted from beads with reducing sample buffer followed by SDS-PAGE. Immunoblots were analyzed via a LI-COR imaging system (LI-COR Biosciences).
PEG-maleimide modification assay
HEK293T cells were transfected with EDEM1 constructs and incubated overnight. The next day (∼20 h later), a subset of the samples was pretreated with 5 mm DTT for 1 h. Cells were lysed with sample buffer (30 mm Tris (pH 6.8), 9% SDS, 15% glycerol) containing 5 mm PEG-maleimide or 20 mm NEM. Lysates were incubated for 1 h. The step-by-step protocol is published in Braakman et al. (46).
CRISPR/Cas9-targeted KO man1a1/1a2/1b1/1c1 in CHO cells
The CRISPR/Cas9 system was used for gene KO in CHO cells as described previously (47). CHOZN GS−/− cells with stable expression of human α-galactosidase A were used as the parental clone for KO man1a1/1a2/1b1/1c1. Cells were maintained as suspension cultures in EX-CELL CHO CD Fusion serum-free medium (Sigma, catalog number 14365C) in a 50-ml TPP TubeSpin® and incubated at 37 °C and 5% CO2. Cells were seeded at 0.5 × 106 cells/ml in a T25 flask (Nunc, Denmark) 1 day prior to transfection. Electroporation was conducted with 2 × 106 cells with a DNA mixture of 1 μg of Cas9-GFP plasmid and 1 μg of guide RNA plasmid (U6GRNA, Addgene plasmid number 68370) using Amaxa kit V and program U24 with Amaxa Nucleofector 2B (Lonza, Switzerland). 48 h after nucleofection, the 10–15% highest GFP-expressing pool of cells were enriched by FACS, and after 1 week cultured cells were single cell–sorted by FACS into 96 wells. Gene disruption was identified by Indel Detection by Amplicon Analysis (IDAA) as described (48) and further verified by Sanger sequencing.
Author contributions
L. L., M. E. O., W. T., H. C., and D. N. H. conceptualization; L. L., M. E. O., Z. Y., and D. N. H. resources; L. L., M. E. O., T. T., W. T., and H. C. data curation; L. L., M. E. O., T. T., W. T., H. C., and D. N. H. formal analysis; L. L. and D. N. H. supervision; L. L., H. C., and D. N. H. funding acquisition; L. L., M. E. O., T. T., and D. N. H. validation; L. L., M. E. O., T. T., W. T., Z. Y., H. C., and D. N. H. investigation; L. L., M. E. O., and D. N. H. visualization; L. L., M. E. O., T. T., W. T., Z. Y., H. C., and D. N. H. methodology; L. L. and M. E. O. writing-original draft; L. L., M. E. O., and D. N. H. project administration; L. L., M. E. O., T. T., W. T., Z. Y., H. C., and D. N. H. writing-review and editing.
Supplementary Material
Acknowledgments
We acknowledge Dr. J. C. Sunryd and J. B. Graham for helpful discussions and critical reading of the manuscript.
This work was supported by National Institutes of Health Grant GM086874 (to D. N. H.), National Institutes of Health Chemistry-Biology Interface Program Training Grant T32GM008515 (to L. L.), and Danish National Research Foundation Grant DNRF107 (to H. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health
This article contains Figs. S1–S5 and supporting methods.
- ER
- endoplasmic reticulum
- EDEM
- ER degradation-enhancing α-mannosidase–like protein
- MLD
- mannosidase-like domain
- ERAD
- ER-associated protein degradation
- IDR
- intrinsically disordered region
- ERQC
- ER quality control
- CNX
- calnexin
- CRT
- calreticulin
- PDI
- protein-disulfide isomerase
- A1AT
- α1-antitrypsin
- HBS
- HEPES-buffered saline
- PEG-maleimide
- methoxy polyethylene glycol maleimide
- NEM
- N-ethylmaleimide
- FL
- full length
- PNGase F
- peptide:N-glycosidase F
- Endo H
- endoglycosidase H
- NOG
- no glycans
- DMEM
- Dulbecco's modified Eagle's medium
- FBS
- fetal bovine serum
- P/S
- penicillin and streptomycin
- HEK
- human embryonic kidney
- CHO
- Chinese hamster ovary
- KO
- knockout
- GS
- glutamine synthetase
- TX
- Triton X-100
- CH
- CHAPS
- BiP
- binding immunoglobulin protein
- NHK
- null Hong Kong
- SSH
- Sonic Hedgehog.
