

Abstract
INTRODUCTION
Our objective was to investigate the effect of sex on cognitive decline within the context of β-amyloid (Aβ) burden and apolipoprotein (APOE) genotype.
METHODS
We analyzed sex-specific effects on Aβ-PET, APOE and rates of change on the Preclinical Alzheimer Cognitive Composite-5 (PACC-5) across three cohorts, ADNI, AIBL and HABS (n=755; clinical dementia rating (CDR)=0; Age(SD)=73.6(6.5); Female=55%). Mixed-effects models of cognitive change by sex, Aβ-PET and APOEε4 were examined with quadratic time-effects over a median of 4 years of follow-up.
RESULTS
APOEε4 prevalence and Aβ burden did not differ by sex. Sex did not directly influence cognitive decline. Females with higher Aβ exhibited faster decline than males. Post-hoc contrasts suggested that females who were Aβ and APOEε4 positive declined faster than their male counterparts.
DISCUSSION
Although Aβ did not differ by sex, cognitive decline was greater in females with higher Aβ. Our findings suggest sex may play a modifying role on risk of AD-related cognitive decline.
Keywords: Preclinical Alzheimer’s disease, amyloid, APOE, sex, gender, cognitive decline
1. INTRODUCTION
Investigating the extent to which the early pathophysiology and cognitive decline vary by sex is critical for understanding the course of Alzheimer’s disease (AD) dementia [1]. Epidemiological studies suggest higher incidence rates for women relative to men for AD dementia in older ages [2], however, this is not always supported [3–5]. Studies are mixed with regard to the effect of sex on cognitive decline in clinically-normal older adults [6, 7]. Steeper decline in delayed recall [8], performance IQ and executive function [9] has been reported in female apolipoprotein ε4 (APOEε4) carriers, however, this finding is not supported by other studies [6, 10]. In addition, some studies suggest males show steeper decline than females in areas of speed, integration and visuospatial ability [11]. A recent meta-analysis found that APOEε4 carriers exhibit particular vulnerability for progression to Alzheimer’s disease (AD) dementia relative to males and female APOEε4 non-carriers between the ages 65-75 years [12]. Carrying the APOEε4 allele confers higher risk for abnormal levels of β-amyloid (Aβ) burden [13] and accumulation [14], and results in steeper cognitive decline when accompanied by high Aβ burden [15, 16]. Hence, it is possible that APOEε4 genetic risk in the presence of Aβ may impart particular susceptibility for females to AD clinical symptoms.
It is unclear whether sex differences in AD dementia risk in those with APOEε4 is exacerbated by higher Aβ burden, whether APOEε4 and Aβ play independent roles, or whether there are other “downstream” mechanisms that account for the increased vulnerability to dementia risk. Disentangling sex-specific effects with respect to Aβ and APOE is particularly relevant during the preclinical stage, given that abnormal levels of Aβ start to accrue decades before the onset of clinical symptoms [17], and that greater focus is being placed on prevention trials [18]. If Aβ burden is differentially associated with cognitive decline in males and females, this will have implications for recruitment and treatment practices in clinical trials.
Our first aim was to ascertain whether sex differences exist in relation to APOE and Aβ burden as estimated by positron emission tomography (PET). Our second aim was to examine the effect of sex on cognitive decline, and whether this was influenced by APOEε4 carriage, abnormal Aβ or both APOE and Aβ. To improve our ability to detect the extent to which females could increase risk for AD biomarkers and cognitive decline, we harmonized data from three well-characterized, longitudinal datasets: the Alzheimer’s Disease Neuroimaging Initiative (ADNI), the Australian Imaging, Biomarker and Lifestyle (AIBL) study of ageing, and the Harvard Aging Brain Study (HABS). This allows us sufficient statistical power to determine potentially small magnitude relationships between sex and AD risk.
2. METHODS
2.1. Participants
Cohort-specific inclusion criteria for recruitment have been published previously [19–21]. For the current study, the baseline was considered to be an individual’s first Aβ-PET scan. Participants were all required to be clinically-normal at baseline (Global clinical dementia rating (CDR) score = 0, MMSE ≥ 24); ADNI’s subjective cognitive decline (SCD) group was included in the current study, given that these participants attained a CDR score of 0. Participants were included if their baseline Aβ-PET scan was within 1 year of a neuropsychological testing session (either before or after the scan), and they had at least 2 follow-up neuropsychological assessments after their baseline visit. We excluded participants who carried APOEε2/ε4 and APOEε2/ε2 (total < 2.9%), given that the effect of these genotypes on AD risk are unclear. For analysis, 755 participants (ADNI, n=330; AIBL, n=161; HABS, n=268) formed the final participant group. We conducted the procedures for this study under the ethical guidelines stipulated by the Partners Human Research Committee, which is the Institutional Review Board for the Massachusetts General Hospital and Brigham and Women’s Hospital.
2.2. Cognitive outcome
We examined cognitive decline using the Preclinical Alzheimer Cognitive Composite score with an additional semantic processing component (PACC-5) [22]. This composite modifies the PACC that was developed as a sensitive measure of Aβ-related cognitive decline [23]. We used the PACC-5 as recent findings suggest the inclusion of a semantic component, specifically the Categories task, adds unique variance associated with Aβ-related cognitive decline beyond that provided by the original PACC components [22]. Each study used a version of the PACC-5 that has been previously published [23–25]. In each study, the PACC-5 includes some overlapping tests (Mini-Mental State Examination [MMSE] and Logical Memory Delayed Recall) and some non-overlapping tests (for ADNI: ADAS-Cog Word Recall, Trails B, and Categories (Animals); for AIBL: the California Verbal Learning Test (second edition), Digit Symbol substitution, and Categories (Animals/Names); for HABS: the Free and Cued Selective Reminding Test, and Categories (Animals/Vegetables/Fruit). It is important to note that all three studies had two overlapping tests in the PACC-5 (MMSE and Logical Memory), however, the other three tests were unique to each study. All test scores were standardized within their own study according to the baseline mean and standard deviation of CDR=0 participants from the respective cohort. The PACC-5 was formed by averaging these z-scores. Baseline and longitudinal slopes for the PACC-5 were compared across the three studies to determine whether means and variances were similar (see Appendix A for cross-cohort distributions). ADNI and HABS participants completed these tests approximately every year, whereas AIBL participants underwent testing every 1.5 years. All available testing sessions following the analysis-defined baseline session were used (for ADNI: 324 participants completed 3 visits, 248 completed 4 visits, 166 completed 5 visits, and 28 completed 6 visits; for AIBL: 155 completed 3 visits, 73 completed 4 visits; for HABS: 244 completed 3 visits, 234 completed 4 visits, 183 completed 5 visits, 116 completed 6 visits). In order to adjust for baseline performance in our models, our cognitive outcome variable was PACC-5 change from baseline (with baseline cognitive performance as a covariate).
2.3. Aß positron emission tomography (PET)
ADNI uses the 18F-AV45 (Florbetapir or FBP) Aβ-PET tracer, while AIBL and HABS use the 11C-Pittsburgh Compound-B (PiB) Aβ-PET tracer. The PET acquisition parameters for each study have been published previously [21, 26, 27]. In brief, ADNI and AIBL’s PET acquisition time was 50–70 minutes post-injection (http://adni.loni.usc.edu/), while for HABS, PiB-PET data were collected 40–60 minutes post-injection. All raw Aβ-PET data were processed with a standard pipeline. For this pipeline, PET data underwent reconstruction and attenuation correction, were evaluated for head motion, and were co-registered/normalized to a PET template in MNI space using the SPM12 unified segmentation, normalization routine, which applies a rigid body registration, followed by an affine registration, and a nonlinear mapping that fits the image to pre-specified 6-class tissue probability map. Summary measures for regions of interest (ROIs) were computed from a probabilistic GTM-Seg atlas in MNI space (Freesurfer v6.0 [28]) as standard uptake value ratios (SUVrs). The following ROIs that have been validated as AD regions of interest in previous publications [16] were aggregated: the frontal, lateral, and retrosplenial (FLR) regions. Values were normalized against the whole cerebellum to yield an Aβ FLR SUVr for each participant. To ensure cross-tracer equivalency, we applied a novel nonlinear transformation mapping approach (NLTM; further details in Appendix B). With this equating method, we extracted equivalent FBP SUVrs (FBPequiv) for all PiB data so that they conformed to the FBP distribution. Although the NTLM is bidirectional, we chose to conform PiB SUVrs to the FBP distribution due to its more limited dynamic range and scale (Appendix B shows the pre- and post- distributions after applying NLTM in comparison with raw values and a linear-only transformation). This equivalence allowed for combined analysis of continuous Aβ FLR SUVr. For a post-hoc analysis, we also dichotomized Aβ using a Gaussian mixture modelling procedure [16], which gave an FBPequiv cut-off of 1.082 for Aβ+ (referred to as Aβstatus to differentiate between the continuous Aβ measure).
