

Abstract
Fucosylated glycosaminoglycan (FG), a structurally complex glycosaminoglycan found up to now exclusively in sea cucumbers, has distinct anticoagulant properties, notably a strong inhibitory activity of intrinsic factor Xase complex (FXase). Knowledge of the FG structures could facilitate the development of a clinically effective intrinsic FXase inhibitor for anticoagulant drugs. Here, a new fucosylated glycosaminoglycan was obtained from the widely traded sea cucumber Bohadschia argus. The precise structure was deduced as {→4)-[l-Fuc3S4S-α-(1→3)-]-d-GlcA-β-(1→3)-d-GalNAc4S6S-β-(1} through analysis of its chemical properties and homogeneous oligosaccharides purified from its β-eliminative depolymerized products. The B. argus FG with mostly 3,4-di-O-sulfated fucoses expands our knowledge on FG structural types. This β-elimination process, producing oligosaccharides with well-defined structures, is a powerful tool for analyzing the structure of complex FGs. Among these oligosaccharides, an octasaccharide displayed potent FXase inhibitory activity. Compared with oligosaccharides with various degrees of polymerization (3n and 3n − 1), our analyses reveal that the purified octasaccharide is the minimum structural unit responsible for the potent selective FXase inhibition, because the d-talitol in the nonsaccharide is unnecessary. The octasaccharide with 2,4-di-O-sulfated fucoses is more potent than that of one with 3,4-di-O-sulfated fucoses. Thus, sulfation patterns can play an important role in the inhibition of intrinsic factor Xase complex.
Keywords: natural product, carbohydrate chemistry, carbohydrate structure, chemical modification, drug discovery, oligosaccharide, thrombosis
Introduction
Fucosylated glycosaminoglycan (FG)3 is a complex glycosaminoglycan found exclusively so far in sea cucumbers (Echinodermata, Holothuroidea), which are widely traded in Asian dried seafood markets. This FG has attracted considerable attention because of its distinctive structure and extensive bioactivities, especially the potent inhibition of the intrinsic factor Xase complex (FXase), which is involved in blood clotting (1). However, natural FG itself has not directly been developed into an anticoagulant drug, partly because of its adverse effects such as factor XII (FXII) activation and platelet aggregation (2). Currently, depolymerization of FG is considered to be an effective method for reducing these side effects (3). Studies have reported the relationships between the structures and anticoagulant activities of depolymerized FG and its derivatives (4–8). However, these studies offer conflicting conclusions (5, 8), particularly the effects of its sulfation patterns in the fucose side chains on anticoagulation, partly because of the inaccessibility of FG fragments with well-defined structures. The structure–activity relationships of the compounds thus still remain to be further clarified in detail.
FG has been recognized to possess a backbone of chondroitin sulfate consisting of the disaccharide repeating units of glucuronic acid (GlcA) and GalNAc residues, and fucose (Fuc) branches linked to the O-3 of GlcA (1). Controversy remains on the sulfation patterns and/or glycosidic linkage types of the branch, which may vary among different sea cucumber species from which the products were obtained (6, 9–11).
As a biomacromolecule, the structure of FG is difficult to elucidate because of the variety of structures and the range and intricate signals of its NMR spectra. Hence, the structures are susceptible to misinterpretation. Researchers have therefore lacked consensus on the structure of FG derived from the same sea cucumber (10, 12, 13). Degradation and analysis of the resulting fragments is considered to be an effective method to resolve the complicated structure. However, except for the deacetylation-deaminative depolymerization (14), few degradation methodologies with minimal damage to the essential structure have been confirmed (15).
Our previous study found that nonasaccharide from the deacetylation-deaminative depolymerized products was the minimum fragment responsible for the potent selective inhibition of intrinsic factor Xase complex. According to the depolymerization reaction, the degree of polymerization (dp) of the depolymerized products was deduced as 3n, and the shorter sequence than nonasaccharide was hexosaccharide (14), where n is the natural number. Nonetheless, one question is whether the oligosaccharides between the two sequences retain inhibition activity comparable to that of nonasaccharide? Additionally, its fucose side chains play an important role in its anticoagulant activity (1–8). Recently, we have found that an FG-derived octasaccharide with 2,4-di-O-sulfated fucose (Fuc2S4S) exhibits strong factor Xase complex inhibitory activity (16). However, the effects of its other sulfation patterns on the inhibition of intrinsic factor Xase complex remain unresolved because oligosaccharides with different sulfation patterns have not yet been purified or synthetized.
In this study, an FG from the tropical sea cucumber Bohadschia argus was selected from abundant representative FGs. This sea cucumber is relatively common in dried seafood markets in mainland China. The structure of the native FG was characterized by analysis of its monosaccharide composition, ratio of sulfate to carboxyl group, infrared spectroscopy, and 1H NMR spectrum. For our purpose of identifying the structure of B. argus FG and getting a shorter sequence than nonasaccharide, a chemical β-eliminative depolymerization method was employed to produce low molecular weight products of FG with certain structural characteristics (17). This method selectively cleaved the β-(1→4) bonds between GalNAc and GlcA residues, and Δ4,5-unsaturated GlcA was formed at the nonreducing termini. The product also showed characteristic UV absorption at 232 nm. This was applied as a simple, rapid, and specific detection method for chromatography analysis and purification. The β-eliminative depolymerized procedure reacts under alkaline conditions in which the peeling reaction probably occurs and results in the stepwise loss of the monosaccharide residue from the reducing end of carbohydrate polymers (18, 19). The resulting fragments are remarkably different from those of deacetylation-deaminative depolymerization and used for a study of structure–activity relationships. Further, anticoagulant activity of these oligosaccharides and their effects on coagulation factors and cofactors were evaluated to clarify the structure–activity relationship, which is a precursor for the discovery of novel anticoagulants.
Results
Purification and characterization of FG
Crude polysaccharides from B. argus were isolated by the methods of papain enzymolysis, alkaline hydrolysis, and ethanol precipitation (20). The crude polymers were further purified by ethanol precipitation and strong anion-exchange FPA98 chromatography. The fraction collected by elution with 2.0 m NaCl solution was dialyzed and lyophilized to obtain FG (Fig. 1A). This product did not have any UV absorption at ∼260 or ∼280 nm as detected by an UV detector, indicating that it contained no contaminants of nucleotide or protein/peptide. The yield of the native FG was ∼0.85% by dry weight of the body ball of the sea cucumber.
Figure 1.