References
- 1. Appenzeller-Herzog C., and Ellgaard L. (2008) The human PDI family: versatility packed into a single fold. Biochim. Biophys. Acta. 1783, 535–548 10.1016/j.bbamcr.2007.11.010 [DOI] [PubMed] [Google Scholar]
- 2. Tannous A., Pisoni G. B., Hebert D. N., and Molinari M. (2015) N-linked sugar-regulated protein folding and quality control in the ER. Semin. Cell Dev. Biol. 41, 79–89 10.1016/j.semcdb.2014.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Parodi A., Cummings R. D., and Aebi M. (2015) Glycans in glycoprotein quality control, in Essentials of Glycobiology (Varki A., Cummings R. D., Esko J. D., Stanley P., Hart G. W., Aebi M., Darvill A. G., Kinoshita T., Packer N. H., Prestegard J. H., Schnaar R. L., and Seeberger P. H., eds) 3rd Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY: http://www.ncbi.nlm.nih.gov/books/NBK453081/ (accessed April 15, 2018) [Google Scholar]
- 4. Lamriben L., Graham J. B., Adams B. M., and Hebert D. N. (2016) N-Glycan-based ER molecular chaperone and protein quality control system: the calnexin binding cycle. Traffic 17, 308–326 10.1111/tra.12358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nakatsukasa K., and Brodsky J. L. (2008) The recognition and retrotranslocation of misfolded proteins from the endoplasmic reticulum. Traffic 9, 861–870 10.1111/j.1600-0854.2008.00729.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hebert D. N., and Molinari M. (2007) In and out of the ER: protein folding, quality control, degradation, and related human diseases. Physiol. Rev. 87, 1377–1408 10.1152/physrev.00050.2006 [DOI] [PubMed] [Google Scholar]
- 7. Frenkel Z., Gregory W., Kornfeld S., and Lederkremer G. Z. (2003) Endoplasmic reticulum-associated degradation of mammalian glycoproteins involves sugar chain trimming to Man6–5GlcNAc2. J. Biol. Chem. 278, 34119–34124 10.1074/jbc.M305929200 [DOI] [PubMed] [Google Scholar]
- 8. Ermonval M., Kitzmüller C., Mir A. M., Cacan R., and Ivessa N. E. (2001) N-glycan structure of a short-lived variant of ribophorin I expressed in the MadIA214 glycosylation-defective cell line reveals the role of a mannosidase that is not ER mannosidase I in the process of glycoprotein degradation. Glycobiology 11, 565–576 10.1093/glycob/11.7.565 [DOI] [PubMed] [Google Scholar]
- 9. Ushioda R., Hoseki J., Araki K., Jansen G., Thomas D. Y., and Nagata K. (2008) ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science 321, 569–572 10.1126/science.1159293 [DOI] [PubMed] [Google Scholar]
- 10. Cormier J. H., Tamura T., Sunryd J. C., and Hebert D. N. (2009) EDEM1 recognition and delivery of misfolded proteins to the SEL1L-containing ERAD complex. Mol. Cell 34, 627–633 10.1016/j.molcel.2009.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hosokawa N., Wada I., Hasegawa K., Yorihuzi T., Tremblay L. O., Herscovics A., and Nagata K. (2001) A novel ER α-mannosidase-like protein accelerates ER-associated degradation. EMBO Rep. 2, 415–422 10.1093/embo-reports/kve084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Molinari M., Calanca V., Galli C., Lucca P., and Paganetti P. (2003) Role of EDEM in the release of misfolded glycoproteins from the calnexin cycle. Science 299, 1397–1400 10.1126/science.1079474 [DOI] [PubMed] [Google Scholar]
- 13. Oda Y., Hosokawa N., Wada I., and Nagata K. (2003) EDEM as an acceptor of terminally misfolded glycoproteins released from calnexin. Science 299, 1394–1397 10.1126/science.1079181 [DOI] [PubMed] [Google Scholar]