2.4. Statistical analysis
Analyses were performed using R version 3.3.2. To determine sex differences in Aβ burden and APOEε4 we ran group comparisons using Wilcoxon/Mann-Whitney and chi-square (χ2) tests, respectively. To investigate sex-specific effects on cognitive decline in association with Aβ and APOEε4 status, we ran a series of hierarchical linear mixed models with subject-specific random intercept nested within cohort. We also modeled cohort as a fixed effect. Covariates were age, years of education, and baseline cognitive performance. As PACC-5 change is best modeled with quadratic time [29, 30], interaction terms with this time effect and all other covariates were included in the models. Only quadratic time terms were modeled, as the global extremum (vertex of parabola) of PACC-5 performance passed through zero at baseline, and an assessment of goodness-of-fit parameters suggested adequate fit against models including the linear terms. We compared goodness-of-fit of increasingly complex models using a log-likelihood ratio test. Multiple independent comparisons (n=5) were accounted for according to a Sidak correction of α = 0.01.
The following hierarchy of models were run:
PACC-5change ~ sex*time + covariates*time
PACC-5change ~ [Aβ OR APOEε4]*sex*time + covariates*time
PACC-5change ~ Aβ*APOEε4*sex*time + covariates*time
PACC-5change ~ Aβ*sex*Age*time + covariates*time
where PACC-5change is the change in PACC-5 from baseline, OR indicates different terms used in models A and B, Aβ is the continuous FBPequiv SUVr, and the covariates were age, years of education and PACC-5 performance at baseline. Note that all lower-order terms were included in each model.
To further explore interactions in Model C between the dichotomized variables Aβstatus (+/−), APOEε4 (+/−) and sex (M/F), pairwise comparisons were performed for the groups: (Aβ−/APOEε4−/M (n=191), Aβ−/APOEε4−/F (n=220), Aβ−/APOEε4+/M (n=41), Aβ−/APOEε4+/F (n=62), Aβ+/APOEε4−/M (n=46), Aβ+/APOEε4−/F (n=59), Aβ+/APOEε4+/M (n=46), and Aβ+/APOEε4+/F (n=54)).
We also ran post-hoc analyses within each study to determine whether patterns of findings were consistent with the combined-cohort results.
3. RESULTS
3.1. Cohort characteristics
We examined 755 clinically-normal individuals with a median of 4 years of follow-up in their respective study (range = 3 – 7 years across the combined group), as summarized in Table 1. ADNI had the shortest follow-up duration (6.08 years), whereas AIBL had the longest follow-up duration (6.98 years). There were no significant differences in follow-up length by sex (t = 0.25, p = 0.80), Aβ status (t = 0.05, p = 0.96), APOE (t = −0.70, p = 0.48), sex*Aβ status (F = 1.61, p = 0.21), or by sex*APOE (F = 0.92, p = 0.34). AIBL participants were significantly younger, and had fewer years of education than the other studies. There were no cohort-level differences in the frequencies of APOEε4 carriers, females/males, or those with Aβ+ status. Differences also did not exist in baseline cognitive performance or individual cognitive slopes between the cohorts (cognitive slopes were extracted from ordinary least squares regression models).
Table 1.
Comparison between the studies on demographics and cognitive performance
| ADNI (n = 330) Mean (SD) |
AIBL (n = 161) Mean (SD) |
HABS (n=268) Mean (SD) |
F, χ2 | Effect size (ηp2 or Cramer’s V) |
p | |
|---|---|---|---|---|---|---|
| Follow-up (yrs) | 3.29 (1.2) | 4.99 (1.3) | 4.56 (1.3) | 129.1 | 0.26 | <0.001 |
| Age | 74.6 (6.5) | 71.6 (6.8)* | 73.4 (6.0) | 11.3 | 0.03 | <.001 |
| Females (n/%) | 173 (52%) | 86 (53%) | 159 (59%) | 3.1 | 0.06 | 0.21 |
| Years of | 16.4 (2.7) | 13.7 (2.2)* | 15.8 (3.0) | 53.8 | 0.12 | <0.001 |
| Education | ||||||
| APOEε4+ (n/%) | 88 (27%) | 49 (32%) | 67 (27%) | 1.13 | 0.04 | 0.56 |
| Aβ SUVr (FBPequiv) | 1.04 (0.2) | 1.05 (0.2) | 1.04 (0.2) | 0.23 | 0.0006 | 0.79 |
| Aβ + status (n/%) | 96 (29%) | 46 (29%) | 76 (28%) | 0.01 | 0.004 | 0.99 |
| MMSEbaseline | 29 (1.2) | 29 (1.2) | 29 (1.1) | 0.37 | 0.001 | 0.69 |
| PACCbaseline | 0.03 (0.6) | 0.07 (0.6) | 0.02 (0.7) | 0.34 | 0.001 | 0.71 |
| PACCslope | −0.05 (0.2) | −0.03 (0.2) | −0.03 (0.2) | 1.01 | 0.003 | 0.36 |
Note: ADNI = Alzheimer’s Disease Neuroimaging Initiative; Australian Imaging, Biomarker and Lifestyle (AIBL) study; HABS = Harvard Aging Brain Study (HABS); SD = standard deviation; FBPequiv = Florebetapir equivalent score; APOE = apolipoprotein
3.2. Sex differences in Aβ burden and APOE carrier status
Females did not exhibit greater median Aβ burden (Diff = −0.006, CI 95% [−0.023, 0.011], p = 0.48; see Figure 1A and cohort-level findings in Appendix D). We also did not find sex differences according to Aβ status (χ2 = 0.80, Cramer’s V = .04, p = 0.37). Females were not more likely be APOEε4 carriers (χ2 = 0.39, Cramer’s V = .02, p = 0.53; see Figure 1B). Neither were female APOEε4 carriers more likely to exhibit greater median Aβ than male APOEε4 carriers (Diff = 0.010, CI 95% [−0.045, 0.066], p = 0.76).
Figure 1.
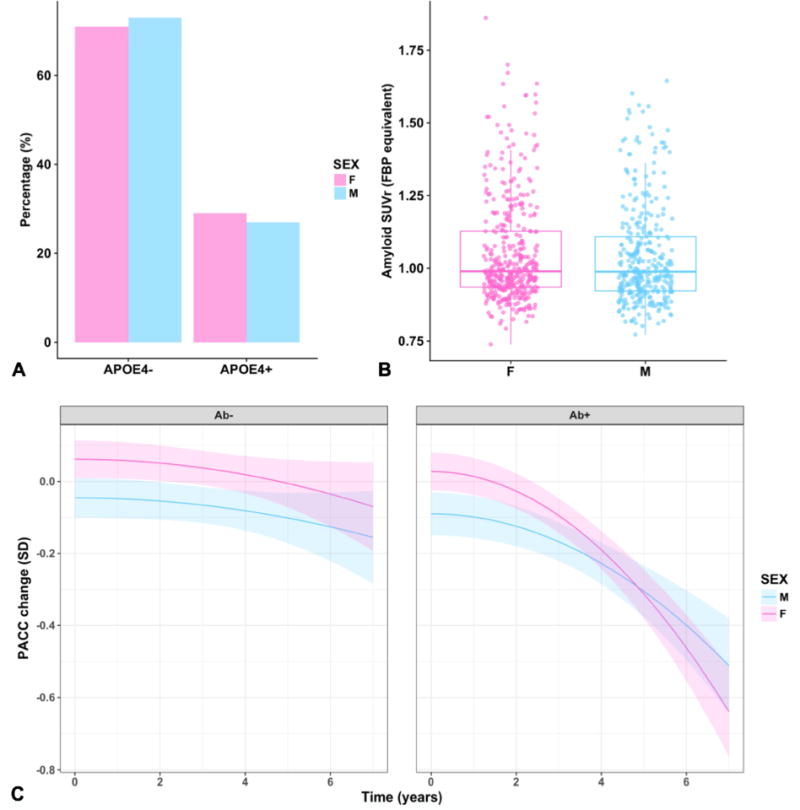
(A) Females and males display equal proportions of APOEε4 carrier status and (B) Aβ burden. (C) Represents decline in global cognition (as measured by the PACC-5) by Aβ and sex. These are model estimates from a continuous model of Aβ; high and low Aβ are represented by the first quartile (on the left) and third quartile (on the right) of Aβ along the continuous spectrum. Each line extends to the longest follow-up period within that group.
3.3. Longitudinal change in cognition
For the first set of mixed-effect models which looked at main effects of sex over time, a main effect of sex did not associate with cognitive decline after adjusting for covariates (p = 0.05; see Table 2 for model estimates of terms of interest, with full models to be found in Appendix D).
Table 2.
