

Abstract
Although dissolution dynamic nuclear polarization is a robust technique to significantly increase magnetic resonance signal, the short T1 relaxation time of most 13C-nuclei limits the timescale of hyperpolarized experiments. To address this issue, we have characterized a non-synthetic approach to extend the hyperpolarized lifetime of 13C-nuclei in close proximity to solvent-exchangeable protons. Protons exhibit stronger dipolar relaxation than deuterium, so dissolving these compounds in D2O to exchange labile protons with solvating deuterons results in longer-lived hyperpolarization of the 13C-nucleus 2-bonds away. 13C T1 and T2 times were longer in D2O versus H2O for all molecules in this study. This phenomenon can be utilized to improve hyperpolarized signal-to-noise ratio as a function of longer T1, and enhanced spectral and imaging resolution via longer T2.
Keywords: DNP, lifetime, T1, T2
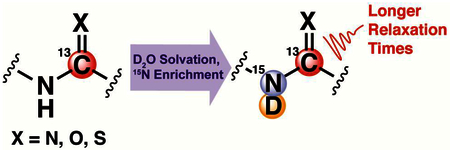
1. Introduction
A major limitation to magnetic resonance imaging (MRI) is low sensitivity, resulting in long acquisition times.1 Thus, most MRI acquisitions are restricted to endogenous 1H-nuclei in water, taking advantage of the prevalence of water in biological tissues and the high natural abundance (99.98%) and sensitivity of 1H, which scales with the nuclear gyromagnetic ratio (γ)2. This is in stark contrast to the MRI-sensitive isotope of carbon, 13C (natural abundance: 1.11%, γ13C / γ1H ≈1/4). Nuclear hyperpolarization addresses the low sensitivity inherent to MRI and facilitates detection of non-1H nuclei by increasing the net alignment of nuclear spins with an external magnetic field. Dissolution dynamic nuclear polarization is the preferred method of hyperpolarization for biological applications, in which non-equilibrium nuclear spin populations are achieved through spin polarization transfer from unpaired electrons to atomic nuclei at <1K.3,4 Dissolution is performed after sufficient hyperpolarization is attained, during which superheated buffer dissolves and ejects the hyperpolarized (HP) compound for experimental use. This technique typically yields 105-fold signal enhancement, facilitating real-time detection of biochemical processes on exogenously administered HP compounds. HP MRI has been applied to various pathologies5–7 but [1-13C]-pyruvate is the only clinically translated probe to date,8–10 partially due to the long spin-lattice relaxation time (T1) of the carbon-1 resonance which dictates the HP lifetime.11
Protium (1H) is a source of dipolar relaxation12,13 and previous reports demonstrate that protium can drastically shorten 13C/15N T1 relaxation times from up to 3-bonds away.14–19 Dipolar relaxation can be reduced by replacing protium with deuterium (2H), usually involving the synthesis of deuterium labeled analogues that can be expensive, laborious, and impractical for most laboratories. Addressing this limitation, other groups have extended 15N and 31P T1 times by exchanging directly-bound or 2-bond protons with deuterons by simply dissolving the compound in D2 O,20,21 but these nuclei have limited applications in biochemical systems.
To facilitate in vitro and in vivo visualization of additional 13C-labeled HP probes, our laboratory has characterized a non-synthetic method to significantly extend the T1 and T2 of 13C-nuclei adjacent to nitrogen atoms with solvent-exchangeable protons, and we demonstrate that this technique can be used to improve in vivo SNR as a function of increased T1. Here, we replace solvent-exchangeable protons with deuterons and investigate its effect on 13C-relaxation across 2-bonds. We expect the relative dipolar relaxation rate on 13C to scale with γ,19 and we hypothesize that this exchange creates an environment with weaker dipolar relaxation (γ 1H / γ2H ≈6.5) that increases the spin-lattice and spin-spin (T2) relaxation times of nearby 13C-nuclei.
2. Materials and Methods
Chemicals
[5-13C]-glutamine and [6-13C]-arginine were purchased from Cambridge Isotope Laboratories (USA), while [13C]-urea and [13C,15N2]-urea were purchased from Sigma-Aldrich (USA). [5-13C,15N]-glutamine and [6-13C,15N3]-arginine were synthesized in-house and the synthetic scheme was adapted from previously reported methods.14,22
Buffers
14.1T thermal equilibrium measurements and hyperpolarized dissolutions were performed with a D2O- or H2O-based buffer. Each buffer consisted of 100mM Tris base and 1mM ethylenediaminetetraacetic acid (Fisher Scientific, USA). H2O-based buffers were adjusted to pH 7.4 with conc. HCl and 10N NaOH (Fisher Scientific, USA) and D2O-based buffers were adjusted to pD 7.4 with conc. DCl and 10N NaOD (Sigma-Aldrich, USA). pH and pD of H2O- and D2O-based buffers were measured with a Ag/AgCl electrode (Fisher Scientific, USA). Since the ionization constants of H2O and D2O (13.99 and 14.96)23 and reduction potentials of H+ and D+ (0V and −0.044V)24 are similar, we assumed that an Ag/AgCl pH electrode could be used to roughly approximate pD.
