

Abstract
Ski-related oncogene SnoN (SnoN or SKIL) regulates multiple signaling pathways in a tissue- and developmental stage–dependent manner and has broad functions in embryonic angiogenesis, mammary gland alveologenesis, cancer, and aging. Here, we report that SnoN also plays a critical role in white adipose tissue (WAT) development by regulating mesenchymal stem cell (MSC) self-renewal and differentiation. We found that SnoN promotes MSC differentiation in the adipocyte lineage by antagonizing activin A/Smad2, but not TGFβ/Smad3 signaling. Mice lacking SnoN or expressing a mutant SnoN defective in binding to the Smads were protected from high-fat diet–induced obesity and insulin resistance, and MSCs lacking a functional SnoN exhibited defective differentiation. We further demonstrated that activin, via Smad2, appears to be the major regulator of WAT development in vivo. We also noted that activin A is abundantly expressed in WAT and adipocytes through an autocrine mechanism and promotes MSC self-renewal and inhibits adipogenic differentiation by inducing expression of the gene encoding the homeobox transcription factor Nanog. Of note, SnoN repressed activin/Smad2 signaling and activin A expression, enabling expression of adipocyte-specific transcription factors and promoting adipogenic differentiation. In conclusion, our study has revealed that SnoN plays an important in vivo role in adipocyte differentiation and WAT development in vivo by decreasing activity in the activin/Smad2 signaling pathway.
Keywords: activin, SMAD transcription factor, transforming growth factor β (TGF-β), obesity, adipocyte, differentiation, adipose tissue, SKI like proto-oncogene, SKIL, Smad2, SnoN, adipose tissue
Introduction
Adipocytes play a critical role in energy homeostasis and metabolism, and dysregulation of adipogenesis leads to human diseases, including obesity and diabetes (1). Adipocytes are derived from mesenchymal stem cells (MSCs),2 which initially undergo lineage commitment to differentiate into pre-adipocytes. Hormonal cues stimulate further differentiation and maturation of pre-adipocytes by inducing expression of transcription factors such as CCAAT/enhancer-binding protein α (C/EBPα) and peroxisome proliferator-activated receptor γ (PPARγ) required for differentiation, which in turn stimulate the expression of many genes involved in adipocyte functions such as adipocyte fatty acid-binding protein (αFABP), fatty-acid synthase (FAS), and leptin (1–3).
Several members of the transforming growth factor-β (TGFβ) family, including TGFβ, activin, bone morphogenetic protein 4 (BMP4) or BMP7, and growth differentiation factor 3 (GDF3), have been shown to regulate various aspects of adipogenic differentiation, glucose metabolism, and energy homeostasis (4, 5). Among them, BMP4 has been shown to promote mouse MSC commitment to the white adipose lineage (6), whereas BMP7 preferentially supports brown adipogenesis in various human and mouse cell types and animal models (5). In contrast, activin, through the action of Smad2, inhibits differentiation of adipocyte progenitor cells (7, 8), and TGFβ, through Smad3, represses the differentiation of pre-adipocyte cell lines by suppressing C/EBPs, leading to a decrease in the expression of several adipocyte marker genes, including PPARγ and leptin (9–11). Consistent with the results from these in vitro studies, transgenic mice overexpressing TGFβ in adipose tissues showed severely reduced white adipose tissues (WAT) and brown adipose tissues (BAT) (12), suggesting that TGFβ may inhibit the development of both BAT and WAT in vivo. A more prominent role of TGFβ/Smad3 signaling in regulation of glucose metabolism and energy homeostasis has been proposed recently based on the findings that elevated circulating TGFβ1 levels correlate positively with morbid obesity in humans and mice (13–17), and loss of Smad3 protected mice from diet-induced obesity and diabetes (17). Interestingly, this protection from obesity is accompanied by the “browning” of WAT as well as increased mitochondrial biogenesis in Smad3−/− mice (17), suggesting an increased transition from WAT to BAT. Finally, GDF3 signaling is regulated by different nutrient conditions to control beige adipogenesis (18).
The TGF-β family of ligands binds to their receptors and signals through the Smad family of proteins (19, 20). BMPs phosphorylate and activate Smad1/5/8, and TGFβ/activin/nodal/growth and differentiation factors signal through Smad2 or Smad3 (21–25). Once phosphorylated, these activated Smad proteins form complexes with Smad4 and regulate the expression of many transcription factors and adipocyte-specific genes, such as C/EBPs, PPARγ, αFABP, and leptin (4, 5). A key regulator of these Smad complexes is SnoN. SnoN is ubiquitously expressed in early embryos and adult tissues, and its expression is induced during specific stages of development and tissue morphogenesis and by cellular stress signals and certain cytokines such as TGFβ (26–28). SnoN has been shown to regulate the activity of several important signaling pathways in a tissue-specific manner. It acts as a potent negative feedback regulator of TGFβ/ALK5 by directly binding to Smad2, Smad3, and Smad4 to repress their transactivation activity. Overexpression of SnoN blocks the growth-inhibitory responses to TGFβ. However, in endothelial cells, in addition to repressing TGFβ signaling, SnoN also enhances activation of Smad1/5 by ALK1 in response to BMP9/10 to promote endothelial cell proliferation and migration (29). In response to cellular stress signals, SnoN has been reported to interact with p53 and promyelocytic leukemia in the promyelocytic leukemia nuclear bodies to induce p53 activation, leading to premature senescence and inhibition of tumorigenesis (30). In the mammary gland, SnoN enhances prolactin/Stat5 signaling to enable expansion of mammary epithelium and onset of lactation (31) and promotes TAZ stability to facilitate malignant progression of breast cancer cells (32). Thus, SnoN cross-talks with several signaling pathways to perform tissue-specific or context-specific functions.
Consistent with these signaling activities, homozygous mice lacking SnoN (SnoN−/−) (33) or expressing a mutant SnoN defective in binding to the Smads (SnoNm/m) display a diverse array of abnormalities in development, including yolk sac and embryonic angiogenesis (29), mammary gland alveologenesis, and onset of lactation (31). The SnoNm/m mice also have a shortened life span and exhibited phenotypes associated with premature aging as well as increased resistance to tumorigenesis (34). In particular, we noticed that these mice consistently exhibited smaller body size and less body weight than the control mice shortly after birth. These mice appeared to have reduced subcutaneous adipose tissues. We therefore asked whether SnoN may also regulate adipose development. Here, we report that SnoN is critical for white adipocyte differentiation from both MSC and pre-adipocytes by repressing activin/Smad2 signaling. Both SnoN−/− and SnoNm/m mice are protected from diet-induced obesity and diabetes. Our results demonstrate for the first time that during physiological development, SnoN targets activin, not TGFβ signaling to regulate WAT development.
Results
SnoN−/− and SnoNm/m mice are resistant to high-fat diet (HFD)-induced obesity
Both SnoN−/− and SnoNm/m mice expressing a mutant SnoN defective in binding to all Smad proteins exhibited progressively smaller body size and less body weight than the control mice after birth (Fig. 1A). In particular, they appeared to be leaner and had less epididymal fat than wildtype (WT) littermates (Fig. 1B), and the white adipocytes appeared to exhibit a smaller size than WT cells (Fig. 1C). These observations prompted us to ask whether these mice might have defects in adipogenesis. To do that, the WT mice, SnoNm/m (KI) mice, and SnoN−/− (KO) mice were subjected to a high-fat diet (HFD: 60 kcal % fat) for 16 weeks, and body weight and metabolic activities were monitored. As shown in Fig. 1D, although WT littermates gained significant body weight under this regime, the weight gains in the SnoNm/m and SnoN−/− mice were more limited, suggesting that these mice were resistant to HFD-induced obesity. The resistance to obesity could be due to less food intake or higher energy expenditure. We therefore monitored the metabolic activities of these mice using a comprehensive laboratory animal monitoring system (CLAMS). We found the amount of food intake was similar among the three groups of mice (Fig. 1E), but the SnoNm/m and SnoN−/− mice showed significantly increased physical activities and higher oxygen consumption in a 12-h light/dark cycle when normalized to their whole-body weight (Fig. 1F), indicating a heightened energy expenditure. Given that oxygen consumption and physical activities are indicators of metabolic rate, which is regulated by circulating adipokines, and reflect the functionality of the adipose tissue, we next focused on the activity of adipose tissue and adipocytes.