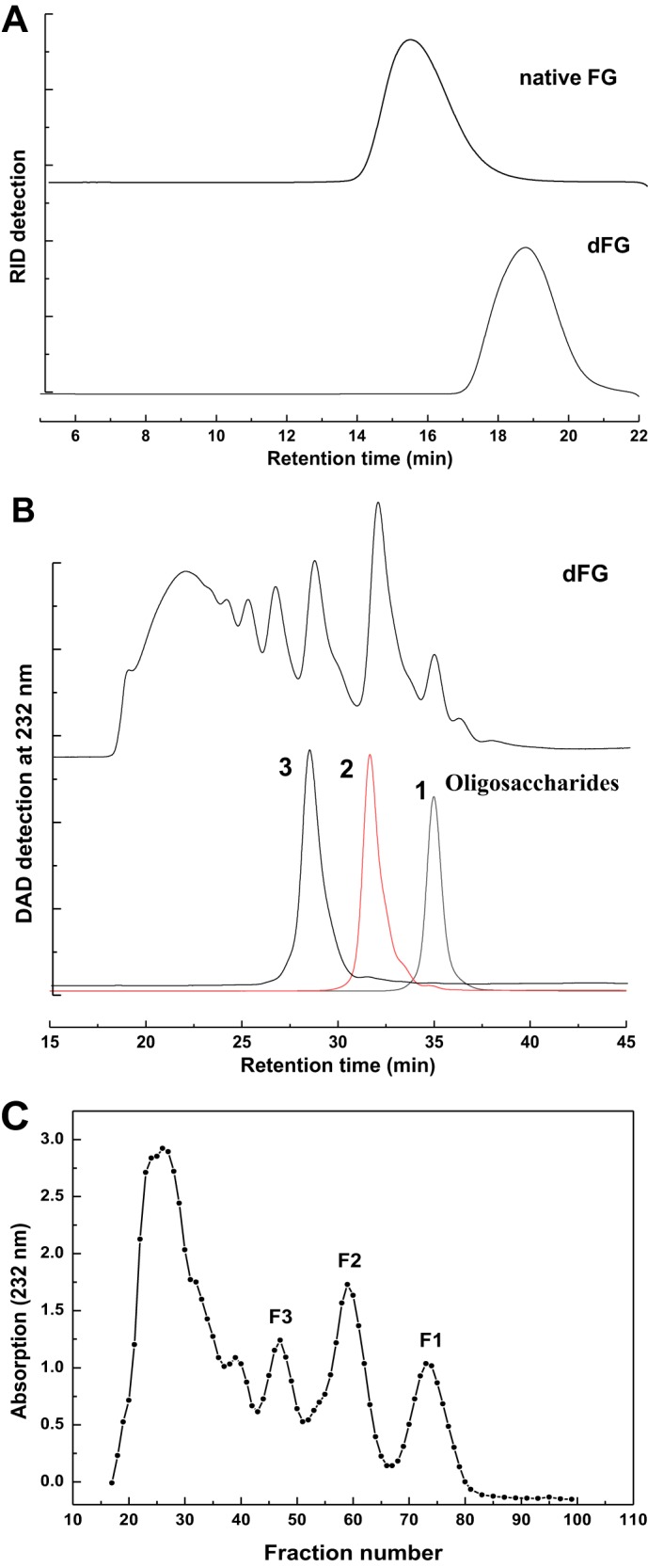
HPGPC profiles of the native FG and dFG (A, Shodex OHpak SB-804 HQ column eluted with 0.1 m NaCl solution under the differential refractive index detector (RID)), and dFG, compounds 1–3 (B, Superdex peptide 10/300 GL column eluted with 0.2 m NaCl solution with DAD detection at 232 nm), and the elution curve of dFG (C, by GPC using the Bio-Gel P10 column eluted with 0.2 m NaCl solution and monitored by UV-visible absorption at 232 nm).
The weight average and number average of the B. argus FG were 70,130 and 60,230 Da, respectively, as determined by multiangle laser light scattering, respectively. These are similar to those of another FG from the sea cucumber Stichopus herrmanni (20). The FG from B. argus contained three monosaccharides, namely d-glucuronic acid (GlcA), GalNAc, and l-fucose (Fuc) as qualitatively identified by HPLC according to 1-phenyl-3-methyl-5-pyrazolone (PMP) derivatization procedures (Fig. S1). Additionally, classical conductimetric analysis showed that the sulfate (SO4) content of the FG was 36.7 ± 0.51% and the molar ratio of sulfate ester to carboxyl group was 3.90 ± 0.05:1 (Fig. S2A).
GalNAc and Fuc of the native FG were readily confirmed by protons with the distinctive values of 1.98 ppm (COCH3) and 1.28 ppm (CH3) in the 1H NMR spectrum, respectively (Fig. S3). The integral ratio of the two signals was ∼1:1, indicating that the two monosaccharide residues, GalNAc and Fuc, were in equal amounts in moles. The two single signals at 5.27 and 5.60 ppm observed in the region of ∼5.0–5.7 ppm could be assigned to anomeric protons of α-l-fucose residues, i.e. 3,4-di-O-sulfated fucose (Fuc3S4S) (∼95%) and minor 2,4-di-O-sulfated fucose (Fuc2S4S) (∼5%) according to previous studies (21, 22). Other signals, especially in the region of 3.40–4.80 ppm, were broad and overlapped, thus hindering the elucidation of the precise structure of the native B. argus FG.
Purification and structural characterization of oligosaccharides
The depolymerized FG (dFG) with molecular mass of 4005 Da, which was much smaller than that of the native FG (Fig. 1A), was obtained by a chemical β-eliminative depolymerization method (17). In brief (Scheme 1), sodium salt of FG was firstly transformed to benzethonium salt, and benzethonium salt was then esterified by benzyl chloride and treated with EtONa/EtOH. After saponification to hydrolyze its benzyl ester and reduction of reducing ends to its alcoholic hydroxyl using NaBH4, the structure of the depolymerized product (dFG) with a yield of ∼65% based on the starting material FG was analyzed (Fig. 1). The sulfate (SO4) content of the dFG was 37.7 ± 0.10%, and the molar ratio of sulfate ester to carboxyl group was 3.86 ± 0.09:1 (Fig. S2B), both of which are similar to those of its native FG (Fig. S2A). Additionally, the characteristic infrared spectroscopy absorptions of the dFG are similar to those of the native FG (Fig. S4).
Scheme 1.

The procedure of β-eliminative depolymerization of native FG. Detailed reaction conditions are described under “Experimental procedures.”
In contrast to the native FG, the signals of 1H NMR spectrum of dFG were much clearer (Fig. S5). In the 1D NMR spectra of dFG, Fuc and GalNAc residues were reconfirmed by the characteristic values of 1.24, 1.32 (H-6), and 2.00 ppm (H-8) in the 1H NMR spectrum. These values corresponded to the signals of 18.56, 18.83, 18.79 (C-6), 25.26 ppm, 25.29, and 25.37 ppm (C-8) in the 13C NMR spectrum. Signals between 5.20 ppm and 5.30 ppm were identified as anomeric protons of 3,4-di-O-sulfated fucose residues. A new signal located at 5.68 ppm, which was absent in the analysis of native FG, could be assigned to H-4 of Δ4,5 unsaturated glucuronic acids (dU), corresponding to the signal at 109.48 ppm assigned to C-4 of dU. Further, the signal at 149.64 ppm was assigned to olefinic C-5 of the newly forming double bond. Detailed analysis was performed by virtue of 2D NMR spectra (Fig. S5). We could roughly resolve the NMR data (Fig. S5 and Table S1) based on our previous results from other sea cucumber species (14, 20), although the signals of this product in the region of 3.5–4.8 ppm still overlapped. The structural information of dFG deduced from NMR data and chemical characteristics may be insufficient to provide direct evidence for the structure of native FG. The dFG is not homogenous but instead is composed of oligosaccharides with special nonreducing ends and reducing termini, so the NMR signals can be complicated.