Thermal Equilibrium T1 and T2 Measurements at 14.1T
A 14.1T NMR spectrometer (Bruker, USA) was used to measure thermal-equilibrium 13C T1 and T2 values at 20°C. Samples were dissolved in either a 95:5 v/v mixture of the H2O and D2O-based buffers, or 100% of the D2O-based buffer. 5% D2O was added to the H2O buffer to facilitate deuterium locking and minimize B0 drift. Urea variants were dissolved to a final concentration of 100mM, whereas glutamine and arginine variants were dissolved to a final concentration of 50mM. T1 values were measured using a standard inversion recovery sequence, and 15 different delay times between the π and π/2 pulses (spanning 3T1) were sampled. Each spectrum was an average of 3 to 4 scans, and a 5T1 to 6T1 pre-scan wait time was implemented to allow re-polarization of carbon nuclear spins between scans. T2 values were measured with a standard Carr-Purcell-Meiboom-Gill (CPMG) sequence, TR = 2τCPMG = 20ms (or 10ms for [13C]-urea). At least 13 different total echo times spanning 3T2 were sampled. Each spectrum was an average of 2 to 6 scans, and a 5T1 to 6T1 pre-scan wait time was implemented to allow re-polarization of carbon nuclear spins between scans. For T1- and T2-relaxation curve fitting, the area under the curve (AUC) of the carbon resonance of interest was integrated for each spectrum. AUC values were plotted against time and fit to a mono-exponential recovery or decay function to calculate T1 or T2, respectively.
Hyperpolarization of 13C-Enriched Molecules
To maximize polarization efficiency, all compounds in this study were dissolved in high molarity in the appropriate solvent to facilitate glass formation upon freezing. Each compound was prepared with a different formulation, as follows: Glutamine variants were mixed with 1.1 equivalent conc. HCl and dissolved to a final concentration of 1.5M using a 35:65 DMSO:H2O (v/v) solution. Urea variants were dissolved to a final concentration of 6M in glycerol. Arginine HCl variants were mixed with 1 equivalent HCl and dissolved to a final concentration of 3.2M in H2O. Each of these preparations contained OX063 radical (General Electric, USA) dissolved to a final concentration of 15mM. All samples were polarized in a SpinLab Polarizer (General Electric, USA) for at least 1h. Following polarization, the hyperpolarized substrate was ejected from the polarizer via dissolution with a large excess of superheated D2O or H2O-based buffer. The dissolution was collected in a pre-chilled glass flask (−20°C), which equilibrated to a final temperature of 25–35°C.
Hyperpolarized T1 Measurements at 1T
Following polarization, 500μL of the hyperpolarized dissolution was added to an NMR tube and transferred to a 1T Spinsolve 13C NMR spectrometer (Magritek, NZ) as fast as possible, typically ranging between 10s-30s post-dissolution. For all compounds, NMR spectra were acquired every 3s with a 10° excitation over a timescale >3T1. The area under the curve (AUC) of the carbon resonance of interest was integrated for each spectrum and plotted as a function of time, after which it was fit to a mono-exponential decay function (corrected for magnetization loss from each excitation pulse) to calculate T1. All reported measurements are averages of at least triplicate dissolutions, and the final concentration of the hyperpolarized compound following dissolution ranged from 5mM to 120mM. The substrate concentration in each dissolution was measured by 13C-NMR at 14.1T in the presence of 1 mM Gd-DOTA and a 10mM [13C]-bicarbonate or [13C]-urea standard. The integral of the resonance of interest was compared to that of the standard to calculate concentrations.
Hyperpolarized and Thermal Equilibrium T2 Measurements at 1T
Thermal equilibrium and hyperpolarized CPMG-type T2 was measured on [13C,15N2]-urea at 1T with a Spinsolve 13C NMR spectrometer (Magritek, NZ) by a single echo train of on-resonance π pulses (66μs), following an initial π/2 pulse (33μs). Pulse lengths were measured on a 6M [1-13C]-acetate sample in H2O, and shimming was performed on an H2O sample. Signal was accrued for 2 × 1ms between each pulse. The total inter-pulse delay was 4ms and the total number of pulses was 65536 for a ~260s acquisition time.