Figure 1.

SnoN−/− and SnoNm/m mice are resistant to HFD-induced obesity. A–C, SnoN−/− and SnoNm/m male mice displayed lower body weight (A), less epididymal fat mass (B), and smaller adipocyte size (C) than age-matched SnoN+/+ male mice. n = 11 mice/group. Epididymal fat pads were collected from these mice and weighed at 6 months of age. Values are expressed as percentage of body weight (n = 5 mice/group). H&E staining was performed on paraffin-embedded sections of these fat tissues (scale bar, 100 μm). D, SnoN−/− and SnoNm/m male mice gained less body weight than control male mice on HFD (n = 4 mice/group). E, SnoN+/+, SnoN−/−, and SnoNm/m male mice took in similar calories, as monitored by weekly food intake for 16 weeks during HFD. F, physical activity (left) and oxygen consumption (right) were determined for SnoN+/+, SnoN−/−, and SnoNm/m male mice on a 16-week HFD by metabolic chamber assay that was monitored through a 12-h light and dark cycle. G–I, EchoMRI analysis of lean (G), WAT mass (H), and percentage of WAT mass over whole-body weight (I) on HFD mice. J, H&E stain of epididymal and subcutaneous adipose tissue sections showed that the sizes of adipocytes from SnoN−/− and SnoNm/m mice were smaller than that of SnoN+/+ mice after 16 weeks of HFD (scale bar, 100 μm). Leptin levels (K) and serum triglyceride levels (L) were lower in SnoN−/− and SnoNm/m mice than in WT littermates after 16 weeks on HFD. The serum was collected after fasting for 6 h (n = 3 mice/group). All data are represented as means ± S.D. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To assess whether the difference in body weights between the control littermates and SnoN mutant mice arose from the difference in adipose mass, EchoMRI was employed to measure total lean mass and fat mass. Although there was no detectable difference in lean mass among the three mouse strains (Fig. 1G), both SnoNm/m and SnoN−/− mice exhibited significantly less WAT mass and body fat content than WT control littermates (Fig. 1, H and I). Indeed, the relative weight of the epididymal and subcutaneous WAT in the SnoNm/m and SnoN−/− mice was significantly decreased (Fig. S1A). These data suggest that the decrease in body weight in SnoN mutant mice was due primarily to a decrease in fat mass, but not to a reduction in the overall body size or lean muscle mass. Finally, in agreement with the notion that adipocytes expand both in size and number to accommodate increased energy storage in obesity, WAT in WT mice contained markedly enlarged adipocytes, whereas those from SnoNm/m and SnoN−/− mice remained small (Fig. 1J).
Serum leptin levels have been reported to correlate with the percentage of body fat in humans and rodents (35, 36). Reflecting the decrease in adiposity, the circulating leptin levels were markedly reduced in SnoNm/m and SnoN−/− mice (Fig. 1K). Similar decreases in the serum triglyceride levels were also detected (Fig. 1L). However, the levels of free fatty acids (FFA), adiponectin, and insulin were not significantly different between the WT and SnoN-deficient mice (Fig. S1, B–D). Taken together, these findings indicate that SnoN−/− and SnoNm/m mice are protected from HFD-induced obesity.
SnoN−/− and SnoNm/m mice are resistant to HFD-induced diabetes
Diet-induced obesity is typically associated with reduced ability to metabolize glucose and increased risk of diabetes. Consistent with its resistance to HFD-induced obesity, SnoNm/m and SnoN−/− mice displayed a lower baseline glucose level than WT mice under fasting conditions (Fig. 2A). In an intraperitoneal glucose tolerance test (GTT), these mice exhibited a lower blood glucose level over the entire time course of the GTT (Fig. 2B). In an insulin tolerance test (ITT), in response to insulin, SnoNm/m and SnoN−/− mice showed a deeper and long-lasting fall in blood glucose levels, whereas WT mice showed little reduction in glucose levels (Fig. 2C). As shown in Fig. 2D, a HFD also induced significant lipid accumulation in the liver of WT mice, as evidenced by the enlarged size and greasy pink appearance (top panels), the extensive vacuolation in H&E-stained liver section (middle panels), and massive lipid droplets by Oil Red O stain (bottom panels). In contrast, SnoN−/− and SnoNm/m animals displayed significantly less fat accumulation in the liver (Fig. 2D), suggesting they are resistant to diet-induced hepatic steatosis.
Figure 2.

SnoN−/− and SnoNm/m mice are resistant to HFD-induced insulin resistance. A, SnoN−/− and SnoNm/m mice showed significantly lower glucose levels compared with SnoN+/+ mice fed with either a ND or HFD. The glucose levels were monitored after fasting for 6 h. B and C, SnoN−/− and SnoNm/m mice displayed improved glucose tolerance (B) and insulin tolerance (C) compared with SnoN+/+ mice. D, SnoN−/− and SnoNm/m mice were resistant to HFD-induced hepatic steatosis. Top panels, macroscopic view of liver tissues; middle panels, H&E staining of paraffin-embedded liver sections; bottom panels, Oil Red O staining of frozen liver sections (scale bar, 100 μm). E and F, SnoN−/− and SnoNm/m mice exhibited significantly reduced levels of inflammatory M1 macrophage-specific transcripts (E) and increased levels of protective M2 macrophage-specific transcripts (F) upon HFD as assayed by qRT-PCR. All data are represented as means ± S.D., n = 4 mice/group. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Obesity is widely viewed as a chronic low-grade inflammatory disease that is accompanied by macrophage infiltration into the adipose tissue (37, 38). This adipose tissue macrophage (ATM) infiltration and the switching from the protective M2 ATMs to the inflammatory M1 phenotype in obese mice are often associated with the development of insulin resistance (39–42). Indeed, HFD-fed SnoN−/− and SnoNm/m mice exhibited reduced levels of inflammatory M1 macrophage-specific genes and increased levels of protective M2 macrophage-specific transcripts (Fig. 2, E and F). Thus, SnoN deficiency strengthens glucose metabolism and protects mice from HFD-induced insulin resistance.
SnoN deficiency suppresses adipocyte differentiation in vitro
We next investigate the mechanism of SnoN regulation of adipocyte differentiation using in vitro culture models. Primary mouse embryonic fibroblasts (MEFs) express many pluripotent MSC markers and can be induced to differentiate into multiple mesenchymal lineages, including that of adipocyte, bone, chondrocyte, and muscle (43). When isolated from E13.5-day embryos, WT MEFs readily differentiated into adipocytes as shown by Oil Red O staining (Fig. 3A). In contrast, SnoNm/m and SnoN−/− MEFs exhibited a drastically reduced ability to differentiate into adipocytes (Fig. 3A).
Figure 3.