Summary of linear mixed models for PACC-5 change (only terms of interest included)
| Estimate | Std Error | t value | p value | |
|---|---|---|---|---|
| Model A: Sex | ||||
| Sex | 0.11 | 0.04 | 2.73 | 0.006 |
| Sex*time2 | − 0.003 | 0.002 | − 1.98 | 0.05 |
| Model B1: Aβ*Sex | ||||
| Aβ*time2 | − 0.04 | 0.01 | − 5.09 | < 0.001 |
| Sex*time2 | 0.03 | 0.01 | 2.58 | 0.01 |
| Aβ*Sex*time2 | − 0.03 | 0.01 | − 2.96 | 0.003 |
| Model B2: APOEε4*Sex | ||||
| APOEε4*time2 | − 0.01 | 0.003 | − 3.95 | < 0.001 |
| Sex*time2 | − 0.005 | 0.002 | − 2.30 | 0.02 |
| APOEε4*Sex*time2 | − 0.006 | 0.005 | 1.32 | 0.18 |
| Model C: Aβ*Sex*APOEε4 | ||||
| APOEε4*Sex*time2 | − 0.03 | 0.02 | − 1.43 | 0.15 |
| Aβ*Sex*time2 | − 0.04 | 0.02 | − 2.44 | 0.01 |
| Aβ*Sex*APOEε4*time2 | 0.04 | 0.02 | 1.37 | 0.17 |
| Model D: Aβ*Sex*Age | ||||
| Aβ*Sex*time2 | 0.33 | 0.12 | 2.70 | 0.007 |
| Aβ*Age*time2 | 0.002 | 0.001 | 1.54 | 0.12 |
| Sex*Age*time2 | 0.005 | 0.002 | 2.70 | 0.007 |
| Age*Aβ*Sex | 0.008 | 0.03 | 0.25 | 0.80 |
| Aβ*Sex*Age*time2 | − 0.005 | 0.002 | − 2.80 | 0.005 |
| Model: Comparisons across Aβstatus/APOEε4/Sex groups | ||||
| Aβ−/APOEε4−/M*Time2|Aβ−/APOEε4−/F*Time2 | 0.001 | 0.002 | 0.36 | 0.72 |
| Aβ−/APOEε4+/M*Time2|Aβ−/APOEε4+/F*Time2 | − 0.004 | 0.005 | − 0.78 | 0.43 |
| Aβ+/APOEε4−/M*Time2|Aβ+/APOEε4−/F*Time2 | − 0.009 | 0.005 | − 1.85 | 0.06 |
| Aβ+/APOEε4+/M*Time2|Aβ+/APOEε4+/F*Time2 | − 0.009 | 0.005 | − 2.04 | 0.04 |
Note: Aβ is the FBP SUVrequiv. Aβ+/− is formed according to the Aβ SUVr cut-off = 1.082. Esimates are unstandardized and the reference group for sex is female
For the next set of models looking at Aβ*sex and APOE*sex interactions over time on cognitive decline, females with elevated Aβ exhibited steeper cognitive decline than males with elevated Aβ over time (p = 0.003; see Figure 1C). A comparison between the Aβ main effects model and Aβ*sex interaction model showed that the latter model fit significantly better than the former, Log Likelihood Ratio (15, 19) = 185.57, p <0.001. By contrast, we did not find an interaction effect of sex and APOE on cognitive decline (p = 0.19).
For the final interaction model between Aβ*sex*APOE over time, a significant interaction did not exist on cognitive decline (p = 0.17). Post-hoc contrasts, however, suggested a weak effect of female APOEε4 carriers with high Aβstatus (Aβ+/APOEε4+/F) having steeper cognitive decline in comparison with male APOEε4 carriers with high Aβstatus (p = 0.04; see Figure 3). This effect, however, did not survive multiple comparison adjustment.
Figure 3.
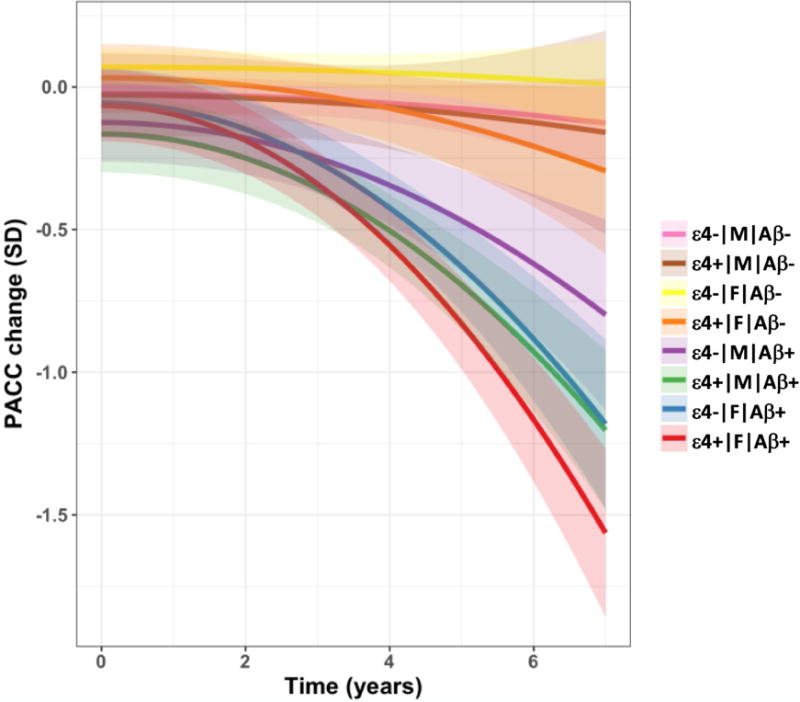
Decline in global cognition (as measured by the PACC-5) by APOEε4 status, sex and Aβ status (see legend for colours). These are model estimates from a factorial model with Aβ status represented by cut-off 1.082. Each line extends to the longest follow-up period within that group.
We next examined the effect of age on the interaction between Aβ and sex on cognitive decline, and found that females with higher Aβ displayed steeper cognitive decline after approximately 80 years of age in comparison with males (see Figure 2). This model did not fit better than a simpler model of Aβ*sex, Log Likelihood Ratio (17, 23) = 4.55, p = 0.60, and as such, we interpreted this finding with caution. We did not consider an interaction between sex, amyloid and education, as our models were not statistically powered to provide reliable estimates (given that only 15 males and 28 females with high Aβ had 12 or less years of education).
Figure 2.
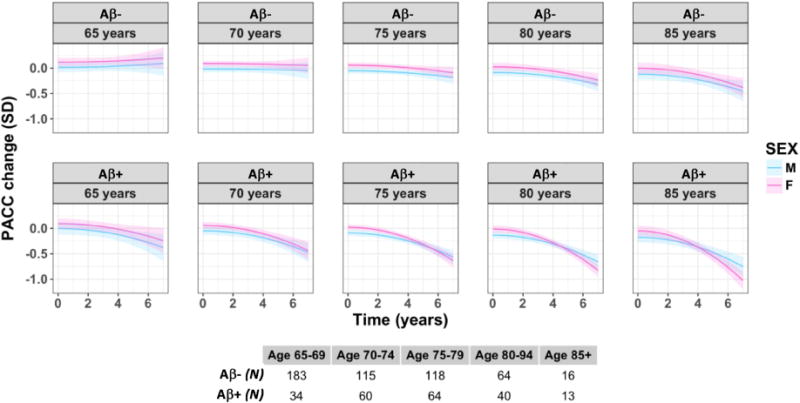
Decline in global cognition (as measured by the PACC-5) by Aβ and sex across the age span (females = pink, males = blue). These are model estimates from a continuous model of Aβ; high and low Aβ is represented by the first quartile (on the top) and third quartiles (on the bottom) of Aβ along the continuous spectrum. Age is also treated continuously in the model, with this visualization showing model estimates at the following ages: 65, 70, 75, 80, 85 years. Each line extends to the longest follow-up period within that group.
When considering each of the three studies in isolation, the same pattern of effects existed for the Aβ*sex*time interaction on cognitive decline. Due to statistical power constraints, however, many of these models did not meet the conventional threshold for statistical significance (see Appendix D). Models of Aβ*sex*time interactions were also conducted on each z-scored test within the PACC-5 (see Appendix A). We found that decline on list-learning delayed recall was significantly steeper in females than males for a given level of high Aβ (estimate = −0.07, SE = 0.02, t = −4.07, p < 0.001), with performance on Logical Memory delayed recall trending (estimate = −0.03, SE = 0.02, t = −1.91, p = 0.05). As list-learning tests were different across the cohorts (ADNI = ADAS-Cog Word Recall, AIBL = CVLT, HABS = FCSRT), we argue that these exploratory analyses warrant replication.