This low-molecular-weight dFG was further separated into several independent signal peaks by high-performance gel permeation chromatography (HPGPC) analysis on a Superdex Peptide 10/300 GL column (Fig. 1B), implying the possibility that several homogeneous oligosaccharides could be obtained by gel chromatography. Therefore, the oligosaccharides were further purified from the low-molecular-weight dFG by gel permeation chromatography (GPC) using a Bio-Gel P10 column. The eluted fractions and the elution curve were monitored by UV-visible absorption at 232 nm (Fig. 1C). The sample marked as peak F1 was purified by Bio-Gel P4 to obtain compound 1, and the samples marked as peaks F2 and F3 were purified by Bio-Gel P10 to obtain compounds 2 and 3, respectively. Each compound was desalted on a Sephadex G10 column and lyophilized. Compounds 1-3 were clearly identified by NMR spectra and ESI–Q-TOF–MS (Fig. 2 and Figs. S6–S11).
Figure 2.

1H/13C NMR spectra of compounds 1 (A and D), 2 (B and E), and 3 (C and F).
Through detailed NMR analysis, we proved that Compound 1 is a trisaccharide. The distinct signal at 5.72 ppm could be assigned to H-4 of Δ4,5 unsaturated glucuronic acids (dU), corresponding to the signal at 109.05 ppm assigned to C-4 of dU (Fig. 2, A and D). The carbon signal at 148.70 ppm was assigned to olefinic C-5 of the double bond. This was in agreement with the β-eliminative degradation mechanism (17). According to the COSY and TOCSY spectra, three spin systems were clearly observed (Figs. S6A and S7A). Characteristic proton values of 1.23 and 1.93 ppm in the 1H NMR spectrum were assigned as CH3 of Fuc and COCH3 of GalNAc-ol (rA), respectively. The alcoholic hydroxyl group at δC 62.11 ppm, which was assigned as C-1 of rA, was produced by reduction with NaBH4 during the depolymerization process. This avoided the intricate sets of signals because of the α/β configuration at the reducing end (23). The obvious downfield shifts of H-3/H-4 of Fuc, H-4/H-6 of rA indicated that Fuc was sulfated at both the C-3 and C-4 positions and rA was sulfated at both the C-4 and C-6 positions (24). Further analysis of HMBC and ROESY spectra revealed that Fuc was attached to the O-3 of dU, and dU was glycosylated at O-3 of rA (Figs. S8A and S10A). The small H–H coupling constants (4.08 Hz) indicated that the anomer proton of Fuc possessed an α configuration (14). Moreover, an α linkage between dU and rA but not β was deduced because the Δ4,5 unsaturated glucuronic acid was formed by β-elimination reaction at the nonreducing end (25, 26). Additionally, the molecular formula was determined to be C20H28 O27NS4Na5 by negative ESI–Q-TOF–MS ion peak at m/z 955.9251 [M − H]− (calculated as 955.9147) and m/z 933.9481 [M − Na]− (calculated as 933.9328) (Fig. S11). Thus, compound 1 was confirmed to be a trisaccharide with the structure of l-Fuc3S4S-α-(1→3)-l-Δ4,5GlcA-α-(1→3)-d-GalNAc4S6S-ol (Fig. 3). The assignments are provided in Table S2.
Figure 3.

Structures of compounds 1–3, which are a trisaccharide, a pentasaccharide, and an octasaccharide, respectively.
Compound 2 showed more signals in its 1D NMR spectrum (Fig. 2, B and E). Apart from the characteristic signals of a double bond, the ratio of CH3 and COCH3 changed to 2:1. Combining with the four carbons at ∼100 ppm, a pentasaccharide was deduced. According to COSY and TOCSY spectra, five spin systems were confirmed (Figs. S6B and S7B). The reducing-end residue was determined to be GlcA-ol. The glycosylated binding sites were confirmed via HMBC and ROESY spectra (Figs. S8B and S10B). Specifically, fucoses (the fucose linked to dU at the nonreducing end and the sulfated fucose linked to the GlcA-ol) were both glycosylated at O-3 of GlcA residues, dU was linked to O-3 of GalNAc, and GalNAc was attached to O-4 of reducing termini GlcA-ol. The sites of sulfation at the C-3 and C-4 positions of fucoses and at the C-4 and C-6 positions of GalNAc were revealed by the downfield shifts of their respective 1D NMR values. Combining with the H–H coupling constants and the analysis of 1, compound 2 was confirmed to own the structure of l-Fuc3S4S-α-(1→3)-l-Δ4,5GlcA-α-(1→3)-d-GalNAc4S6S-β-(1→4)-[l-Fuc3S4S-α-(1→3)]-d-GlcA-ol (Fig. 3). Moreover, ESI–Q-TOF–MS analysis revealed an m/z of 729.4685 for [M − 2Na]2− (calculated as 729.4465) and m/z of 478.6516 for [M − 3Na]3− (calculated as 478.6346) (Fig. S11), confirming the molecular formula of C32H43O43NS6Na8. The assignments are given in Table S2.
The structure of compound 3 was deduced using the same methods as for compounds 1 and 2. Detailed analysis (Fig. 2, C and F) revealed that compound 3 possessed a trisaccharide additional to that of compound 2. Linkages between different residues and the sulfated positions were the same as compound 2. Its molecular formula of C52H69O70N2S10Na13 was determined by the negative ESI–Q-TOF–MS ion peak at m/z 1206.9280 [M − 2Na]2− (calculated as 1206.8997), m/z 796.9594 [M − 3Na]3− (calculated as 796.9367) and m/z 591.9741 [M − 4Na]4− (calculated as 591.9552) (Fig. S11). Therefore, compound 3 was deduced as an octasaccharide with the structure of l-Fuc3S4S-α-(1→3)-l-Δ4,5GlcA-α-(1→3)-d-GalNAc4S6S-β-(1→4)-[l-Fuc3S4S-α-(1→3)-]-d-GlcA-β-(1→3)-d-GalNAc4S6S-β-(1→4)-[l-Fuc3S4S-α-(1→3)-]-d-GlcA-ol (Fig. 3). The assignments are given in Table 1. Further analysis of the NMR spectra indicates that minor Fuc2S4S residues existing in these purified oligosaccharides are also α-(1→3)–linked to GlcA (Figs. S6–S10).
Table 1.