SnoN deficiency inhibits adipocyte differentiation. A, SnoN−/− and SnoNm/m MEFs exhibited a markedly reduced ability to differentiate into adipocytes. MEFs isolated from E13.5 embryos were induced to differentiate in vitro. Oil Red O staining of MEF was performed at day 10 of differentiation. B, SnoN protein was induced during adipocyte differentiation. C3H10T1/2 cells (top panels) or 3T3-L1 cells (bottom panels) were induced to differentiate, and total cell lysates prepared from cells harvested before (day 0) or at the indicated times following differentiation were analyzed by Western blotting with anti-SnoN. Tubulin was used as a loading control. C, CRISPR/Cas9-based approach was used to generate homozygous SnoN KO in C3H10T1/2 cells. Western blotting was performed to examine the expression of SnoN in these cells. D, SnoN KO led to a significant reduction in adipocyte differentiation. Oil Red O staining was performed on day 7 of differentiation. E, expression of adipogenic marker genes was decreased in SnoN−/− cells. mRNA levels of Pparγ, Cebpβ, αFabp, and adipsin were measured by qRT-PCR at day 7 following differentiation. F–H, SnoN KD led to a significant reduction in adipocyte differentiation of 3T3-L1 cells. The protein levels of SnoN in 3T3L1 parental cells or two shSnoN stable clones shSnoN-1 or shSnoN-2 were examined by Western blotting (F). Oil Red O staining was performed at day 7 of differentiation (G). mRNA levels of Pparγ and αFabp were measured by qRT-PCR at day 7 following differentiation (H). I, SnoN KO affected the osteoblast differentiation of C3H10T1/2 cells. Osteogenesis of these cells was evaluated by staining of alkaline phosphatase (ALP) activity. Scale bar, 100 μm in A, D, and G. Data in E and H are represented as means ± S.D. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
In the adipocyte lineage, MSC initially differentiate into a pre-adipocyte stage and, in the presence of hormonal cues, the pre-adipocytes then proliferate by mitotic clonal expansion and further differentiate into mature adipocytes (4). To determine whether SnoN regulates the lineage commitment of MSC to pre-adipocytes or the proliferation and differentiation of pre-adipocytes to mature adipocytes, we employed C3H10T1/2 pluripotent MSC model and 3T3-L1 pre-adipocyte model to mimic the two steps in adipocyte differentiation, respectively. Interestingly, differentiation of both C3H10T1/2 and 3T3-L1 cells into the adipocyte lineage was accompanied by an induction of SnoN protein after 1 or 2 days of induction (Fig. 3B), suggesting that high levels of SnoN may be important for the differentiation process. To test this, we performed a CRISPR/Cas9-based knockout of SnoN expression in C3H10T1/2 cells (Fig. S2 and Fig. 3C) or an shRNA-based knockdown (KD) of SnoN in 3T3-L1 cells (Fig. 3F), and we examined the ability of these cells to differentiate into adipocytes. As shown in Fig. 3, D and G, elimination of SnoN expression in both C3H10T1/2 and 3T3-L1 cells led to a significant reduction in adipocyte differentiation. Accompanying the reduced differentiation, the expression of adipogenic marker genes, Pparγ, Cebpβ, and αFabp, as well as adipsin, was also markedly decreased in SnoN KO or KD cells (Fig. 3, E and H). Thus, SnoN is essential for both lineage commitment and pre-adipocyte differentiation.
Because MSCs are capable of differentiating into other lineages, including osteoblasts and chondrocytes, in addition to adipocytes, we next asked whether SnoN also affected the differentiation of MSCs to these lineages. Interestingly, deletion of SnoN also dramatically impaired the differentiation of C3H10T1/2 cells to osteoblasts (Fig. 3I), a process that is also normally inhibited by TGFβ and activin signaling. Thus, SnoN also affects the differentiation of MSCs to other lineages, indicating that SnoN likely suppresses the self-renewal of MSCs and promotes lineage commitment.
SnoN promotes adipocyte differentiation by antagonizing Smad2 but not Smad3
The SnoNm/m mice express a mutant SnoN that cannot bind to the Smad proteins and therefore cannot antagonize Smad signaling (34). Because SnoNm/m mice were similar to the SnoN−/− mice in resistance to HFD-induced obesity and because both SnoNm/m and SnoN−/− MEFs displayed similar defects in adipocyte differentiation, SnoN likely regulates adipocyte differentiation through a Smad-dependent mechanism. Indeed, as shown in Fig. S3, A and B, although re-expression of WT SnoN in C3H10T1/2 SnoN KO cells partially restored their ability to differentiate into adipocytes, overexpression of mutant SnoN defective in Smad binding (mSnoN) failed to rescue the impaired adipogenic differentiation. The inability of reintroduced WT SnoN to fully restore adipogenic differentiation ability is mostly likely due to the lower expression level of the ectopically expressed WT SnoN (Fig. S3A).
Smad proteins signal downstream of several TGFβ superfamily members, including TGFβ, activins, and BMPs, all of which have been reported to regulate adipocyte differentiation (4, 5). In particular, TGFβ, through the action of Smad3 (9), and activin, via Smad2 (7), have been shown to inhibit adipocyte differentiation in vitro. BMP4 and BMP7 promote the development of WAT and BAT, respectively (6, 44, 45), whereas GDF3 suppresses the differentiation of beige adipocytes (18).
We first employed the type I receptor kinase inhibitor SB-431542 targeting TGFβ and activin signaling to assess whether SnoN regulation of adipocyte differentiation involved TGFβ/activin signaling. As shown in Fig. 4A, treatment of SnoN−/− cells with SB-431542 readily rescued the adipocyte differentiation, suggesting that indeed the elevated TGFβ or activin signaling in SnoN−/− MSCs was likely responsible for the defective adipocyte differentiation. To further distinguish between the contribution of TGFβ and activin signaling, we reduced Smad2 or Smad3 levels in the SnoN−/− cells by siRNA, respectively (Fig. 4B). Surprisingly, Smad2 and Smad3 appeared to exert distinct effects on SnoN-dependent adipocyte differentiation. While reducing Smad2 rescued the impaired adipocyte differentiation, reducing Smad3 had little effect (Fig. 4C). The differential role of Smad2 and Smad3 in adipocyte differentiation is further substantiated by the significant difference in Smad2 and Smad3 expression levels. In both WAT in vivo (Fig. 4D) and in 10T1/2 cells (Fig. S4A, right panel), the Smad3 level was significantly lower than that of Smad2, strongly suggesting that Smad3 may not play a significant role in adipose development in vivo.
Figure 4.

SnoN regulates adipocyte differentiation by antagonizing Smad2. A, SB-431542 rescued the adipocyte differentiation in SnoN−/− cells. C3H10T1/2 SnoN−/− cells were treated with the differentiation-inducing agents together with 10 μm SB-431542 or DMSO. At day 7, cells were stained with Oil Red O for lipid visualization (scale bar, 100 μm). B and C, reducing Smad2, but not Smad3, in the SnoN−/− cells by siRNA rescued the adipocyte differentiation. Control siRNA (sictrl), siSmad2, or siSmad3 was transfected into C3H10T1/2 SnoN−/− cells. B, knockdown efficiency was confirmed by Western blotting. C, adipogenic differentiation was assessed by Oil Red O staining. D, levels of Smad2 and Smad3 expression in WATs were measured by qRT-PCR. E, mRNA levels of Pparγ and αFabp in various C3H10T1/2-derived cell lines were quantified by qRT-PCR on day 7 following differentiation. F, reducing Smad2, but not Smad3, expression down-regulated Nanog induction in SnoN−/− cells. The levels of Nanog mRNA were measured by qRT-PCR. G, SnoN−/− cells showed a prolonged activation of Smad2 during adipocyte differentiation. The levels of phospho-Smad2, phospho-Smad3, and total Smad proteins at different days of adipogenic differentiation were examined by Western blotting. Tubulin was used as a loading control. H, levels of Nanog expression were examined by qRT-PCR. Data in D–F and H are represented as means ± S.D. *, p < 0.05; **, p < 0.01; ***, p < 0.001. Scale bar, 100 μm in A and C.
Consistent with the morphological changes, expression of adipogenic marker genes such as Pparγ and αFabp also increased in SnoN−/− cells with reduced Smad2 but not with reduced Smad3 (Fig. 4E). Similar results were also observed in 3T3-L1 SnoN KD cells (Fig. S3, E–G). Nanog is required for the maintenance of pluripotency and self-renewal of MSC and has been previously shown to be a Smad2 target gene (46). We found that expression of Nanog was up-regulated in SnoN−/− cells (Fig. 4F), in agreement with the defective differentiation of these cells. Knocking down Smad2 reduced nanog expression, whereas Smad3 KD further enhanced its expression, suggesting that SnoN mainly regulates Smad2 signaling during adipocyte differentiation.