4. DISCUSSION
In a large cross-cohort dataset of clinically-normal older adults, females with elevated Aβ were found to decline in cognition more rapidly than males with a comparable level of Aβ. This mirrors recent work in ADNI reporting an interactive effect of sex and CSF Aβ42 on episodic memory decline [31]. We did not find an interactive effect of sex and APOE on cognitive decline, however, there was a weak effect of female APOEε4 carriers with higher Aβ demonstrating faster rates of cognitive decline in comparison with their male counterparts. There was no main effect of sex on cognitive decline, although females exhibited better cognitive performance at baseline, supporting the notion that females outperform males on tests of verbal memory tasks [32], which are key components of the PACC-5 [22].
The mechanism explaining the interaction between sex and Aβ remains unclear. In line with previous studies [33–35], sex was not associated with Aβ burden at baseline (as either a continuous or dichotomous variable), even in APOEε4 carriers. It is important to note that sex differences in amyloid load have been reported in relation to family history; for instance, maternal history of sporadic AD dementia influences Aβ-PET retention in normal subjects [36], and closer proximity to one’s parental estimated year of onset of sporadic AD is associated with elevated amyloid burden in female, but not male, subjects [37]. We did not measure family history in the current study, however, it is possible that co-varying for proximity to parental age at onset may highlight differences in amyloid burden between males and females.
An alternative possibility is that sex effects in relation to Aβ burden do exist, but perhaps at earlier ages, such as during menopause, which was not examined in the current study. Postmortem work shows extensive senile plaque build-up in women who are in the neurofibrillary stages I, II and III compared with men in similar stages, and particularly in those with APOEε4 [38]. Animal studies have also reported elevated risk for greater Aβ burden in females than males in a range of transgenic mouse models [39, 40]; much more so than sex differences in tauopathy [41]. Estrogen is often implicated in findings of sex differences in animal models [42], with ovariectomies increasing Aβ burden in female mice [43], and estrogen replacement reducing the risk of Aβ burden [44]. These findings mirror human studies suggesting that higher estradiol in females is associated with better memory performance [45], oophorectomies that occur prior to menopause increases risk of cognitive impairment in females [46], and epidemiological studies that show chronic (10+ years) use of hormone replacement therapy close to menopause may be a protective factor for AD risk [47]. Clinical trials of estrogen replacement, however, have been disappointing to date [48], highlighting the complex role that sex hormones may play in AD dementia risk [1]. We did not examine menopausal onset in the current study, nor did we measure use of hormone replacement therapy. It will be important for future studies to assess cognitive changes in relation to Aβ, APOE and sex prior to and during the full menopause phase.
Other interpretations should also be considered. If similar levels of high Aβ burden lead to steeper cognitive decline in women, this may imply a greater sensitivity in women to Aβ burden relative to men, which may be mediated by greater levels of tau and/or neurodegeneration in Aβ positive females. It is also possible that, despite similar levels of Aβ burden at baseline, females may accumulate Aβ at a faster rate, however, the literature has not comprehensively investigated this supposition. In one recent study, proximity to parental estimated year of onset for sporadic AD corresponded with higher Aβ burden cross-sectionally in females relative to males, but no evidence was found of sex differences in Aβ accumulation longitudinally [37].
As mentioned above, downstream mechanisms of Aβ burden, such as tauopathy and neurodegeneration, may occur to a greater extent in Aβ positive females than males, that would mirror our finding of greater cognitive decline among the same group. Mounting cross-sectional evidence suggests that females, particularly those with APOEε4, exhibit greater levels of tau pathology and neurodegeneration [33, 35, 49, 50]. Female APOEε4 carriers show greater levels of CSF total tau [33], hippocampal atrophy [51], cortical thinning [50], and lower intrinsic connectivity in the default network [49] in comparison with male carriers. However, other work, by Jack and colleagues [35] showed that in normal subjects, males have smaller hippocampal volumes in comparison with females, and Koran and colleagues [31] did not find an interaction between sex and CSF total tau on episodic memory decline in a model including normal, MCI and AD dementia patients. As such, further research should focus on the question of whether sex differences in tauopathy or neurodegeneration might drive risk for AD-related cognitive decline.
Unlike previous studies on clinical progression to mild cognitive impairment (MCI) or AD dementia [12, 33, 52], we did not find a sex-by-APOE interaction effect on cognitive decline. It is possible that previous APOE findings represent proxies for Aβ and/or neurodegeneration effects that are not feasibly measurable in epidemiological cohorts. Previous studies report that risk for clinical progression in female APOEε4 carriers exists largely within the specific 65-75 age range [12, 52], suggesting that cognitive decline would need to occur prior to this age. We were unable to accurately estimate cognitive decline in those below 65 years of age due to small sample sizes in that range, and so we may have failed to capture sex-APOE effects on cognition. A weak three-way sex-APOE-Aβ interaction was found, but this finding did not survive multiple comparison adjustment and thus requires replication.
Another departure from previous studies relates to the age at which sex effects appeared; we found cognitive decline was more likely at later decades, unlike epidemiological studies that report effects in those between 65-75 years [12]. It is possible that our finding represents a ‘survivor bias’ effect [53], such that sex-related cognitive decline in the oldest-old is a ‘second-wave’ risk. Individuals who have survived past factors that reduce the age of onset to AD dementia, such as APOE [54], and other factors that increase mortality, such as early male death from cardiovascular disease [55] may result in differential vulnerability to sex effects. As followup time did not differ in our study by sex it is unlikely that our findings are simply driven by the fact that women were followed for longer. Our age interactive model did not significantly explain more variance than simpler models, and so any interpretation of age effects should be considered with this caveat in mind.
Our study has several limitations. We investigated three cohorts of convenience: participants are primarily educated, of higher socioeconomic status, and are not very racially diverse compared to the general population. In addition, the AIBL study is enriched for APOEε4 carriers, thus increasing the proportion of carriers over the population prevalence rate [20]. As such, these cohorts may not reflect sex effects in the general population. In addition, we assumed similarity between cohorts, although demographically they were slightly different at baseline. In order to account for this, we processed Aβ-PET data with the same PET-pipeline, and scrutinized baseline and longitudinal cognitive performance to ensure similarity of cognitive performance. We also accounted for random cohort effects in our linear mixed effects models, and ran analyses within each cohort to confirm that the pattern of results was similar across the cohorts. As such, we argue that the advantage of combining cohorts allows idiosyncratic noise from within each cohort to be smoothed out. Nevertheless, these findings require replication in other large datasets, such as Mayo Clinic, the Framingham Heart Study, or even meta-analysis datasets such as the Amyloid Biomarker Study [56].
It is important to note that harmonizing across cohorts is a complex endeavor [57], particularly with regard to equating across neuropsychological test performance. As the PACC-5 composite included both overlapping and non-overlapping tests, we found similar baseline and slope variability across the cohorts. This does not negate the fact that biases may be introduced by including three non-overlapping cognitive tests to form the PACC-5 across the three cohorts. Our future aim will be to further harmonize across neuropsychological tests in order to probe within cognitive domains (e.g. episodic memory, executive function, processing speed, etc). Neuropsychological tests within each cognitive domain largely do not overlap between the cohorts, and as such, it will be necessary to implement item-response theory equating methods, similar to those applied in other cohort-harmonization studies [58]. We found that memory, in this case, performance from list-learning and story learning tests from the PACC-5, may be the domain most affected by the sex*Aβ interaction, however, as these tests do not overlap between the studies, our analyses are highly exploratory and need to be replicated in other cohorts.
One strength of our study involves our treatment of the different Aβ-PET tracers across the studies. In order to address differences in dynamic range, sensitivity and scaling between PiB and FBP Aβ-PET tracers, we employed a non-linear transformation mapping approach. We used this method to account for non-linearities that exist in the extreme ends of the PiB and FBP SUVr distributions. An assessment of cumulative distribution functions between raw and equivalent SUVrs showed that non-linear mapping preserves the distributional properties of the original SUVr much better than linear transformations (see Appendix B). Regardless, methods to accurately translate SUVrs across Aβ tracers is complex, and will require further refinement and testing in out-of-sample cohorts.
Determining sex-specific effects on rates of cognitive decline in the context of genetic risk and AD pathology will aid in more accurate detection of individuals most vulnerable to AD pathology, and further clarify recruitment and treatment approaches for AD clinical trials with reference to sex. We found that sex differences, both as a main effect and interacted with APOEε4, were not apparent on cognitive decline, however, females with high Aβ burden did show steeper cognitive decline trajectories in comparison to males with similarly high Aβ burden. Taken together, these findings suggest that sex effects may be salient in preclinical AD, when Aβ burden is apparent [1]. The mechanism underlying sex-specific sensitivity to Aβ, however, is yet to be elucidated. Taken together, these findings imply that the influence of sex and sex-specific risk factors on AD risk should be considered with respect to the cascade of events thought to underlie the development of AD dementia, and that the impact of sex is present during the preclinical stage of the disease. The effects observed in this study do, however, highlight the benefit of harmonizing across smaller datasets of clinically normal older adults to detect complex modifying effects in preclinical AD, which will become important at the scalable-level of large clinical trials, such as Anti-Amyloid Treatment in Asymptomatic Alzheimer’s Disease (A4) [18].