For hyperpolarized measurements, 200μl hyperpolarized [13C,15N2]-urea was placed in a Shigemi tube immediately following dissolution to reduce errors due to B1 inhomogeneity during CPMG acquisition. Dissolution concentrations ranged from 5–8mM [13C,15N2]-urea. 3 experiments with hyperpolarized [13C,15N2]-urea in D2O and 2 experiments in H2O were performed from single dissolutions. The first experiment for each dissolution was diluted 1:10 in the appropriate buffer to avoid oversaturation of the receiver. Fewer experiments were performed in H2O due to the shorter T1 of [13C,15N2]-urea in H2O. Decay curves were fit to a mono-exponential decay function to calculate T2. Discarding the first 1024 points to remove potential errors due to excitation of residual z-magnetization also revealed no significant effect in T2 fit values.
In vivo Perfusion Imaging
All animal experiments were approved by the Institutional Animal Care and Use Committee at Memorial Sloan Kettering Cancer Center (protocol number 13–12–019). Two different samples of [13C,15N2]-urea were prepared for polarization in a SpinLab polarizer, one for dissolution with the D2O-based buffer and the other with the H2O based buffer. The D2O and H2O samples were polarized for 3h16m and 3h, respectively. Meanwhile, a 1/3mm diameter, 28cm long catheter (Braintree Scientific, USA) was placed in the lateral tail vein of a female Balb/c mouse (12 weeks old), and a 10U/mL heparin in saline solution was used to prevent coagulation and blockage of the catheter. The mouse was anesthetized with 1L/min room air, 1.5% isoflurane and loaded in a Bruker 3T MRI equipped with a dual tune 1H/13C coil. A 6M 13C-urea phantom was placed besides the mouse, and the mouse was positioned such that its kidneys were centered within the coil. An axial 1H T1-weighted GRE containing the mouse kidneys and phantom was acquired on a 40×40mm field of view with 128×128 resolution and 2 mm slice thickness.
For hyperpolarized MRI experiments, the hyperpolarized urea dissolution was held above a small magnetic field (60G) for 25s following dissolution and prior to in vivo injection to simulate QC time. At 25s post dissolution, the mouse was injected with 100 μL of the dissolution (in addition to the 100 μL of dead volume in the catheter) over the course of 10 seconds. At the start of injection, an axial 2D 13C EPI pulse sequence was initiated. The acquisition was localized to the same region as the anatomic T1-weighted image and acquired with a 36×36mm field of view (16×16 resolution), 10mm slice thickness, 15° excitation, 3s repetition time (40 repetitions), and ~5ms readout time for each time point. This procedure was performed with the D2O-based dissolution, the mouse was kept in the MRI under anesthesia for 10 minutes, and the procedure was repeated with the second dissolution in H2O on the same mouse.
[13C,15N2]-urea concentrations in each dissolution was measured by 13C NMR at 14.1T in the presence of a 20mM 2-13C-glycine standard and 1 mM Gd-DOTA. Concentrations were 27.6 mM [13C,15N2]-urea in H2 O, and 38.0 mM [13C,15N2]-urea in D2O.
3. Results and Discussion
To test this hypothesis, we characterized the relaxation properties of [5-13C]-glutamine, [13C]-urea, and [6-13C]-arginine, which contain one, two, or three nitrogen residues covalently bonded to the sp2 hybridized carbon, respectively (Figure 1). All reported T1 and T2 values were measured for the 13C-enriched position in H2O or D2O. Since 14N is a quadrupole nucleus (natural abundance: 99.63%, I=1), scalar and dipolar mechanisms both exhibit a dominant contribution to 13C relaxation times.25 This is in contrast to 15N (I=½) for which scalar relaxation is significantly less pronounced, so 15N-enriched variants were included in this study to investigate the effect of scalar relaxation in both solvent systems.
Figure 1.

Chemical structures of all compounds included in this study.
Thermal equilibrium 13C T1 and T2 relaxation times at 14.1T were measured for all compounds in Figure 1 with a standard inversion recovery or Carr-Purcell-Meiboom-Gill (CPMG) pulse sequence, respectively (Table 1).12 With respect to [13C]-urea, proton-to-deuteron exchange extends 13C T1 from 31.01s to 57.24s at 14.1T. Similar values and trends were measured for [13C,15N2]-urea, suggesting 14N-mediated scalar relaxation is not a dominant contribution to T1 relaxation at 14.1T (Figure 2A). Contrary to T1, T2 times were sensitive to 15N-enrichment at 14.1T. 13C T2 only marginally increased for [13C]-urea when dissolved in D2O, but T2 times were significantly longer for [13C,15N2]-urea in either solvent. In addition, a large increase in T2 was appreciated for [13C,15N2]-urea when dissolved in D2O, increasing from 20.12s to 34.61s (Figure 2B). The pronounced effect of 14N-mediated scalar relaxation on 13C T2 and its negligible effect on T1 has been previously demonstrated with urea at 3T.25,26 The field dependence of 14N-mediated scalar relaxation on T1 has also been reported,25 as it is a dominant relaxation mechanism at near-zero field (~10G) but becomes negligible at higher fields. The trends regarding 15N-enrichment in Table 1 are consistent with previous results, but to our knowledge this has not been demonstrated in the setting of D2O and H2O solvation. Importantly, the trends observed for urea held for all compounds in this study and we postulate that this phenomenon can be generalized to other compounds with similar structures. In addition, though [13C,15N2]-urea exhibits the greatest absolute increase in relaxation times when dissolved in D2O, the arginine variants exhibits a similar or greater decrease in relaxation rate via D2O solvation (R1 = 1/T1, R2 = 1/T2) when compared to urea. This is somewhat expected given that arginine has more exchangeable protons and since proton for deuteron exchange degreases dipolar relaxation rate as a function of decreased γ. However, this is a simplistic interpretation of the results and a thorough analysis of the relaxation mechanisms in each spin system is necessary to explain the change in relaxation rate between H2O and D2O solvation.