- 14. Kosmaoglou M., Kanuga N., Aguilà M., Garriga P., and Cheetham M. E. (2009) A dual role for EDEM1 in the processing of rod opsin. J. Cell Sci. 122, 4465–4472 10.1242/jcs.055228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Olivari S., Cali T., Salo K. E., Paganetti P., Ruddock L. W., and Molinari M. (2006) EDEM1 regulates ER-associated degradation by accelerating de-mannosylation of folding-defective polypeptides and by inhibiting their covalent aggregation. Biochem. Biophys. Res. Commun. 349, 1278–1284 10.1016/j.bbrc.2006.08.186 [DOI] [PubMed] [Google Scholar]
- 16. Hosokawa N., Tremblay L. O., Sleno B., Kamiya Y., Wada I., Nagata K., Kato K., and Herscovics A. (2010) EDEM1 accelerates the trimming of 1,2-linked mannose on the C branch of N-glycans. Glycobiology 20, 567–575 10.1093/glycob/cwq001 [DOI] [PubMed] [Google Scholar]
- 17. Ninagawa S., Okada T., Sumitomo Y., Kamiya Y., Kato K., Horimoto S., Ishikawa T., Takeda S., Sakuma T., Yamamoto T., and Mori K. (2014) EDEM2 initiates mammalian glycoprotein ERAD by catalyzing the first mannose trimming step. J. Cell Biol. 206, 347–356 10.1083/jcb.201404075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ninagawa S., Okada T., Sumitomo Y., Horimoto S., Sugimoto T., Ishikawa T., Takeda S., Yamamoto T., Suzuki T., Kamiya Y., Kato K., and Mori K. (2015) Forcible destruction of severely misfolded mammalian glycoproteins by the non-glycoprotein ERAD pathway. J. Cell Biol. 211, 775–784 10.1083/jcb.201504109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tamura T., Cormier J. H., and Hebert D. N. (2011) Characterization of early EDEM1 protein maturation events and their functional implications. J. Biol. Chem. 286, 24906–24915 10.1074/jbc.M111.243998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shenkman M., Groisman B., Ron E., Avezov E., Hendershot L. M., and Lederkremer G. Z. (2013) A shared endoplasmic reticulum-associated degradation pathway involving the EDEM1 protein for glycosylated and nonglycosylated proteins. J. Biol. Chem. 288, 2167–2178 10.1074/jbc.M112.438275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Marin M. B., Ghenea S., Spiridon L. N., Chiritoiu G. N., Petrescu A.-J., and Petrescu S.-M. (2012) Tyrosinase degradation is prevented when EDEM1 lacks the intrinsically disordered region. PLoS One 7, e42998 10.1371/journal.pone.0042998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tang H.-Y., Huang C.-H., Zhuang Y.-H., Christianson J. C., and Chen X. (2014) EDEM2 and OS-9 are required for ER-associated degradation of non-glycosylated sonic hedgehog. PLoS One 9, e92164 10.1371/journal.pone.0092164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Saeed M., Suzuki R., Watanabe N., Masaki T., Tomonaga M., Muhammad A., Kato T., Matsuura Y., Watanabe H., Wakita T., and Suzuki T. (2011) Role of the endoplasmic reticulum-associated degradation (ERAD) pathway in degradation of hepatitis C virus envelope proteins and production of virus particles. J. Biol. Chem. 286, 37264–37273 10.1074/jbc.M111.259085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xue B., Dunbrack R. L., Williams R. W., Dunker A. K., and Uversky V. N. (2010) PONDR-FIT: a meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta 1804, 996–1010 10.1016/j.bbapap.2010.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ward J. J., McGuffin L. J., Bryson K., Buxton B. F., and Jones D. T. (2004) The DISOPRED server for the prediction of protein disorder. Bioinformatics 20, 2138–2139 10.1093/bioinformatics/bth195 [DOI] [PubMed] [Google Scholar]