Research in context.
Systematic review: The authors reviewed the literature using Google Scholar and PubMed sources. Sex effects on cognitive decline within the context of Alzheimer’s disease biomarkers are not yet as widely published, however, there have been some publications on sex differences in dementia incidence and AD biomarker risk which the authors referred to. These relevant citations are appropriately cited.
Interpretation: Although the sexes did not differ on amyloid burden or APOE carrier status, our findings suggest that females decline faster in cognition for a given level of amyloid in comparison with males.
Future directions: In order to examine sex-related amyloid effects on cognition, it will be important to measure neurodegenerative and tauopathy associations. Further, cognitive domains should be investigated; it is possible that certain cognitive domains may be more affected in females than males for a given level of amyloid, however, this remains to be investigated.
Highlights.
Females are not more likely to exhibit steeper cognitive decline
Females are not more likely to exhibit high amyloid or carry APOEε4 than males
For a given level of amyloid-PET, females exhibit steeper cognitive decline than males
For a given level of APOE risk, females do not exhibit steeper decline than males
Acknowledgments
Some data used in the preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). The ADNI was launched in 2003 as a public-private partnership, led by Principal Investigator Michael W. Weiner, MD. The primary goal of ADNI has been to test whether serial magnetic resonance imaging (MRI), positron emission tomography (PET), other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of mild cognitive impairment (MCI) and early Alzheimer’s disease (AD). For up-to-date information, see www.adni-info.org.
For funding sources, RFB is funded with the NHMRC Dementia Research Fellowship (APP1105576). JR is funded by the Canadian Institutes of Health Research Postdoctoral Fellowship. PM is a full-time employee of Cogstate Ltd. SML receives financial support from the CRC for Mental Health. The Cooperative Research Centre (CRC) program is an Australian Government Initiative. HJ received funding from Alzheimer Nederland [WE.15-2014-06]. TH is funded by NIH (K01 040197). BJH is funded by the Belgian American Education Foundation (BAEF); Belgian National Fund for Scientific Research (FNRS); and Saint-Luc Foundation. YYL is funded with the NHMRC Dementia Research Fellowship (APP1111603). This work was supported with funding from the National Institutes of Health, including P01 AG036694 (Sperling and Johnson), P50 AG005134 (Sperling, Johnson, Hedden), K23 EB019023 (Sepulcre), K23 AG049087 (Chhatwal), K24 AG035007 (Sperling), K01 040197 (Hedden). This research was carried out in part at the Athinoula A. Martinos Center for Biomedical Imaging at the Massachusetts General Hospital, using resources provided by the Center for Functional Neuroimaging Technologies, P41EB015896, a P41 Biotechnology Resource Grant supported by the National Institute of Biomedical Imaging and Bioengineering (NIBIB), National Institutes of Health. This work also involved the use of instrumentation supported by the NIH Shared Instrumentation Grant Program and/or High-End Instrumentation Grant Program; specifically, grant numbers S10RR021110, S10RR023401, and S10RR023043. For ADNI, data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Financial disclosures
APS has been a paid consultant for Janssen Pharmaceuticals and Biogen. KVP has been a paid consultant for Biogen. Dr Rentz served as a consultant for Eli Lilly, Biogen Idec, Lundbeck Pharmaceuticals, and serves as a member of the Scientific Advisory Board for Neurotrack. Dr Johnson has served as paid consultant for Bayer, GE Healthcare, Janssen Alzheimer’s Immunotherapy, Siemens Medical Solutions, Genzyme, Novartis, Biogen, Roche, ISIS Pharma, AZTherapy, GEHC, Lundberg, and Abbvie. He is a site coinvestigator for Lilly/Avid, Pfizer, Janssen Immunotherapy, and Navidea. He has spoken at symposia sponsored by Janssen Alzheimer’s Immunotherapy and Pfizer. K. Johnson receives funding from NIH grants R01EB014894, R21 AG038994, R01 AG026484, R01 AG034556, P50 AG00513421, U19 AG10483, P01 AG036694, R13 AG042201174210, R01 AG027435, and R01 AG037497 and the Alzheimer’s Association grant ZEN-10-174210. Dr Sperling has served as a paid consultant for Abbvie, Biogen, Bracket, Genentech, Lundbeck, Roche, and Sanofi. She has served as a co- investigator for Avid, Eli Lilly, and Janssen Alzheimer Immunotherapy clinical trials. She has spoken at symposia sponsored by Eli Lilly, Biogen, and Janssen. R. Sperling receives research support from Janssen Pharmaceuticals, and Eli Lilly and Co. These relationships are not related to the content in the manuscript. She also receives research support from the following grants: P01 AG036694, U01 AG032438, U01 AG024904, R01 AG037497, R01 AG034556, K24 AG035007, P50 AG005134, U19 AG010483, R01 AG027435, Fidelity Biosciences, Harvard NeuroDiscovery Center, and the Alzheimer’s Association. Dr Chhatwal is funded by NIH (K23 AG049087)
APPENDIX A: Density plots of baseline PACC performance and longitudinal PACC slopes for each cohort

APPENDIX B: Visualisation of the Amyloid-PET SUVr distributions
Visualisation of the Amyloid-PET SUVr distributions of (A) raw SUVrs across the three cohorts (AIBL and HABS use PiB, while ADNI uses Florebetapir), (B) linearly transformed SUVrs, and (C) our method of non-linearly transformed SUVrs of PiB to be equated with the Florbetapir distribution

Non-linear method:
We utilize baseline Aβ-PET datasets of clinically-normal participants from ADNI (FBP), AIBL (PIB), and HABS (PIB). Summary measures were computed via a PET only pipeline with direct spatial normalization of PET data to MNI space. Measurements were made as SUVr using a cortical composite (FLR) with a whole-cerebellum reference region. Cross-sample mapping was performed via 10,000 bootstrapped samples of PiB and FBP matched for age, sex and apolipoprotein ε4 (APOEε4) status. A transfer function for each bootstrapped sample was generated via smoothed cumulative distribution functions. An SUVr equivalency map and confidence intervals were extracted across these 10,000 fits using sliding-window PCA.