To examine whether Smad2 activity is altered during adipocyte differentiation, we next examined the levels of phospho-Smad2 in C3H10T1/2 and SnoN−/− cells during differentiation. As shown in Fig. 4G, undifferentiated C3H10T1/2 cells displayed a basal level of phospho-Smad2, and this was moderately induced within the 1st day of differentiation, but was significantly down-regulated after day 3. Surprisingly, the SnoN−/− cells showed a prolonged activation of Smad2. Consistent with this, an enhanced expression of Nanog was also observed in these SnoN−/− cells, both under the undifferentiated state and upon differentiation (Fig. 4H), indicating SnoN may not only function as a co-repressor of Smad2 but also as a negative regulator of its activation. In contrast to the changes in Smad2 phosphorylation, no consistent changes in the phosphorylation of Smad3 or its expression were detected (Fig. 4G). Finally, re-expression of human Smad2, but not Smad3, reversed the adipocyte differentiation induced by siSmad2 (Fig. S3, C, D, H, and I). These data suggest that SnoN is an important negative regulator of Smad2, but not Smad3, during adipocyte differentiation and promote MSC differentiation by repressing nanog expression and MSC self-renewal.
SnoN suppresses activin A expression both in vitro and in vivo
The activation and phosphorylation of Smad2 can be induced by either TGFβ or activin (19). To determine which of the signaling pathways is regulated by SnoN during adipogenesis, we treated C3H10T1/2 cells with activin A or TGFβ1 and examined their ability to inhibit adipocyte differentiation. As shown in Fig. 5A, activin A potently inhibited adipogenesis, whereas TGFβ1 had little effect, suggesting that activin might be the predominant regulator of adipogenesis. Consistent with this idea, in WAT isolated from WT mice, activin A was expressed at a much higher level than TGFβ1, and this difference was further enlarged in WAT from SnoN−/− and SnoNm/m mice following the HFD (Fig. 5B). In contrast, the levels of types I and II receptors for activin A were comparable with that for TGFβ1 in WATs (Fig. 5C). Correlating with this high activin A level, the levels of phospho-Smad2 (Fig. 5D) and nanog expression (Fig. 5E) in WAT from SnoN−/− and SnoNm/m mice were also elevated.
Figure 5.

SnoN suppresses activin A expression both in vitro and in vivo. A, activin A, but not TGFβ, inhibited adipocyte differentiation. Confluent C3H10T1/2 cells were pre-treated with 7.7 nm activin A or 100 pm TGFβ and subjected to adipogenic inducers in the presence of activin A or TGFβ. Adipogenic differentiation was monitored by Oil Red O staining at day 7. B, WATs expressed more activin A than TGFβ1, and this expression was further elevated in WATs from SnoN-deficient mice. The mRNA levels of activin A and TGFβ1 in WAT from HFD-fed mice were examined by qRT-PCR. C, WATs expressed comparable levels of types I and II receptors for activin A and TGFβ1, as measured by qRT-PCR. D, phospho-Smad2 levels were markedly up-regulated in WAT from HFD-fed SnoN−/− and SnoNm/m mice. Western blotting was performed to measure pSmad2, pSmad3, Smad2, and Smad3 levels in extracts prepared from WATs. Tubulin was used as a loading control. E, mRNA levels of Nanog in WATs were determined by qRT-PCR. F, activin A levels were markedly up-regulated in SnoN−/− cells. Media from C3H10T1/2 and SnoN−/− cultures were collected at different days during differentiation as indicated. Activin A or TGFβ1 concentration was measured by ELISA. G, activin A neutralizing antibody promoted adipocyte differentiation of SnoN−/− cells. SnoN−/− cells were pre-treated with or without 1 μg/ml anti-activin A antibody for 1 day and then subjected to the differentiation agents. Oil Red O staining was performed 7 days later. H, activin A expression is regulated by a Smad2-dependent autocrine mechanism. Control siRNA (sictrl), siSmad2, or siSmad3 was transfected into either C3H10T1/2 or SnoN−/− cells. The mRNA levels of activin A and TGFβ were quantified by qRT-PCR. Data in B, C, E, and H are represented as means ± S.D. *, p < 0.05; **, p < 0.01; ***, p < 0.001. Scale bars = 100 μm in A and G.
Similarly, in the C3H10T1/2 culture model, levels of auto-secreted activin A were significantly higher than that of TGFβ1 and were further induced in SnoN−/− cells (Fig. 5F), consistent with the prolonged phospho-Smad2 observed in these cells (Fig. 4G). In contrast, the levels of TGFβ1 remained low in both WT and SnoN−/− cells (Fig. 5F). Interestingly, the level of activin type I receptor Alk4 was significantly higher than that of Alk5 (Fig. S4A), suggesting that the entire activin A signaling pathway may be more abundant than that of TGFβ1 in C3H10T1/2 cells. To further prove that elevated activin A is the key repressor of adipogenesis, we employed a specific activin A antibody to neutralize secreted activin A in SnoN−/− cells. As shown in Fig. 5G, treatment of cells with the activin A neutralizing antibody resulted in a robust adipocyte differentiation. These data suggest that activin A is the major TGFβ family member in MSCs and adipose tissues and plays a prominent role in adipogenesis.
The observation that the activin A level was significantly up-regulated in SnoNm/m and KO tissues and cells suggests that activin A expression itself might be regulated by the Smad pathway. Indeed, reducing expression of Smad2, but not Smad3, by siRNA suppressed the up-regulation of activin A in SnoN−/− cells (Fig. 5H). In contrast, TGFβ expression was not significantly altered in cells lacking SnoN nor was it affected by altering Smad2 expression (Fig. 5H). These data suggest that in WAT and adipocyte progenitor cells, activin A expression is regulated in a positive feedback manner by its own Smad2 pathway, and deletion of SnoN leads to increased Smad2 signaling and increased activin A expression. Our data have provided direct evidence to support the model that activin A is regulated by an autocrine mechanism in the adipogenic progenitor cells and plays a critical role in adipose development.
Discussion
Multiple members of the TGFβ superfamily, including TGFβs, activins, and BMPs, have been implicated in adipogenesis in vitro and in vivo (4, 5). However, the physiological contexts under which each member exerts their activities in adipogenesis in vivo have not been clearly defined. Our study revealed for the first time that under physiological conditions in vivo, the activin–SnoN signaling axis appears to be the major regulator of WAT development. Activin A is expressed in the undifferentiated adipocyte progenitor cells at a high concentration through an autocrine mechanism and is suppressed during adipocyte differentiation (7). Signaling through Smad2, activin A inhibits adipocyte differentiation by promoting Nanog expression to maintain self-renewal and the undifferentiated state of MSCs, by repressing the expression of adipogenic transcription factors Cebpβ and Pparγ and also by further enhancing the expression of its own ligand. During adipogenesis, this inhibitory activity of activin on adipogenesis is significantly dampened by SnoN. We showed that SnoN expression is markedly induced during adipocyte differentiation, and this elevated SnoN suppresses the expression of activin A and the activities of activin/Smad2 to promote MSC differentiation into the adipocyte lineage and to further enhance pre-adipocyte differentiation (Fig. 6). Thus, SnoN is a key positive regulator of WAT development in vivo through its ability to repress activin/Smad2 signaling and that activin/Smad2, not TGFβ/Smad3, is the physiological target of SnoN during WAT development in vivo.
Figure 6.
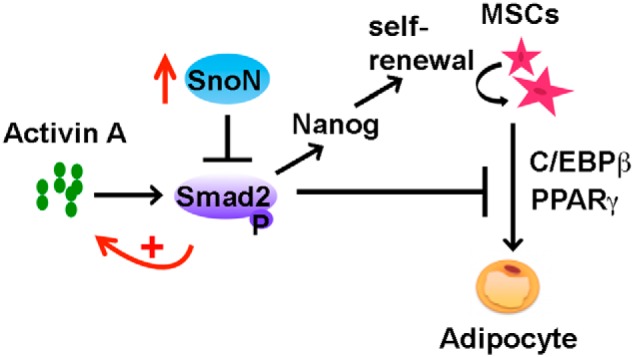
Model of SnoN regulation of adipogenesis through antagonizing the activities of activin A/Smad2 signaling.