1H/13C NMR chemical shift assignments of compound 3 (δ, ppm; J, Hz)
The data (δ) were measured in HOD (D 99.9%). The assignments were based on HSQC, COSY, TOCSY, ROESY, and HMBC experiments.
| Octasaccharide (3) |
||||||||
|---|---|---|---|---|---|---|---|---|
| dF | dU | dA | Fuc | GlcA | rA | rF | rU | |
| H1 | 5.20 J (1,2) = 4.32 | 4.85 J (1,2) = 8.64 | 4.53 J (1,2) = 8.56 | 5.28 J (1,2) = 4.16 | 4.43 J (1,2) = 7.60 | 4.63 J (1,2) = 7.60 | 5.04 J (1,2) = 4.08 | 3.73 J (1,1′) = 11.76 |
| 3.69 (H-1′) J (1/1′,2) = 3.92, 6,72 | ||||||||
| C1 | 100.91 | 105.74 | 102.38 | 101.99 | 106.34 | 104.17 | 104.04 | 65.33 |
| H2 | 3.88 J (2,3) = 10.40 | 3.83 J (2,3) = 8.32 | 4.08 J (2,3) = 9.52 | 3.87 J (2,3) = 10.48 | 3.55 J (2,3) = 8.36 | 3.98 | 3.89 J (2,3) = 10.56 | 4.07 J (2,3) = 3.92 |
| C2 | 69.18 | 72.99 | 54.27 | 69.12 | 76.20 | 54.27 | 69.34 | 72.56 |
| H3 | 4.53 J (3,4) = 2.96 | 4.42 J (3,4) = 2.72 | 4.10 J (3,4) = 2.52 | 4.46 J (3,4) = 2.88 | 3.63 J (3,4) = 8.72 | 3.97 J (3,4) = 0.80 | 4.55 J (3,4) = 3.36 | 4.02 J (3,4) = 3.92 |
| C3 | 77.97 | 79.09 | 78.88 | 78.11 | 82.00 | 78.47 | 77.97 | 82.10 |
| H4 | 4.83 | 5.68 | 4.91 | 4.96 | 3.95 J (4,5) = 9.32 | 4.75 | 4.83 | 4.01 J (4,5) = 4.08 |
| C4 | 81.70 | 109.42 | 78.98 | 82.10 | 77.95 | 78.98 | 81.66 | 84.20 |
| H5 | 4.27 J (5,6) = 6.72 | 4.01 J (5,6/6′) = 5.28, 8.56 | 4.88 J (5,6) = 6.48 | 3.64 | 4.01 J (5,6/6′) = 4.56, 8.72 | 4.36 J (5,6) = 6.56 | 4.25 | |
| C5 | 69.24 | 149.61 | 74.74 | 69.00 | 79.91 | 75.04 | 69.55 | 74.64 |
| H6 | 1.23 | 4.32 J (6,6′) = 11.96 | 1.34 | 4.22 J (6,6′) = 10.96 | 1.24 | |||
| 4.22 (H-6′) | 4.12 (H-6′) | |||||||
| C6 | 18.56 | 171.62 | 69.99 | 18.79 | 177.73 | 70.50 | 18.83 | 180.09 |
| H7 | ||||||||
| C7 | 177.72 | 177.72 | ||||||
| H8 | 1.99 | 2.00 | ||||||
| C8 | 25.26 | 25.29 | ||||||
Structural deduction of the native FG
Because the double bond of unsaturated GlcA was formed by the β-elimination reaction and the alcoholic hydroxyl was formed by NaBH4 (16, 17), the sequences of these oligosaccharides all contained the repeating trisaccharide unit {[l-Fuc3S4S-α-(1→3)-]-d-GlcA-β-(1→3)-d-GalNAc4S6S-β-(1→4)-}. We employed a “bottom up” strategy (Fig. 4), combined with the physicochemical property and NMR analysis of the native polysaccharide and its depolymerized product (Figs. S1–S5 and Table S1). This approach allowed us to finally construct the precise structure of native B. argus FG as {[l-Fuc3S4S-α-(1→3)-]-d-GlcA-β-(1→3)-d-GalNAc4S6S-β-(1→4)-}n. The highly regular FG comprises a backbone of chondroitin sulfate E, branched by mostly Fuc3S4S (∼95%) and minor Fuc2S4S (∼5%) as a monosaccharide Fuc side chain. The side chains exist only at O-3 of GlcA but not at O-4 or O-6 of GalNAc (Fig. 4). Apart from another highly regular FG with major Fuc2S4S (16, 20), the B. argus FG with mostly 3,4-di-O-sulfated fucoses expands the knowledge on FG structural types.
Figure 4.
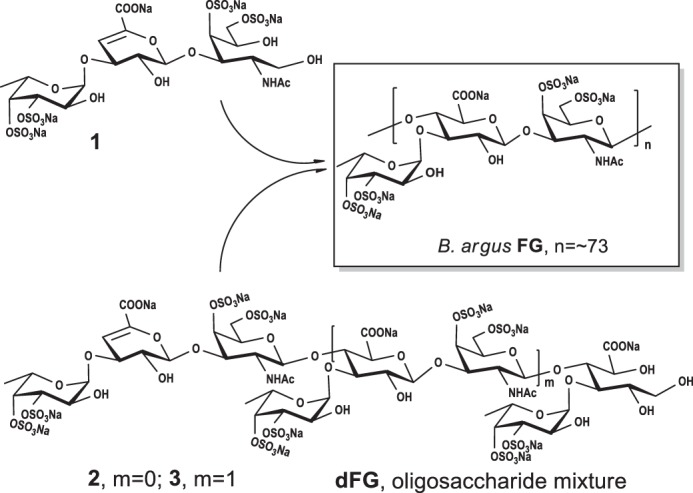
The deduction of native FG from the sea cucumber B. argus.
Analysis of structure–activity relationship
Because the peeling reaction occurred during β-eliminative depolymerization (16, 19), compounds 2 and 3 are a pentasaccharide and octasaccharide, respectively (Figs. 3 and 5). Their structures exhibit certain differences from those of the hexasaccharide and nonasaccharide derived by deacetylation-deaminative depolymerization (Fig. 5) (14). This conveniently allows us to examine more detailed structure–activity relationships.
Figure 5.

Structural comparison between compounds 3, R1, and R2.
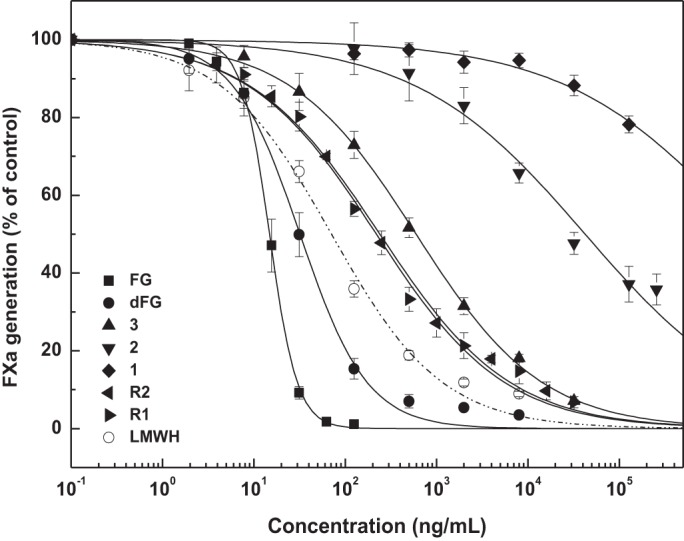
To assess the anticoagulant activities, the native FG from B. argus, its depolymerized product dFG and compounds 1–3 were evaluated and compared with the nonasaccharide (R2) (14) and octasaccharide (R1) (16) from our previous reports. The native FG and dFG exhibited strong APTT prolonging activities in a dose-dependent manner. Specifically, concentrations required to double the APTT were 4.13 and 12.04 μg/ml, respectively (Table 2). The two compounds did not affect PT of human plasma at the concentrations tested (1280 μg/ml). As expected, LMWH displayed strong anticoagulant activities.
Table 2.