APPENDIX C. Model estimates for each mixed-effects model in the combined group, followed by the model estimates within each cohort for the sex*amyloid model
| Estimate | Std Error | t value | p value | |
|---|---|---|---|---|
| Model: Sex | ||||
| Time2 | 0.03 | 0.01 | 2.70 | <0.001 |
| Baseline PACC-5 | −0.21 | 0.03 | −6.18 | <0.001 |
| Age | −0.01 | 0.003 | −3.07 | 0.01 |
| Education | 0.01 | 0.007 | 1.95 | 0.07 |
| Sex | 0.11 | 0.04 | 2.74 | 0.003 |
| Cohort | −0.03 | 0.02 | −1.28 | 0.42 |
| Cohort*time2 | 0.02 | 0.001 | 9.26 | <0.001 |
| Baseline PACC-5*time2 | 0.003 | 0.002 | 1.68 | 0.06 |
| Age*time2 | −0.001 | 0.0001 | −6.25 | <0.001 |
| Education*time2 | 0.0004 | 0.0003 | 1.12 | 0.24 |
| Sex*time2 | −0.003 | 0.002 | −1.99 | 0.05 |
| Model: Aβ*Sex | ||||
| Time2 | 0.07 | 0.01 | 4.59 | <0.001 |
| Baseline PACC-5 | −0.22 | 0.01 | −6.62 | <0.001 |
| Age | −0.009 | 0.003 | −2.90 | 0.02 |
| Education | 0.02 | 0.007 | 2.32 | 0.02 |
| Aβ | −0.31 | 0.17 | −1.79 | 0.07 |
| Sex | 0.02 | 0.24 | 0.06 | 0.95 |
| Cohort | −0.03 | 0.02 | −1.29 | 0.41 |
| Cohort*time2 | 0.01 | 0.001 | 8.90 | <0.001 |
| Baseline PACC-5*time2 | 0.001 | 0.002 | 0.82 | 0.41 |
| Age*time2 | −0.0008 | 0.0002 | −4.95 | <0.001 |
| Education*time2 | 0.0002 | 0.0004 | 0.62 | 0.54 |
| Aβ*Sex | 0.10 | 0.23 | 0.44 | 0.66 |
| Aβ*time2 | −0.04 | 0.01 | −5.09 | <0.001 |
| Sex*time2 | 0.03 | 0.01 | 2.58 | 0.01 |
| Aβ*Sex*time2 | −0.03 | 0.01 | −2.96 | 0.003 |
| Model: APOEε4*Sex | ||||
| Time2 | 0.05 | 0.01 | 3.51 | <0.001 |
| Baseline PACC-5 | −0.21 | 0.03 | −5.98 | <0.001 |
| Age | −0.01 | 0.003 | −2.97 | 0.003 |
| Education | 0.02 | 0.01 | 2.11 | 0.04 |
| APOEε4 | −0.03 | 0.07 | −0.48 | 0.63 |
| Sex | 0.11 | 0.05 | 2.24 | 0.03 |
| Cohort | −0.03 | 0.02 | −1.36 | 0.40 |
| Cohort*time2 | 0.01 | 0.001 | 9.24 | <0.001 |
| Baseline PACC-5*time2 | 0.003 | 0.002 | 1.59 | 0.11 |
| Age*time2 | −0.001 | 0.0001 | −6.66 | <0.001 |
| Education*time2 | 0.0003 | 0.0003 | 0.83 | 0.40 |
| APOEε4*time2 | −0.01 | 0.004 | −3.95 | 0.001 |
| Sex*time2 | −0.006 | 0.003 | −2.30 | 0.02 |
| APOEε4*Sex*time2 | 0.006 | 0.005 | 1.32 | 0.19 |
| Model: Aβ*Sex*APOEε4 | ||||
| Time2 | 0.06 | 0.02 | 3.36 | <0.001 |
| Baseline PACC-5 | −0.16 | 0.03 | −5.47 | <0.001 |
| Age | −0.009 | 0.003 | −3.15 | 0.002 |
| APOEε4 | 0.45 | 0.34 | 1.31 | 0.19 |
| Education | 0.01 | 0.006 | 2.04 | 0.04 |
| Aβ | −0.03 | 0.21 | −0.13 | 0.90 |
| Sex | 0.31 | 0.29 | 1.09 | 0.28 |
| Cohort | −0.03 | 0.02 | −1.23 | 0.43 |
| Cohort*time2 | 0.01 | 0.001 | 8.33 | <0.001 |
| Baseline PACC-5*time2 | 0.0003 | 0.002 | 0.15 | 0.88 |
| Age*time2 | −0.0007 | 0.0002 | −4.42 | <0.0001 |
| APOEε4*time2 | 0.02 | 0.02 | 0.99 | 0.32 |
| Education*time2 | 0.0003 | 0.0003 | 0.91 | 0.36 |
| Aβ*time2 | −0.03 | 0.01 | −2.24 | 0.03 |
| Sex*time2 | 0.04 | 0.02 | 2.30 | 0.02 |
| Aβ*Sex*time2 | −0.04 | 0.02 | −2.44 | 0.01 |
| Aβ*APOEε4*time2 | −0.02 | 0.02 | −1.17 | 0.23 |
| Sex*APOEε4*time2 | −0.03 | 0.02 | −1.43 | 0.15 |
| APOEε4*Aβ*Sex | 0.42 | 0.41 | 1.03 | 0.30 |
| Aβ*Sex*APOEε4*time2 | 0.03 | 0.02 | 1.37 | 0.17 |
| Model: Aβ*Sex*Age | ||||
| Time2 | 0.19 | 0.09 | 2.14 | 0.03 |
| Baseline PACC-5 | −0.16 | 0.03 | −5.73 | <0.001 |
| Age | −0.001 | 0.02 | −0.04 | 0.96 |
| Education | 0.01 | 0.006 | 2.10 | 0.04 |
| Aβ | 0.42 | 1.79 | 0.24 | 0.81 |
| Sex | 0.51 | 2.49 | 0.20 | 0.84 |
| Cohort | −0.03 | 0.02 | −1.30 | 0.42 |
| Cohort*time2 | 0.01 | 0.001 | 8.89 | <0.001 |
| Baseline PACC-5*time2 | 0.0009 | 0.002 | 0.52 | 0.60 |
| Age*time2 | −0.002 | 0.001 | −1.91 | 0.06 |
| Education*time2 | 0.0001 | 0.0003 | 0.45 | 0.65 |
| Aβ*time2 | −0.17 | 0.09 | −2.41 | 0.02 |
| Sex*time2 | −0.31 | 0.13 | −2.41 | 0.01 |
| Aβ*Sex*time2 | 0.32 | 0.12 | 2.60 | 0.009 |
| Aβ*Age *time2 | 0.002 | 0.001 | 1.54 | 0.12 |
| Sex*Age*time2 | 0.004 | 0.002 | 2.57 | 0.01 |
| Age*Aβ*Sex | 0.008 | 0.03 | 0.25 | 0.80 |
| Aβ*Sex*Age* time2 | −0.005 | 0.002 | −2.80 | 0.005 |
| Model: Comparisons across Aβ status/APOEε4/Sex groups | ||||
| Aβ−/APOEε4−/M*Time2 vs Aβ−/APOEε4−/F*Time2 | 0.001 | 0.002 | 0.36 | 0.72 |
| Aβ−/APOEε4+/M*Time2 vs Aβ−/APOEε4+/F*Time2 | −0.004 | 0.005 | −0.78 | 0.43 |
| Aβ+/APOEε4−/M*Time2 vs Aβ+/APOEε4−/F*Time2 | −0.009 | 0.005 | −1.85 | 0.06 |
| Aβ+/APOEε4+/M*Time2 vs Aβ+/APOEε4+/F*Time2 | −0.009 | 0.005 | −2.04 | 0.04 |
Note: Aβ is the FBP SUVrequiv. Aβ+/− is formed according to the Aβ SUVr cut off = 1.082
Comparison of Aβ-amyloid*sex model within each cohort
| Coefficients |
Study
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| ADNI | AIBL | HABS | |||||||
| Estimate | Conf. Int. | p-value | Estimate | Conf. Int. | p-value | Estimate | Conf. Int. | p-value | |
| Fixed Parts | |||||||||
| Intercept | 0.69 | −0.11-1.49 | .090 | 0.52 | −0.66-1.71 | .390 | 0.77 | −0.13-1.67 | .095 |
| I(timc_yrs^2) | 0.12 | 0.05-0.19 | .001 | 0.04 | 0.00-0.08 | .030 | 0.12 | 0.08-0.16 | <.001 |
| PACC_bl | −0.20 | −0.29-−0.10 | <.001 | −0.21 | −0.37-−0.06 | .008 | −0.12 | −0.21-−0.03 | .013 |
| YrsEd | 0.01 | −0.01-0.03 | .321 | 0.01 | −0.04-0.05 | .795 | 0.01 | −0.01-0.03 | .195 |
| Age | −0.01 | −0.02-−0.00 | .005 | −0.01 | −0.02-0.01 | .417 | −0.01 | −0.01-0.00 | .245 |
| SEXF | 0.08 | −0.52-0.68 | .796 | 0.18 | −0.91-1.27 | .749 | −0.03 | −0.72-0.67 | .940 |
| Amyloid | 0.02 | −0.42-0.46 | .923 | −0.26 | −0.93-0.41 | .451 | −0.66 | −1.20-−0.12 | .017 |
| I(timc_yrs^2):PACC_bl | 0.01 | 0.00-0.02 | .046 | −0.00 | −0.01-0.00 | .633 | −0.00 | −0.00-0.00 | .994 |
| I(timc_yrs^2):YrsEd | 0.00 | −0.00-0.00 | .126 | 0.00 | 0.00-0.00 | .015 | −0.00 | −0.00-0.00 | .096 |
| I(timc_yrs^2):Agc | −0.00 | −0.00-−0.00 | .004 | −0.00 | −0.00-0.00 | .269 | −0.00 | −0.00-−0.00 | <.001 |
| I(timc_yrs^2):SEXF | 0.01 | −0.04-0.06 | .665 | 0.01 | −0.02-0.05 | .550 | 0.02 | −0.01-0.05 | .140 |
| I(timc_yrs^2):Amyloid | −0.08 | −0.12-−0.04 | <.001 | −0.05 | −0.07-−0.02 | <.001 | −0.03 | −0.05-−0.01 | .005 |
| SEXF:Amyloid | 0.00 | −0.57-0.57 | .999 | −0.08 | −1.11-0.95 | .883 | 0.14 | −0.52-0.80 | .682 |
| I(timc_yrs^2): SEXF: Amyloid | −0.00 | −0.05-0.04 | .855 | −0.01 | −0.04-0.02 | .550 | −0.03 | −0.06-−0.00 | .045 |
| Random Parts | |||||||||
| σ2 | 0.214 | 0.131 | 0.143 | ||||||
| τ00,ID | 0.116 | 0.189 | 0.119 | ||||||
| NID | 330 | 160 | 265 | ||||||
| ICCID | 0.351 | 0.590 | 0.455 | ||||||
|
| |||||||||
| Observations | 1102 | 510 | 1164 | ||||||
| R2/Ω02 | .613/.591 | .761/.749 | .653/.642 | ||||||
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu) and Australian Imaging Biomarkers and Lifestyle study of ageing database (aibl.loni.usc.edu). As such, the investigators within the ADNI and AIBL contributed to the design and implementation of ADNI and AIBL and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI and AIBL investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf and https://aibl.csiro.au/about/aibl-research-team/
References
- 1.Mielke MM, Vemuri P, Rocca WA. Clinical epidemiology of Alzheimer’s disease: assessing sex and gender differences. Clin Epidemiol. 2014;6:37–48. doi: 10.2147/CLEP.S37929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andersen K, Launer LJ, Dewey ME, Letenneur L, Ott A, Copeland JRM, et al. Gender differences in the incidence of AD and vascular dementia: The EURODEM Studies. Neurology. 1999;53:1992. doi: 10.1212/wnl.53.9.1992. [DOI] [PubMed] [Google Scholar]