Although activin A/Smad2 has been show to inhibit differentiation of human adipose progenitors, a direct examination of the role of activin/Smad2 in adipogenesis in vivo has not been possible due to early embryonic or early neonatal lethality of the mice lacking activin ligands, receptors, or Smad2 (47–55). Our results here confirm that activin A is a primary player in WAT development in vivo. Activin A is secreted by the undifferentiated human adipocyte progenitors at a much higher level than that of TGFβ, and differentiation of these adipocyte progenitor cells is potently inhibited by activin A, but not TGFβ. In these cells, phosphorylation of Smad2 is reduced significantly during differentiation, correlating with the gradual decrease of activin A expression. Finally, silencing of Smad2, but not Smad3, in SnoN−/− cells fully rescued the adipocyte differentiation, indicating that the adipogenic defects of SnoN−/− cells are mediated by Smad2.
TGFβ signaling, mainly through Smad3, has also been shown to inhibit pre-adipocyte differentiation in several cell line models (9–11, 56, 57). Overexpression of WT Smad3 in 3T3-F442A pre-adipocytes suppressed adipocyte differentiation, whereas expression of dominant-negative Smad3 enhanced the process (9). Consistent with this view, transgenic mice expressing TGFβ1 in WAT and BAT led to reduced WAT and BAT (12). However, studies of the Smad3 knockout mice suggest that under physiological conditions in vivo, TGFβ/Smad3 signaling mainly regulates energy homeostasis and glucose metabolism (17, 58). Smad3 loss protects against diet-induced obesity and insulin resistance and is accompanied by the browning of WAT adipocytes, leading to a marked increase in mitochondrial biogenesis and basal respiration (17). Thus, TGFβ/Smad3 plays an important and negative role in beige or brown fat development, and its effect on white fat development may be indirect. These phenotypes differ from that displayed by the SnoN-deficient mice in that no browning of WAT has been detected. In addition, reducing Smad3 in SnoN-deficient adipocyte progenitor cells did not rescue the impaired differentiation, suggesting that SnoN did not target TGFβ/Smad3 signaling in WAT in vivo. Thus, although SnoN is capable of binding to Smad2, Smad3, and Smad4 and represses their ability to mediate TGFβ, activin, and Nodal signaling in vitro, which of these pathways are also targeted by SnoN under physiological conditions in vivo may vary from tissue to tissue. In particular, it depends on the availability and activity of the individual ligand, its receptor, and downstream Smad proteins that are present in a given tissue. Indeed, our earlier studies suggest that in endothelial cells, SnoN regulates both TGFβ pathways through the ALK5/Smad3 branch and BMP9/10 pathway via ALK1/Smad1/5 (29). This report is another demonstration that during WAT development the target of SnoN is activin/Smad2 signaling.
The differentiation of MSCs to adipocytes is a two-step process that starts with lineage commitment to generate pre-adipocytes, followed by further maturation of pre-adipocytes to adipocytes. We showed here that SnoN is capable of blocking both steps of adipogenesis in an activin A-dependent manner. Functional deletion of SnoN not only inhibits pre-adipocyte maturation and the expression of the adipocyte markers Pparγ and Cebpβ, but it also results in elevated expression of Nanog in MSCs, promoting self-renewal of MSCs and inhibiting its differentiation. These data indicate that SnoN may facilitate lineage commitment of MSCs. Because MSCs can also differentiate into mesenchymal lineages such as osteoblasts, SnoN is predicted to also affect differentiation to those lineages. Indeed, we found that loss of SnoN markedly inhibited osteoblast differentiation (Fig. 3I), suggesting that SnoN plays a more global role in mesenchymal differentiation. Given that activin or TGFβ is a potent regulator of osteoblast differentiation, it is likely that this effect of SnoN is also Smad-dependent.
In addition to TGFβ/Smad signaling, SnoN can also regulate the activity of several other important signaling pathways, such as p53, prolactin/STAT5, and Hippo/TAZ (28, 32). Although these pathways have all been reported to be involved in regulation of adipogenesis either positively or negatively (59–61), they are unlikely to mediate the effects of SnoN on adipogenesis because introduction of a SnoN mutant that is defective in Smad binding but still retains its interaction with these pathways failed to rescue the impaired differentiation. This suggests that SnoN regulates adipogenesis mainly through modulating activin/Smad2 signaling. However, in our in vivo feeding studies, the defects in adipogenesis displayed by the SnoN KI mice were always less severe than those exhibited by the SnoN KO mice, indicating that other SnoN-regulated pathways, in addition to the Smad pathway, may also have a minor contribution.
In summary, our study has revealed an important role of SnoN in adipocyte differentiation and WAT development in vivo by regulating the activity of the activin/Smad2 signaling. The ability of SnoN to regulate multiple signaling pathways suggests that it may play an important role in regulating tissue development, homeostasis, and diseases.
Experimental procedures
Mice, cell lines, and antibodies
The SnoNm/m mice expressing a mutant SnoN gene containing point mutations that alter amino acid residues 88–92 and 267–277 to alanine have been described previously (29). The SnoN−/− mice were obtained from S. Pearson-White (Social & Scientific Systems, Inc., Silver Spring, MD).
293T and C3H10T1/2 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 100 units/ml penicillin/streptomycin (all from Invitrogen). The 3T3-L1 pre-adipocytes were cultured in DMEM (high glutamine) supplemented with 10% calf serum and penicillin/streptomycin. Primary MEF cells were cultured in high-glucose DMEM supplemented with 10% FBS and 100 units/ml penicillin/streptomycin (all from Invitrogen).
Anti-SnoN (H317) and anti-Smad3 (FL425) were purchased from Santa Cruz Biotechnology. Anti-Smad2 was from BD Biosciences. Anti-phospho-Smad3 (Ser-423/425) was from Cell Signaling Technology. Anti-phospho-Smad2 antisera (rabbit polyclonal antibody against a peptide (KKK-SSpMSp) containing two phosphoserine residues at the C terminus of Smad2) was a gift from A. Moustakas (Ludwig Institute for Cancer Research, Uppsala, Sweden). Anti-activin A was from R&D Systems. Antibody against tubulin was from Calbiochem.
Mouse siSmad2 Silencer® Select (s69493) and mouse siSmad3 Silencer® Select (s69494) were purchased from Life Technologies, Inc. Scrambled non-targeting siRNA pool 2 was purchased from Dharmacon. The sequence of mouse shSnoN is 5′-ctccattctgcagaggaag-3′.
Metabolic studies
All mice were housed with a 12-h light/dark cycle and given ad libitum access to food and water. Mice were fed either with normal diet (ND: 13.5% fat; LabDiet) or high-fat diet (HFD: 60% fat; D12492, Research Diets) starting at 6 weeks of age. Food was changed, and food intake and body weight were measured at the same time every week for 16 weeks. The same amount of food was given to all the mice. At the end of the 16-week HFD, oxygen consumption, CO2, heat production, activity/locomotion, and food and water intake were simultaneously determined in metabolic chambers (CLAMS, Columbus Instruments) with four mice per experiment for a maximum of 24 h. Individual mice were placed in a chamber with an airflow of 0.6 liters/min, and one reading per mouse was taken at 10-min intervals over 24 h. Body composition was determined using an EchoMRI body composition analyzer (EchoMRITM). GTT and ITT were performed on mice fasted for 6 h by intraperitoneal injection of 2.5 g of dextrose/kg body weight (ThermoFisher Scientific) or 0.75 units/kg insulin (Santa Cruz Biotechnology), respectively. Blood glucose concentrations were measured at 0, 30, 60, and 120 min after dextrose injection or at 0, 15, 30, 45, and 60 min after insulin injection by using a clinical grade glucometer (OneTouch Ultra, LifeScan, Inc.). Blood was collected from the fasted mice for serum analyses as follows: serum triglyceride levels were measured by InfinityTM triglycerides assay kit (ThermoFisher Scientific); serum leptin levels were measured by mouse leptin ELISA kit (Crystal Chem); serum adiponectin levels were measured by mouse adiponectin ELISA kit (Crystal Chem); and plasma levels of insulin were measured by ELISA (ALPCO). Plasma FFA levels were measured enzymatically (WAKO Chemicals).