Anticoagulant activities of FG-derived oligosaccharides and their effects of on coagulation factors and cofactors
| Molecular mass | APTTa | Anti-FXase (IC50)b | Anti-FIIa/AT (IC50)b | Anti-FXa/AT (IC50)b | |
|---|---|---|---|---|---|
| Da | μg/ml | ng/ml | ng/ml | ng/ml | |
| FG | 70,130c | 4.13 ± 0.05 | 14.83 ± 0.49 | 530.8 ± 23.08 | 3341 ± 142 |
| dFG | 4005c | 12.03 ± 0.04 | 31.99 ± 2.33 | >100,000 | >100,000 |
| 3 | 2459 (dp8) | 47.40 ± 0.20 | 597.1 ± 60.10 | >100,000 | >100,000 |
| 2 | 1504 (dp5) | 334.3 ± 7.5 | >10,000 | >100,000 | >100,000 |
| 1 | 956 (dp3) | >1000 | >100,000 | >100,000 | >100,000 |
| R1d | 2459 (dp8) | 25.09 ± 0.31 | 239.7 ± 24.55 | >100,000 | >100,000 |
| R2d | 2805 (dp9) | 27.58 ± 0.89 | 219.4 ± 22.36 | >100,000 | >100,000 |
| LMWHe | 4500–5500 | 10.21 ± 0.09 | 70.94 ± 6.68 | 38.00 ± 0.96 | 75.57 ± 2.80 |
a Concentration required to double the APTT of human plasma.
b The IC50 value is the concentration of each agent required to inhibit 50% of protease activity.
c The molecular mass was determined by multiangle laser light scattering.
e LMWH, a low-molecular-weight heparin (enoxaparin), was used as a positive control from Sanofi–Aventis.
APTT-prolonging activities of homogeneous oligosaccharides 3, R1, and R2 were 47.43, 25.11, and 27.57 μg/ml, respectively (Table 2). These results indicated potent intrinsic anticoagulant activities of these compounds. Notably, potency of compound 3 with Fuc3S4S residues for APTT-prolonging was approximately twice as weak as that of R1 with Fuc2S4S residues. The difference in anticoagulant activities is attributed to their sulfation patterns because they are both octasaccharides with similar chemical structures (Fig. 5).
As for PT and TT, no significant influences were observed in these oligosaccharides at concentrations as high as 1280 μg/ml. This result means that they have no or little effect on the extrinsic and common coagulation pathways.
Intrinsic factor Xase complex is the last and rate-limiting target enzyme of the intrinsic coagulation pathway and has been recognized as an ideal target for developing safer anticoagulants inhibitors (27, 28). Structurally novel FG-derived oligosaccharides thus possess selective FXase inhibitory activity, which may make them of special interest as potential candidate drugs. We evaluated the effects of these oligosaccharides on coagulation factors and cofactors (Fig. 6 and Table 2). The B. argus FG, dFG, and compound 3 along with R1 and R2 exhibited potent inhibitory activities for intrinsic factor Xase complex, comparable to positive control LMWH. Among these homogeneous oligosaccharides, compound 3, R1, and R2 displayed much stronger anti-FXase activity. This was higher than their anti-FXa and anti-FIIa activities in the presence of AT, indicating that the anticoagulant mechanisms are significantly different from those of heparin-like drugs (29).
Figure 6.
Anti-FXase activities of FG, dFG, compounds 1–3, R1, and R2.
Compared with chemical structures of these oligosaccharides (Fig. 5), we found that at least three trisaccharide repeating units might be required both for the strong intrinsic anticoagulant activity and the potent selective inhibition of intrinsic factor Xase complex. Notably, we observed some differences in the activity. The structure–activity relationships could be described according to comparison between two octasaccharides and a nonsaccharide. Octasaccharides (3 and R1) without GalNAc at the reducing end revealed that the anhydro-d-talitol in the nonsaccharide (R2) obtained from deacetylation-deaminative depolymerized FG is unnecessary. The minimum size for strong factor Xase complex inhibition is probably an octasaccharide. Additionally, the double bond at the nonreducing end in the octasaccharide might not be required. Based on the two octasaccharides with different sulfated fucose patterns, the degree of polymerization is essential for the activity. However, the difference in 2,4-di-O-sulfated and 3,4-di-O-sulfated fucose residues account for the potency of intrinsic factor Xase complex inhibition. The octasaccharide with 2,4-di-O-sulfated fucoses is 2.5 times more potent than that of one with 3,4-di-O-sulfated fucoses (Table 2). Considering both the existence of the 4-O-sulfated fucose residue in the two types of octasaccharides and the lack of a 4-mono-O-sulfated fucose residue of FG derivatives, the importance of the 4-O-sulfated ester in their activity remains uncertain.
Discussion
As a biomacromolecule, the structural elucidation of FG is challenging because of its heterogeneous and complex chemical properties. These include the sulfation patterns and glycosidic linkage types of side chains, especially the marked variation differing in a species-specific manner. With the exception of deacetylation-deaminative depolymerization, few degradation methodologies imparting minimal damage on the essential structure have been confirmed.
The present study further confirms that the chemical β-eliminative depolymerization is highly selectively to cleave the β-(1→4) bonds between GalNAc and GlcA residues. The structures of the obtained oligosaccharides exhibited a high regularity (Fig. 3), providing a direct evidence on the chemical structure of FG. Additionally, the significant and characteristic UV absorption at 232 nm is important for chromatography analysis and purification. β-Eliminative depolymerization produces FG oligosaccharides with well-defined structures retaining their essential characteristics, with the exception of the double bond of unsaturated GlcA formed by the β-elimination reaction and the alcoholic hydroxyl formed by NaBH4. Thus, the chemical structure of the complex FG inferred from structures of oligosaccharides is highly reliable. This method should be applicable for analyzing the structures of other FGs from various sea cucumber species.
The purified oligosaccharides can be used not only to clarify the structure of the parent compound FG, but also to study their structure–activity relationships in a more detailed manner. For some oligosaccharides (dp, 3n) from the deacetylation-deaminative depolymerized products and (dp, 3n − 1) from the chemical β-eliminative depolymerized products, structure–activity relationship analysis showed that octasaccharide is the minimum size responsible for the potent selective FXase inhibition. Their sulfation patterns also result in different anticoagulant activities and intrinsic factor Xase complex inhibition. Nevertheless, these structurally novel FG-derived octasaccharides possess potent selective intrinsic factor Xase inhibitory activity, conferring special interest as potential candidate drugs.
Experimental procedures
Materials
Dried sea cucumber from the tropical species B. argus was purchased in seafood markets in Guangzhou city of Guangdong province in China. Amberlite FPA98Cl ion-exchange resin was purchased from the Rohm and Haas Company. The monosaccharides including l-fucose, d-glucuronic acid, d-galactose, and N-acetyl-d-galactosamine were purchased from Alfa Aesar. PMP (99%) was purchased from Sigma. Bio-Gel P4/P10 and Sephadex G10/G25 were from Bio-Rad Laboratories and GE Healthcare Life Sciences, respectively. Benzyl chloride was from Aladdin Chemistry Co. Ltd. Benzethonium chloride was from Yangzhou Aoxin Chemical Factory (China). Deuterium oxide (99.9% atom D) was obtained from Sigma–Aldrich. LMWH (enoxaparin, 0.4 ml × 4000 AXaIU) was from Sanofi–Aventis (France). The activated partial thromboplastin time (APTT) kits, CaCl2, standard human plasma (Teco Medical, Niederbayern, Germany), Biophen FVIII: C kit, Biophen anti-Xa kits, and anti-IIa kits, were all from Hyphen Biomed (France). Human factor VIII was from Bayer HealthCare LLC. All other chemicals were of reagent grade and obtained commercially.