- 3.Chêne G, Beiser A, Au R, Preis SR, Wolf PA, Dufouil C, et al. Gender and incidence of dementia in the Framingham Heart Study from mid-adult life. Alzheimer’s & Dementia. 2015;11:310–20. doi: 10.1016/j.jalz.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fiest KM, Roberts JI, Maxwell CJ, Hogan DB, Smith EE, Frolkis A, et al. The Prevalence and Incidence of Dementia Due to Alzheimer’s Disease: a Systematic Review and Meta-Analysis. Canadian Journal of Neurological Sciences/Journal Canadien des Sciences Neurologiques. 2016;43:S51–S82. doi: 10.1017/cjn.2016.36. [DOI] [PubMed] [Google Scholar]
- 5.Perera G, Pedersen L, Ansel D, Alexander M, Arrighi HM, Avillach P, et al. Dementia prevalence and incidence in a federation of European Electronic Health Record databases: The European Medical Informatics Framework resource. Alzheimer’s & Dementia. 2018;14:130–9. doi: 10.1016/j.jalz.2017.06.2270. [DOI] [PubMed] [Google Scholar]
- 6.Barnes L, Wilson R, Schneider J, Bienias J, Evans D, Bennett D. Gender, cognitive decline, and risk of AD in older persons. Neurology. 2003;60:1777–81. doi: 10.1212/01.wnl.0000065892.67099.2a. [DOI] [PubMed] [Google Scholar]
- 7.Ferreira L, Ferreira Santos-Galduróz R, Ferri CP, Fernandes Galduróz JC. Rate of cognitive decline in relation to sex after 60 years-of-age: A systematic review. Geriatrics & gerontology international. 2014;14:23–31. doi: 10.1111/ggi.12093. [DOI] [PubMed] [Google Scholar]
- 8.Hyman BT, Gomez-Isla T, Briggs M, Chung H, Nichols S, Kohout F, et al. Apolipoprotein E and cognitive change in an elderly population. Annals of Neurology. 1996;40:55–66. doi: 10.1002/ana.410400111. [DOI] [PubMed] [Google Scholar]
- 9.Mortensen EL, Høgh P. A gender difference in the association between APOE genotype and age-related cognitive decline. Neurology. 2001;57:89–95. doi: 10.1212/wnl.57.1.89. [DOI] [PubMed] [Google Scholar]
- 10.Swan GE, Lessov-Schlaggar CN, Carmelli D, Schellenberg GD, Rue AL. Apolipoprotein E ε4 and Change in Cognitive Functioning in Community-Dwelling Older Adults. Journal of Geriatric Psychiatry and Neurology. 2005;18:196–201. doi: 10.1177/0891988705281864. [DOI] [PubMed] [Google Scholar]
- 11.McCarrey AC, An Y, Kitner-Triolo MH, Ferrucci L, Resnick SM. Sex differences in cognitive trajectories in clinically normal older adults. Psychology and aging. 2016;31:166. doi: 10.1037/pag0000070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Neu SC, Pa J, Kukull W, Beekly D, Kuzma A, Gangadharan P, et al. Apolipoprotein E Genotype and Sex Risk Factors for Alzheimer Disease: A Meta-analysis. JAMA neurology. 2017;74:1178–89. doi: 10.1001/jamaneurol.2017.2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jack CR, Wiste HJ, Weigand SD, Knopman DS, Mielke MM, Vemuri P, et al. Different definitions of neurodegeneration produce similar amyloid/neurodegeneration biomarker group findings. Brain. 2015 doi: 10.1093/brain/awv283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lim YY, Mormino EC, Initiative FtAsDN APOE genotype and early β-amyloid accumulation in older adults without dementia. Neurology. 2017;89:1028–34. doi: 10.1212/WNL.0000000000004336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lim YY, Ellis KA, Ames D, Darby D, Harrington K, Martins RN, et al. Aβ amyloid, cognition, and APOE genotype in healthy older adults. Alzheimer Dem. 2013;9:538–45. doi: 10.1016/j.jalz.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 16.Mormino EC, Betensky RA, Hedden T, Schultz AP, Ward A, Huijbers W, et al. Amyloid and APOE ε4 interact to influence short-term decline in preclinical Alzheimer disease. Neurology. 2014;82:1760–7. doi: 10.1212/WNL.0000000000000431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Villemagne VL, Burnham S, Bourgeat P, Brown B, Ellis KA, Salvado O, et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. 2013;12:357–67. doi: 10.1016/S1474-4422(13)70044-9. [DOI] [PubMed] [Google Scholar]
- 18.Sperling RA, Rentz DM, Johnson KA, Karlawish J, Donohue M, Salmon DP, et al. The A4 Study: Stopping AD Before Symptoms Begin? Science Translational Medicine. 2014;6:228fs13. doi: 10.1126/scitranslmed.3007941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aisen PS, Petersen RC, Donohue MC, Gamst A, Raman R, Thomas RG, et al. Clinical Core of the Alzheimer’s Disease Neuroimaging Initiative: progress and plans. Alzheimer’s & Dementia. 2010;6:239–46. doi: 10.1016/j.jalz.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ellis KA, Bush A, Darby D, De Fazio D, Foster J, Hudson P, et al. The Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging: methodology and baseline characteristics of 1112 individuals recruited for a longitudinal study of Alzheimer’s disease. Int Psychogeriatr. 2009;21:672–87. doi: 10.1017/S1041610209009405. [DOI] [PubMed] [Google Scholar]
- 21.Dagley A, LaPoint M, Huijbers W, Hedden T, McLaren DG, Chatwal JP, et al. Harvard Aging Brain Study: Dataset and accessibility. NeuroImage. 2017;144:255–8. doi: 10.1016/j.neuroimage.2015.03.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Papp KV, Rentz DM, Orlovsky I, Sperling RA, Mormino EC. Optimizing the preclinical Alzheimer’s cognitive composite with semantic processing: The PACCC5. Alzheimer’s and Dementia: Translational Research and Clinical Interventions. 2017 doi: 10.1016/j.trci.2017.10.004. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Donohue MC, Sperling RA, Salmon DP, Rentz DM, Raman R, Thomas RG, et al. The preclinical alzheimer cognitive composite: Measuring amyloid-related decline. JAMA Neurology. 2014;71:961–70. doi: 10.1001/jamaneurol.2014.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burnham SC, Bourgeat P, Doré V, Savage G, Brown B, Laws S, et al. Clinical and cognitive trajectories in cognitively healthy elderly individuals with suspected non-Alzheimer’s disease pathophysiology (SNAP) or Alzheimer’s disease pathology: a longitudinal study. The Lancet Neurology. 2016;15:1044–53. doi: 10.1016/S1474-4422(16)30125-9. [DOI] [PubMed] [Google Scholar]
- 25.Mormino EC, Papp KV, Rentz DM, et al. Heterogeneity in suspected non–alzheimer disease pathophysiology among clinically normal older individuals. JAMA Neurology. 2016;73:1185–91. doi: 10.1001/jamaneurol.2016.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rowe CC, Ellis K, Rimajova M, Bourgeat P, Pike KE, Jones G, et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiology of Aging. 2010;31:1275–83. doi: 10.1016/j.neurobiolaging.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 27.Landau SM, Mintun MA, Joshi AD, Koeppe RA, Petersen RC, Aisen PS, et al. Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Annals of neurology. 2012;72:578–86. doi: 10.1002/ana.23650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fischl B, Sereno MI, Tootell RB, Dale AM. High-resolution intersubject averaging and a coordinate system for the cortical surface. Human brain mapping. 1999;8:272–84. doi: 10.1002/(SICI)1097-0193(1999)8:4<272::AID-HBM10>3.0.CO;2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Donohue MC, Sperling RA, Petersen R, Sun C-K, Weiner MW, Aisen PS. Association Between Elevated Brain Amyloid and Subsequent Cognitive Decline Among Cognitively Normal Persons. JAMA. 2017;317:2305–16. doi: 10.1001/jama.2017.6669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Buckley RF, Schultz AP, Hedden T, Papp KV, Hanseeuw BJ, Marshall G, et al. Functional network integrity presages cognitive decline in preclinical Alzheimer disease. Neurology. 2017;89:29–37. doi: 10.1212/WNL.0000000000004059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koran MEI, Wagener M, Hohman TJ. Sex differences in the association between AD biomarkers and cognitive decline. Brain Imaging and Behavior. 2017;11:205–13. doi: 10.1007/s11682-016-9523-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van Hooren S, Valentijn A, Bosma H, Ponds R, Van Boxtel M, Jolles J. Cognitive functioning in healthy older adults aged 64–81: a cohort study into the effects of age, sex, and education. Aging, Neuropsychology, and Cognition. 2007;14:40–54. doi: 10.1080/138255890969483. [DOI] [PubMed] [Google Scholar]