Table 1.
13C T1 and T2 relaxation times at 14.1T
| Compound | Labeling | T1 (s) | T2 (s) | ||
|---|---|---|---|---|---|
| H2O | D2O | H2O | D2O | ||
| Glutamine | 13C | 10.66 ± 0.34 | 12.34 ± 0.21 | 0.354 ± 0.003a | 0.455 ± 0.004a |
| Glutamine | 13C, 15N | 10.79 ± 0.34 | 12.29 ± 0.32 | 3.35 ± 0.21 | 4.66 ± 0.67 |
| Urea | 13C | 31.01 ± 1.18 | 57.24 ± 3.95 | 0.089 ± 0.001a | 0.100 ± 0.002a |
| Urea | 13C, 15N2 | 31.97 ± 1.38 | 57.34 ± 2.04 | 20.12 ± 0.17 | 34.61 ± 0.64 |
| Arginine | 13C | 6.62 ± 0.22 | 9.74 ± 0.32 | 0.184 ± 0.003a | 0.234 ± 0.004a |
| Arginine | 13C, 15N3 | 6.83 ± 0.19 | 9.71 ± 0.26 | 0.82 ± 0.02 | 2.68 ± 0.03 |
T2 values of compounds without 15N-enrichment reported to 3 decimal points to demonstrate measurement accuracy.
Figure 2.

(A) Thermal equilibrium 13C inversion recovery (B) and CPMG decay curves at 14.1T, acquired on [13C]- and [13C,15N2]-urea in H2O or D2O, pH or pD 7.4, 20°C. (C) Nuclear spin quantum numbers (I) and gyromagnetic ratio ([γ] = MHz T−1) of each nucleus in each urea variant, in H2O and D2O.
Though dipolar relaxation decreases via proton-to-deuteron exchange, scalar relaxation is introduced through the addition of deuterium (I=1). From Table 1, scalar relaxation at 14.1T can only be appreciated when measuring T2. Thus, we hypothesize that scalar relaxation from exchangeable deuterons is weak and does not offset the reduction in dipolar relaxation from proton-to-deuteron exchange, as solvation in D2O increases T2 for all compounds studied. This is reasonable considering scalar relaxation scales with J-coupling,12 and we postulate that 2JCD is small since carbon-deuterium peak splitting was not apparent in our 13C-spectra. In addition, 2JCH for covalently bound protons ranges from −10Hz to 15Hz,12 and we roughly approximate 2JCD falls within −1.5Hz to 2.3Hz since coupling scales with γ. 2JCD is likely even smaller for the compounds in this study due to rapid deuteron solvent exchange.27 Therefore, we also expect minimal scalar relaxation from deuterium on 13C T1 at near-zero field, which is an important consideration for HP experiments since samples are briefly exposed to low magnetic fields in the moments following dissolution.
To test whether the thermal equilibrium trends at 14.1T also apply to the HP state, HP T1 relaxation times were measured for these compounds at 1T. [6-13C]-arginine in H2O was omitted from this study because it contains three 14N quadrupole nuclei that rapidly relax 6-13C hyperpolarization in near-zero field immediately following dissolution, making accurate HP measurements challenging. The remaining compounds were polarized in a SpinLab (General Electric) and dissolution was performed using either buffered H2O or D2O. Pulse-and-acquire 13C-NMR spectra were acquired with a 1T NMR spectrometer (Magritek) and fit to a mono-exponential decay function to calculate T1 (Table 2)28. Dissolution of the HP substrate in D2O resulted in prolonged HP 13C T1 times at 1T, illustrated for [13C,15N2]-urea in Figure 3A. 15N-enrichment did not significantly affect HP T1 values in either solvent for all compounds, similar to thermal equilibrium measurements at 14.1T, suggesting 1T is strong enough to mitigate scalar relaxation on T1. However, spectra for hyperpolarized compounds without 15N-enrichment exhibited reduced SNR due to decreased T1 in near-zero field,25 resulting in an increased rate polarization loss during transfer of the sample from the polarizer to the spectrometer. These spectra also exhibited increased line-broadening as a result of shorter T2 at 1T. The T1 increase in D2O relative to H2O ranged from 1.3 to 2.4-fold and loosely scaled with number of directly bonded nitrogen residues. Compared to other studies investigating deuterium-enrichment at 2-bond covalently bound hydrogen, our observed HP T1 increase is comparable to or larger than that of other studies,14,17 though our measurements were recorded at a lower field strength. HP T2 at 1T was also measured with a CPMG acquisition on [13C,15N2]-urea, which increased from 32.6±0.8s in H2O to 67.4±1.9s in D2O (Figure 3B). These results demonstrate that this method can also be utilized to significantly extend HP T2, so long as the molecule is 15N-enriched to remove quadrupolar relaxation. Since these results are in line with the trends observed at 14.1T, we believe the HP T2 at 1T of the other compounds also exhibit a similar trend.