Isolation and purification of fucosylated glycosaminoglycan
FG was isolated using a previously described procedure with minor modifications (20). Briefly, 300 g of dried body wall of B. argus was pulverized and dissolved in deionized water (100 mg/ml). 1.0% papain was added to release the protein from the proteoglycans at 50 °C for 6 h, followed by 0.25 m sodium hydroxide for 2 h. Then the extract was fractionally precipitated with different concentrations of ethanol, and crude fucosylated glycosaminoglycan was obtained at EtOH/H2O of 4:6 (v/v). Further purification was carried out by strong anion exchange resin (FPA98 OH form, 4.5 × 45 cm), eluted with distilled water followed by NaCl solutions of increasing concentration (0.5, 1.0, 2.0, and 3.0 m) with 2 bed volume, respectively. Each fraction eluted by water and solutions of gradient NaCl concentration was collected, dialyzed by a dialysis bag with molecular mass cutoff of 3 kDa (Spectrum Laboratories Inc.) and lyophilized.
Analysis of physicochemical properties
The purity and weight-average molecular mass of the samples were examined by HPGPC using an Agilent Technologies 1260 series (Agilent Co., USA) apparatus with differential refraction detector and multiangle laser light-scattering detector (DAWN HELEOS-II), equipped with a Shodex OH-pak SB-806 HQ column (8 mm × 300 mm). The elution solvent was 0.1 m NaCl solution. The flow rate was 0.5 ml/min, and the temperature of the column was 40 °C.
Monosaccharide composition of FG was analyzed by reverse-phase HPLC according to PMP derivatization procedures (30). The dried polysaccharide (3 mg) was dissolved in 1 ml of 2 m TFA, then sealed, and incubated at 110 °C for 4 h in a heating block. The reaction mixture was then evaporated to remove residual TFA with methanol. The above samples were reconstituted in 500 μl of H2O. Then 100 μl of the sample solution, 200 μl of 0.5 m PMP in methanol, and 100 μl of 0.6 m sodium hydroxide were mixed and incubated at 70 °C for 30 min. After adjusting the pH to 7, 500 μl of chloroform was added to extract PMP three times. The top aqueous layer was collected for HPLC analysis. Analysis of the PMP-labeled polysaccharides was carried out using an Agilent Technologies 1260 series apparatus equipped with DAD detectors and an Agilent Eclipse XDB C18 (4.6 mm ×150 mm).
The ratios of sulfate to carboxyl group of the FG and dFG were calculated by a conductive method (31). Approximately 5 mg of each sample was dissolved in 1 ml of H2O and placed on the strong cation exchange column (Dowex 50w × 8 50–100H, 1 × 15 cm). 30 ml of the solution eluted by distilled water was collected and titrated by 0.02 m NaOH.
Chemical β-eliminative depolymerization of FG
The procedure of β-eliminative depolymerization was employed (Scheme 1). In brief, benzethonium chloride solution (1500 mg in 24 ml of H2O) was slowly dropped into the FG solution (600 mg in 8.89 ml H2O) and stirred at room temperature, and the mixed solution was centrifuged (4000 rpm × 15 min). The white precipitate was dried under reduced pressure at 40 °C, and the yield was 1508 mg. Then the dry FG-benzethonium salt was dissolved in 7.54 ml of dimethyl formamide, and 360 μl of benzyl chloride was added. After heating at 35 °C for 24 h, 2.63 ml of freshly prepared 0.08 m EtONa/EtOH was added at room temperature, and the reaction was kept for 30 min at 300 rpm. Then 10.53 ml of saturated sodium chloride solution and 42.12 ml of EtOH were added, the precipitate was collected after centrifuging (4000 rpm × 15 min), and the above treatment was performed three times. To remove the benzyl groups, 0.34 ml of freshly prepared 6 m NaOH and 151.32 mg of sodium borohydride were added to the resulting precipitate diluted in 40 ml of H2O at 25 °C for 30 min. Subsequently, the solution was neutralized with HCl and concentrated to desalt by Sephadex G25 and lyophilized to obtain the depolymerized product dFG.
Purification of oligosaccharides
390 mg of depolymerized product dFG was dissolved in 3.5 ml of H2O and filtered through a membrane with a 0.45-μm pore size. The solution was then subjected to a Bio-Gel P10 column (medium, 2 cm × 200 cm) and eluted with 0.2 m NaCl solution. Approximately 2.5 ml of fractions per tube were collected and monitored by UV absorption (232 nm). Fractions exhibiting the same signal peak were pooled, and the resulting oligosaccharides were further purified by Bio-Gel P10 or P4. These were determined analytically by HPGPC with a Superdex Peptide 10/300 GL column with 0.2 m NaCl solution as the eluent at a flow rate of 0.4 ml/min and monitored by a DAD detector at 232 nm. Each oligosaccharide was desalted on a Sephadex G10 column and lyophilized.
NMR and ESI–Q-TOF–MS analysis
NMR spectroscopy was performed at 298 K with a Bruker Advance 600M/800M spectrometer equipped with a 13C/1H dual probe in FT mode. Each sample was dissolved in 0.5 ml of 99.9% D2O at a concentration of 10–20 g/liter. 1H-1H COSY, TOCSY, ROESY, 1H-13C HSQC, and HMBC spectra were recorded using state–time proportional phase incrementation for quadrature detection in the indirect dimension. All samples were previously dissolved in deuterium oxide (99.9% D) and lyophilized three times to replace exchangeable protons with D2O.
Negative-ion ESI-MS was conducted on a micrOTOF-QII mass spectrometer (Bruker Daltonik, Berlin, Germany). The data were analyzed using Bruker Compass Data-Analysis 4.0 software (Bruker Daltonik). The MS spectrometric conditions were as follows: ESI in negative ion mode, capillary voltage of 3500 V, nebulizer pressure of 1.5 bar, and drying gas flow rate of 4.0 liters/min. The mass spectra of the oligosaccharides were acquired in scan mode (m/z scan range 50–3000).
Determination of anticoagulant activities
APTT, PT, and TT were determined with a coagulometer (Teco MC-4000) using APTT, PT, and TT reagents and standard human plasma as previously described (14, 16).
Inhibition of intrinsic factor Xase complex
The inhibition of intrinsic factor Xase complex was determined using a previously described method (5). Different concentrations of samples were incubated with 2 IU/ml FVIII (30 μl), 60 nm FIXa (30 μl), 47 nm FIIa in 0.32 mg/ml synthetic phospholipids, 20 mm Tris-HCl, and 12 mm CaCl2 in a final volume of 90 μl for 2 min at 37 °C. The reaction was initiated by the addition of 30 μl of 50 nm FX containing 20 mm Tris-HCl, 0.3% fibrin polymerization inhibitor, and 0.1% PEG-8000 (pH 7.5) for 1 min at 37 °C. A volume of 30 μl of 8.40 mm chromogenic substrate S-2765 was added. The amount of FXa formed was determined by comparing the rate of S-2765 substrate hydrolysis to a standard curve at a wavelength of 405 nm using a Bio-Tek Microplate Reader (ELx 808, USA) at 37 °C.