- 33.Altmann A, Tian L, Henderson VW, Greicius MD. Sex modifies the APOE-related risk of developing Alzheimer disease. Annals of neurology. 2014;75:563–73. doi: 10.1002/ana.24135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mielke MM, Wiste HJ, Weigand SD, Knopman DS, Lowe VJ, Roberts RO, et al. Indicators of amyloid burden in a population-based study of cognitively normal elderly. Neurology. 2012;79:1570–7. doi: 10.1212/WNL.0b013e31826e2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jack CR, Wiste HJ, Weigand SD, Knopman DS, Vemuri P, Mielke MM, et al. Age, sex, and APOE ε4 effects on memory, brain structure, and β-amyloid across the adult life span. JAMA Neurology. 2015;72:511–9. doi: 10.1001/jamaneurol.2014.4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maye JE, Betensky RA, Gidicsin CM, Locascio J, Becker JA, Pepin L, et al. Maternal dementia age at onset in relation to amyloid burden in non-demented elderly offspring. Neurobiology of aging. 2016;40:61–7. doi: 10.1016/j.neurobiolaging.2015.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Villeneuve S, Vogel JW, Gonneaud J, Binette AP, Rosa-Neto P, Gauthier S, et al. Proximity to Parental Symptom Onset and Amyloid-β Burden in Sporadic Alzheimer Disease. JAMA neurology. 2018 doi: 10.1001/jamaneurol.2017.5135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Corder EH, Ghebremedhin E, Taylor MG, Thal DR, Ohm TG, Braak H. The Biphasic Relationship between Regional Brain Senile Plaque and Neurofibrillary Tangle Distributions: Modification by Age, Sex, and APOE Polymorphism. Annals of the New York Academy of Sciences. 2004;1019:24–8. doi: 10.1196/annals.1297.005. [DOI] [PubMed] [Google Scholar]
- 39.Callahan MJ, Lipinski WJ, Bian F, Durham RA, Pack A, Walker LC. Augmented Senile Plaque Load in Aged Female β-Amyloid Precursor Protein-Transgenic Mice. The American Journal of Pathology. 2001;158:1173–7. doi: 10.1016/s0002-9440(10)64064-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Howlett DR, Richardson JC, Austin A, Parsons AA, Bate ST, Davies DC, et al. Cognitive correlates of Aβ deposition in male and female mice bearing amyloid precursor protein and presenilin-1 mutant transgenes. Brain Research. 2004;1017:130–6. doi: 10.1016/j.brainres.2004.05.029. [DOI] [PubMed] [Google Scholar]
- 41.Hirata-Fukae C, Li H-F, Hoe H-S, Gray AJ, Minami SS, Hamada K, et al. Females exhibit more extensive amyloid, but not tau, pathology in an Alzheimer transgenic model. Brain Research. 2008;1216:92–103. doi: 10.1016/j.brainres.2008.03.079. [DOI] [PubMed] [Google Scholar]
- 42.Yue X, Lu M, Lancaster T, Cao P, Honda S-I, Staufenbiel M, et al. Brain estrogen deficiency accelerates Aβ plaque formation in an Alzheimer’s disease animal model. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:19198–203. doi: 10.1073/pnas.0505203102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zheng H, Xu H, Uljon S, Gross R, Hardy K, Gaynor J, et al. Modulation of Aβ peptides by estrogen in mouse models. Journal of neurochemistry. 2002;80:191–6. doi: 10.1046/j.0022-3042.2001.00690.x. [DOI] [PubMed] [Google Scholar]
- 44.Huang J, Guan H, Booze RM, Eckman CB, Hersh LB. Estrogen regulates neprilysin activity in rat brain. Neuroscience Letters. 2004;367:85–7. doi: 10.1016/j.neulet.2004.05.085. [DOI] [PubMed] [Google Scholar]
- 45.Rentz DM, Weiss BK, Jacobs EG, Cherkerzian S, Klibanski A, Remington A, et al. Sex differences in episodic memory in early midlife: Impact of reproductive aging. Menopause. 2017;24:400–8. doi: 10.1097/GME.0000000000000771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rocca WA, Bower JH, Maraganore DM, Ahlskog JE, Grossardt BR, de Andrade M, et al. Increased risk of cognitive impairment or dementia in women who underwent oophorectomy before menopause. Neurology. 2007;69:1074–83. doi: 10.1212/01.wnl.0000276984.19542.e6. [DOI] [PubMed] [Google Scholar]
- 47.Shao H, Breitner JCS, Whitmer RA, Wang J, Hayden K, Wengreen H, et al. Hormone therapy and Alzheimer disease dementia: New findings from the Cache County Study. Neurology. 2012;79:1846–52. doi: 10.1212/WNL.0b013e318271f823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Henderson VW. Alzheimer’s disease: Review of hormone therapy trials and implications for treatment and prevention after menopause. The Journal of Steroid Biochemistry and Molecular Biology. 2014;142:99–106. doi: 10.1016/j.jsbmb.2013.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Damoiseaux JS, Seeley WW, Zhou J, Shirer WR, Coppola G, Karydas A, et al. Gender Modulates the APOE ε4 Effect in Healthy Older Adults: Convergent Evidence from Functional Brain Connectivity and Spinal Fluid Tau Levels. The Journal of Neuroscience. 2012;32:8254–62. doi: 10.1523/JNEUROSCI.0305-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sampedro F, Vilaplana E, De Leon MJ, Alcolea D, Pegueroles J, Montal V, et al. APOE-by-sex interactions on brain structure and metabolism in healthy elderly controls. Oncotarget. 2015;6:26663. doi: 10.18632/oncotarget.5185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Holland D, Desikan RS, Dale AM, McEvoy LK. Higher Rates of Decline for Women and Apolipoprotein E ε4 Carriers. American Journal of Neuroradiology. 2013;34:2287–93. doi: 10.3174/ajnr.A3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Farrer LA, Cupples L, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein e genotype and alzheimer disease: A meta-analysis. JAMA. 1997;278:1349–56. [PubMed] [Google Scholar]
- 53.Domingue BW, Belsky DW, Harrati A, Conley D, Weir DR, Boardman JD. Mortality selection in a genetic sample and implications for association studies. International Journal of Epidemiology. 2017;46:1285–94. doi: 10.1093/ije/dyx041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Blacker D, Haines JL, Rodes L, Terwedow H, Go RCP, Harrell LE, et al. ApoE-4 and Age at Onset of Alzheimer’s Disease: The NIMH Genetics Initiative. Neurology. 1997;48:139–47. doi: 10.1212/wnl.48.1.139. [DOI] [PubMed] [Google Scholar]
- 55.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Heart disease and stroke statistics—2016 update. Circulation. 2016;133:e38–e360. doi: 10.1161/CIR.0000000000000350. [DOI] [PubMed] [Google Scholar]
- 56.Jansen WJ, Ossenkoppele R, Tijms BM, Fagan AM, Hansson O, Klunk WE, et al. Association of Cerebral Amyloid-β Aggregation With Cognitive Functioning in Persons Without Dementia. JAMA psychiatry. 2018;75:84–95. doi: 10.1001/jamapsychiatry.2017.3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gross AL, Sherva R, Mukherjee S, Newhouse S, Kauwe JSK, Munsie LM, et al. Calibrating Longitudinal Cognition in Alzheimer’s Disease Across Diverse Test Batteries and Datasets. Neuroepidemiology. 2014;43:194–205. doi: 10.1159/000367970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gross AL, Hassenstab JJ, Johnson SC, Clark LR, Resnick SM, Kitner-Triolo M, et al. A classification algorithm for predicting progression from normal cognition to mild cognitive impairment across five cohorts: The preclinical AD consortium. Alzheimer’s & Dementia: Diagnosis, Assessment & Disease Monitoring. 2017;8:147–55. doi: 10.1016/j.dadm.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]