Table 2.
HP 13C T1 relaxation times at 1T
| Compound | Labeling | H2Oa | D2Oa |
|---|---|---|---|
| Glutamine | 13C | 21.8 ± 1.8 | 34.1 ± 2.5 |
| Glutamine | 13C, 15N | 23.6 ± 2.1 | 31.2 ± 3.8 |
| Urea | 13C | 51.6 ± 5.4 | 123.8 ± 19.6 |
| Urea | 13C, 15N2 | 53.3 ± 2.6 | 115.9 ± 8.2 |
| Arginine | 13C | ------------ | 32.1 ± 4.5 |
| Arginine | 13C, 15N3 | 13.3 ± 0.8 | 31.2 ± 6.8 |
T1 values reported in seconds as the average and standard deviation of at least 3 fits. Unpaired two-tailed t-test demonstrate that all T1 increases in D2O were statistically significant (P<0.05) except for [5-13C,15N]-glutamine (P=0.053).
Figure 3.

(A) 1T 13C-NMR spectra (3s repetition, 10° pulse-angle, every 3rd spectrum shown) of HP [13C,15N2]-urea dissolved in H2O or D2O, pH or pD 7.4, 25–35°C. (B) T2 decay curve for HP [13C,15N2]-urea in H2O or D2O at 25–35°C.
All HP decay curves in D2O exhibited mono-exponential behavior, implying proton-todeuteron exchange was complete by the time of signal acquisition (>15s post-dissolution). Proton exchange rates in H2O for all compounds in this study have been previously measured, with urea and glutamine exhibiting the slowest exchange rates (~1s−1).29–31 Since the concentration of all HP molecules following dissolution were 3–4 orders of magnitude lower than the deuteron concentration in D2O (~102 M), we assume 1s−1 is sufficiently fast to achieve isotopic equilibrium within a few seconds, as reflected in our data.
We also demonstrate that dissolution in D2O can benefit in vivo experiments and facilitate clinical translation of new probes, despite the in vivo environment being primarily H2O. A key difference between pre-clinical and clinical imaging studies is the addition of a quality control (QC) process. This delays administration of the HP solution by upwards of one minute, during which polarization decays as a function of T1.8 We hypothesized that dissolution in D2O rather than H2O can extend 13C T1 during QC, translating to higher 13C polarization upon intravenous injection. [13C,15N2]-urea was chosen for this proof-of-concept experiment, which has been extensively studied as a pre-clinical perfusion imaging agent and its prospects of clinical translation are promising.26,32–35
Two samples of [13C,15N]-urea were polarized for ~3 hours and dissolved to similar final concentrations in either D2O or H2O. Dissolutions were held in a 60G field for 25s following dissolution to simulate QC and subsequently intravenously administered to a healthy mouse. At the start of injection, an axial 2D 13C echo-planar imaging (EPI) sequence localized to the kidneys was initiated with a 3s repetition time (Figure 4). Both dissolutions were injected into the same mouse, spaced 10 minutes apart, and no obvious morbidity was observed as a result of the injections. The images demonstrate that dissolution in D2O increases HP signal in the kidneys, and the results reaffirm our hypothesis that deuterium mediated scalar relaxation is weak since the D2O sample still exhibited higher signal than its H2O counterpart despite being held at low field for 25s. In addition, assuming T1 values at 60G are similar to 1T, the signal enhancement observed in Figure 4C is more than expected based on differences in T1 relaxation during QC alone (which only yields a 33% SNR increase in D2O after 25s). This implies a possible in vivo lengthening of the HP state, suggesting D2O is not immediately diluted into the blood and stays as a bolus with the HP substrate.
Figure 4.

(A) Axial 1H T1-weighted gradient echo of a healthy female Balb/c mouse. Scale bar: 5mm. (B) Axial 13C-EPI with the same field of view (FOV) following injection of HP [13C,15N2]-urea in H2O or D2O. Intensity values are the sum of the total intensity in time, normalized to the same scale. (C) Total 13C signal in the FOV at each timepoint following injection with HP [13C,15N2]-urea, normalized to [13C,15N2]-urea concentration in each dissolution.