Tissues were harvested for RNA, protein analysis, or histological analysis. All protocols were approved by the Animal Care and Use Committee of the University of California, Berkeley.
Generation of SnoN KO cell line
The SnoN-null C3H10T/12 cells were generated using the CRISPR/Cas9 system (62). The positive SnoN KO clones were identified by sequencing, which detected homozygous frameshift mutations in the first exon of the snoN gene (Fig. S2). Loss of SnoN protein expression was verified by Western blotting. The sequence for mouse SnoN sgRNA1_F was CACCGCGCTTGGCGTGCTCGCCATC and for mouse SnoN sgRNA1_R was AAACGATGGCGAGCACGCCAAGCGC.
Transfection, infection, and Western blotting
siRNAs were transiently transfected into C3H10T1/2 or 3T3-L1 cells using Lipofectamine® RNAiMAX transfection reagent (Invitrogen) according to the manufacturer's protocol. For retroviral infection, cDNAs or shRNAs in retroviral vectors were co-transfected with packaging plasmids (pCMV-gag-pol and pBS-VSVG) into 293T cells to generate a high-titer viral supernatant, which was used subsequently to infect the C3H10T1/2 cells. After 48 h, infected cells were selected with 1.5 μg/ml puromycin. Pools of puromycin-resistant cells were analyzed in various experiments. Western blotting was performed as described previously (63).
Differentiation of MSCs
Primary MEF cells were isolated from E13.5 embryos of SnoN+/+, SnoN−/−, and SnoNm/m mice. Briefly, embryos were separated from maternal tissues and yolk sac, finely minced, and digested with 0.25% trypsin, 1 mm EDTA for 15 min at 37 °C. The suspension was transferred to a new tube and centrifuged. The pellet was resuspended in culture medium and plated in a p50 dish (passage No. 0). All adipocyte differentiation experiments were carried out using MEFs at passage no. 3–4. MEFs were cultured to confluence in a 6-well cluster plate. Two days later, these cells were treated with differentiation-inducing medium containing 0.5 mm 3-isobutyl-1-methylxanthine (IBMX), 1 μm dexamethasone, 10 μg/ml insulin, 10 μm troglitazone, and 10% FBS for 4 days followed by culturing in insulin medium for another 6 days, with the medium being renewed every other day.
For adipogenic differentiation of C3H10T1/2 cells, cells at 2 days post-confluence were treated with induction medium containing 1 μm dexamethasone, 0.5 μm rosiglitazone, 0.5 mm IBMX, 5 μg/ml insulin, and 10% FBS for 3 days followed by insulin plus rosiglitazone for an additional 4 days.
For adipogenic differentiation of 3T3-L1 cells, cells at 2 days post-confluence (in DMEM with 10% calf serum) were changed to induction medium containing 1 μm dexamethasone, 0.5 mm IBMX, 10 μg/ml insulin, and 10% FBS for 2 days followed by insulin medium (10 μg/ml insulin and 10% FBS in DMEM) for an additional 4 days.
For osteogenic differentiation, C3H10T1/2 cells were cultured in growth medium to confluency, followed by treatment with 50 μg/ml ascorbic acid, 10 mm β-glycerophosphate, and 50 ng/ml BMP2. Osteoblastic phenotypes were observed over a 2–3-week period, and the intensity of alkaline phosphatase activity per dish of cells was evaluated by BCIP/NBT staining (BCIP®/NBT Liquid Substrate System, Sigma).
Oil Red O staining
Frozen liver sections of 8 mm thickness were fixed in formalin, washed with water, rinsed with 60% isopropyl alcohol, and stained with freshly prepared Oil Red O working solution as described previously (64). To stain adipocytes, cells were fixed in 4% paraformaldehyde for 30 min, rinsed, and incubated with Oil Red O working solution for 50 min. All images were visualized under a Lumar version 12 epifluorescence stereoscope (Carl Zeiss) or scanned on an Epson Perfection 4490 photoscanner.
qRT-PCR
Total RNA was extracted from cells using the RNeasy mini kit (Qiagen). RNA extraction from WAT was performed using TRIzol reagent (Life Technologies, Inc.) according to the manufacturer's instructions. cDNA was produced by reverse transcription (SuperScript III; Invitrogen). qRT-PCR was performed with the ABI 7300 (Applied Biosystem) per the manufacturer's instruction. All PCRs were performed in technical duplicate in three independent experiments. Primers used for qRT-PCR are listed in Table S1.
Statistical analysis
All data are presented as means ± S.D. Statistical significance between groups was determined using the two-tailed unpaired Student's t test or one-way analysis of variance (Newman-Keuls multiple comparison test) with Prism 5 software. p values are shown when relevant (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
Author contributions
Q. Z. and K. L. conceptualization; Q. Z. formal analysis; Q. Z. validation; Q. Z., A. C., and A. X. investigation; Q. Z. visualization; Q. Z. methodology; Q. Z. writing-original draft; K. L. resources; K. L. data curation; K. L. supervision; K. L. funding acquisition; K. L. project administration; K. L. writing-review and editing.
Supplementary Material
Acknowledgments
We thank Hei Sook Sul, Andreas Stahl, and Danica Chen for assistance, cell lines, and helpful discussions. We are grateful to Xiaodan Ji for the contribution and assistance with two panels in Fig. S3. We also thank to Kevin Tharp and Zhifang Zheng for the help and advice on metabolic chamber assays; Michael Wendland at LKS Small Animal Imaging Facilities at UC-Berkeley for assistance with EchoMRI; Steve Ruzin and Denise Schichnes at CNR Biological imaging facility at UC-Berkeley for assistance with microscopy; and Pathology Services Inc. for histology services.
This work was supported by Department of Defense CDMRP BCRP Grant W81XWH-15-1-0051 (to K. L.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S4 and Table S1.
- MSC
- mesenchymal stem cell
- WAT
- white adipose tissue
- HFD
- high-fat diet
- qRT-PCR
- quantitative RT-PCR
- TGFβ
- transforming growth factor-β
- BAT
- brown adipose tissue
- HFD
- high-fat diet
- αFABP
- adipocyte fatty acid-binding protein
- C/EBP
- CCAAT/enhancer-binding protein
- PPARγ
- peroxisome proliferator-activated receptor γ
- BMP
- bone morphogenetic protein
- MEF
- mouse embryonic fibroblast
- FBS
- fetal bovine serum
- DMEM
- Dulbecco's modified Eagle's medium
- IBMX
- 3-isobutyl-1-methylxanthine
- CLAMS
- comprehensive laboratory animal monitoring system
- KI
- knockin
- GTT
- glucose tolerance test
- ITT
- insulin tolerance test
- ND
- normal diet
- ATM
- adipose tissue macrophage
- FFA
- free fatty acid.