- 26. Sickmeier M., Hamilton J. A., LeGall T., Vacic V., Cortese M. S., Tantos A., Szabo B., Tompa P., Chen J., Uversky V. N., Obradovic Z., and Dunker A. K. (2007) DisProt: the Database of Disordered Proteins. Nucleic Acids Res. 35, D786–D793 10.1093/nar/gkl893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Prilusky J., Felder C. E., Zeev-Ben-Mordehai T., Rydberg E. H., Man O., Beckmann J. S., Silman I., and Sussman J. L. (2005) FoldIndex: a simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics 21, 3435–3438 10.1093/bioinformatics/bti537 [DOI] [PubMed] [Google Scholar]
- 28. Le Fourn V., Gaplovska-Kysela K., Guhl B., Santimaria R., Zuber C., and Roth J. (2009) Basal autophagy is involved in the degradation of the ERAD component EDEM1. Cell. Mol. Life Sci. 66, 1434–1445 10.1007/s00018-009-9038-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gülow K., Bienert D., and Haas I. G. (2002) BiP is feed-back regulated by control of protein translation efficiency. J. Cell Sci. 115, 2443–2452 [DOI] [PubMed] [Google Scholar]
- 30. van der Lee R., Buljan M., Lang B., Weatheritt R. J., Daughdrill G. W., Dunker A. K., Fuxreiter M., Gough J., Gsponer J., Jones D. T., Kim P. M., Kriwacki R. W., Oldfield C. J., Pappu R. V., Tompa P., et al. (2014) Classification of intrinsically disordered regions and proteins. Chem. Rev. 114, 6589–6631 10.1021/cr400525m [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gnann A., Riordan J. R., and Wolf D. H. (2004) Cystic fibrosis transmembrane conductance regulator degradation depends on the lectins Htm1p/EDEM and the Cdc48 protein complex in yeast. Mol. Biol. Cell 15, 4125–4135 10.1091/mbc.e04-01-0024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sokołowska I., Piłka E. S., Sandvig K., Wěgrzyn G., and Słomińska-Wojewódzka M. (2015) Hydrophobicity of protein determinants influences the recognition of substrates by EDEM1 and EDEM2 in human cells. BMC Cell Biol. 16, 1 10.1186/s12860-015-0047-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ronzoni R., Berardelli R., Medicina D., Sitia R., Gooptu B., and Fra A. M. (2016) Aberrant disulphide bonding contributes to the ER retention of α1-antitrypsin deficiency variants. Hum. Mol. Genet. 25, 642–650 10.1093/hmg/ddv501 [DOI] [PubMed] [Google Scholar]
- 34. Vallée F., Lipari F., Yip P., Sleno B., Herscovics A., and Howell P. L. (2000) Crystal structure of a class I α1,2-mannosidase involved in N-glycan processing and endoplasmic reticulum quality control. EMBO J. 19, 581–588 10.1093/emboj/19.4.581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jansen G., Määttänen P., Denisov A. Y., Scarffe L., Schade B., Balghi H., Dejgaard K., Chen L. Y., Muller W. J., Gehring K., and Thomas D. Y. (2012) An interaction map of endoplasmic reticulum chaperones and foldases. Mol. Cell. Proteomics 11, 710–723 10.1074/mcp.M111.016550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jessop C. E., Watkins R. H., Simmons J. J., Tasab M., and Bulleid N. J. (2009) Protein disulphide isomerase family members show distinct substrate specificity: P5 is targeted to BiP client proteins. J. Cell Sci. 122, 4287–4295 10.1242/jcs.059154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Khodayari N., Marek G., Lu Y., Krotova K., Wang R. L., and Brantly M. (2017) Erdj3 has an essential role for Z variant α-1-antitrypsin degradation. J. Cell. Biochem. 