4. Conclusion
This study demonstrates that exchange of nitrogen-bound protons with deuterons by solvation in D2O reduces dipolar relaxation on 2-bond 13C-nuclei at the cost of a minimal increase in scalar relaxation through the introduction of deuterium to the spin system. Given the dramatic increase in T2 for 13C,15N-enriched molecules in D2O (>300% in the case of arginine), higher spatial and spectral resolution can be acquired for in vitro HP experiments in D2O. This method of improving T1 and T2 is not only limited to the three compounds in this study, but should be applicable to compounds with similar structures such as thiourea36, amino acid derivatives,37–40 and small peptides,41 all previously studied with HP MR. However, the reported phenomenon appears to be somewhat specific for amide-like carbons, as less significant increases were observed for sp3 hybridized carbons in glucose when 2-bond hydroxyl protons are exchanged with deuterons.18 We hypothesize that bond lengths and angles play a role in mediating this effect, as the discussed relaxation mechanisms are J-coupling and distance dependent, and this will be investigated in future studies. Ultimately, this non-synthetic approach provides a means of extending the HP lifetime and has the potential to enhance in vitro biochemical assays and in vivo MRI. In future experiments, we intend to investigate whether in vivo T1 and T2 are indeed longer when HP compounds are administered with a D2O bolus. In vivo lengthening of T2would be particularly interesting, as it will accommodate longer readout times and improve image resolution and signal intensity. As this method is independent of polarization technique, we believe this technique can be applied to the broader field of HP MR and used with PHIP-, SABER-, and optically-polarized molecules, so long as they are structurally similar to the compounds in this study.
Solvation of molecules with amide-like groups in D2O extends amide-carbon T1 & T2.
This is due to a decrease in dipolar relaxation via proton to deuteron exchange.
This phenomenon applies to the thermal equilibrium and hyperpolarized states.
Decreased relaxation improves in vivo SNR in hyperpolarized MRI experiments.
Acknowledgements
We would like to thank Dr. George Sukenick for his helpful advice with NMR experiments. This work was supported by the National Institutes of Health [F30 CA225174 (AC), T32 GM007739 (AC and KK), S10 OD016422 (KK) and P30 CA008748 (KK)], the Ludwig Center for Basic and Translational Immunology (KK), the Geoffrey Beene Cancer Research Center (KK), and the Thompson Family Foundation (KK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- (1).Terreno E; Castelli DD; Viale A; Aime S Challenges for Molecular Magnetic Resonance Imaging. Chem. Rev 2010, 110, 3019–3042. [DOI] [PubMed] [Google Scholar]
- (2).Graaf R. A. De. In Vivo NMR Spectroscopy - 2nd Edition: Principles and Techniques, 2nd ed; John Wiley & Sons Ltd, 2007. [Google Scholar]
- (3).Nikolaou P; Goodson BM; Chekmenev EY NMR Hyperpolarization Techniques for Biomedicine. Chem. - A Eur. J 2015, 21, 3156–3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Comment A; Merritt ME Hyperpolarized Magnetic Resonance as a Sensitive Detector of Metabolic Function. Biochemistry 2014, 53, 7333–7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Kurhanewicz J; Vigneron DB; Brindle K; Chekmenev EY; Comment A; Cunningham CH; Deberardinis RJ; Green GG; Leach MO; Rajan SS; et al. Analysis of Cancer Metabolism by Imaging Hyperpolarized Nuclei: Prospects for Translation to Clinical Research. Neoplasia 2011, 13, 81–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Cho A; Lau JYC; Geraghty BJ; Cunningham CH; Keshari KR Noninvasive Interrogation of Cancer Metabolism with Hyperpolarized 13C MRI. J. Nucl. Med 2017, 58, 1201–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Schroeder MA; Clarke K; Neubauer S; Facc F Hyperpolarized Magnetic Resonance : A Novel Technique for the In Vivo Assessment of Cardiovascular Disease. Circulation 2012, 124, 1580–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Nelson