References
- 1. Rosen E. D., and Spiegelman B. M. (2014) What we talk about when we talk about fat. Cell 156, 20–44 10.1016/j.cell.2013.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tang Q. Q., and Lane M. D. (2012) Adipogenesis: from stem cell to adipocyte. Annu. Rev. Biochem. 81, 715–736 10.1146/annurev-biochem-052110-115718 [DOI] [PubMed] [Google Scholar]
- 3. Farmer S. R. (2006) Transcriptional control of adipocyte formation. Cell Metab. 4, 263–273 10.1016/j.cmet.2006.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Grafe I., Alexander S., Peterson J. R., Snider T. N., Levi B., Lee B., and Mishina Y. (2017) TGF-β family signaling in mesenchymal differentiation. Cold Spring Harb. Perspect. Biol. 10, a022202 10.1101/cshperspect.a022202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zamani N., and Brown C. W. (2011) Emerging roles for the transforming growth factor-β superfamily in regulating adiposity and energy expenditure. Endocr. Rev. 32, 387–403 10.1210/er.2010-0018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tang Q. Q., Otto T. C., and Lane M. D. (2004) Commitment of C3H10T1/2 pluripotent stem cells to the adipocyte lineage. Proc. Natl. Acad. Sci. U.S.A. 101, 9607–9611 10.1073/pnas.0403100101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zaragosi L. E., Wdziekonski B., Villageois P., Keophiphath M., Maumus M., Tchkonia T., Bourlier V., Mohsen-Kanson T., Ladoux A., Elabd C., Scheideler M., Trajanoski Z., Takashima Y., Amri E. Z., Lacasa D., et al. (2010) Activin a plays a critical role in proliferation and differentiation of human adipose progenitors. Diabetes 59, 2513–2521 10.2337/db10-0013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hirai S., Yamanaka M., Kawachi H., Matsui T., and Yano H. (2005) Activin A inhibits differentiation of 3T3-L1 preadipocyte. Mol. Cell. Endocrinol. 232, 21–26 10.1016/j.mce.2005.01.001 [DOI] [PubMed] [Google Scholar]
- 9. Choy L., Skillington J., and Derynck R. (2000) Roles of autocrine TGF-β receptor and Smad signaling in adipocyte differentiation. J. Cell Biol. 149, 667–682 10.1083/jcb.149.3.667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Choy L., and Derynck R. (2003) Transforming growth factor-β inhibits adipocyte differentiation by Smad3 interacting with CCAAT/enhancer-binding protein (C/EBP) and repressing C/EBP transactivation function. J. Biol. Chem. 278, 9609–9619 10.1074/jbc.M212259200 [DOI] [PubMed] [Google Scholar]
- 11. Ignotz R. A., and Massagué J. (1985) Type β transforming growth factor controls the adipogenic differentiation of 3T3 fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 82, 8530–8534 10.1073/pnas.82.24.8530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Clouthier D. E., Comerford S. A., and Hammer R. E. (1997) Hepatic fibrosis, glomerulosclerosis, and a lipodystrophy-like syndrome in PEPCK-TGF-β1 transgenic mice. J. Clin. Invest. 100, 2697–2713 10.1172/JCI119815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lin Y., Nakachi K., Ito Y., Kikuchi S., Tamakoshi A., Yagyu K., Watanabe Y., Inaba Y., Kazuo Tajima, and Jacc Study Group. (2009) Variations in serum transforming growth factor-β1 levels with gender, age and lifestyle factors of healthy Japanese adults. Dis. Markers 27, 23–28 10.1155/2009/529253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fain J. N., Tichansky D. S., and Madan A. K. (2005) Transforming growth factor β1 release by human adipose tissue is enhanced in obesity. Metabolism 54, 1546–1551 10.1016/j.metabol.2005.05.024 [DOI] [PubMed] [Google Scholar]
- 15. Alessi M. C., Bastelica D., Morange P., Berthet B., Leduc I., Verdier M., Geel O., and Juhan-Vague I. (2000) Plasminogen activator inhibitor 1, transforming growth factor-β1, and BMI are closely associated in human adipose tissue during morbid obesity. Diabetes 49, 1374–1380 10.2337/diabetes.49.8.1374 [DOI] [PubMed] [Google Scholar]
- 16. Samad F., Yamamoto K., Pandey M., and Loskutoff D. J. (1997) Elevated expression of transforming growth factor-β in adipose tissue from obese mice. Mol. Med. 3, 37–48 [PMC free article] [PubMed] [Google Scholar]
- 17. Yadav H., Quijano C., Kamaraju A. K., Gavrilova O., Malek R., Chen W., Zerfas P., Zhigang D., Wright E. C., Stuelten C., Sun P., Lonning S., Skarulis M., Sumner A. E., Finkel T., and Rane S. G. (2011) Protection from obesity and diabetes by blockade of TGF-β/Smad3 signaling. Cell Metab. 14, 67–79 10.1016/j.cmet.2011.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shen J. J., Huang L., Li L., Jorgez C., Matzuk M. M., and Brown C. W. (2009) Deficiency of growth differentiation factor 3 protects against diet-induced obesity by selectively acting on white adipose. Mol. Endocrinol. 23, 113–123 10.1210/me.2007-0322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Massagué J., Seoane J., and Wotton D. (2005) Smad transcription factors. Genes Dev. 19, 2783–2810 10.1101/gad.1350705 [DOI] [PubMed] [Google Scholar]
- 20. Feng X. H., and Derynck R. (2005) Specificity and versatility in TGF-β signaling through Smads. Annu. Rev. Cell Dev. Biol. 21, 659–693 10.1146/annurev.cellbio.21.022404.142018 [DOI] [PubMed] [Google Scholar]
- 21. ten Dijke P., and Hill C. S. (2004) New insights into TGF-β-Smad signalling. Trends Biochem. Sci. 29, 265–273 10.1016/j.tibs.2004.03.008 [DOI] [PubMed] [Google Scholar]
- 22. Massagué J. (2012) TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 13, 616–630 10.1038/nrm3434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Morikawa M., Derynck R., and Miyazono K. (2016) TGF-β and the TGF-β family: context-dependent roles in cell and tissue physiology. Cold Spring Harb. Perspect. Biol. 8, a021873 10.1101/cshperspect.a021873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moustakas A., and Heldin C. H. (2009) The regulation of TGFβ signal transduction. Development 136, 3699–3714 [DOI] [PubMed] [Google Scholar]
- 25. Guo X., and Wang X. F. (2009) Signaling cross-talk between TGF-β/BMP and other pathways. Cell Res. 19, 71–88 10.1038/cr.2008.302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jahchan N. S., and Luo K. (2010) SnoN in mammalian development, function and diseases. Curr. Opin. Pharmacol. 10, 670–675 10.1016/j.coph.2010.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Deheuninck J., and Luo K. (2009) Ski and SnoN, potent negative regulators of TGF-β signaling. Cell Res. 19, 47–57 10.1038/cr.2008.324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhu Q., and Luo K. (2012) SnoN in regulation of embryonic development and tissue morphogenesis. FEBS Lett. 586, 1971–1976 10.1016/j.febslet.2012.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhu Q., Kim Y. H., Wang D., Oh S. P., and Luo K. (2013) SnoN facilitates ALK1-Smad1/5 signaling during embryonic angiogenesis. J. Cell Biol. 202, 937–950 10.1083/jcb.201208113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pan D., Zhu Q., and Luo K. (2009) SnoN functions as a tumour suppressor by inducing premature senescence. EMBO J. 28, 3500–3513 10.1038/emboj.2009.250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jahchan N. S., Wang D., Bissell M. J., and Luo K. (2012) SnoN regulates mammary gland alveologenesis and onset of lactation by promoting prolactin/STAT5 signaling. Development 139, 3147–3156 10.1242/dev.079616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhu Q., Le Scolan E., Jahchan N., Ji X., Xu A., and Luo K. (2016) SnoN antagonizes the Hippo kinase complex to promote TAZ signaling during breast carcinogenesis. Dev. Cell 37, 399–412 10.1016/j.devcel.2016.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shinagawa T., Dong H.-D., Xu M., Maekawa T., and Ishii S. (2000) The sno gene, which encodes a component of the histone deacetylation complex, acts as a tumor suppressor in mice. EMBO J. 19, 2280–2291 10.1093/emboj/19.10.2280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pan D., Zhu Q., Conboy M. J., Conboy I. M., and Luo K. (2012) SnoN activates p53 directly to regulate aging and tumorigenesis. Aging Cell 11, 902–911 10.1111/j.1474-9726.2012.00857.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Maffei M., Halaas J., Ravussin E., Pratley R. E., Lee G. H., Zhang Y., Fei H., Kim S., Lallone R., and Ranganathan S. (1995) Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat. Med. 1, 1155–1161 10.1038/nm1195-1155 [DOI] [PubMed] [Google Scholar]