118, 3090–3101 10.1002/jcb.26069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yu S., Ito S., Wada I., and Hosokawa N. (2018) ER-resident protein 46 (ERp46) triggers the mannose-trimming activity of ER degradation-enhancing α-mannosidase-like protein 3 (EDEM3). J. Biol. Chem. 293, 10663–10674 10.1074/jbc.RA118.003129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mast S. W., Diekman K., Karaveg K., Davis A., Sifers R. N., and Moremen K. W. (2005) Human EDEM2, a novel homolog of family 47 glycosidases, is involved in ER-associated degradation of glycoproteins. Glycobiology 15, 421–436 10.1093/glycob/cwi014 [DOI] [PubMed] [Google Scholar]
- 40. Merulla J., Fasana E., Soldà T., and Molinari M. (2013) Specificity and regulation of the endoplasmic reticulum-associated degradation machinery. Traffic 14, 767–777 10.1111/tra.12068 [DOI] [PubMed] [Google Scholar]
- 41. Fredrickson E. K., Rosenbaum J. C., Locke M. N., Milac T. I., and Gardner R. G. (2011) Exposed hydrophobicity is a key determinant of nuclear quality control degradation. Mol. Biol. Cell 22, 2384–2395 10.1091/mbc.e11-03-0256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Reed B. J., Locke M. N., and Gardner R. G. (2015) A conserved deubiquitinating enzyme uses intrinsically disordered regions to scaffold multiple protein interaction sites. J. Biol. Chem. 290, 20601–20612 10.1074/jbc.M115.650952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jang B.-Y., Ryoo H. D., Son J., Choi K.-C., Shin D.-M., Kang S.-W., and Kang M.-J. (2015) Role of Drosophila EDEMs in the degradation of the α-1-antitrypsin Z variant. Int. J. Mol. Med. 35, 870–876 10.3892/ijmm.2015.2109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hirao K., Natsuka Y., Tamura T., Wada I., Morito D., Natsuka S., Romero P., Sleno B., Tremblay L. O., Herscovics A., Nagata K., and Hosokawa N. (2006) EDEM3, a soluble EDEM homolog, enhances glycoprotein endoplasmic reticulum-associated degradation and mannose trimming. J. Biol. Chem. 281, 9650–9658 10.1074/jbc.M512191200 [DOI] [PubMed] [Google Scholar]
- 45. Timms R. T., Menzies S. A., Tchasovnikarova I. A., Christensen L. C., Williamson J. C., Antrobus R., Dougan G., Ellgaard L., and Lehner P. J. (2016) Genetic dissection of mammalian ERAD through comparative haploid and CRISPR forward genetic screens. Nat. Commun. 7, 11786 10.1038/ncomms11786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Braakman I., Lamriben L., van Zadelhoff G., and Hebert D. N. (2017) Analysis of disulfide bond formation. Curr. Protoc. Protein Sci. 90, 14.1.1–14.1.21 10.1002/cpps.43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schulz M. A., Tian W., Mao Y., Van Coillie J., Sun L., Larsen J. S., Chen Y.-H., Kristensen C., Vakhrushev S. Y., Clausen H., and Yang Z. (2018) Glycoengineering design options for IgG1 in CHO cells using precise gene editing. Glycobiology 28, 542–549 10.1093/glycob/cwy022 [DOI] [PubMed] [Google Scholar]
- 48. Yang Z., Steentoft C., Hauge C., Hansen L., Thomsen A. L., Niola F., Vester-Christensen M. B., Frödin M., Clausen H., Wandall H. H., and Bennett E. P. (2015) Fast and sensitive detection of indels induced by precise gene targeting. Nucleic Acids Res. 43, e59 10.1093/nar/gkv126 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.