SJ; Kurhanewicz J; Vigneron DB; Larson PEZ; Harzstark AL; Ferrone M; van Criekinge M; Chang JW; Bok R; Park I; et al. Metabolic Imaging of Patients with Prostate Cancer Using Hyperpolarized [1–13C]Pyruvate. Sci. Transl. Med 2013, 5, 198ra108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Cunningham CH; Lau JYC; Chen AP; Geraghty BJ; Perks WJ; Roifman I; Wright GA; Connelly KA Hyperpolarized 13C Metabolic MRI of the Human Heart. Circ. Res 2016, 119, 1177–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Miloushev VZ; Granlund KL; Boltyanskiy R; Lyashchenko SK; DeAngelis LM; Mellinghoff IK; Brennan CW; Tabar V; Yang TJ; Holodny AI; et al. Metabolic Imaging of the Human Brain with Hyperpolarized 13C Pyruvate Demonstrates 13C Lactate Production in Brain Tumor Patients. Cancer Res. 2018, DOI: 10.1158/0008-5472.CAN-18-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Keshari KR; Wilson DM Chemistry and Biochemistry of 13C Hyperpolarized Magnetic Resonance Using Dynamic Nuclear Polarization. Chem. Soc. Rev 2014, 43, 1627–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).van de Ven FJM Multidimensional NMR in Liquids: Basic Principles and Experimental Methods; Wiley-VCH: New York City, NY, 1995. [Google Scholar]
- (13).Kowalewski J; Effemey M; Jokisaari J Dipole-Dipole Coupling Constant for a Directly Bonded CH Pair—A Carbon-13 Relaxation Study. J. Magn. Reson 2002, 157, 171–177. [DOI] [PubMed] [Google Scholar]
- (14).Qu W; Zha Z; Lieberman BP; Mancuso A; Stetz M; Rizzi R; Ploessl K; Wise D; Thompson C; Kung HF Facile Synthesis [5-13C-4-2H2]-L-Glutamine for Hyperpolarizd MRS Imaging of Cancer Metabolism. Acad. Radiol 2011, 18, 932–939. [DOI] [PubMed] [Google Scholar]
- (15).Kumagai K; Kawashima K; Akakabe M; Tsuda M; Abe T; Tsuda M Synthesis and Hyperpolarized 15N NMR Studies of 15N-Choline-d13. Tetrahedron 2013, 69, 3896–3900. [Google Scholar]
- (16).Nonaka H; Hirano M; Imakura Y; Takakusagi Y; Ichikawa K; Sando S Design of a 15N Molecular Unit to Achieve Long Retention of Hyperpolarized Spin State. Sci. Rep 2017, 7, 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Taglang C; Korenchan DE; von Morze C; Yu J; Najac C; Wang S; Blecha JE; Subramaniam S; Bok R; VanBrocklin HF; et al. Late-Stage Deuteration of 13C-Enriched Substrates for T1 Prolongation in Hyperpolarized 13C MRI. Chem. Commun 2018, 54, 5233–5236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Mishkovsky M; Anderson B; Karlsson M; Lerche MH; Sherry AD; Gruetter R; Kovacs Z; Comment A Measuring Glucose Cerebral Metabolism in the Healthy Mouse Using Hyperpolarized 13C Magnetic Resonance. Sci. Rep 2017, 7, 4–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Maltseva TV; Földesi A; Chattopadhyaya J T1 and T2 Relaxations of the 13C Nuclei of Deuterium-Labeled Nucleosides. Magn. Reson. Chem 1998, 36, 227–239. [Google Scholar]
- (20).Harris T; Gamliel A; Uppala S; Nardi-Schreiber A; Sosna J; Gomori JM; Katz-Brull R Long-Lived 15N Hyperpolarization and Rapid Relaxation as a Potential Basis for Repeated First Pass Perfusion Imaging - Marked Effects of Deuteration and Temperature. ChemPhysChem 2018, DOI: 10.1002/cphc.201800261. [DOI] [PubMed] [Google Scholar]
- (21).Nardi-Schreiber A; Gamliel A; Harris T; Sapir G; Sosna J; Gomori JM; Katz-Brull R Biochemical Phosphates Observed Using Hyperpolarized 31P in Physiological Aqueous Solutions. Nat. Commun 2017, 8, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Hamilton DJ; Sutherland A A Flexible Approach for the Synthesis of Selectively Labelled L-Arginine. Tetrahedron Lett. 2004, 45, 5739–5741. [Google Scholar]
- (23).Shoesmith DW; Lee W The Ionization Constant of Heavy Water (D2O) in the Temperature Range 298 to 523K. Can. J. Chem 1976, 54, 3553–3558. [Google Scholar]
- (24).CRC Handbook of Chemistry and Physics, 55th ed; Weast RC, Astle MJ, Beyer WH, Eds.; CRC Press: Boca Raton, FL, 1973. [Google Scholar]
- (25).Chiavazza E; Kubala E; Gringeri CV; Düwel S; Durst M; Schulte RF; Menzel MI Earth’s Magnetic Field Enabled Scalar Coupling Relaxation of 13C Nuclei Bound to Fast-Relaxing Quadrupolar 14N in Amide Groups. J. Magn. Reson 2013, 227, 35–38. [DOI] [PubMed] [Google Scholar]