- 36. Frederich R. C., Hamann A., Anderson S., Löllmann B., Lowell B. B., and Flier J. S. (1995) Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat. Med. 1, 1311–1314 10.1038/nm1295-1311 [DOI] [PubMed] [Google Scholar]
- 37. Weisberg S. P., McCann D., Desai M., Rosenbaum M., Leibel R. L., and Ferrante A. W. Jr. (2003) Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest. 112, 1796–1808 10.1172/JCI200319246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lumeng C. N., Deyoung S. M., Bodzin J. L., and Saltiel A. R. (2007) Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes 56, 16–23 10.2337/db06-1076 [DOI] [PubMed] [Google Scholar]
- 39. Xu H., Barnes G. T., Yang Q., Tan G., Yang D., Chou C. J., Sole J., Nichols A., Ross J. S., Tartaglia L. A., and Chen H. (2003) Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Invest. 112, 1821–1830 10.1172/JCI200319451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Castoldi A., Naffah de Souza C., Câamara N. O., and Moraes-Vieira P. M. (2015) The macrophage switch in obesity development. Front. Immunol. 6, 637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lumeng C. N., Bodzin J. L., and Saltiel A. R. (2007) Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Invest. 117, 175–184 10.1172/JCI29881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Olefsky J. M., and Glass C. K. (2010) Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 72, 219–246 10.1146/annurev-physiol-021909-135846 [DOI] [PubMed] [Google Scholar]
- 43. Yusuf B., Gopurappilly R., Dadheech N., Gupta S., Bhonde R., and Pal R. (2013) Embryonic fibroblasts represent a connecting link between mesenchymal and embryonic stem cells. Dev. Growth Differ. 55, 330–340 10.1111/dgd.12043 [DOI] [PubMed] [Google Scholar]
- 44. Tseng Y. H., Kokkotou E., Schulz T. J., Huang T. L., Winnay J. N., Taniguchi C. M., Tran T. T., Suzuki R., Espinoza D. O., Yamamoto Y., Ahrens M. J., Dudley A. T., Norris A. W., Kulkarni R. N., and Kahn C. R. (2008) New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature 454, 1000–1004 10.1038/nature07221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Seale P., Kajimura S., Yang W., Chin S., Rohas L. M., Uldry M., Tavernier G., Langin D., and Spiegelman B. M. (2007) Transcriptional control of brown fat determination by PRDM16. Cell Metab. 6, 38–54 10.1016/j.cmet.2007.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sakaki-Yumoto M., Liu J., Ramalho-Santos M., Yoshida N., and Derynck R. (2013) Smad2 is essential for maintenance of the human and mouse primed pluripotent stem cell state. J. Biol. Chem. 288, 18546–18560 10.1074/jbc.M112.446591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Matzuk M. M., Kumar T. R., Vassalli A., Bickenbach J. R., Roop D. R., Jaenisch R., and Bradley A. (1995) Functional analysis of activins during mammalian development. Nature 374, 354–356 10.1038/374354a0 [DOI] [PubMed] [Google Scholar]
- 48. Matzuk M. M., Kumar T. R., and Bradley A. (1995) Different phenotypes for mice deficient in either activins or activin receptor type II. Nature 374, 356–360 10.1038/374356a0 [DOI] [PubMed] [Google Scholar]
- 49. Nomura M., and Li E. (1998) Smad2 role in mesoderm formation, left-right patterning and craniofacial development. Nature 393, 786–790 10.1038/31693 [DOI] [PubMed] [Google Scholar]
- 50. Waldrip W. R., Bikoff E. K., Hoodless P. A., Wrana J. L., and Robertson E. J. (1998) Smad2 signaling in extraembryonic tissues determines anterior-posterior polarity of the early mouse embryo. Cell 92, 797–808 10.1016/S0092-8674(00)81407-5 [DOI] [PubMed] [Google Scholar]
- 51. Weinstein M., Yang X., Li C., Xu X., Gotay J., and Deng C. X. (1998) Failure of egg cylinder elongation and mesoderm induction in mouse embryos lacking the tumor suppressor smad2. Proc. Natl. Acad. Sci. U.S.A. 95, 9378–9383 10.1073/pnas.95.16.9378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Heyer J., Escalante-Alcalde D., Lia M., Boettinger E., Edelmann W., Stewart C. L., and Kucherlapati R. (1999) Postgastrulation Smad2-deficient embryos show defects in embryo turning and anterior morphogenesis. Proc. Natl. Acad. Sci. U.S.A. 96, 12595–12600 10.1073/pnas.96.22.12595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gu Z., Nomura M., Simpson B. B., Lei H., Feijen A., van den Eijnden-van Raaij J., Donahoe P. K., and Li E. (1998) The type I activin receptor ActRIB is required for egg cylinder organization and gastrulation in the mouse. Genes Dev. 12, 844–857 10.1101/gad.12.6.844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ferguson C. A., Tucker A. S., Christensen L., Lau A. L., Matzuk M. M., and Sharpe P. T. (1998) Activin is an essential early mesenchymal signal in tooth development that is required for patterning of the murine dentition. Genes Dev. 12, 2636–2649 10.1101/gad.12.16.2636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Song J., Oh S. P., Schrewe H., Nomura M., Lei H., Okano M., Gridley T., and Li E. (1999) The type II activin receptors are essential for egg cylinder growth, gastrulation, and rostral head development in mice. Dev. Biol. 213, 157–169 10.1006/dbio.1999.9370 [DOI] [PubMed] [Google Scholar]
- 56. Torti F. M., Torti S. V., Larrick J. W., and Ringold G. M. (1989) Modulation of adipocyte differentiation by tumor necrosis factor and transforming growth factor β. J. Cell Biol. 108, 1105–1113 10.1083/jcb.108.3.1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jeoung D. I., Tang B., and Sonenberg M. (1995) Mitogenic response to TGF-β in 3T3-F442A cells. Biochem. Biophys. Res. Commun. 216, 964–969 10.1006/bbrc.1995.2714 [DOI] [PubMed] [Google Scholar]
- 58. Tan C. K., Leuenberger N., Tan M. J., Yan Y. W., Chen Y., Kambadur R., Wahli W., and Tan N. S. (2011) Smad3 deficiency in mice protects against insulin resistance and obesity induced by a high-fat diet. Diabetes 60, 464–476 10.2337/db10-0801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hong J. H., Hwang E. S., McManus M. T., Amsterdam A., Tian Y., Kalmukova R., Mueller E., Benjamin T., Spiegelman B. M., Sharp P. A., Hopkins N., and Yaffe M. B. (2005) TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science 309, 1074–1078 10.1126/science.1110955 [DOI] [PubMed] [Google Scholar]
- 60. Minamino T., Orimo M., Shimizu I., Kunieda T., Yokoyama M., Ito T., Nojima A., Nabetani A., Oike Y., Matsubara H., Ishikawa F., and Komuro I. (2009) A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat. Med. 15, 1082–1087 10.1038/nm.2014 [DOI] [PubMed] [Google Scholar]
- 61. Stewart W. C., Pearcy L. A., Floyd Z. E., and Stephens J. M. (2011) STAT5A expression in Swiss 3T3 cells promotes adipogenesis in vivo in an athymic mice model system. Obesity 19, 1731–1734 10.1038/oby.2011.66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., and Zhang F. (2013) Genome engineering using the CRISPR/Cas9 system. Nat. Protoc. 8, 2281–2308 10.1038/nprot.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhu Q., Krakowski A. R., Dunham E. E., Wang L., Bandyopadhyay A., Berdeaux R., Martin G. S., Sun L., and Luo K. (2007) Dual role of SnoN in mammalian tumorigenesis. Mol. Cell. Biol. 27, 324–339 10.1128/MCB.01394-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kim K. A., Kim J. H., Wang Y., and Sul H. S. (2007) Pref-1 (preadipocyte factor 1) activates the MEK/extracellular signal-regulated kinase pathway to inhibit adipocyte differentiation. Mol. Cell. Biol. 27, 2294–2308 10.1128/MCB.02207-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.