Inhibition of human FIIa and FXa in the presence of AT
The anti-FIIa and anti-FXa activities in the presence of AT were measured using Biophen heparin anti-IIa kits and Biophen heparin anti-FXa kits (5, 14). A mixture containing 30 μl of FG or its depolymerized product or oligosaccharide and 30 μl of 1 IU/ml AT was incubated at 37 °C for 2 min, and then 30 μl of 24 NIH/ml FIIa (or 8 μg/ml bovine FXa) was added. After incubation for 2 min (or 1 min for FXa), the residual thrombin or FXa activity was measured by the addition of 30 μl of 1.25 mm thrombin chromogenic substrate CS-01 or 1.2 mm FXa chromogenic substrate SXa-11. The absorbance of the reaction mixture was read at 405 nm.
Author contributions
R. Y., Z. L., and L. Zhao data curation; R. Y., N. G., M. W., and J. Z. supervision; R. Y. and L. Zhou investigation; R. Y. writing-original draft; R. Y., M. W., and J. Z. writing-review and editing; N. G., L. Zhao, and J. Z. methodology; L. Zhao, F. S., and M. W. resources.
Supplementary Material
Acknowledgment
We thank Steven W. Purcell (National Marine Science Centre, Southern Cross University, Lismore, Australia) for the suggestions and revision of this manuscript.
This work was supported in part by National Natural Science Foundation of China Grants 81673330 and 81773737, Yunnan Provincial Science and Technology Department Grants 2016FA050 and 2010CI116, Kunming Institute of Botany Grant KIB2017011, and Chinese Academy of Sciences Youth Innovation Promotion Association Grant 2017435. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Tables S1 and S2 and Figs. S1–S11.
- FG
- fucosylated glycosaminoglycan
- GAG
- glycosaminoglycan
- LMWH
- low molecular weight heparin
- dU
- Δ4,5 unsaturated glucuronic acid
- GlcA
- glucuronic acid
- rA
- GalNAc-ol or GalNAc linked to GlcA-ol at the reducing end
- GalNAc4S6S
- 4,6-di-O-sulfated GalNAc
- Fuc
- fucose
- Fuc3S4S
- 3, 4-di-O-sulfated fucose
- Fuc2S4S
- 2, 4-di-O-sulfated fucose
- APTT
- activate partial thromboplastin time
- PT
- prothrombin time
- TT
- thrombin time
- AT
- antithrombin
- FXase
- intrinsic factor Xase complex
- FIIa
- thrombin
- FXa
- factor Xa
- dp
- degree of polymerization
- PMP
- 1-phenyl-3-methyl-5-pyrazolone
- dFG
- depolymerized FG
- GPC
- gel permeation chromatography
- HPGPC
- high-performance GPC
- ESI
- electrospray ionization
- Q-TOF
- quantitative time-of-flight
- TOCSY
- total correlation spectroscopy
- HSQC
- heteronuclear single quantum coherence
- HMBC
- heteronuclear multiple bond correlation
- ROESY
- rotating-frame overhause effect spectroscopy
- DAD
- diode array detector.
References
- 1. Pomin V. H. (2014) Holothurian fucosylated chondroitin sulfate. Marine Drugs 12, 232–254 10.3390/md12010232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fonseca R. J., Oliveira S. N., Pomin V. H., Mecawi A. S., Araujo I. G., and Mourão P. A. (2010) Effects of oversulfated and fucosylated chondroitin sulfates on coagulation: challenges for the study of anticoagulant polysaccharides. Thromb. Haemost. 103, 994–1004 10.1160/TH09-10-0734 [DOI] [PubMed] [Google Scholar]
- 3. Kitazato K., Kitazato K. T., Nagase H., and Minamiguchi K. (1996) DHG, a new depolymerized holothurian glycosaminoglycan, exerts an antithrombotic effect with less bleeding than unfractionated or low molecular weight heparin, in rats. Thromb. Res. 84, 111–120 10.1016/0049-3848(96)00166-1 [DOI] [PubMed] [Google Scholar]
- 4. Wu M., Huang R., Wen D., Gao N., He J., Li Z., and Zhao J. (2012) Structure and effect of sulfated fucose branches on anticoagulant activity of the fucosylated chondroitin sulfate from sea cucumber Thelenata ananas. Carbohydr. Polymers 87, 862–868 10.1016/j.carbpol.2011.08.082 [DOI] [PubMed] [Google Scholar]
- 5. Wu M., Wen D., Gao N., Xiao C., Yang L., Xu L., Lian W., Peng W., Jiang J., and Zhao J. (2015) Anticoagulant and antithrombotic evaluation of native fucosylated chondroitin sulfates and their derivatives as selective inhibitors of intrinsic factor Xase. Eur. J. Med. Chem. 92, 257–269 10.1016/j.ejmech.2014.12.054 [DOI] [PubMed] [Google Scholar]
- 6. Chen S., Xue C., Yin L., Tang Q., Yu G., and Chai W. (2011) Comparison of structures and anticoagulant activities of fucosylated chondroitin sulfates from different sea cucumbers. Carbohydr. Polymers 83, 688–696 10.1016/j.carbpol.2010.08.040 [DOI] [Google Scholar]
- 7. Chen S., Li G., Wu N., Guo X., Liao N., Ye X., Liu D., Xue C., and Chai W. (2013) Sulfation pattern of the fucose branch is important for the anticoagulant and antithrombotic activities of fucosylated chondroitin sulfates. Biochim. Biophys. Acta 1830, 3054–3066 10.1016/j.bbagen.2013.01.001 [DOI] [PubMed] [Google Scholar]
- 8. Santos G. R., Glauser B. F., Parreiras L. A., Vilanova E., and Mourão P. A. (2015) Distinct structures of the alpha-fucose branches in fucosylated chondroitin sulfates do not affect their anticoagulant activity. Glycobiology 25, 1043–1052 10.1093/glycob/cwv044 [DOI] [PubMed] [Google Scholar]
- 9. Vieira R. P., Mulloy B., and Mourão P. A. (1991) Structure of a fucose-branched chondroitin sulfate from sea cucumber: evidence for the presence of 3-O-sulfo-β-d-glucuronosyl residues. J. Biol. Chem. 266, 13530–13536 [PubMed] [Google Scholar]
- 10. Kariya Y., Watabe S., Kyogashima M., Ishihara M., and Ishii T. (1997) Structure of fucose branches in the glycosaminoglycan from the body wall of the sea cucumber Stichopus japonicus. Carbohydr. Res. 297, 273–279 10.1016/S0008-6215(96)00258-3 [DOI] [PubMed] [Google Scholar]
- 11. Ustyuzhanina N. E., Bilan M. I., Dmitrenok A. S., Tsvetkova E. A., Shashkov A. S., Stonik V. A., Nifantiev N. E., and Usov A. I. (2016) Structural characterization of fucosylated chondroitin sulfates from sea cucumbers Apostichopus japonicus and Actinopyga mauritiana. Carbohydr. Polymers 153, 399–405 10.1016/j.carbpol.2016.07.076 [DOI] [PubMed] [Google Scholar]