- (26).Reed GD; von Morze C; Bok R; Koelsch BL; Van Criekinge M; Smith KJ; Shang Hong; Larson PEZ; Kurhanewicz J; Vigneron DB High Resolution 13C MRI With Hyperpolarized Urea: In Vivo T2 Mapping and 15N Labeling Effects. IEEE Trans. Med. Imaging 2014, 33, 362–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Yavari I; Roberts JD Differential Rates of Proton Exchange for the Guanidinium Nitrogens of L-Arginine Determined by Natural-Abundance Nitrogen-15 Nuclear Magnetic Resonance Spectrosopy. Biochem. Biophys. Res. Commun 1978, 83, 635–640. [DOI] [PubMed] [Google Scholar]
- (28).Puckeridge M; Pagès G; Kuchel PW Simultaneous Estimation of T1 and the Flip Angle in Hyperpolarized NMR Experiments Using Acquisition at Non-Regular Time Intervals. J. Magn. Reson 2012, 222, 68–73. [DOI] [PubMed] [Google Scholar]
- (29).Liepinsh E; Otting G Proton Exchange Rates from Amino Acid Side Chains- Implications for Image Contrast. Magn. Reson. Med 1996, 35, 30–42. [DOI] [PubMed] [Google Scholar]
- (30).Vold RL; Daniel ES; Chan SO Magnetic Resonance Measurements of Proton Exchange in Aqueous Urea. J. Am. Chem. Soc 1970, 92, 6771–6776. [Google Scholar]
- (31).Krishna NR; Sarathy KP; Huang DH; Stephens RL; Glickson JD; Smith CW; Walter R Primary Amide Hydrogen Exchange in Model Amino Acids: Asparagine, Glutamine, and Glycine Amides. J. Am. Chem. Soc 1982, 104, 5051–5053. [Google Scholar]
- (32).Von Morze C; Larson PEZ; Hu S; Keshari K; Wilson DM; Ardenkjaer-Larsen JH; Goga A; Bok R; Kurhanewicz J; Vigneron DB Imaging of Blood Flow Using Hyperpolarized [13C]Urea in Preclinical Cancer Models. J. Magn. Reson. Imaging 2011, 33, 692–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Lau AZ; Miller JJ; Robson MD; Tyler DJ Cardiac Perfusion Imaging Using Hyperpolarized 13C Urea Using Flow Sensitizing Gradients. Magn. Reson. Med 2016, 75, 1474–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Nielsen PM; Szocska Hansen ES; Nørlinger TS; Nørregaard R; Bonde Bertelsen L; Stødkilde Jørgensen H; Laustsen C Renal Ischemia and Reperfusion Assessment with Three-Dimensional Hyperpolarized 13C,15N2-Urea. Magn. Reson. Med 2016, 76, 1524–1530. [DOI] [PubMed] [Google Scholar]
- (35).Hansen ESS; Stewart NJ; Wild JM; Stødkilde-Jørgensen H; Laustsen C Hyperpolarized 13C,15N2-Urea MRI for Assessment of the Urea Gradient in the Porcine Kidney. Magn. Reson. Med 2016, 76, 1895–1899. [DOI] [PubMed] [Google Scholar]
- (36).Wibowo A; Park JM; Liu S-C; Khosla C; Spielman DM Real-Time in Vivo Detection of H2O2 Using Hyperpolarized 13C-Thiourea. ACS Chem. Biol 2017, 12, 1737–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Hata R; Nonaka H; Takakusagi Y; Ichikawa K; Sando S Design of a Hyperpolarized Molecular Probe for Detection of Aminopeptidase N Activity. Angew. Chemie - Int. Ed 2016, 55, 1765–1768. [DOI] [PubMed] [Google Scholar]
- (38).Wilson DM; Hurd RE; Keshari K; Van Criekinge M; Chen AP; Nelson SJ; Vigneron DB; Kurhanewicz J Generation of Hyperpolarized Substrates by Secondary Labeling with [1,1-13C] Acetic Anhydride. Proc Natl Acad Sci USA 2009, 106, 5503–5507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Flavell RR; Morze C. Von; Blecha JE; Korenchan DE; Criekinge M. Van; Sriram R; Gordon JW; Chen H; Subramaniam S; Bok RA; et al. Application of Good’s Buffers to PH Imaging Using Hyperpolarized 13C MRI. Chem. Commun 2015, 51, 14119–14122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Hundshammer C; Düwel S; Ruseckas D; Topping G; Dzien P; Müller C; Feuerecker B; Hövener JB; Haase A; Schwaiger M; et al. Hyperpolarized Amino Acid Derivatives as Multivalent Magnetic Resonance PH Sensor Molecules. Sensors 2018, 18, DOI: 10.3390/s18020600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Jamin Y; Gabellieri C; Smyth L; Reynolds S; Robinson SP; Springer CJ; Leach MO; Payne GS; Eykyn TR Hyperpolarized 13C Magnetic Resonance Detection of Carboxypeptidase G2 Activity. Magn. Reson. Med 2009, 62, 1300–1304. [DOI] [PubMed] [Google Scholar]