- 12. Kariya Y., Watabe S., Hashimoto K., and Yoshida K. (1990) Occurrence of chondroitin sulfate E in glycosaminoglycan isolated from the body wall of sea cucumber Stichopus japonicus. J. Biol. Chem. 265, 5081–5085 [PubMed] [Google Scholar]
- 13. Yoshida K. I., Minami Y., Nemoto H., Numata K., and Yamanaka E. (1992) Structure of DHG, a depolymerized glycosaminoglycan from sea cucumber, Stichopus japonicus. Tetrahedron Lett. 33, 4959–4962 10.1016/S0040-4039(00)61245-5 [DOI] [Google Scholar]
- 14. Zhao L., Wu M., Xiao C., Yang L., Zhou L., Gao N., Li Z., Chen J., Chen J., Liu J., Qin H., and Zhao J. (2015) Discovery of an intrinsic tenase complex inhibitor: pure nonasaccharide from fucosylated glycosaminoglycan. Proc. Natl. Acad. Sci. U.S.A. 112, 8284–8289 10.1073/pnas.1504229112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Santos G. R., Porto A. C., Soares P. A., Vilanova E., and Mourão P. A. (2017) Exploring the structure of fucosylated chondroitin sulfate through bottom-up nuclear magnetic resonance and electrospray ionization-high-resolution mass spectrometry approaches. Glycobiology 27, 625–634 10.1093/glycob/cwx031 [DOI] [PubMed] [Google Scholar]
- 16. Shang F., Gao N., Yin R., Lin L., Xiao C., Zhou L., Li Z., Purcell S. W., Wu M., and Zhao J. (2018) Precise structures of fucosylated glycosaminoglycan and its oligosaccharides as novel intrinsic factor Xase inhibitors. Eur. J. Med. Chem. 148, 423–435 10.1016/j.ejmech.2018.02.047 [DOI] [PubMed] [Google Scholar]
- 17. Gao N., Lu F., Xiao C., Yang L., Chen J., Zhou K., Wen D., Li Z., Wu M., Jiang J., Liu G., and Zhao J. (2015) β-Eliminative depolymerization of the fucosylated chondroitin sulfate and anticoagulant activities of resulting fragments. Carbohydr. Polymers 127, 427–437 10.1016/j.carbpol.2015.04.002 [DOI] [PubMed] [Google Scholar]
- 18. Murase T., and Kajihara Y. (2010) Unique cleavage of 2-acetamido-2-deoxy-d-glucose from the reducing end of biantennary complex type oligosaccharides. Carbohydr. Res. 345, 1702–1707 10.1016/j.carres.2010.05.020 [DOI] [PubMed] [Google Scholar]
- 19. Huang Y., Mao Y., Zong C., Lin C., Boons G. J., and Zaia J. (2015) Discovery of a heparan sulfate 3-O-sulfation specific peeling reaction. Anal. Chem. 87, 592–600 10.1021/ac503248k [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li X., Luo L., Cai Y., Yang W., Lin L., Li Z., Gao N., Purcell S. W., Wu M., and Zhao J. (2017) Structural elucidation and biological activity of a highly regular fucosylated glycosaminoglycan from the edible sea cucumber Stichopus herrmanni. J. Agric. Food Chem. 65, 9315–9323 10.1021/acs.jafc.7b03867 [DOI] [PubMed] [Google Scholar]
- 21. Luo L., Wu M., Xu L., Lian W., Xiang J., Lu F., Gao N., Xiao C., Wang S., and Zhao J. (2013) Comparison of physicochemical characteristics and anticoagulant activities of polysaccharides from three sea cucumbers. Marine Drugs 11, 399–417 10.3390/md11020399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mourão P. A., Pereira M. S., and Pavão M. S., Mulloy B., Tollefsen D. M., Mowinckel M. C., and Abildgaard U. (1996) Structure and anticoagulant activity of a fucosylated chondroitin sulfate from Echinoderm: sulfated fucose branches on the polysaccharide account for its high anticoagulant action. J. Biol. Chem. 271, 23973–23984 10.1074/jbc.271.39.23973 [DOI] [PubMed] [Google Scholar]
- 23. Yu F., Wolff J. J., Amster I. J., and Prestegard J. H. (2007) Conformational preferences of chondroitin sulfate oligomers using partially oriented NMR spectroscopy of 13C-labeled acetyl groups. J. Am. Chem. Soc. 129, 13288–13297 10.1021/ja075272h [DOI] [PubMed] [Google Scholar]
- 24. Ustyuzhanina N. E., Bilan M. I., Dmitrenok A. S., Nifantiev N. E., and Usov A. I. (2017) Two fucosylated chondroitin sulfates from the sea cucumber Eupentacta fraudatrix. Carbohydr. Polymers 164, 8–12 10.1016/j.carbpol.2017.01.034 [DOI] [PubMed] [Google Scholar]
- 25. Philips E. D., Zemlicka J., and Horwitz J. P. (1973) Unsaturated sugars I. Decarboxylative elimination of methyl 2,3-di-O-benzyl-α-d-glucopyranosiduronic acid to methyl 2,3-di-O-benzyl-4-deoxy-β-l-threo-pent-4-enopyranoside. Carbohydr. Res. 30, 281–286 10.1016/S0008-6215(00)81814-5 [DOI] [PubMed] [Google Scholar]
- 26. Mizumoto S., Murakoshi S., Kalayanamitra K., Deepa S. S., Fukui S., Kongtawelert P., Yamada S., and Sugahara K. (2013) Highly sulfated hexasaccharide sequences isolated from chondroitin sulfate of shark fin cartilage: insights into the sugar sequences with bioactivities. Glycobiology 23, 155–168 10.1093/glycob/cws137 [DOI] [PubMed] [Google Scholar]
- 27. Rand M. D., Lock J. B., van't Veer C., Gaffney D. P., and Mann K. G. (1996) Blood clotting in minimally altered whole blood. Blood 88, 3432–3445 [PubMed] [Google Scholar]
- 28. Sheehan J. P., and Walke E. N. (2006) Depolymerized holothurian glycosaminoglycan and heparin inhibit the intrinsic tenase complex by a common antithrombin-independent mechanism. Blood 107, 3876–3882 10.1182/blood-2005-07-3043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kurachi K., Fujikawa K., Schmer G., and Davie E. W. (1976) Inhibition of bovine factor IXa and factor Xa β by antithrombin III. Biochemistry 15, 373–377 10.1021/bi00647a021 [DOI] [PubMed] [Google Scholar]
- 30. Liu J., Shang F., Yang Z., Wu M., and Zhao J. (2017) Structural analysis of a homogeneous polysaccharide from Achatina fulica. Int. J. Biol. Macromol. 98, 786–792 10.1016/j.ijbiomac.2017.01.149 [DOI] [PubMed] [Google Scholar]
- 31. Casu B., and Gennaro U. (1975) A conductimetric method for the determination of sulphate and carboxyl groups in heparin and other mucopolysaccharides. Carbohydr. Res. 39, 168–176 10.1016/S0008-6215(00)82654-3 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.